

Abstract
Patients with the genetic disease xeroderma pigmentosum (XP) lack the capacity to carry out a specific type of DNA repair process called nucleotide excision repair (NER). The NER pathway plays a critical role in the repair of DNA damage resulting from UV radiation. A subset of XP patients develop a profound neurodegenerative condition known as XP neurological disease. Robbins and colleagues (PNAS 75:1984–88, 1978) hypothesized that since UV light cannot reach into the human brain, XP neurological disease results from some form of endogenous DNA damage that is normally repaired by the NER pathway. In the absence of NER, the damage accumulates, causing neuronal death by blocking transcription. In this manuscript, I consider the evidence that a particular class of oxidative DNA lesions, the 8, 5’-cyclopurine-2’-deoxynucleosides, fulfills many of the criteria expected of neurodegenerative DNA lesions in XP. Specifically, these lesions are chemically stable, endogenous DNA lesions that are repaired by the NER pathway but not by any other known process, and strongly block transcription by RNA polymerase II in cells from XP patients. A similar set of criteria might be used to evaluate other candidate DNA lesions responsible for neurological diseases resulting from defects in other DNA repair mechanisms as well.
Keywords: oxidative stress, transcription, aging, DNA repair, dopamine, hydrogen peroxide, substantia nigra
Introduction
The DNA in our cells is constantly being damaged by endogenous sources such as oxygen radicals, as well as by environmental factors such as mutagenic chemicals and radiation. Fortunately, all cells are protected from these types of damages by an array of DNA repair pathways. A priori, it would be assumed that the major medical outcome of hereditary deficiencies in DNA repair processes would be an increased risk of cancer. Indeed there is an increased risk of cancer in several known DNA repair deficiencies including xeroderma pigmentosum (XP), Fanconi anemia, and hereditary non-polyposis colon cancer (Friedberg et al., 2005). However, it is now clear that the other major clinical manifestation of hereditary DNA repair deficiency is neurological disease, as discussed by other contributors to this Special Issue.
The focus of this manuscript is to discuss the neurological disease in patients with xeroderma pigmentosum (XP), the first example of a neurological disease associated with defective DNA repair (Andrews et al., 1978). I will first review the clinical and neuropathological characteristics of XP neurological disease, as well as the evidence that it results from defective nucleotide excision repair (NER). I will then discuss the evidence that a specific class of oxidatively-induced DNA lesions, the 8, 5’-cyclopurine-2’-deoxynucleosides (cyclopurine-deoxynucleosides), are currently the best candidates for endogenous DNA lesions that are responsible for neurological disease in XP patients, and the clinical implications of this hypothesis. Finally, I will briefly consider the relevance of cyclopurine-deoxynucleosides in individuals with normal NER.
Xeroderma Pigmentosum (XP) and XP Neurological Disease
XP is a rare genetic disorder characterized by extreme sensitivity of the skin to sunlight, and a dramatically increased risk of skin cancer on some exposed areas of the body. XP can result from mutations in any of eight genes, denoted XPA-G and V. Patients with mutations in an individual XP gene are considered to be in a complementation group. For example, patients with mutations in the XPA gene are said to be in complementation group A. Cells from patients in complementation groups A through G are defective in nucleotide excision repair (NER), the main DNA repair pathway for ultraviolet (UV) light-induced DNA damage, whereas group V patients lack a specific type of DNA polymerase that can bypass mutagenic DNA damage resulting from UV light (Friedberg et al., 2005, Wattendorf and Kraemer, 2005).
Approximately 20% of XP patients worldwide develop a pattern of neurological abnormalities referred to as XP neurological disease (Robbins et al., 1991, Rapin et al., 2000). In the past, any XP patient with neurologic abnormalities was described as having DeSanctis-Cacchione syndrome (DCS) (De Sanctis and Cacchione, 1932). At present, however, the term DCS is reserved for XP patients with severe neurologic disease as well as dwarfism and immature sexual development (Wattendorf and Kraemer, 2005; see also below).
The signs and symptoms of XP neurological disease include peripheral neuropathy, sensorineural deafness, microcephaly, cerebral dysfunction, ventricular dilation, cortical atrophy, basal ganglia and cerebellar disturbances (Robbins et al., 1991). Progressive loss of tendon stretch reflexes and deteriorating hearing are required for a diagnosis of XP neurological disease (Rapin et al., 2000). The earliest signs of XP neurological disease are reduced tendon reflexes, most likely resulting from degeneration of the peripheral nervous system, and ataxia. As the disease progresses, the ataxia becomes worse and other motor abnormalities occur, eventually resulting in the patient becoming wheelchair bound. The patients subsequently experience progressive cognitive decline and dementia.
Robbins has classified several different forms of XP neurological disease, depending upon the age of onset (Robbins et al., 1991), which is in turn dependent on the specific mutation(s) that the patient inherited. In Japanese XPA patients, the onset and severity of neurological disease, including the age at which the patient will lose the ability to walk, can be accurately predicted from the patients’ mutations (Maeda et al., 1995).
Neuropathology in XP Neurological Disease
The results of several neuropathological examinations of patients with XP neurological disease have been published (Yano, 1950, Reed et al., 1965, Roytta and Antitnen, 1986, Itoh et al., 1999, Hayashi et al., 2004), and the description below is a summary of these studies.
At the gross level, the dominant feature of XP neurological disease is atrophy of the brain, spinal cord, and peripheral nervous system. The atrophy can be very severe, with loss of up to 40% of the brain tissue mass. At the microscopic level, the dominant observation is that of neuronal loss in different regions of the brain, as described below. Because this loss of neurons occurs in the absence of any other obvious causative processes, such as amyloid plaques, or Lewy bodies, it is considered to be a primary neurodegeneration.
While neuronal loss is widespread in the brains of XP patients, it is not uniform. Neuronal loss is prominent in cerebral cortex, where large neurons appear to be more strongly affected than smaller neurons. The number of large neurons is also reduced in the basal forebrain (i.e. the nucleus basalis of Meynert and substantia innominata). Neuronal loss also occurs in the hippocampus, and is prominent in the striatum (particularly the caudate nucleus) where the number of large neurons is reduced more than smaller neurons. While neuronal loss was observed in the anterior thalamic nucleus, other thalamic and hypothalamic nuclei were reported to appear normal, both in size and number (Yano, 1950), although other authors (Roytta and Antitnen, 1986) describe a light accumulation of lipofuschin granules in the neurons in these areas.
In the midbrain, all authors have observed that neurons in the substantia nigra and locus coeruleus are very severely affected. Yano describes the locus coeruleus as “macroscopically indistinguishable” in the brains of the XP patients he examined. Consistent with these observations, the levels of the dopamine metabolite homovanillic acid (HVA) in the cerebrospinal fluid (CSF) from an XP patient were reduced by over 95% (Roytta and Antitnen, 1986) . These observations indicate that monoamine producing neurons are particularly susceptible to degeneration in XP. Possible reasons for this are discussed below (see Figure 3).
Figure 3.
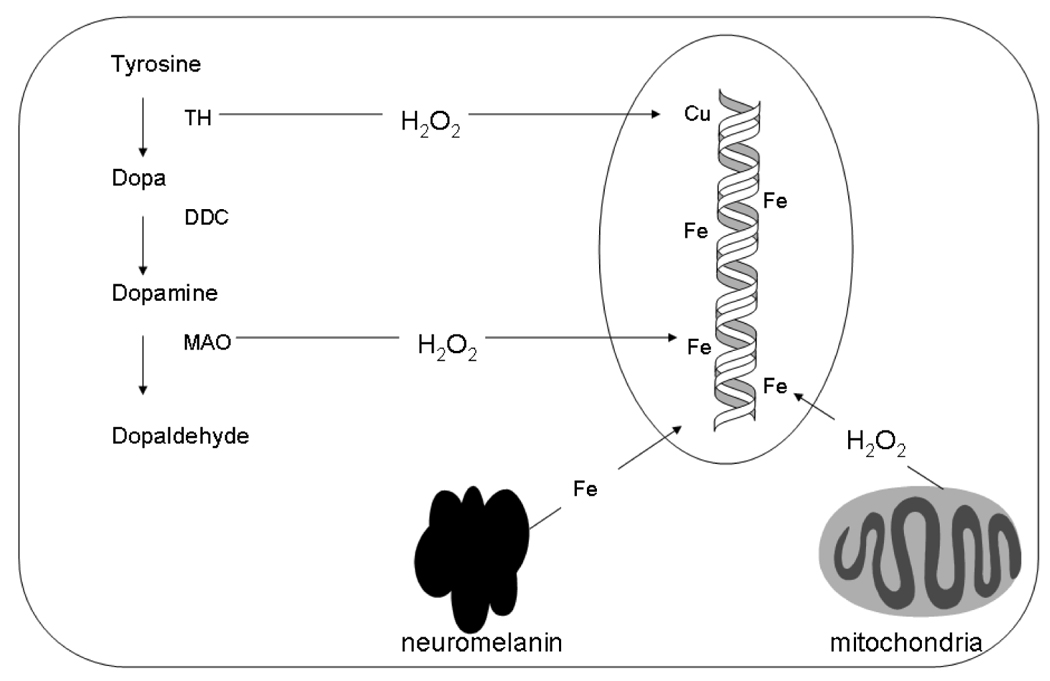
Potential sources of elevated oxidatively-induced DNA damage in catecholamine neurons that could increase result in increased cyclopurine-deoxynucleoside formation. Abbreviations: TH; tyrosine hydroxylase, DDC; dopa decarboxylase; MAO; monoamine oxidase. Note that both H2O2 and Fe could react to form the hydroxyl radical outside the nucleus, but such radicals would never reach the genomic DNA and are therefore not relevant to the mechanisms of XP neurological disease.
