

Abstract
Combined treatment with quercetin and TRAIL induced cytotoxicity and enhanced annexin V staining and poly (ADP-ribose) polymerase (PARP) cleavage in human prostate cancer cell lines DU-145 and PC-3. These indicators of apoptosis resulted from the activation of caspase-8, -9, and -3. Although the expression levels of FLIPs, cIAP1, cIAP2, and the Bcl-2 family were not changed in quercetin-treated cells, significant downregulation of survivin occurred. Knockdown survivin by siRNA significantly increased TRAIL-induced apoptosis. We hypothesized that quercetin-induced activation of MAPK (ERK, p38, JNK) is responsible for downregulation of survivin gene expression. To test this hypothesis, we selectively inhibited MAPK during treatment with quercetin. Our data demonstrated that inhibitor of ERK (PD98059), but not p38 MAPK (SB203580) or JNK (SP600125), significantly maintained the intracellular level of survivin during treatment with quercetin. Interestingly, PD98059 also prevented quercetin-induced deacetylation of histone H3. Data from survivin promoter activity assay suggest that the Sp1 transcription factor binds to the survivin promoter region and quercetin inhibits its binding activity through deacetylation of histone H3. Quercetin-induced activation of the ERK-MSK1 signal transduction pathway may be responsible for deacetylation of histone H3. Taken together, our findings suggest that quercetin enhances TRAIL induced apoptosis by inhibition of survivin expression, through ERK-MSK1-mediated deacetylation of H3.
Keywords: Quercetin, TRAIL, Apoptosis, Survivin, ERK
1. Introduction
Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL), a member of the tumor necrosis factor (TNF) superfamily, triggers apoptosis in a variety of malignant cells without marked toxicity to normal cells [1]. Apoptosis is initiated by binding of TRAIL to the death domains (DD) of death receptors DR4 and/or DR5, which are widely expressed in most human cells. TRAIL can also interact with two non-functional decoy receptors, DcR1 and DcR2, without eliciting cell death [1]. Upon TRAIL-mediated activation of the death receptor, its intracellular death domain attracts the Fas-associated death domain adaptor molecule [2], which further recruits the initiators caspase-8 and caspase-10 to the death receptor to form the death-inducing signaling complex. This process ultimately results in the activation of the terminal executioners caspase-3, caspase-6, and caspase-7, thereby leading to cell death [3].
Epidemiological studies have shown that the consumption of vegetables, fruits and tea, of which quercetin is a constituent, lowers the risk of cancer [4]. Quercetin has been shown to inhibit the enzymes involved in cell proliferation and in the cell signal transduction pathway including protein kinase C [5], tyrosine kinase [6], cdc25 phosphatase [7], PI-3 kinase [8], P-Akt [9], DNA topoisomerase II [10], and c-Jun N-terminal kinase (JNK) [11]. Quercetin has a wide range of biological activities including inhibition of mutant p53 expression [12], and androgen receptor expression and function in LNCaP cells [13]. Quercetin-mediated apoptosis may result from the induction of stress proteins including the heat shock proteins, disruption of microtubules and mitochondrial release of cytochrome c, and activation of caspases [14, 15, 16, 17]. These activities of quercetin make it a promising candidate for treatment and prevention of various cancers.
In this study, we investigated whether quercetin can promote TRAIL-induced apoptotic death. We observed that downregulation of survivin is responsible for quercetin-promoted TRAIL-induced apoptosis in human prostate cancer cells. ERK-mediated deacetylation of histone H-3 results in downregulation of survivin through inhibiting Sp1 binding activity and subsequently enhances TRAIL cytotoxicity.
2. Materials and methods
2.1. Cell culture and survival assay
Human prostate cancer cell lines DU-145 and PC-3 were obtained from the American Type Culture Collection (Manassas, VA, USA). DU-145 cells were cultured in DMEM medium (Gibco BRL, Gaithersburg, MD, USA) containing 10% fetal bovine serum (HyClone, Logan, UT, USA) and 26 μM sodium bicarbonate for monolayer cell culture. The dishes containing cells were kept in a 37°C humidified incubator with a mixture of 95% air and 5% CO2. At 1 day prior to the experiment, cells were plated into 60-mm dishes. For trypan blue exclusion assay [18], trypsinized cells were pelleted and resuspended in 0.2 ml of medium, 0.5 ml of 0.4% trypan blue solution, and 0.3 ml of phosphate-buffered saline solution (PBS). The samples were mixed thoroughly, incubated at room temperature for 15 min and examined under a light microscope. At least 300 cells were counted for each survival determination.
2.2. Drug treatment
Quercetin and H-89 were obtained from Sigma Chemical Co. (St Louis, MO, USA). SP600125, SB203580, and PD98059 were purchased from Calbiochem (San Diego, CA, USA). A stock solution was prepared in DMSO.
