

Abstract
A procedure has been developed for the construction of 7,5-fused ring systems through ring expansion of silyl enol ethers. This method has been applied to the synthesis of an intermediate en route to the natural product, tricholomalide A.
The bicyclo[5.3.0]decane ring system (hydroazulene) appears to be a common structural motif in various natural products.1 Despite ongoing efforts, the development of effective and useful methods for the construction of these substructures remains a challenge to the synthetic chemist.2 We recently reported a route which leads to the 7,5-membered bicyclic ring motif of the guanacastepenes, via a so-called ‘homo-Robinson annulation’ (Figure 1).3 In this process, we demonstrated that the 6-membered enone ring of hydrindane derivative 3 could be expanded into its corresponding 7-membered dienone 5 through a four-step sequence involving: formation of silyl enol ether, cyclopropanation, oxidative ring opening, and dehydrohalogenation. This protocol appeared to allow for access to the important hydroazulene intermediate, 5, in the early stage of the guanacastepene synthesis.4 This intermediate cannot be reached via direct Robinson annulation.
Figure 1.
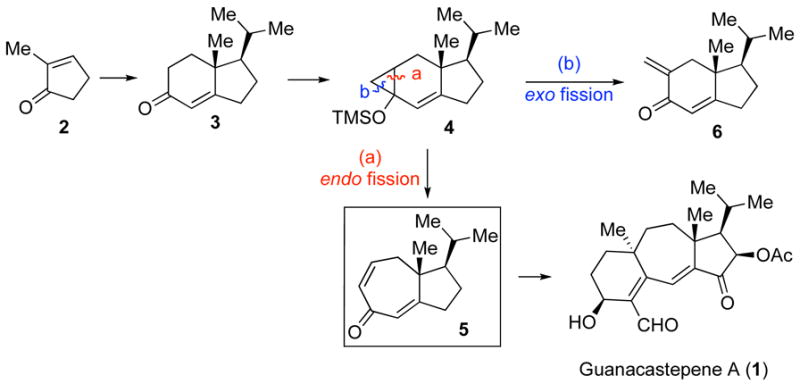
Previously reported Homo-Robinson annulation route to 5.
Subsequently, we launched a program directed toward the total synthesis of the neurotrophic diterpene, tricholomalide A (7, Figure 2), isolated from the mushroom Tricholoma sp.5 We envisioned that building block 5 could serve as a highly adaptable intermediate en route to tricholomalide A, due to its useful dienone functionality. However, in attempting to repeat the reaction procedure described in our earlier report, we were unable to obtain 5 in useful quantities. This surprising result led us to explore this reaction further. Mechanistically, there are two possible pathways of silyloxycyclopropane opening. Thus, cleavage of the endo bond proceeds to give the desired ring-expanded enone, which is subsequently advanced to the corresponding dienone 5 (path a, Figure 1). Alternatively, exo cleavage would afford an α-halomethyl ketone, which upon elimination, would generate the exo methylene compound 6 (path b, Figure 1).
Figure 2.
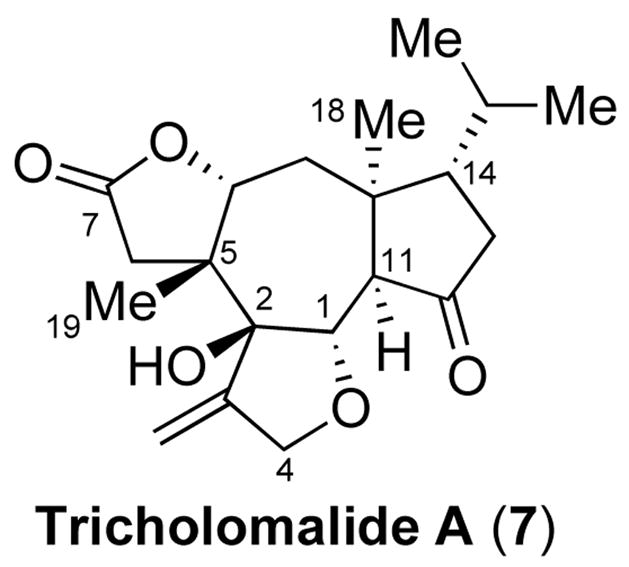
Tricholomalide A (7).