Neuronal loss is severe in some brainstem nuclei but not in others (Hayashi et al., 2004). In the cerebellum, Purkinje cells are significantly affected whereas granule neurons are largely spared. Large neurons are preferentially affected in the spinal cord, and in dorsal root ganglia. The pathology in the spinal cord has been described as similar to that of patients with Friedreichs ataxia (Yano, 1950).
Differences in Neuropathology between XP and Cockayne Syndrome
Cockayne syndrome (CS) is a rare neurodevelopmental disorder characterized by sun sensitivity, as well as abnormal “bird-like” facies, severe cachectic dwarfism, and signs of premature aging (Rapin et al., 2000, Friedberg et al., 2005). Pure CS, i.e. CS without the additional clinical features of XP, results from mutations in the CSA or CSB genes, whereas certain mutations in the XPB, XPD, or XPG genes result in a disease with the combined features of CS and XP (Friedberg et al., 2005). For further discussion of XP and CS, see the contribution by Kraemer in this volume.
XP and CS are often grouped together as related diseases, due to the overlapping sun sensitivity phenotypes, as well as the role of the CS proteins in transcription-coupled NER (Friedberg et al., 2005). However, it is important to emphasize that even though patients with both diseases show neurological abnormalities, the specific nature of the neuropathologies are qualitatively different. In particular, the brains of CS patients show an unusual type of white matter loss that has been characterized as dysmyelination or tigroid leukodystrophy (Brumback et al., 2004). This specific type of pathology is not observed in XP, or in any of the other neurologic disease associated with defective DNA repair. In addition, calcification of the basal ganglia and other regions of the brain is observed in CS, but not in XP (Leech et al., 1985, Rapin et al., 2000).
Can DeSanctis-Cacchione Syndrome Result From Mutations in the CSB Gene?
As noted above, the term DCS refers to a very severe form of XP neurological disease (De Sanctis and Cacchione, 1932). In light of this, the report that two brothers who were described as having DCS (Greenhaw et al., 1992) had inherited mutations in the CSB gene (Colella et al., 2000) was surprising. However, the clinical description of these patients raises some questions about the classification of these patients as having DCS.
First, one of the siblings had optic atrophy, and both had leukodystrophy. Both of these are common features of CS pathology, but are not observed in patients with XP neurological disease (Rapin et al., 2000). In particular, leukodystrophy is the most characteristic neuropathological feature of CS (Brumback et al., 2004). Second, levels of CSF HVA in the two siblings examined by Greenhaw et al (1992) were reported to be normal. In contrast, as noted above, CSF HVA levels were previously shown to be greatly reduced in a patient with XP neurological disease (Roytta and Antitnen, 1986), and several authors have observed that central dopamine neurons undergo profound degeneration in the brains of patients with XP neurological disease. The normal levels of CSF HVA in the siblings examined by Greenhaw et al (1992) are instead more consistent with the reported absence of degeneration of the substantia nigra dopamine neurons in CS patients (Itoh et al., 1999). Finally, while the patients described by Greenhaw et al (1992) were noted to have reduced CSF levels of the serotonin metabolite 5-HIAA, this has also been observed in a CS patient (Ellaway et al., 2000),
For these reasons, the clinical and neuropathological information about the siblings reported by Greenhaw et al (1992) is more consistent with a diagnosis of CS rather than XP neurological disease. A diagnosis of CS would also obviously be consistent with the inherited mutations in the CSB gene observed in the patients. The apparent absence of detectable basal ganglia calcification in the siblings studied by Greenhaw et al (1992) is notable, but other CS patients without basal ganglia calcification have been described in the literature (Ozdirim et al., 1996).
The Mechanistic Basis of XP Neurological Disease
XP Neurological Disease Results from Defective NER Repair
Robbins and colleagues (Andrews et al., 1978) compared the relative survival of cells from different XP patients with or without XP neurological disease to different doses of UV-radiation. These authors observed a strong correlation between reduced cell survival after UV and the severity of XP neurological disease. It is worth emphasizing that the correlation was between neurological disease and cell survival after UV radiation as opposed to other measures of DNA repair such as unscheduled DNA synthesis. The most likely explanation for this is that cell survival requires the repair of transcription blocking lesions from the transcribed strands of genes.
Notably, the correlation between cell survival after UV radiation and XP neurological disease involves patients from three NER deficient complementation groups (A, C, and D). If XP patients from only one complementation group developed neurological disease, one might argue that this was the result of some function of the protein other than NER. Indeed, the XPD protein is part of the transcription factor TFIIH, a protein complex that functions in both transcription initiation and NER (Egly, 2001). However, genotype-phenotype analyses have shown that the XP neurological disease is associated with XPD mutations that specifically affect the NER function of XPD (Lehmann, 2001). In contrast, the XPA protein is not involved in basal transcription; it has no other known function aside from an essential role in all types of NER, yet the most severe cases of XP neurological disease result from mutations in the XPA gene. Based on these considerations, there is compelling evidence that XP neurological disease results from defective NER.
How Does Defective NER Cause XP Neurological Disease?
To explain the neurodegeneration observed in XP patients, Robbins and colleagues (Andrews et al., 1978) hypothesized that because the DNA damaging wavelengths of UV light cannot reach the human brain, XP neurological disease must result from the accumulation of some type of endogenous of DNA damage that is normally repaired by the NER pathway. In the neurons of XP patients, the damage accumulates as a result of the absence of NER, leading to neuronal death by blocking transcription.
According to this hypothesis, the criteria for DNA lesions which are responsible for neurodegeneration in XP patients, which I will refer to as neurodegenerative DNA lesions, are as follows: 1. The lesions should be substrates for NER, but not for other DNA repair pathways. 2. The lesions should block transcription. 3. The lesions should result from an endogenous process. 4. The lesions should accumulate in cells of XP patients.
Though not explicitly stated in the original hypothesis, another criterion should be mentioned. Since the neurodegenerative disease in XP patients progresses slowly, over years or decades, it follows that the lesions that cause the neurodegeneration should also be stable in DNA over such periods of time. DNA itself undergoes degradation over time as a result of its inherent chemical properties, and some DNA lesions are particularly unstable even in the absence of repair (Lindahl, 1993).
These criteria are the framework of the search for neurodegenerative DNA lesions in XP. A similar set of criteria could be applied to the search of DNA lesions responsible for some of the other neurodegenerative diseases resulting from DNA repair defects that are discussed by other authors in this Special Issue.
Oxidative Stress as a Possible Source of Neurodegenerative DNA Lesions in XP
An important clue into the mechanisms of XP neurologic disease came from a study by Lindahl and colleagues (Satoh et al., 1993). These authors used the in vitro NER assay (Wood et al., 1988) to show that exposure of DNA to oxygen radicals generated a class of DNA lesion(s) that are normally repaired by the NER, and proposed that oxidative stress could be the source of the DNA damage that causes neuronal death in XP patients. In considering the possible identities of oxidatively-induced lesions that are repaired by NER, Satoh et al suggested “putative intrastrand purine dimers” detected using the 32P-postlabeling method (Carmichael et al., 1992; see also Randerath et al., 1991) as well as cyclopurine-deoxynucleosides. The possible role of cyclopurine-deoxynucleosides in XP neurological disease had also been considered by Robbins and colleagues (Dizdaroglu et al., 1987).
Cyclopurine-Deoxynucleosides: Candidate Neurodegenerative DNA Lesions in XP
Cyclopurine-deoxynucleosides were originally discovered by radiation chemists studying the effects of ionizing radiation on DNA (Keck, 1968, Mariaggi et al., 1976, Raleigh et al., 1976). Both cyclo-dA and cyclo-dG lesions can be formed, and can exist in either of two diastereomeric forms, 5’R and 5’S (figure 1). In contrast to 8-oxo-dG, which can be generated by exposing DNA to singlet oxygen, the formation of cyclopurine-deoxynucleosides specifically requires the hydroxyl radical.