2.3. Antibodies
Polyclonal anti-Bc1-XL, anti-caspase-3, anti-caspase-9, anti-DR5, anti-DR4, and anti-DcR2 antibodies were purchased from Santa Cruz (Santa Cruz, CA, USA), anti-cIAP-1, anti-cIAP-2, anti-survivin from R&D Systems (Minneapolis, MN, USA), anti-phospho-Akt, anti-phospho-JNK, anti-phospho-ERK, anti-Akt, anti-JNK, anti-ERK, anti-acetyl H3, anti-acetyl H4 from Cell Signaling, and anti-FLIP from Calbiochem. Monoclonal antibodies were purchased from the following companies: anti-caspase-8 from Upstate Biotechnology, anti-PARP from Biomol Research Laboratory (Plymouth Meeting, PA, USA), anti-Bcl-2 from Santa Cruz (Santa Cruz, CA, USA), and anti-actin from ICN (Costa Mesa, CA, USA).
2.4. Production of recombinant TRAIL
A human TRAIL cDNA fragment (amino acids 114-281) obtained by RT-PCR was cloned into a pET-23d (Novagen, Madison, WI, USA) plasmid, and His-tagged TRAIL protein was purified using the Qiagen express protein purification system (Qiagen, Valencia, CA, USA).
2.5. Protein extracts and PAGE
Cells were lysed with 1 × Laemmli lysis buffer (2.4 M glycerol, 0.14 M Tris, pH 6.8, 0.21 M SDS, 0.3 mM bromophenol blue) and boiled for 10 min. Protein content was measured with BCA Protein Assay Reagent (Pierce, Rockford, IL, USA). The samples were diluted with 1 × lysis buffer containing 1.28 M β-mercaptoethanol, and equal amounts of protein were loaded on 12% SDS-polyacrylamide gels. SDS-PAGE analysis was performed according to Laemmli [19] using a Hoefer gel apparatus.
2.6. Immunoblot analysis
Proteins were separated by SDS-PAGE and electrophoretically transferred to nitrocellulose membrane. The nitrocellulose membrane was blocked with 5% nonfat dry milk in PBS-Tween-20 (0.1%, v/w) at 4°C overnight. The membrane was incubated with primary antibody (diluted according to the manufacturer's instructions) for 2 h. Horseradish peroxidase-conjugated anti-rabbit or anti-mouse IgG was used as the secondary antibody. Immunoreactive protein was visualized by the chemiluminescence protocol (ECL, Amersham, Arlington Heights, IL, USA). Quantitation of X-ray film was carried out by scanning densitometer (Personal Densitometer, Molecular Dynamics, Sunnyvale, CA, USA) using area integration.
2.7. Annexin V analysis
For the analysis of apoptosis, DU-145 cells were stimulated with quercetin and/or TRAIL. Resulting, cultures with or without and apoptotic cell death was measured by flow cytometry using FITC-conjugated annexin V and PI (PharMingen, San Diego, CA, USA) according to the manufacturer's instructions.
2.8. HDAC enzyme activity assay
HDAC activity was measured using the HDAC assay kit (colorimetric detection) according to the instructions from the manufacturer (Upstate, Lake Placid, NY, USA). Briefly, 15 μL nuclear fraction was diluted to 25 μL with the assay buffer. Following the addition of colorimetric HDAC substrate, the reaction mixture was incubated for 30 minutes at 37°C, and stopped by 60 minutes with HDAC activator solution. An activator solution was then added to release the coloriphore from the deacetylated substrates, and colorimetric of the reaction mixture was monitored at absorbance read at 405 nm using a Plate reader.
2.9. Transfections of Sp1 and luciferase assays
A plasmid encoding human Sp1, pCAGGS, was kindly provided by Dr. Ferruccio Galbiati (University of Pittsburgh, PA, USA). Transfections were performed using LipofectAmine Plus reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's protocol. Briefly, 1 × 105 DU-145 cells were cultured in 6-well culture plates in 2 ml of DMEM supplemented with 10% FBS, and incubated for 24 h. After washing the cells with OPTI-MEM medium (Invitrogen) two times, 800 μl of OPTI-MEM medium and 200 μl of LipofectAmine reagent containing 0.1 to 1 μg of Sp1 were added. After 4h of incubation, medium was changed to 2 ml of DMEM supplemented with 10 % FBS, and cells were incubated for an additional 24 h. For the transfection of Sp1 expression vector, pCAGGS-Sp1 was transfected into DU-145 cells. We purchased survivin promoter vectors (pLUC-1430, -649, -230, -123, -108, -89, -42) from Health Research Inc. (Buffalo, NY, USA). In brief, cells were plated onto 6-well plates at a density of 1 × 105 cells/well and grown overnight. Cells were cotransfected with 1 μg of various plasmid constructs and 1 μg of the pCMV-β-galactosidase plasmid for 4 h by the LipofectAmine method. After transfection, cells were cultured in 10% FBS medium with a vehicle (DMSO) or quercetin for 24 h. Luciferase and β-galactosidase activities were assayed according to the manufacturer's protocol (Promega, Madison, WI, USA). Luciferase activity was normalized for β-galactosidase activity in the cell lysate and expressed as an average of three independent experiments.