Upon revisiting this protocol, we now found that, contrary to our previous claim, the major product of the ring expansion sequence starting with 4 is actually 6. In addition, upon re-examination, other substrates described in the original report were also found to have preferentially undergone exo-fission, to produce exo-methylene type adducts as the major reaction products. The experimental details of these studies may be found in the accompanying Supporting Information. In this Communication, we disclose the reinvestigation of our Homo-Robinson annulation strategy and provide a procedure for the construction of the 7,5-fused ring structure of tricholomolide A (7) through a major modification of the original protocol.
In light of these findings, we sought to modify the originally reported procedure so as to promote formation of the desired isomeric product. In order to ensure that our ring opening process would occur through a radical intermediate, as proposed by Saegusa,6,7 we used iron (III) nitrate as the oxidant, to effect cyclopropane ring opening, and diphenyldisulfide to trap the ensuing radical intermediate (Scheme 1).8 The resulting mixture of sulfides 8a,b was reduced by Raney nickel to provide a 6.9:1:1 inseparable mixture of 9, 6, and 10.9 Thus, the oxidative ring cleavage had again predominantly occurred in an exo fashion to give the exo methylene compound as the major product.
Scheme 1.
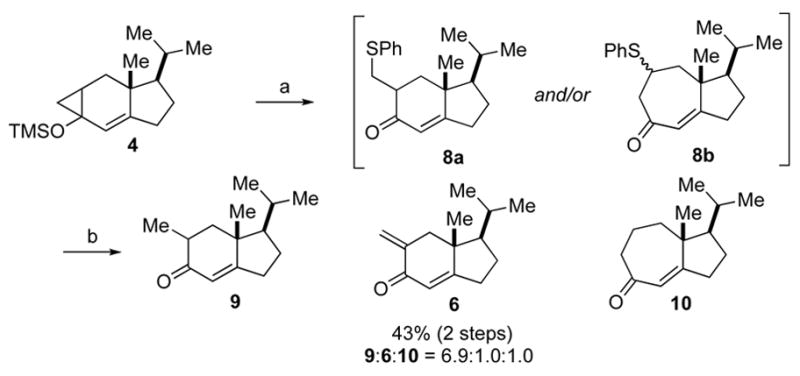
Ring expansion of 4 using Fe(NO)3. Reagents and conditions: (a) Fe(NO3)3, PhSSPh, DMF, 0 °C to 23 °C. (b) Raney Ni, EtOH, 23 °C, 43% (9:6:10 = 6.9:1.0:1.0,2 steps)
Recognizing that the previously disclosed oxidative ring opening procedure would not serve as a viable route to our target system, tricholomalide A, we pursued a different ring opening approach that would not proceed through a radical intermediate. Indeed, we postulated that, by forming a halocyclopropane intermediate, we could conceivably trigger the ring fission in the desired way through exposure to acid or silver cation.10 Moreover, depending on the substrate, i.e. dihalo or methyl halocyclopropane, the ultimate product could be either an α-halo or an α-methyl enone. The latter could be a more synthetically useful intermediate toward tricholomalide A. The execution of this plan is illustrated in Scheme 2. Thus, enone 311 was treated with LDA and TMSC1 to generate the silyl enol ether, which was then converted to the dibromocyclopropane 12 through addition of dibromocarbene. Happily, silver nitrate promoted the successful ring opening of 11 to afford the desired bromodienone 12 in 59% overall yield (3 steps). The structure of 12 was confirmed by NMR analysis of the reduced compound 5, which was identical to the compound published in the literature (the yield of the reduction of 12 was not optimized). Following an analogous procedure, we synthesized the chloromethylcyclopropane 13, which was also treated with silver nitrate to produce methyl dienone 14. In this case, a slightly elevated temperature was required and a small amount of ethylidene enone, resulting from exo cleavage, was generated as a side product. It is noteworthy that we have successfully gained access to useful quantities of these bicyclic tricholomalide A building blocks (i.e. 12 and 14) through modification of our original protocol.
Scheme 2.