Figure 1.
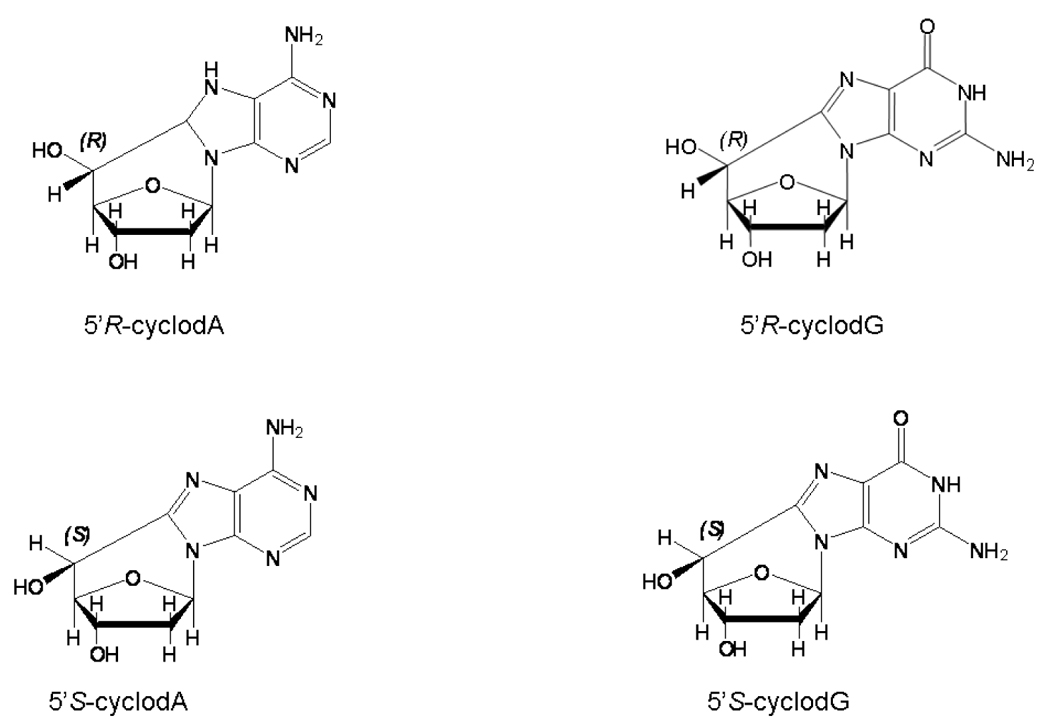
Structures of the cyclopurine-deoxynucleoside lesions.
The ability to synthesize oligonucleotides containing single cyclopurine-deoxynucleoside lesions (Romieu et al., 1999, Brooks et al., 2000) made it possible to directly investigate the repair pathways for these lesions in human cells. As a result, in 2000, two groups independently published the results of such studies, and these are considered below.
Criterion 1: Cyclopurine-Deoxynucleosides Are Repaired by Nucleotide Excision Repair
The first criterion for a candidate neurodegenerative DNA lesion in XP is that the lesion is a substrate for NER. To test this possibility, we (Brooks et al., 2000) utilized a 140 nucleotide long duplex containing a single 5’(S) cyclo-dA lesion in a central location. Incubation of the substrate with whole-cell extracts from Chinese hamster ovary cells resulted in the release of oligonucleotides between 24 to 32 nucleotides in length containing the cyclo-dA lesion, which is diagnostic for NER. Complementation studies using extracts from different NER-deficient cell lines were used to conclusively demonstrate that the excision was due to NER.
Kuraoka et al., (2000) created duplex closed-circular plasmids containing single cyclopurine-deoxynucleosides (both the 5’R and 5’S forms) and demonstrated that incubation of such constructs with human cell extracts resulted in NER synthesis and excision of oligonucleotides containing the cyclopurine-deoxynucleoside lesion. The excision was prevented by a neutralizing anti-XPA antibody, but restored by addition of recombinant XPA protein. The 5’R form of cyclo-dA was found to be a 3–4 times better substrate for NER than the 5’S form. Interestingly, the efficiency of repair of cyclopurine-deoxynucleosides was found to be 40–150 fold lower than a 1, 3 intrastrand cisplatin crosslink which is a good substrate for NER.
Thus both groups, using different sources of lesion-containing DNA substrates (produced by entirely different synthetic methods), as well as different biochemical methods, reached the same conclusion: cyclopurine-deoxynucleosides are substrates for nucleotide excision repair.
Cyclopurine-Deoxynucleosides Are Not Substrates for Other DNA Repair Pathways
To address the possibility that cyclopurine-deoxynucleosides could be repaired by glycosylase-initiated base excision repair (BER) in mammalian cells, we (Brooks et al., 2000) prepared nuclear extracts from rat brain, and incubated these with oligonucleotide duplexes containing a cyclo-dA lesion under conditions in which nucleotide excision repair could not be detected. We found that while glycosylase activity was readily observed on positive control substrates, no evidence of glycosylase activity towards the cyclo-dA lesion could be detected.
Kuraoka et al. (2000) devised a novel assay, based on the ability of cyclopurine-deoxynucleoside lesions to prevent the digestion of DNA by a restriction enzyme, to search for repair of these lesions by either a glycosylase or direct reversal of the lesion. However, they were unable to detect any evidence for such repair pathways in extracts of human cells. Also, Romieu et al. (1999) were unable to detect cleavage of cyclopurine-deoxynucleoside-containing oligonucleotides with either of two bacterial glycosylases.
Taken together, the results of the studies indicate that neither glycosylase-initiated base excision repair, nor any other DNA repair pathway can repair DNA containing cyclopurine-deoxynucleoside lesions. The presence of the covalent 8,5’ bond is the most likely reason for the failure of BER to remove the lesion, because even if a glycosylase could cleave the glycosidic bond, the base would still remain attached to the sugar-phosphate backbone via the 8,5’ bond. Thus the totality of the evidence indicates that the NER pathway is the only known DNA repair pathway that can remove cyclopurine-deoxynucleoside lesions from DNA in human cells. It should be noted however, that we cannot formally rule out the possibility that some cells may possess an as yet unknown mechanism for removing these lesions.
Criterion 2: Cyclopurine-Deoxynucleosides Block Transcription by RNA Polymerase II
An essential criterion for a neurodegenerative DNA lesion in XP is that the lesion should block transcription by RNA polymerase II (Pol II). To address this question in vivo, we (Brooks et al., 2000) introduced the approach of synthesizing covalently-closed plasmid DNA molecules containing a single cyclo-dA lesion on the transcribed strand of a luciferase reporter gene, then transfecting these constructs into either normal or NER deficient cells. Using this approach, we found that a single cyclo-dA lesion strongly reduced (by approximately 75%) luciferase activity in transformed fibroblast cells from two human XPA patients. Interestingly, a single thymine dimer in the same sequence context also strongly reduced luciferase activity to the same extent as the cyclo-dA lesion. The effect of the cyclo-dA lesion on transcription in XPA cells was observed in two different sequence contexts. Importantly, the effect of the lesion on luciferase activity in normal cells was significantly less than in XPA patient cells, consistent with repair of the lesion from the transcribed strand of the gene in vivo.
We also demonstrated that the presence of a cyclopurine-deoxynucleoside lesion in a transcription factor binding site can block transcription factor binding, and inhibit gene expression in XP cells (Marietta et al., 2002), illustrating another mechanism by which these lesions can reduce gene expression.
Criterion 3: Cyclopurine-Deoxynucleosides are Endogenous DNA Lesions
Since cyclopurine-deoxynucleosides can be formed in DNA by ionizing radiation (i.e. the hydroxyl radical) they were likely to be formed in vivo by endogenous oxidative stress as well. However, this remained to be shown definitively. In the early studies by Dizdaroglu and colleagues using GC-MS (Dizdaroglu et al., 1987), cyclopurine-deoxynucleosides were detected in DNA from cells exposed to ionizing radiation. However, the basal levels of cyclopurine-deoxynucleosides, i.e. in the absence of ionizing radiation, were near the limit of detection. In more recent studies, using improved LC-MS and GC-MS instrumentation, Dizdaroglu and colleagues (Dizdaroglu et al., 2001), were able to detect basal levels of cyclopurine-deoxynucleosides on the order of one lesion in 107 nucleotides in unirradiated calf thymus DNA, presenting strong evidence for endogenous formation of these lesions in DNA from mammalian cells.