2.10. Chromatin immunoprecipitation assays
DU-145 cells were left untreated or treated with quercetin (100 μM) for 24 h. Chromatin immunoprecipitation assay was done 48 h after drug stress with the chromatin immunoprecipitation assay kit from Upstate Cell Signaling Solutions (Temecula, CA, USA). Briefly, cells were cross-linked with 1% formaldehyde for 10 minutes at 37°C. The cross-linked chromatin was then extracted, diluted with lysis buffer, and sheared by sonication. After preclearing with a 1:2 mix of protein A/protein G-agarose bead (Upstate), the chromatin was divided into equal samples for immunoprecipitation with anti-Sp1, anti-immunoglobulin G (negative control) polyclonal antibody (Santa Cruz Biotechnology). The immunoprecipitates were pelleted by centrifugation and incubated at 65°C to reverse the protein-DNA cross-linking. The DNA was extracted from the elute by the phenol/chloroform method and then precipitated by ethanol. Purified DNA was subjected to PCR with primers specific for a region (-264 to -31) in the survivin promoter spanning seven putative Sp1 binding sites. The sequences of the PCR primers used are as follows: F1 5′-TTCTTTGAAAAGCAGTCGAGGGG-3′, R1 5′-CGCGATTCAAATCTGGCGGTTA-3′.
3. Results
3.1. Combined treatment with quercetin and TRAIL induces synergistic apoptosis in prostate cancer cells
In search of novel strategies to target tumor cell resistance, we investigated the anti-tumor effect of the chemopreventive, natural compound quercetin on the induction of apoptosis in human prostate cancer DU-145 cells. As shown in Figure 1A, little cytotoxicity was observed with quercetin (10-200 μM) alone. Unlike quercetin, TRAIL alone induced cell death in a concentration-dependent manner; 97, 88, and 80% of the cells survived after exposure to 10, 50, and 100 ng/ml of TRAIL, respectively, for 24 h (Fig. 1B). Given the relatively low cytotoxic activity of quercetin as a single agent, we then tested quercetin in combination treatments. Surprisingly, we found that quercetin strongly cooperated with the death ligand TRAIL to induce apoptosis in DU-145 cells in a dose-dependent manner; 90, 55 and 35% of the cells survived after exposure to 10, 50, and 100 ng/ml of TRAIL in combination with 100 μM quercetin, respectively, for 24 h (Fig. 1B). Similar results were observed by Annexin V and PI staining assay. Data from FACS analysis show that apoptotic death occurred during combined treatment with TRAIL and quercetin (Fig. 1C). Combined treatment of quercetin with TRAIL produced a synergistic cytotoxicity in cells in another prostate cancer cell line, PC-3. Typical morphological changes of apoptotic cell death were observed under phase-contrast microscopy (Fig. 1D) and viability decreased in a dose-dependent manner; 72, 47, 2, and 1% of the cells survived after exposure to 0, 10, 30, and 100 μM of quercetin in combination with 50 ng/ml TRAIL, respectively, for 24 h (Fig. 1E). We observed that the synergy of quercetin with TRAIL in PC-3 cells was greater than that in DU-145 cells. Since both cell lines are tumorigenic and lack wild-type p53, this difference is probably due to an intrinsic difference in TRAIL sensitivity. Nonetheless, all these results clearly indicate that quercetin sensitizes TRAIL-induced apoptosis in prostate cells.
Fig. 1. Effect of quercetin on TRAIL-induced cytotoxicity in human prostate DU-145 and PC-3 cells.