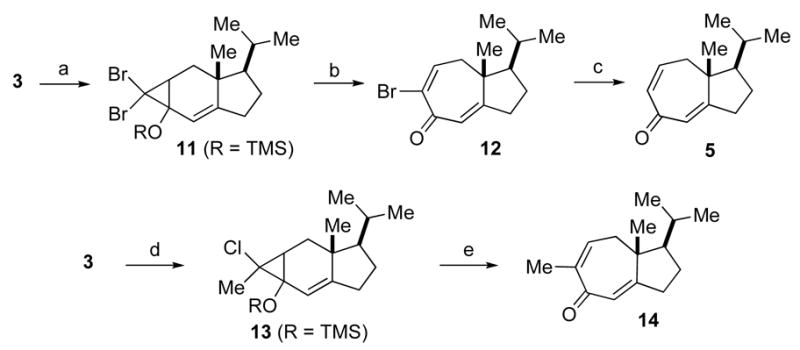
Synthesis of the ring expanded compounds 12 and 14. Reagents and conditions: (a) i) LDA, TMSC1, THF, −78 °C. ii) CHBr3, KOtBu, pentane 0 °C to 23 °C. (b) AgNO3, pyr, EtOH, 23 °C, 1.5 h, 59% (3 steps from 3). (c) BF3·OEt2, NaI, CH3CN, 0 °C, 41%. (d) i) LDA, TMSC1, THF, −78 °C. ii) dichloroethane, nBuLi, THF, −35 °C to 0 °C. (e) AgNO3, pyr, EtOH, 60 °C, 3.5 h, 45% (3 steps from 3).
With these functionalized hydroazulenes in hand, we attempted to address the installation of the C5 quaternary center in tricholomalide A, in which the C19 methyl group is anti to the angular methyl group at the ring juncture of the 7,5-bicyclic system (see 7, Figure 1). We were able to elaborate bromodienone 12 to the α-ethynyl dienone 15 via Sonogashira coupling with ethynylsilane (Scheme 3). However, deconjugative alkylation of 15 preferentially afforded the undesired O-alkylated product 17 although the first deconjugation step had occurred at the desired γ-carbon.
Scheme 3.
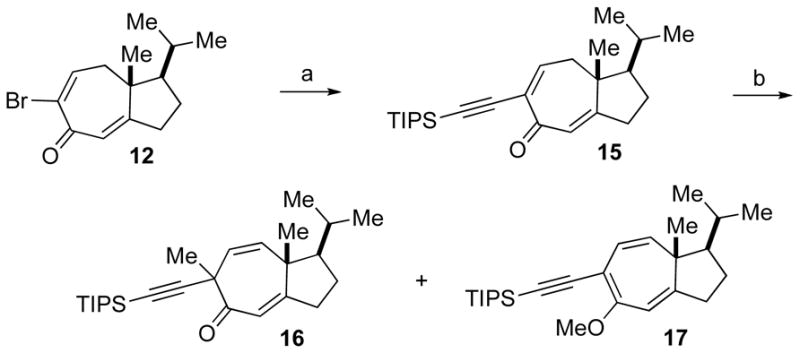
Synthetic approach toward tricholomalide A. Reagents and condtions: (a) TIPS-acetylene, PdCl2(PPh3)2, CuI, iPr2NH, THF, 0 °C to 23 °C, 90%. (b) KOtBu, MeI, THF, 0 °C to 23 °C, 78% (16:17 = 1:3.2).
Alternatively, we envisioned utilizing the resident α-methyl group of 14 to establish the quaternary center by installing the two carbon units via intramolecular cyclopropanation. Deconjugation of α-methyl dienone 14 followed by Luche reduction furnished a 3:1 mixture of two epimeric allylic alcohols, of which the minor alcohol 19 was easily converted into the major alcohol 18 in two steps (Scheme 4). Treatment of 18 with methyl malonyl chloride produced the malonate, which was converted to the diazomalonate 20 via the diazo transfer reaction developed by Davies.12 Upon slow addition of 20 into copper (II) bis(salicylidene-tert-butylamine) at 110 °C,13 intramolecular cyclopropanation proceeded smoothly to provide the cyclopropyl ester 21 as a single isomer in good yield. Next, we explored various nucleophiles, such as halide, phenylsulfide, and alcohol to accomplish the crucial nucleophilic ring opening of activated cyclopropane 21.14 We were ultimately able to obtain lactone 22, along with the hydrolyzed acid congener, through treatment of 21 with lithium chloride and camphor sulfonic acid,15 albeit in low yield. Meanwhile, we indirectly determined the stereochemistry of the newly generated quaternary carbon (C5 of tricholomalide A) in 22 by analysis of the X-ray structure of lactone 24 (Figure 3), prepared from ally lie alcohol 19 through an analogous intramolecular cyclopropanation route.