A series of studies by Randerath and colleagues utilizing the highly sensitive 32P-postlabeling assay demonstrated the existence of a number of endogenous DNA modifications which they refer to as I-compounds (Randerath et al., 1999). A subset of these, referred to as type II I-compounds, had been shown to be stimulated by oxidative stress, and required the presence of adenosine residues in the DNA for their formation (Randerath et al., 1996). In a collaborative study between my laboratory and that of Randerath, we were able to show that a subset of these type-II I-compounds were in fact dinucleotides of the sequence 5’ dN-cyclo-dA 3’, where dN is any of the normal deoxynucleotides (Randerath et al., 2001). The recovery of cyclopurine-deoxynucleosides as dinucleotides in this assay results from the inability of the nucleases used in the post-labeling method (micrococcal nuclease and spleen phosphodiesterase) to cleave the phosphodiester bond 5’ to the cyclo-dA lesion (Randerath et al., 1996; see also Jaruga et al., 2004).
Our results using the 32P postlabeling assay indicate that the “putative intrastrand purine dimers” (Carmichael et al., 1992, Lloyd et al., 1997) which were proposed as candidate neurodegenerative lesions in XP by (Satoh et al., 1993) are also dinucleotides containing cyclo-dA. However, because the postlabeling studies were done in different laboratories, and used different reagents and chromatographic solvents, it is not absolutely certain that the same adducts were being studied in both cases. In any case, it is clear that there are additional endogenous DNA modifications present in genomic DNA that are not cyclopurine-deoxynucleosides, but may have significance for XP neurological disease (see below).
Criterion 4: Do Cyclopurine-Deoxynucleosides Accumulate in XP Cells?
The last criterion for a neurodegenerative lesion in XP is that the lesion should accumulate in cells from XP patients. We have addressed this possibility in several cell culture and animal systems, but have not observed differences in basal cyclopurine-deoxynucleoside levels between normal and XP samples (Theruvathu, Dizdaroglu, and Brooks, unpublished observations). However, we used total DNA samples in these studies, of which only a fraction represents the transcribed strands of active genes that the hypothesis is concerned with. This is a serious limitation in view of the fact terminally differentiated cells have little or no NER in non-transcribed sequences (Nouspikel and Hanawalt, 2002), which is primarily what we are assaying in total DNA samples. A method to accurately measure the low basal levels of cyclopurine-deoxynucleosides specifically on the transcribed strand of active genes remains a technical hurdle that must be addressed to fully understand the role of these lesions in XP neurological disease.
A Detailed Look at the Mechanisms of Cyclopurine-Deoxynucleoside Formation
While cyclopurine-deoxynucleosides are oxidatively-induced lesions, and are formed in DNA by the action of the hydroxyl radical, there are important differences in the mechanisms of formation of these lesions as compared to other oxidatively-induced purine lesions. These differences have mechanistic and practical implications as described below.
The formation of 8-oxo-dA and 8-oxo-dG results from a direct addition of the hydroxyl radical to purine ring (Figure 2). In contrast, the formation of cyclopurine-deoxynucleosides proceeds via a two-step mechanism (Dizdaroglu, 1986, Evans et al., 2004). In the first step, the hydroxyl radical abstracts a hydrogen atom from C5’ of the deoxyribose sugar, resulting in a carbon-centered radical. Then, in the second step, this radical attacks the C8 of the purine ring, resulting in a stable covalent 8, 5’ bond (Figure 2). The second step of the process is relatively slow, and it is also possible for the C5’radical to react with molecular oxygen, which prevents lesion formation. Therefore, cyclopurine-deoxynucleoside formation is actually inhibited by molecular oxygen. One practical implication of this mechanism is that the oxygen present in most buffers used to dissolve DNA in the laboratory protects the DNA from artifactual cyclopurine-deoxynucleoside formation. This is in contrast to other lesions, such as 8-oxo-dG, where artifactual lesion formation during DNA isolation and storage is a potential concern (Jaruga et al., 2004).
Figure 2.
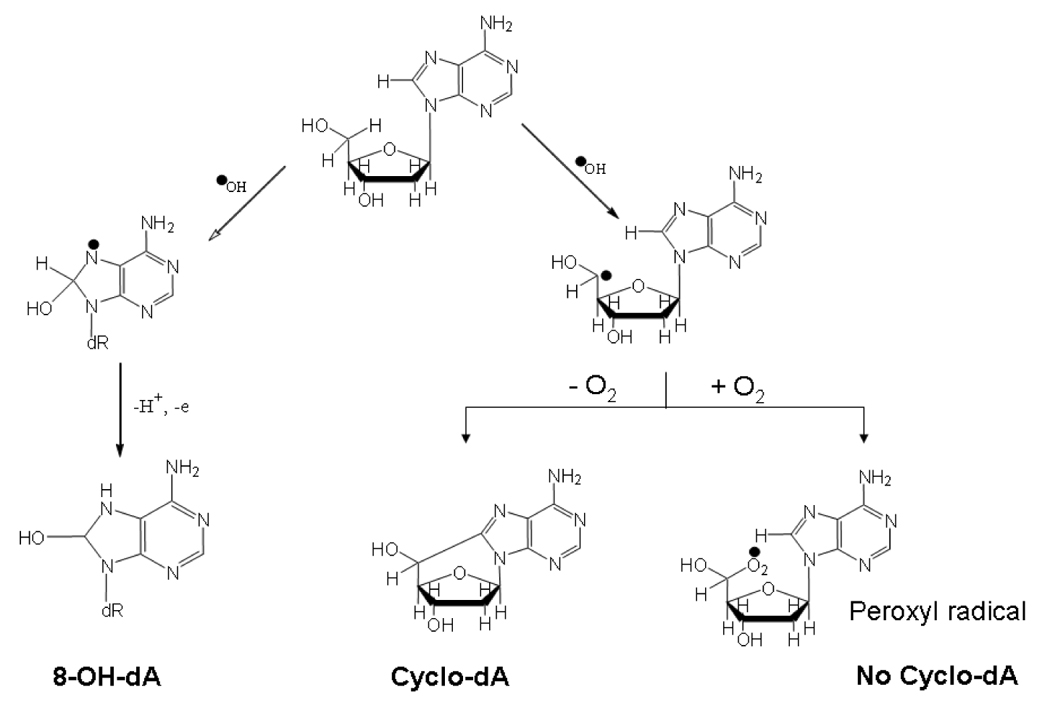
Mechanisms of formation of 8,5’-cyclo-dA versus 8-OH-dA, illustrating the ability of O2 to inhibit cyclopurine-deoxynucleoside formation.
In view of this, how can cyclopurine-deoxynucleoside lesions form in vivo? The answer appears to be that the mammalian cell nucleus is a relatively poorly oxygenated compartment (Joenje, 1989, Lindahl, 1993). Thus the most likely mechanism for endogenous cyclopurine-deoxynucleoside formation would involve the diffusion of hydrogen peroxide (H2O2) into the cell nucleus, where it reacts with metals such as iron or copper that are present in close association with duplex DNA (Henle et al., 1999) via the Fenton reaction. The resulting hydroxyl radical can immediately react with DNA to form the C5’ radical as described above. The low oxygen concentration in the nucleus would then allow at least some of these carbon-centered radicals to form cyclopurine-deoxynucleosides.
The crucial role of H2O2 in cyclopurine-deoxynucleoside formation may also explain the particular susceptibility of catecholamine neurons to degeneration in XP neurological disease. H2O2 formation in cells is thought to result from mitochondrial enzymes such as superoxide dismutase (Cadenas and Davies, 2000) as well as from the action of certain cytosolic enzymes (Imlay, 2003). The enzymes tyrosine hydroxylase (TH), the rate limiting enzyme in catecholamine biosynthesis, and monoamine oxidase (MAO), are particularly relevant in this regard. The degradation of dopamine by MAO generates H2O2 (Jenner and Olanow, 1996) and TH can also produce H2O2 by partial uncoupling of TH with its cofactor (Haavik et al., 1997). Thus MAO and TH in the catecholamine neurons are constitutive sources of H2O2 that could diffuse into the nucleus and cause cyclopurine-deoxynucleoside formation. In addition, dopamine itself can react with copper in a redox cycling reaction that generates H2O2, and ultimately DNA damage (Levay et al., 1997). Finally, neuromelanin in human dopamine neurons can bind iron but may also release it as well (Gerlach et al., 2003). Perhaps for these reasons, basal levels of oxidative stress in the substantia nigra of the human brain are significantly higher than in other brain regions (Floor and Wetzel, 1998).
Based on these considerations, it is therefore likely that the catecholamine-producing cells in the human brain experience higher levels of cyclopurine-deoxynucleoside formation than other neurons, which could provide an explanation for the dramatic loss of catacholaminergic neurons that has been consistently observed in XP neurological disease (Figure 3). For these reasons, the development of drugs for Parkinson’s disease patients that are designed to reduce oxidative stress in human dopamine neurons (Youdim et al., 2005) might also be beneficial for patients with XP neurological disease.