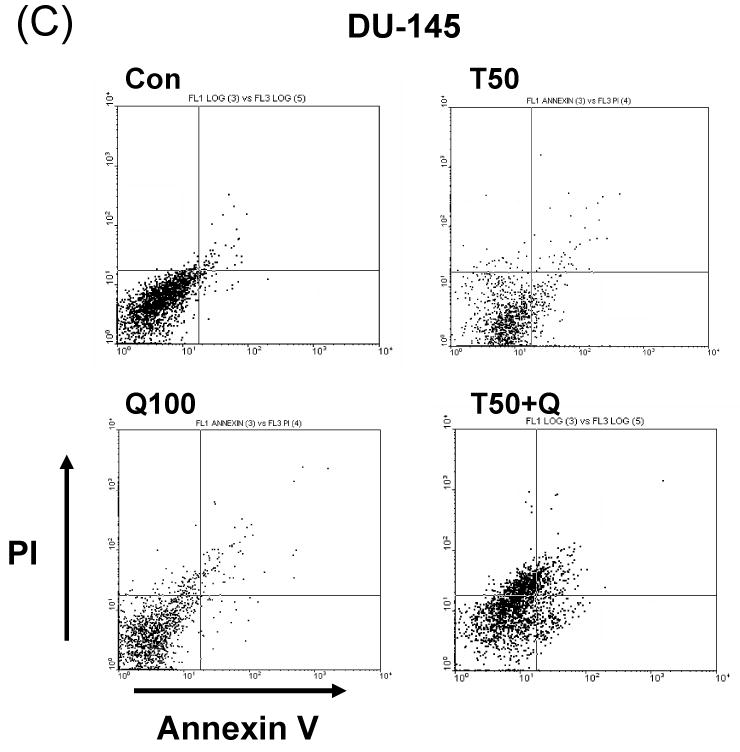
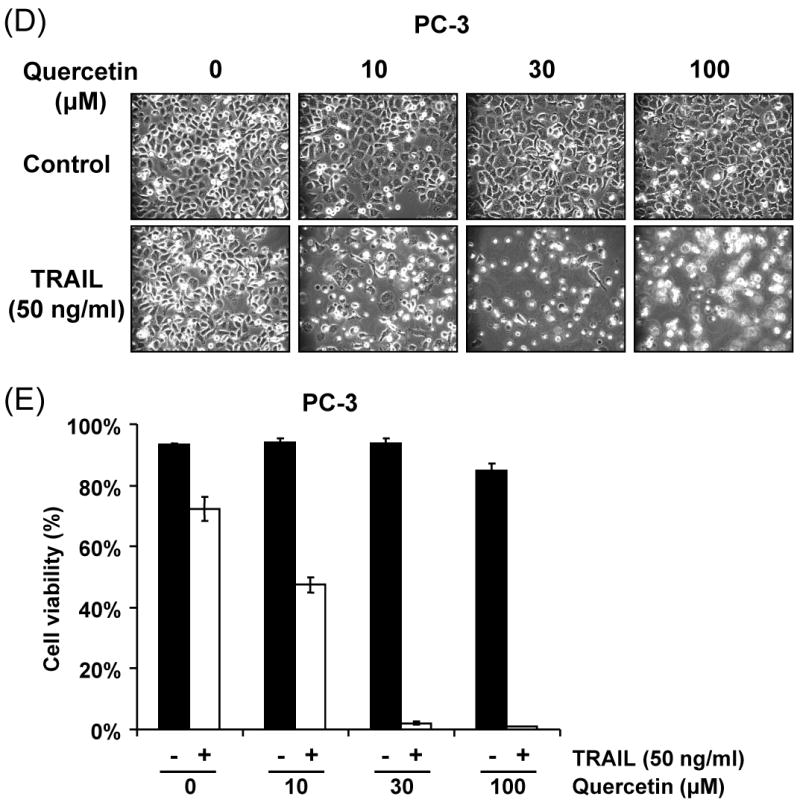
(A) DU-145 cells were treated for 24 h with quercetin (10-200 μM). (B) DU-145 cells were treated for 24 h with various concentrations of TRAIL (10-100 ng/ml) in the presence or absence of 100 μM quercetin. Cell survival was determined by the trypan blue exclusion assay. Error bars represent standard error from the mean (SEM) for three separate experiments. (C) Cells were treated for 4h with TRAIL (50 ng/ml) in the presence or absence of 100 μM quercetin. After treatment, apoptosis was detected by the FACS analysis. Con: control, T50: TRAIL 50ng/ml, Q100: Quercetin 100 μM, T50+Q: TRAIL 50 ng/ml plus Quercetin 100 μM. PC-3 cells were treated with various concentrations of quercetin (10-100 μM) for 24 h in the presence or absence of 50 ng/ml of TRAIL. The morphological features were analyzed with a phase-contrast microscope (200×) (D) and cell survival was determined by the trypan blue exclusion assay (E). Error bars represent standard error from the mean (SEM) for three separate experiments.
3.2. Sensitization to TRAIL-induced apoptosis through quercetin-mediated caspase activation
To confirm the activation of apoptotic signals by combined treatment with quercetin and TRAIL, we performed western blot analysis of PARP and caspase-8, -9, -3 in DU-145 cells treated with quercetin and/or TRAIL (Fig. 2). After treatment of DU-145 cells with TRAIL (50, 100 ng/ml) for different dose of quercetin (10-200 μM), immunoblot analysis of cell lysates demonstrated processed polypeptides for pro-caspase-8, an initiator caspase, a decrease in the major form of pro-caspase-3, an effector caspase, and finally, PARP cleavage. Taken together, TRAIL plus quercetin further enhanced activation of caspases-8, -9, -3 and the associated cleavage of PARP, showing that TRAIL-induced sensitization to quercetin is associated with an increase in the activation of caspases -8, -9, and -3.