Scheme 4.
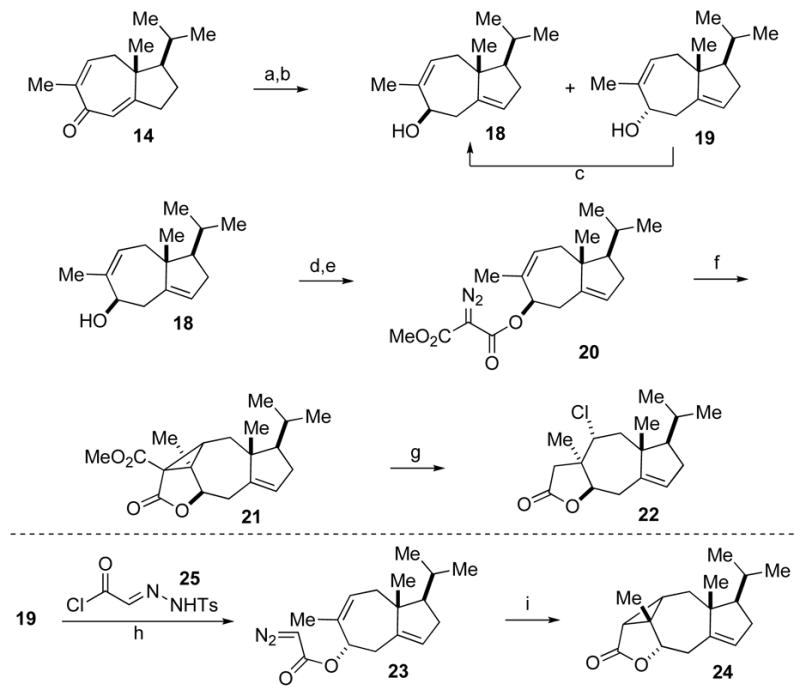
Synthesis of intermediate 22. Reagents and conditions: (a) KHMDS, THF −78 °C; aq. NH4Cl, 65% (BORSM). (b) NaBH4, CeCl3·7H2O, 0 °C, 96% (18:19 = 3.2:1). (c) i) p-NO2BzOH, PPh3, DIAD, toluene, 0 °C, 68%. ii) KOH, THF/MeOH/H2O (2:2:1), 84%. (d) methyl malonyl chloride, Et3N, CH2C12, 0 °C to 23 °C, 84%. (e) p-AcNH-PhSO2N3, DBU, 0 °C, 99%. (f) copper(II) bis(salicylidene-tert-butylamine), toluene, 110 °C, 81%. (g) LiCl, CSA, DMF, 145 °C, 11%. (h) 25, PhNMe2, Et3N, 0 °C to 23 °C, 50%. (i) copper(II) bis(salicylidene-tert-butylamine), toluene, 110 °C, 59%.
Figure 3.
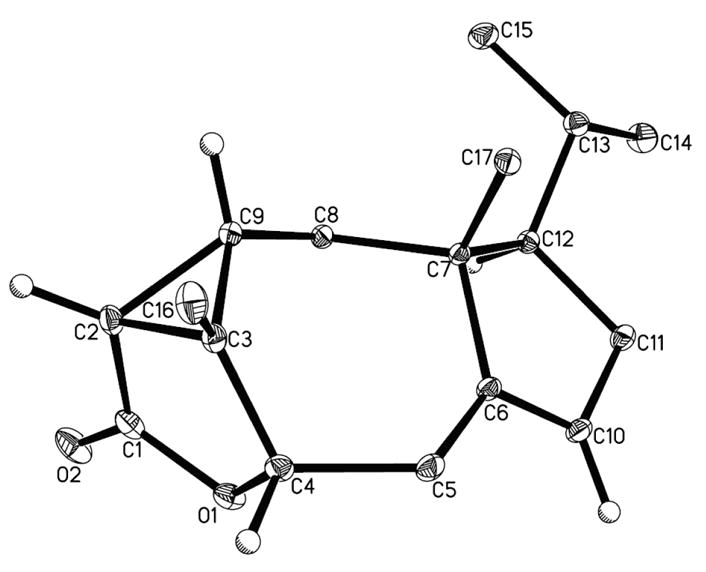
X-ray structure of 24.