Other Potential Neurodegenerative Lesions in XP
“Nonbulky” Oxidatively-Induced DNA Lesions: 8-oxo-dG and Thymine Glycol
The suggestion that “nonbulky” oxidatively-induced lesions might play a role in neurodegeneration in XP was first proposed by Reardon et al. (1997), based on their observation that a single 8-oxo-dG, thymine glycol, or urea residue could be excised from duplex DNA by NER in vitro. While these authors acknowledged that such lesions could also be repaired by base excision repair, they suggested that the capacity for removing such lesions by glycosylase initiated BER might be insufficient in neurons with high levels of oxidative stress, and thus NER may have an important role in repair of such lesions.
One problem with this hypothesis is that even if NER plays a significant role in repair of these lesions in neurons, which remains to be shown, 8-oxo-dG and thymine glycol lesions have minimal effects on transcription by RNA Pol II in vitro (Tornaletti, 2005), and in cells from XP patients (Brooks et al, unpublished observations; see also Larsen et al., 2004). Moreover, Runger et al., (1995) found no difference between cells from XP patients versus normal controls in the repair of plasmids treated with methylene blue plus visible light. This method generates high levels of 8-oxo-dG in DNA via the formation of singlet oxygen (Runger et al., 1995). Cyclopurines are not expected to be formed with this method since, as noted above, singlet oxygen does not generate cyclopurine-deoxynucleosides lesions, and oxygen free radicals are not generated by methylene blue plus visible light (Floyd et al., 1989). Thus, in contrast to the cyclopurine-deoxynucleosides, both 8-oxo-dG and thymine glycol can be repaired by pathways other than NER, and neither lesion has the ability to block transcription by RNA Pol II in cells from XP patients that would be consistent with causing neurodegeneration.
Propano-deoxyguanosine (PdG) Lesions
Other potential neurodegenerative DNA lesions in XP are the PdG adducts. These adducts result from the reaction of aldehydes such as malondialdehyde with DNA (Burcham, 1998). While these aldehydes can result from lipid peroxidation, it should be noted that some of these lesions can form from other mechanisms. In particular, the malondialdehyde-dG adduct can also form as a result of an oxidative process involving the formation of base propenals in DNA (Dedon et al., 1998).
PdG lesions are repaired by NER (Johnson et al., 1997, Choudhury et al., 2004), but apparently not by other processes. Also, some of these lesions can block or reduce transcription by RNA Pol II, at least in vitro (Cline et al., 2004), and are endogenous DNA lesions (Marnett, 1999, Chung et al., 2000). As such, these lesions appear to be better candidates for neurodegenerative lesions in XP than the oxidatively-induced lesions like 8-oxo-dG that are repaired by BER. However, a complicating factor with PdG lesions is that, in contrast to cyclopurine deoxynucleosides, PdG lesions have been shown to undergo a ring-opening reaction when present in double-stranded DNA (Mao et al., 1999) which can result in the conversion of PdG lesions into either DNA-protein crosslinks or DNA intrastrand crosslinks. The repair of DNA interstrand crosslinks does not appear to require NER (Zheng et al., 2006), and while there is some evidence for a role for NER in the repair of DNA-peptide crosslinks from in vitro studies (Minko et al., 2002, Reardon and Sancar, 2006), other evidence indicates that aldehyde-derived DNA-protein crosslinks are unstable in vivo even in the absence of repair (Quievryn and Zhitkovich, 2000). Finally, C-C bonds such as the 8-5’ bond present in cyclopurine deoxynucleotides are more stable than the N-C bonds present in PdG adducts. Therefore, even in the absence of NER, PdG lesions are not as likely to remain stable in the DNA over the long periods of time (years to decades) over which XP neurological disease progresses as are cyclopurine deoxynucleosides.
Other Endogenous DNA Modifications
While the foregoing analysis indicates that the cyclopurine-deoxynucleosides appear to be the best candidates for neurodegenerative DNA adducts in XP that are currently known, it is important to note that our knowledge of the types of DNA adducts that can form in cells in general, and in neurons in particular, is certainly incomplete. The results from postlabeling studies show that there are additional types of DNA modifications in the mammalian genome that are, at present, structurally uncharacterized (Randerath et al., 1999). Some of these vary across tissues, and are likely to vary between individual cell types as well, depending upon the specific biochemical processes that are active and the byproducts they produce (e.g. see above and Figure 3). The possibility that some of these currently uncharacterized DNA lesions may play a crucial role in causing XP neurological disease must therefore be kept in mind.
How Does Blocking Transcription Cause Neurons To Die?
In principle, there are at least two distinct ways in which the accumulation of transcription blocking lesions such as cyclopurine-deoxynucleosides can cause neuronal death in XP. These can be described as either a passive or active mechanism. In the passive mechanism, the accumulation of the lesions on the transcribed strand of active genes would result in a progressive decrease in the amount of the encoded proteins. While the reduction in the level of a few proteins would not be expected to have a significant effect, particularly as there are two functional copies of most genes, as the process continues, the level of more and more proteins would be decreased, to the point that the cell would no longer be able to survive. In essence, this mechanism can be thought of as a kind of molecular “death by a thousand cuts”. The original hypothesis of Robbins and co-workers (Andrews et al., 1978) described this type of mechanism.
In contrast to this passive mechanism, more recent studies indicate that blockage of elongating Pol II could serve as a trigger for apoptosis (Ljungman and Lane, 2004). While most of this work has focused on a P53 -dependent mechanism, the relevance of this model for neurodegeneration is uncertain since the P53 dependent apoptosis after UV exposure in fibroblasts is associated with S-phase (McKay et al., 2002), which does not normally occur in terminally differentiated neurons. However, a recent study has provided evidence for a role for the JNK pathway in triggering apoptosis from unrepaired DNA damage in stationary phase cells (Hamdi et al., 2005). This mechanism may be more relevant for understanding the mechanisms by which unrepaired cyclopurine-deoxynucleosides in transcribed genes could cause neuronal death in XP.
A final possibility, which encompasses aspects of both the passive and active mechanisms, is based on the work of McKay and colleagues (McKay et al., 2004). In essence, these authors pointed out that larger genes would be more likely to sustain a transcription-blocking lesion than smaller genes (assuming DNA lesions are random throughout the genome). They further observed that anti-apoptotic genes are on average larger than proapoptotic genes, leading to the proposal that the passive accumulation of lesions might result in tipping the balance between pro and anti-apoptotic genes in favor of an apoptotic pathway. While McKay and colleagues were focusing on apoptosis follow UV exposure, a similar mechanism might be relevant for XP neurological disease.
Clinical Considerations
At present, there are no therapeutic options available for patients with XP neurological disease. Thus it is worth considering how investigation into neurodegenerative DNA lesions in XP might address this problem. If the specific DNA lesion(s) responsible for neurodegeneration in XP could be identified, then a potential therapeutic strategy would be to reduce the rate of lesion formation. As described above, cyclopurine-deoxynucleosides result from the diffusion of H2O2 into the cell nucleus. Therefore, drugs that would prevent or reduce H2O2 levels in cells would be expected to reduce cyclopurine-deoxynucleoside lesion formation. The class of drugs known as catalase mimetics (Doctrow et al., 2002) would seem promising in this regard.
Another approach is based on understanding the mechanisms of neurodegeneration resulting from unrepaired DNA damage. As suggested above, if neurodegeneration in XP results from an active mechanism such as apoptosis via the P53 or JNK pathways, then drugs targeted to interfere with apoptosis or specific apoptotic pathways might also be expected to have a therapeutic effect (Zhu et al., 2002, Zhang and Zhang, 2005).
In this regard, the development of an animal model that accurately reproduces the neurodegeneration suffered by human XP patients would be of great value. Knockout mice lacking the Xpa gene do not develop any detectable neurodegeneration (de Vries and van Steeg, 1996), in contrast to the human XPA patients. However, neurological abnormalities can be obtained by crossing the Xpa mice into other DNA repair deficient backgrounds (Murai et al., 2001). While the types of neuropathology observed in these animals are somewhat different than in human XP patients, these or other NER deficient mice with neurological abnormalities could be valuable in testing possible therapeutic strategies for XP neurological disease.
Are Cyclopurine-Deoxynucleosides Relevant For Individuals With Normal NER?