Fig. 2. Effect of quercetin on TRAIL-induced proteolytic cleavage of PARP and activation of caspases in DU-145 cells.
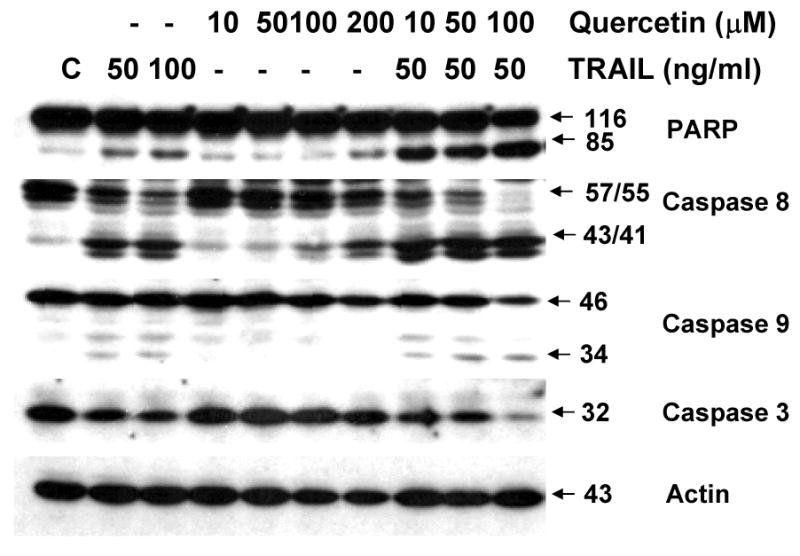
Cells were treated for 24 h with various concentrations of quercetin in the presence or absence of 50 ng/ml TRAIL and then harvested. Cell lysates containing equal amounts of protein (20 μg) were separated by SDS-PAGE and immunoblotted with anti-caspase-8, anti-capase-9, anti-caspase-3, or anti-PARP antibody. Actin was used to confirm the amount of proteins loaded in each lane.
3.3. Modulation of apoptosis regulatory molecules by quercetin
To gain insight into the molecular mechanism linking apoptosis sensitivity to treatment with quercetin, we investigated key molecules of the death receptor or mitochondrial pathway. We examined whether changes in the amounts of antiapoptotic proteins are associated with the promotion of apoptosis by TRAIL in combination with quercetin. DU-145 cells were treated with 50 ng/ml TRAIL in the presence of 0-100 μM quercetin. Data from Western blot analysis reveal that the combined treatment did not significantly alter the levels of FLIP/L, FLIP/s, and Bcl-2 family protein expression (Fig. 3A). As antiapoptotic proteins such as cIAP-1, cIAP-2, XIAP, and survivin have also been implicated in the regulation of TRAIL-induced apoptosis, expression of these proteins was next assessed in the presence of quercetin (10-100 μM). Quercetin Treatment resulted in significant down-regulation of survivin protein expression in a dose-dependent manner (Fig. 3B). But neither cIAP-1, cIAP-2, nor XIAP cellular protein levels were altered by quercetin treatment of DU-145 cells. This quercetin-mediated down-regulation of survivin was also confirmed in another prostate cancer line, PC-3 (Fig. 3C). Taken together, down-regulation of survivin gene expression may be an important fact for sensitization to TRAIL-induced apoptosis by quercetin.
Fig. 3. Intracellular levels of FLIPs, Bcl-2 family, and IAP family proteins during treatment with quercetin in the presence or absence of TRAIL.