In conclusion, we have reported the development of a new and reliable synthetic protocol for the construction of the hydroazulene motif of tricholomalide A. This method involves the cyclopropanation of a silyl enol ether with halocarbene, followed by silver promoted ring opening of the resulting cyclopropane, to produce a highly functionalized hydroazulene framework. In addition, we have developed an accessible route to an advanced intermediate (22) containing the important C5 quaternary center of tricholomalide A. Further application of this method toward the total synthesis of tricholomalide A is in progress and it will be reported in due course.
Supplementary Material
Acknowledgments
Support for this research was provided by the National Institutes of Health (HL25848). We thank Ms. Daniela Buccella for the crystal structure analysis. The NSF (CHE-0619638) is thanked for acquisition of an X-ray diffractometer.
References
- 1.a) Connolly JD, Hill RA, editors. Dictionary of Terpenoids. Chapman and Hall; New York: 1991. [Google Scholar]; b) Fraga BM. Nat Prod Rep. 2005;22:465. doi: 10.1039/b501837b. [DOI] [PubMed] [Google Scholar]; c) Hanson J. Nat Prod Rep. 2005;22:594. doi: 10.1039/b501834j. [DOI] [PubMed] [Google Scholar]
- 2.For a recent review, see: Kantorowski EJ, Kurth MJ. Tetrahedron. 2000;56:4317.
- 3.Yun H, Danishefsky SJ. Tetrahedron Lett. 2005;46:3879. [Google Scholar]
- 4.Mandal M, Yun H, Dudley GB, Lin S, Tan DS, Danishefsky SJ. J Org Chem. 2005;70:10619. doi: 10.1021/jo051470k. [DOI] [PubMed] [Google Scholar]
- 5.Tsukamoto S, Macabalang AD, Nakatani K, Obara Y, Nakahata N, Ohta TJ. Nat Prod. 2003;66:1578. doi: 10.1021/np030140x. [DOI] [PubMed] [Google Scholar]
- 6.Ito Y, Fujii S, Saegusa T. J Org Chem. 1976;41:2073. [Google Scholar]
- 7.It is known that FeCl3 acts as a Lewis acid in some cases. For representative reactions mediated by FeCl3 as Lewis acid, Denmark SE, Habermas KL, Hite GA, Jones TK. Tetrahedron. 1986;42:2821.Matusuda T, Tanino K, Kuwajima I. Tetrahedron Lett. 1989;30:4267.Tietze LF, Beifuss U. Synthesis. 1988:359.
- 8.Booker-Milburn KI, Barker A, Brailsford W, Cox B, Mansley TE. Tetrahedron. 1998;54:1532. [Google Scholar]
- 9.One possible explanation for the modest yield of this reaction is that the ring expanded intermediate 8ab appeared to be unstable as it decomposes during this process. Nevertheless, it is clear that the amount of exo cleaved product is substantial based on the yield of the two step sequences.
- 10.a) Blanco L, Amice P, Conia JM. Synthesis. 1981:289. [Google Scholar]; b) Wenkert E, Arrhenius TS, Bookser B, Guo M, Mancini P. J Org Chem. 1990;55:1185. [Google Scholar]; c) Macdonald TL. J Org Chem. 1978;43:4241. [Google Scholar]; d) Strok G, Nussim M, August B. Tetrahedron. 1966;(Suppl 8 Pt 1):105. [Google Scholar]
- 11.Pierre D, Gilles D, Laurent H, Jean Marie P. J Chem Soc, Perkin Trans 1. 1992:387. [Google Scholar]
- 12.Baum JS, Shook DA, Davies HML. Synth Commun. 1987;17:1709. [Google Scholar]
- 13.Corey EJ, Myers AG. Tetrahedron Lett. 1984;25:3550. [Google Scholar]
- 14.For a review, see: Danishefsky SJ. Acc Chem Res. 1979;12:66.Wong HNC, Hon M-Y, Tse C-W, Yip Y-C. Chem Rev. 1989;89:165.
- 15.Fukuyama T, Chen XJ. Am Chem Soc. 1994;116:3125. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.