Finally, it is reasonable to ask whether cyclopurine-deoxynucleosides lesions, or other lesions repaired by NER, are of any significance in individuals with normal NER. As pointed out by (Lindahl, 1993), there is little evolutionary reason for normal cells to repair DNA lesions that might only have detrimental effects in older (> 40 years of age) individuals. Indeed, the evidence that the NER pathway does not function in non-transcribed DNA in at least some terminally differentiated cells, including neuronal cells (Nouspikel and Hanawalt, 2002), raises the possibility that cyclopurine-deoxynucleosides might accumulate in non-transcribed DNA with aging. We have shown that cyclopurine-deoxynucleoside lesions can prevent transcription factors from binding to their target DNA sequences (Marietta et al., 2002 and manuscript in preparation), illustrating one mechanism by which unrepaired cyclopurine-deoxynucleosides in non-transcribed DNA could impair gene expression and degrade neuronal function over time. Such effects may well be of significance for the decline in neuronal function during human aging (Long et al., 1999). Other possible mechanisms by which accumulated endogenous DNA damage resulting from the lack of NER in non-transcribed DNA sequences could contribute to neuronal dysfunction have been suggested by Nouspikel and Hanawalt (2003) Additional studies on the accumulation of DNA damage in the brain during aging will be important to address these possibilities.
Acknowledgements
I thank Cheryl Marietta, Anoop Patel, Shiva Kambhampati, Tracy Gilman, and Dr. Jacob Theruvathu for comments on the manuscript, and Dr. Jacob Theruvathu for assistance with the preparation of the figures. I also thank Dr. Jay H. Robbins for inviting me to collaborate with him on the cyclopurine-deoxynucleoside studies described herein, as well as for many very helpful discussions regarding XP neurological disease.
Abbreviations
- 5-HIAA
5-hydroxyindole acetic acid
- 8-oxo-dG
8-oxo-2’-deoxyguanosine
- BER
base excision repair
- CS
Cockayne syndrome
- CSF
cerebrospinal fluid
- Cyclo-dA
8, 5’-cyclo-2’-deoxyadenosine
- Cyclo-dG
8,5’-cyclo-2’-deoxyguanosine
- Cyclopurine-deoxynucleoside
8,5’-cyclopurine-2’-deoxynucleoside
- DCS
DeSanctis-Cacchione syndrome
- GC-MS
gas chromatography-mass spectrometry
- HVA
Homovanillic acid
- JNK
c-Jun N-terminal kinase
- LC-MS
liquid chromatography-mass spectrometry
- MAO
monoamine oxidase
- NER
nucleotide excision repair
- Pol II
RNA polymerase II
- PdG
Propano-deoxyguanosine
- TH
tyrosine hydroxylase
- Thymine dimer
(cis-syn) cyclobutane pyrimidine dimer
- XP
xeroderma pigmentosum
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Andrews A, Barrett S, Robbins J. Xeroderma Pigmentosum neurological abnormalities correlate with the colony forming ability after ultraviolet irradiation. Proc Natl Acad Sci USA. 1978;75:1984–1988. doi: 10.1073/pnas.75.4.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks PJ, Wise DS, Berry DA, Kosmoski JV, Smerdon MJ, Somers RL, Mackie H, Spoonde AY, Ackerman EJ, Coleman K, Tarone RE, Robbins JH. The oxidative DNA lesion 8,5'-(S)-cyclo-2'-deoxyadenosine is repaired by the nucleotide excision repair pathway and blocks gene expression in mammalian cells. J Biol Chem. 2000;275:22355–22362. doi: 10.1074/jbc.M002259200. [DOI] [PubMed] [Google Scholar]
- Brumback RA, Brooks PJ, Leech R. Cockayne Syndrome. In: Golden J, Harding B, editors. Pediatric Neuropathology. Pegnitz: International Society of Neuropathology Press; 2004. [Google Scholar]
- Burcham PC. Genotoxic lipid peroxidation products: their DNA damaging properties and role in formation of endogenous DNA adducts. Mutagenesis. 1998;13:287–305. doi: 10.1093/mutage/13.3.287. [DOI] [PubMed] [Google Scholar]
- Cadenas E, Davies KJ. Mitochondrial free radical generation, oxidative stress, and aging. Free Radic Biol Med. 2000;29:222–230. doi: 10.1016/s0891-5849(00)00317-8. [DOI] [PubMed] [Google Scholar]
- Carmichael PL, She MN, Phillips DH. Detection and characterization by 32P-postlabelling of DNA adducts induced by a Fenton-type oxygen radical-generating system. Carcinogenesis. 1992;13:1127–1135. doi: 10.1093/carcin/13.7.1127. [DOI] [PubMed] [Google Scholar]
- Choudhury S, Pan J, Amin S, Chung FL, Roy R. Repair kinetics of trans-4-hydroxynonenal-induced cyclic 1,N2-propanodeoxyguanine DNA adducts by human cell nuclear extracts. Biochemistry. 2004;43:7514–7521. doi: 10.1021/bi049877r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung FL, Nath RG, Ocando J, Nishikawa A, Zhang L. Deoxyguanosine adducts of t-4-hydroxy-2-nonenal are endogenous DNA lesions in rodents and humans: detection and potential sources. Cancer Res. 2000;60:1507–1511. [PubMed] [Google Scholar]
- Cline SD, Riggins JN, Tornaletti S, Marnett LJ, Hanawalt PC. Malondialdehyde adducts in DNA arrest transcription by T7 RNA polymerase and mammalian RNA polymerase II. Proc Natl Acad Sci U S A. 2004;101:7275–7280. doi: 10.1073/pnas.0402252101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colella S, Nardo T, Botta E, Lehmann AR, Stefanini M. Identical mutations in the CSB gene associated with either Cockayne syndrome or the DeSanctis-Cacchione variant of xeroderma pigmentosum. Hum Mol Genet. 2000;9:1171–1175. doi: 10.1093/hmg/9.8.1171. [DOI] [PubMed] [Google Scholar]
- De Sanctis C, Cacchione A. L'Idiozia xerodermica. Riv Sper Freniat. 1932;56:269–292. (translated) [Google Scholar]
- de Vries A, van Steeg H. Xpa knockout mice. Semin Cancer Biol. 1996;7:229–240. doi: 10.1006/scbi.1996.0031. [DOI] [PubMed] [Google Scholar]
- Dedon PC, Plastaras JP, Rouzer CA, Marnett LJ. Indirect mutagenesis by oxidative DNA damage: formation of the pyrimidopurinone adduct of deoxyguanosine by base propenal. Proc Natl Acad Sci U S A. 1998;95:11113–11116. doi: 10.1073/pnas.95.19.11113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dizdaroglu M. Free-radical-induced formation of an 8,5'-cyclo-2'-deoxyguanosine moiety in deoxyribonucleic acid. Biochem J. 1986;238:247–254. doi: 10.1042/bj2380247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dizdaroglu M, Dirksen ML, Jiang HX, Robbins JH. Ionizing-radiation-induced damage in the DNA of cultured human cells. Identification of 8,5-cyclo-2-deoxyguanosine. Biochem J. 1987;241:929–932. doi: 10.1042/bj2410929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dizdaroglu M, Jaruga P, Rodriguez H. Identification and quantification of 8,5'-cyclo-2'-deoxy-adenosine in DNA by liquid chromatography/ mass spectrometry. Free Radic Biol Med. 2001;30:774–784. doi: 10.1016/s0891-5849(01)00464-6. [DOI] [PubMed] [Google Scholar]
- Doctrow SR, Huffman K, Marcus CB, Tocco G, Malfroy E, Adinolfi CA, Kruk H, Baker K, Lazarowych N, Mascarenhas J, Malfroy B. Salen-manganese complexes as catalytic scavengers of hydrogen peroxide and cytoprotective agents: structure-activity relationship studies. J Med Chem. 2002;45:4549–4558. doi: 10.1021/jm020207y. [DOI] [PubMed] [Google Scholar]
- Egly JM. The 14th Datta Lecture. TFIIH: from transcription to clinic. FEBS Lett. 2001;498:124–128. doi: 10.1016/s0014-5793(01)02458-9. [DOI] [PubMed] [Google Scholar]
- Ellaway CJ, Duggins A, Fung VS, Earl JW, Kamath R, Parsons PG, Antony JA, North KN. Cockayne syndrome associated with low CSF 5-hydroxyindole acetic acid levels. J Med Genet. 2000;37:553–557. doi: 10.1136/jmg.37.7.553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans MD, Dizdaroglu M, Cooke MS. Oxidative DNA damage and disease: induction, repair and significance. Mutat Res. 2004;567:1–61. doi: 10.1016/j.mrrev.2003.11.001. [DOI] [PubMed] [Google Scholar]