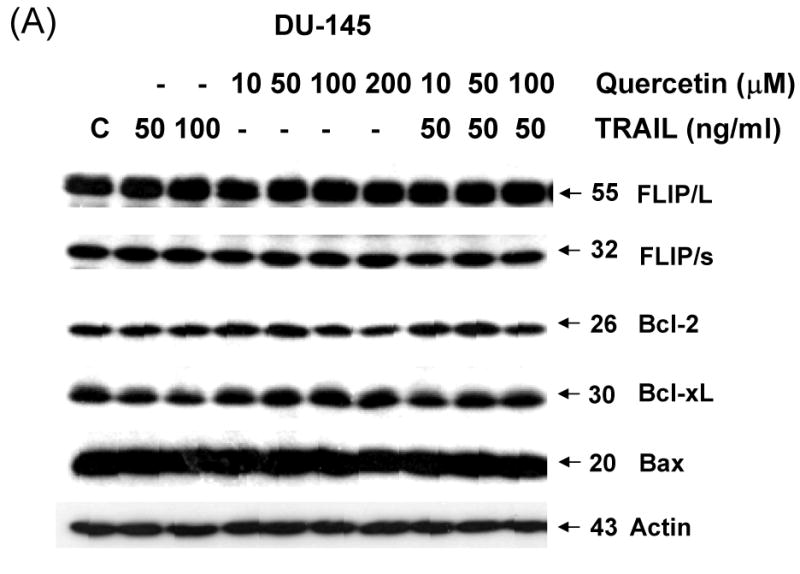
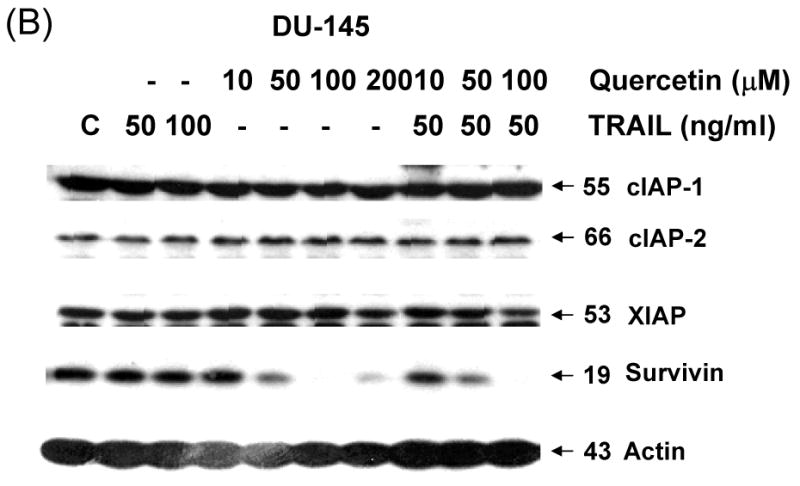
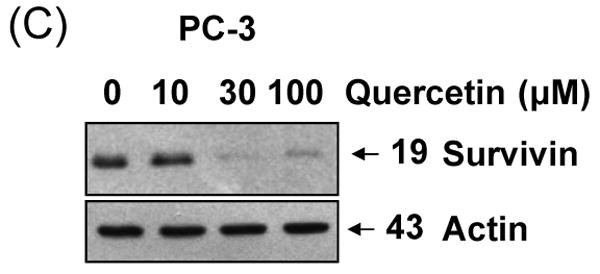
DU-145 cells were treated for 24 h with various concentrations of quercetin (10-100 μM) in the presence or absence of 50 ng/ml TRAIL. Equal amounts of protein (20 μg) were separated by SDS-PAGE and immunoblotted as described in materials and methods. Protein levels of FLIPs and Bcl-2 family (A) or IAP family (B) were analyzed during treatment with quercetin in the presence or absence of TRAIL. (C) PC-3 cells were treated for 24 h with indicated amount of quercetin (10-100 μM) and survivin level was measured by western blot analysis.
3.4. Effect of TRAIL on silenced survivin gene expression
Next, we further analyzed the involvement of survivin in the sensitization effect of quercetin. Figure 3 shows that quercetin and TRAIL induced apoptosis corresponded with down-regulation of survivin. To confirm the functional significance of survivin down-regulation in TRAIL-induced apoptosis, we employed the siRNA duplex against survivin. DU-145 cells transfected with the control siRNA or survivin siRNA were treated with or without TRAIL for 4h and subjected to immunoblot analysis. As shown in Fig. 4A, transfection of siRNA against survivin resulted in enhancement of TRAIL-induced apoptosis in DU-145 cells, compared to the cells tranfected with control siRNA. The results from cell viability assay were consistent with that from PARP cleavage assay. TRAIL-induced cytotoxicity was promoted in survivin knock-down DU-145 cells (Fig. 4B). Taken together, these results suggest that survivin down-regulation is critical for the TRAIL induced apoptosis signal pathway.
Fig. 4. Effect of silenced survivin expression on TRAIL-induced apoptosis in DU-145 cells.
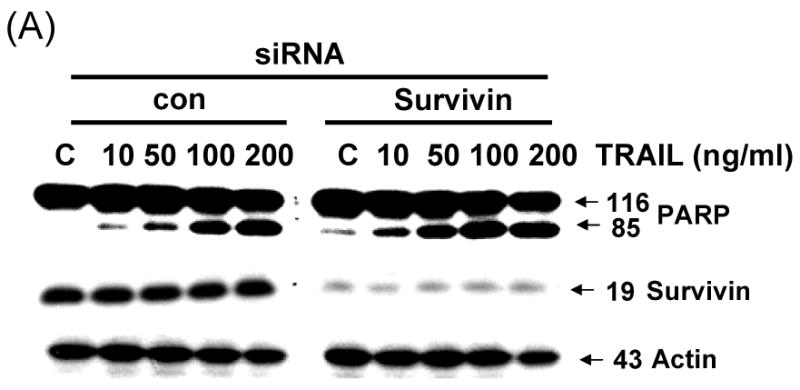

Cells were transfected with a control vector or silenced survivin oligonucleotide DNA for 24 h. Transfected cells were treated for 4 h with various concentrations of TRAIL (10-200 ng/ml). (A) Cell lysates were subjected to immunoblotting for anti-PARP or anti-survivin. Actin was shown as an internal standard. (B) Cell survival was determined by the trypan blue exclusion assay. Error bars represent standard error from the mean (SEM) for three separate experiments.
3.5. Quercetin inhibits transcription from the survivin gene promoter by blocking DNA binding of the Sp1 promoter
Next, we attempted to further clarify the underlying mechanisms of quercetin-induced survivin down-regulation, employing reverse transcription-PCR and the luciferase gene expression system. First, to further elucidate the mechanism responsible for the changes in amount of survivin protein, we determined levels of survivin mRNA by RT-PCR. Treatment with quercetin resulted in marked decreases of survivin mRNA levels in a dose-dependent manner (Fig. 5A). Second, to define the regulatory region of the survivin promoter, we constructed a series of chimeric plasmids to contain progressive 5′ end deletion of the survivin 5′ flanking sequence spliced to the firefly luciferase coding sequence in the promoterless plasmid vector pGL3-basic (Fig. 5B) and transiently transfected these plasmids into DU-145 cells. We measured luciferase levels to reflect promoter activity at 24 h after transfection in DU-145 cells treated or untreated with quercetin (Fig. 5C). The construct pLUC-1430 showed a ∼ 2 fold increase in promoter activity compared with pLUC-42. These results show that potential positive regulatory elements are located between -230 and -123 nucleotide and might be enough to regulate survivin promoter activity. As shown in Fig. 5C quercetin inhibited survivin promoter activity, suggesting that survivin promoter was down-regulated by quercetin. Sp1 protein expression was also decreased during the quercetin treatment (Fig. 8A). Inhibition of Sp1 has been postulated as a mechanism of action of quercetin [21]. Taken together, our results and previous literature suggest that multiple Sp1 binding sites in the proximal promoter region are critical not only for basal transcription of survivin but also for quercetin-mediated inhibition of transactivation of survivin promoter.
Fig. 5. Effect of quercetin on the mRNA levels, the promoter activity of Survivin and Sp1 binding activity in DU-145 cells.
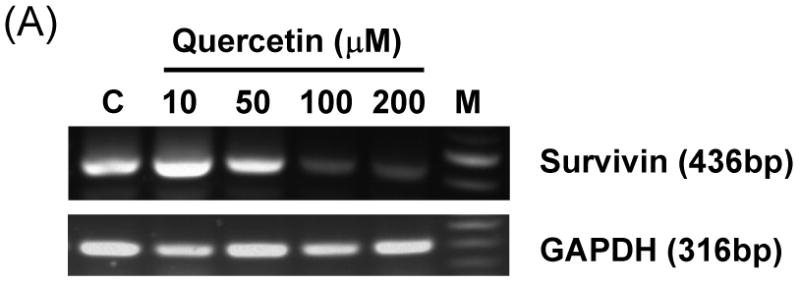

(A) DU-145 cells were treated with the indicated concentration of quercetin. Total RNA was isolated and RT-PCR analysis was performed. A representative study is shown; two additional experiments yielded similar results. (B) Schematic structure of survivin promoter constructs used for testing luciferase activity. (C) Various promoter plasmids were transfected, and treated with quercetin. The cells were lysed and luciferase activity measured.
Fig. 8. Sp1 is associated with quercetin-mediated down-regulation of survivin.