- Floor E, Wetzel MG. Increased Protein Oxidation in Human Substantia Nigra Pars Compacta in Comparison with Basal Ganglia and Prefrontal Cortex Measured with an Improved Dinitrophenylhydrazine Assay. Journal of Neurochemistry. 1998;70:268–275. doi: 10.1046/j.1471-4159.1998.70010268.x. [DOI] [PubMed] [Google Scholar]
- Floyd RA, West MS, Eneff KL, Schneider JE. Methylene blue plus light mediates 8-hydroxyguanine formation in DNA. Arch Biochem Biophys. 1989;273:106–111. doi: 10.1016/0003-9861(89)90167-7. [DOI] [PubMed] [Google Scholar]
- Friedberg E, Walker G, Siede W, Wood R, Schultz R, Ellenberger T. DNA Repair and Mutagenesis. Washington DC: ASM press; 2005. [Google Scholar]
- Gerlach M, Double KL, Ben-Shachar D, Zecca L, Youdim MB, Riederer P. Neuromelanin and its interaction with iron as a potential risk factor for dopaminergic neurodegeneration underlying Parkinson's disease. Neurotox Res. 2003;5:35–44. doi: 10.1007/BF03033371. [DOI] [PubMed] [Google Scholar]
- Greenhaw GA, Hebert A, Duke-Woodside ME, Butler IJ, Hecht JT, Cleaver JE, Thomas GH, Horton WA. Xeroderma pigmentosum and Cockayne syndrome: overlapping clinical and biochemical phenotypes. Am J Hum Genet. 1992;50:677–689. [PMC free article] [PubMed] [Google Scholar]
- Haavik J, Almas B, Flatmark T. Generation of reactive oxygen species by tyrosine hydroxylase: a possible contribution to the degeneration of dopaminergic neurons? J Neurochem. 1997;68:328–332. doi: 10.1046/j.1471-4159.1997.68010328.x. [DOI] [PubMed] [Google Scholar]
- Hamdi M, Kool J, Cornelissen-Steijger P, Carlotti F, Popeijus HE, van der Burgt C, Janssen JM, Yasui A, Hoeben RC, Terleth C, Mullenders LH, van Dam H. DNA damage in transcribed genes induces apoptosis via the JNK pathway and the JNK-phosphatase MKP-1. Oncogene. 2005;24:7135–7144. doi: 10.1038/sj.onc.1208875. [DOI] [PubMed] [Google Scholar]
- Hayashi M, Araki S, Kohyama J, Shioda K, Fukatsu R, Tamagawa K. Brainstem and basal ganglia lesions in xeroderma pigmentosum group A. J Neuropathol Exp Neurol. 2004;63:1048–1057. doi: 10.1093/jnen/63.10.1048. [DOI] [PubMed] [Google Scholar]
- Henle ES, Han Z, Tang N, Rai P, Luo Y, Linn S. Sequence-specific DNA cleavage by Fe2+mediated fenton reactions has possible biological implications. J Biol Chem. 1999;274:962–971. doi: 10.1074/jbc.274.2.962. [DOI] [PubMed] [Google Scholar]
- Imlay JA. Pathways of oxidative damage. Annu Rev Microbiol. 2003;57:395–418. doi: 10.1146/annurev.micro.57.030502.090938. [DOI] [PubMed] [Google Scholar]
- Itoh M, Hayashi M, Shioda K, Minagawa M, Isa F, Tamagawa K, Morimatsu Y, Oda M. Neurodegeneration in hereditary nucleotide repair disorders. Brain Dev. 1999;21:326–333. doi: 10.1016/s0387-7604(99)00033-9. [DOI] [PubMed] [Google Scholar]
- Jaruga P, Theruvathu J, Dizdaroglu M, Brooks PJ. Complete release of (5'S)-8,5'-cyclo-2'-deoxyadenosine from dinucleotides, oligodeoxynucleotides and DNA, and direct comparison of its levels in cellular DNA with other oxidatively induced DNA lesions. Nucleic Acids Res. 2004;32:e87. doi: 10.1093/nar/gnh087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenner P, Olanow CW. Oxidative stress and the pathogenesis of Parkinson's disease. Neurology. 1996;47:S161–S170. doi: 10.1212/wnl.47.6_suppl_3.161s. [DOI] [PubMed] [Google Scholar]
- Joenje H. Genetic toxicology of oxygen. Mutat Res. 1989;219:193–208. doi: 10.1016/0921-8734(89)90001-5. [DOI] [PubMed] [Google Scholar]
- Johnson KA, Fink SP, Marnett LJ. Repair of propanodeoxyguanosine by nucleotide excision repair in vivo and in vitro. J Biol Chem. 1997;272:11434–11438. doi: 10.1074/jbc.272.17.11434. [DOI] [PubMed] [Google Scholar]
- Keck K. Bildung von cyclonudleotiden bei bestrahlung wassiriger losungen von purinenucleotiden. Z Naturforsch. 1968;23b:1034–1043. [PubMed] [Google Scholar]
- Kuraoka I, Bender C, Romieu A, Cadet J, Wood RD, Lindahl T. Removal of oxygen free-radical-induced 5',8-purine cyclodeoxynucleosides from DNA by the nucleotide excision-repair pathway in human cells. Proc Natl Acad Sci USA. 2000;97:3832–3837. doi: 10.1073/pnas.070471597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen E, Kwon K, Coin F, Egly JM, Klungland A. Transcription activities at 8-oxoG lesions in DNA. DNA Repair (Amst) 2004;3:1457–1468. doi: 10.1016/j.dnarep.2004.06.008. [DOI] [PubMed] [Google Scholar]
- Leech RW, Brumback RA, Miller RH, Otsuka F, Tarone RE, Robbins JH. Cockayne syndrome: clinicopathologic and tissue culture studies of affected siblings. J Neuropathol Exp Neurol. 1985;44:507–519. [PubMed] [Google Scholar]
- Lehmann AR. The xeroderma pigmentosum group D (XPD) gene: one gene, two functions, three diseases. Genes Dev. 2001;15:15–23. doi: 10.1101/gad.859501. [DOI] [PubMed] [Google Scholar]
- Levay G, Ye Q, Bodell WJ. Formation of DNA adducts and oxidative base damage by copper mediated oxidation of dopamine and 6-hydroxydopamine. Exp Neurol. 1997;146:570–574. doi: 10.1006/exnr.1997.6560. [DOI] [PubMed] [Google Scholar]
- Lindahl T. Instability and decay of the primary structure of DNA. Nature. 1993;362:709–714. doi: 10.1038/362709a0. [DOI] [PubMed] [Google Scholar]
- Ljungman M, Lane DP. Transcription - guarding the genome by sensing DNA damage. Nat Rev Cancer. 2004;4:727–737. doi: 10.1038/nrc1435. [DOI] [PubMed] [Google Scholar]
- Lloyd DR, Phillips DH, Carmichael PL. Generation of putative intrastrand cross-links and strand breaks in DNA by transition metal ion-mediated oxygen radical attack. Chem Res Toxicol. 1997;10:393–400. doi: 10.1021/tx960158q. [DOI] [PubMed] [Google Scholar]
- Long JM, Mouton PR, Jucker M, Ingram DK. What counts in brain aging? Design-based stereological analysis of cell number. J Gerontol A Biol Sci Med Sci. 1999;54:B407–B417. doi: 10.1093/gerona/54.10.b407. [DOI] [PubMed] [Google Scholar]
- Maeda T, Sato K, Minami H, Taguchi H, Yoshikawa K. Chronological difference in walking impairment among Japanese group A xeroderma pigmentosum (XP-A) patients with various combinations of mutation sites. Clin Genet. 1995;48:225–231. doi: 10.1111/j.1399-0004.1995.tb04094.x. [DOI] [PubMed] [Google Scholar]
- Mao H, Schnetz-Boutaud NC, Weisenseel JP, Marnett LJ, Stone MP. Duplex DNA catalyzes the chemical rearrangement of a malondialdehyde deoxyguanosine adduct. Proc Natl Acad Sci U S A. 1999;96:6615–6620. doi: 10.1073/pnas.96.12.6615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariaggi N, Cadet J, Teoule R. Cyclisation radicalaire de la desoxy-2′-adenosine en solution aqueuse, sous l'effet du rayonnement gamma. Tetrahedron. 1976;32:2385–2387. [Google Scholar]
- Marietta C, Gulam H, Brooks PJ. A single 8,5'-cyclo-2'-deoxyadenosine lesion in a TATA box prevents binding of the TATA binding protein and strongly reduces transcription in vivo. DNA Repair. 2002;1:967–975. doi: 10.1016/s1568-7864(02)00148-9. [DOI] [PubMed] [Google Scholar]
- Marnett LJ. Lipid peroxidation-DNA damage by malondialdehyde. Mutat Res. 1999;424:83–95. doi: 10.1016/s0027-5107(99)00010-x. [DOI] [PubMed] [Google Scholar]
- McKay BC, Becerril C, Spronck JC, Ljungman M. Ultraviolet light-induced apoptosis is associated with S-phase in primary human fibroblasts. DNA Repair (Amst) 2002;1:811–820. doi: 10.1016/s1568-7864(02)00109-x. [DOI] [PubMed] [Google Scholar]
- McKay BC, Stubbert LJ, Fowler CC, Smith JM, Cardamore RA, Spronck JC. Regulation of ultraviolet light-induced gene expression by gene size. Proc Natl Acad Sci U S A. 2004;101:6582–6586. doi: 10.1073/pnas.0308181101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minko IG, Zou Y, Lloyd RS. Incision of DNA-protein crosslinks by UvrABC nuclease suggests a potential repair pathway involving nucleotide excision repair. Proc Natl Acad Sci U S A. 2002;99:1905–1909. doi: 10.1073/pnas.042700399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murai M, Enokido Y, Inamura N, Yoshino M, Nakatsu Y, van der Horst GT, Hoeijmakers JH, Tanaka K, Hatanaka H. Early postnatal ataxia and abnormal cerebellar development in mice lacking Xeroderma pigmentosum Group A and Cockayne syndrome Group B DNA repair genes. Proc Natl Acad Sci U S A. 2001;98:13379–13384. doi: 10.1073/pnas.231329598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nouspikel T, Hanawalt P. DNA repair in terminally differentaied cells. DNA Repair. 2002;1:59–75. doi: 10.1016/s1568-7864(01)00005-2. [DOI] [PubMed] [Google Scholar]
- Nouspikel T, Hanawalt PC. When parsimony backfires: neglecting DNA repair may doom neurons in Alzheimer's disease. Bioessays. 2003;25:168–173. doi: 10.1002/bies.10227. [DOI] [PubMed] [Google Scholar]
- Ozdirim E, Topcu M, Ozon A, Cila A. Cockayne syndrome: review of 25 cases. Pediatr Neurol. 1996;15:312–316. doi: 10.1016/s0887-8994(96)00229-9. [DOI] [PubMed] [Google Scholar]
- Quievryn G, Zhitkovich A. Loss of DNA-protein crosslinks from formaldehyde-exposed cells occurs through spontaneous hydrolysis and an active repair process linked to proteosome function. Carcinogenesis. 2000;21:1573–1580. [PubMed] [Google Scholar]
- Raleigh JA, Kremers W, Whitehouse R. Radiation chemistry of nucleotides: 8,5'-cyclonucleotide formation and phosphate release initiated by hydroxyl radical attack on adenosine monophosphates. Radiat Res. 1976;65:414–422. [PubMed] [Google Scholar]
- Randerath K, Randerath E, Smith CV, Chang J. Structural origins of bulky oxidative DNA adducts (type II I-compounds) as deduced by oxidation of oligonucleotides of known sequence. Chem Res Toxicol. 1996;9:247–254. doi: 10.1021/tx950085v. [DOI] [PubMed] [Google Scholar]
- Randerath K, Randerath E, Zhou GD, Li D. Bulky endogenous DNA modifications (I-compounds) -possible structural origins and functional implications. Mutat Res. 1999;424:183–194. doi: 10.1016/s0027-5107(99)00018-4. [DOI] [PubMed] [Google Scholar]
- Randerath K, Yang P, Danna T, Reddy R, Watson W, Randerath E. Bulky addcuts detected by 32P postlabelling in DNA modifed by oxidative damage in vitro: Comparison with I compounds. Mutation Research. 1991;250:135–144. doi: 10.1016/0027-5107(91)90169-o. [DOI] [PubMed] [Google Scholar]
- Randerath K, Zhou GD, Somers RL, Robbins JH, Brooks PJ. A 32P-postlabeling assay for the oxidative DNA lesion 8,5'-cyclo-2'-deoxyadenosine in mammalian tissues: evidence that four type II I-compounds are dinucleotides containing the lesion in the 3' nucleotide. J Biol Chem. 2001;276:36051–36057. doi: 10.1074/jbc.M105472200. [DOI] [PubMed] [Google Scholar]
- Rapin I, Lindenbaum Y, Dickson DW, Kraemer KH, Robbins JH. Cockayne syndrome and xeroderma pigmentosum. Neurology. 2000;55:1442–1449. doi: 10.1212/wnl.55.10.1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reardon JT, Bessho T, Kung HC, Bolton PH, Sancar A. In vitro repair of oxidative DNA damage by human nucleotide excision repair system: possible explanation for neurodegeneration in xeroderma pigmentosum patients. Proc Natl Acad Sci U S A. 1997;94:9463–9468. doi: 10.1073/pnas.94.17.9463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reardon JT, Sancar A. Repair of DNA-polypeptide crosslinks by human excision nuclease. Proc Natl Acad Sci U S A. 2006;103:4056–4061. doi: 10.1073/pnas.0600538103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed WB, Mary SB, Nickel WR. Xeroderma Pigmentosum With Neurological Complications: The De Sanctis-Cacchione Syndrome. Arch Dermatol. 1965;91:224–226. doi: 10.1001/archderm.1965.01600090032005. [DOI] [PubMed] [Google Scholar]
- Robbins JH, Brumback RA, Mendiones M, Barrett SF, Carl JR, Cho S, Denckla MB, Ganges MB, Gerber LH, Guthrie RA, et al. Neurological disease in xeroderma pigmentosum. Documentation of a late onset type of the juvenile onset form. Brain. 1991;114:1335–1361. doi: 10.1093/brain/114.3.1335. [DOI] [PubMed] [Google Scholar]
- Romieu A, Gasparutto D, Cadet J. Synthesis and characterization of oligonucleotides containing 5',8-cyclopurine 2'-deoxyribonucleosides: (5'R)-5',8-cyclo-2'-deoxyadenosine, (5'S)-5',8-cyclo-2'-deoxyguanosine, and (5'R)-5',8-cyclo-2'-deoxyguanosine. Chem Res Toxicol. 1999;12:412–421. doi: 10.1021/tx9802668. [DOI] [PubMed] [Google Scholar]
- Roytta M, Antitnen A. Xeroderma pigmentosum with neurological abnormalities. A clinical and neuropathological study. Acta Neurol Scand. 1986;73:191–199. doi: 10.1111/j.1600-0404.1986.tb03262.x. [DOI] [PubMed] [Google Scholar]
- Runger TM, Epe B, Moller K. Repair of ultraviolet B and singlet oxygen-induced DNA damage in xeroderma pigmentosum cells. J Invest Dermatol. 1995;104:68–73. doi: 10.1111/1523-1747.ep12613504. [DOI] [PubMed] [Google Scholar]
- Satoh M, Jones C, Wood R, Lindahl T. DNA excision-repair defect of xeroderma pigmentosum prevents removal of a class of oxygen free radical induced base lesions. Proc Nat'l Acad Sci. 1993;90:6335–6339. doi: 10.1073/pnas.90.13.6335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tornaletti S. Transcription arrest at DNA damage sites. Mutat Res. 2005;577:131–145. doi: 10.1016/j.mrfmmm.2005.03.014. [DOI] [PubMed] [Google Scholar]
- Wattendorf D, Kraemer K. GeneReviews. vol. 2005. Seattle: University of Washington, Seattle; 2005. Xeroderma Pigmentosum [Includes: DeSanctis-Cacchione Syndrome] [Google Scholar]
- Wood RD, Robins P, Lindahl T. Complementation of the xeroderma pigmentosum DNA repair defect in cell-free extracts. Cell. 1988;53:97–106. doi: 10.1016/0092-8674(88)90491-6. [DOI] [PubMed] [Google Scholar]
- Yano K. Xeroderma pigmentosum with disturbance of the central nervous system: A Histopathological investigation. Folia Psychiatr Neurol Jpn. 1950;4:143–175. (translated) [PubMed] [Google Scholar]
- Youdim MB, Fridkin M, Zheng H. Bifunctional drug derivatives of MAO-B inhibitor rasagiline and iron chelator VK-28 as a more effective approach to treatment of brain ageing and ageing neurodegenerative diseases. Mech Ageing Dev. 2005;126:317–326. doi: 10.1016/j.mad.2004.08.023. [DOI] [PubMed] [Google Scholar]
- Zhang GY, Zhang QG. Agents targeting c-Jun N-terminal kinase pathway as potential neuroprotectants. Expert Opin Investig Drugs. 2005;14:1373–1383. doi: 10.1517/13543784.14.11.1373. [DOI] [PubMed] [Google Scholar]
- Zheng H, Wang X, Legerski RJ, Glazer PM, Li L. Repair of DNA interstrand cross-links: interactions between homology-dependent and homology-independent pathways. DNA Repair (Amst) 2006;5:566–574. doi: 10.1016/j.dnarep.2006.01.010. [DOI] [PubMed] [Google Scholar]
- Zhu X, Yu QS, Cutler RG, Culmsee CW, Holloway HW, Lahiri DK, Mattson MP, Greig NH. Novel p53 inactivators with neuroprotective action: syntheses and pharmacological evaluation of 2-imino-2,3,4,5,6,7-hexahydrobenzothiazole and 2-imino-2,3,4,5,6,7-hexahydrobenzoxazole derivatives. J Med Chem. 2002;45:5090–5097. doi: 10.1021/jm020044d. [DOI] [PubMed] [Google Scholar]