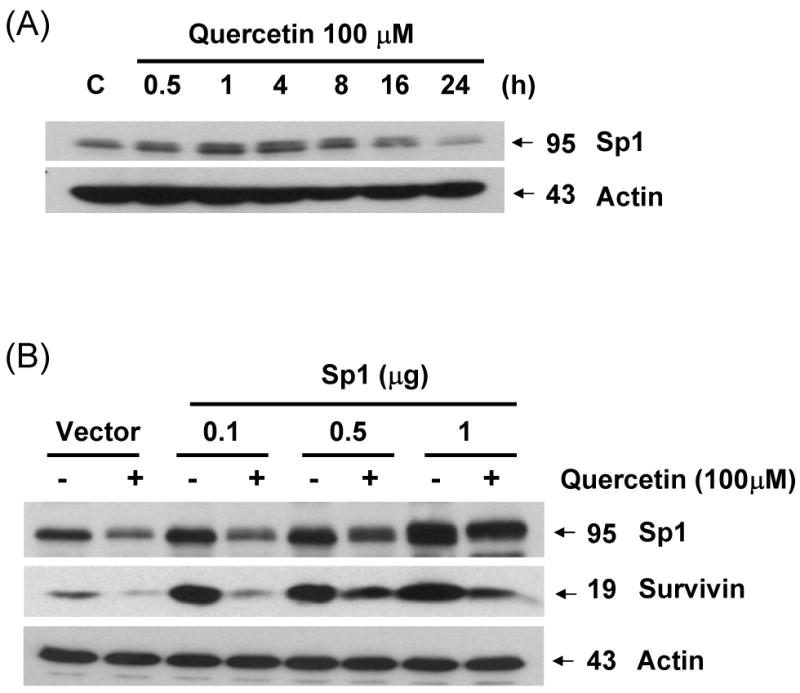
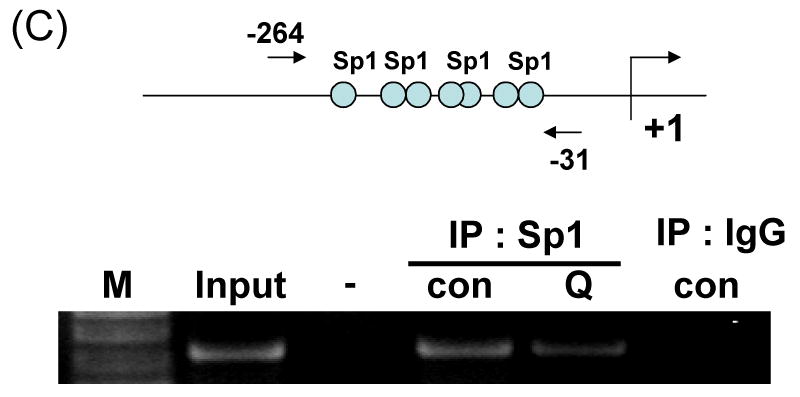
(A) DU-145 cells were incubated for the periods indicated in the presence or absence of quercetin. The intracellular levels of Sp1 were determined by western blot analysis. (B) Effect of ectopic expression of Sp1 on survivin expression. DU-145 cells were transfected with various concentrations of Sp1 cDNA plasmid (0.1-1 μg). The expression levels of Sp1 and survivin protein in transfected cells were determined by western blot analysis. (C) Chromatin immunoprecipitation analyses were performed with anti-Sp1 antibody, as described in material and methods. The chromatins were extracted from sonicated cells (Input), protein A without antibody (-), protein A with antibody (anti-Sp1) with/without quercetin treatment, or with pre-immune serum (anti-IgG). Specific promoter regions of the survivin gene were amplified by PCR, separated on 1.2% agarose gels, and stained with ethidium bromide. Also indicated is the relative position of the PCR product generated in ChIP assay.
3.6. Effect of quercetin on survivin protein expression by MAPK pathways
As the next step in the present study, the activation of MAPK subfamilies by quercetin was examined by using specific antibodies against the phosphorylated (activated) forms of the kinases. The results reveal that all three MAPK families examined are rapidly and strongly activated by the treatment with 100 μM of quercetin, following a time-dependent pattern (Fig. 6A). In particular, JNK MAPK activation displayed a rapid onset within 30 min of treatment, followed by a progressive decline, returning to basal levels after 1 h. Activation of p38 by quercetin was intense, reaching maximum values within 30 min of treatment, decreasing thereafter and reaching control levels at 16 h. Activation of ERKs was also observed at 30 min of quercetin treatment, followed by a consistently strongly activated form within 24 h. The above results indicate that quercetin induces a differential activation of the three well-established MAPK subfamilies, in relation to time of exposure. These MAPKs were activated by quercetin in a dose-dependent manner (10-100 μM) (Fig. 6B). Although phosphorylation of JNK and p38 occurred at 100 μM of quercetin treatment, treatment with 10 μM of quercetin for 1 h was enough to activate ERK. Although MAPK activation during quercetin treatment has been studied in detail in a variety of cell types, the signaling pathways leading to their phosphorylation under such conditions are not yet clarified. To date, there is no evidence how quercetin stress-induced MAPK activation affects survivin gene expression in prostate cancer cells. In order to study these pathways, pharmacological inhibitors of various kinases were used. The results of these experiments show that PD98059 (a specific inhibitor of MEK kinases) abolishes the activation of ERKs (Fig. 6C) producing recovery of survivin down-regulation in dose dependent manner. But SB203580 (a specific inhibitor of p38 kinases) and SP600125 (a specific inhibitor of JNK kinases) have no significant effect on survivin expression level (Figs. 6D and 6E). The inhibitor of MEK, and consequently of ERKs, PD98059 prevented quercetin-mediated down-regulation of survivin (Fig. 6C). The above results demonstrate that quercetin's activation of ERKs regulates survivin gene expression. On the contrary, activation of p38 and JNK-MAPK seems to not affect survivin gene expression. Cell viability assay confirmed the role of ERK in the regulation of survivin gene expression. The inhibition of ERK by PD98059 reduced the cytotoxic effect of combined treatment with quercetin and TRAIL (Fig. 6F).
Fig. 6. Effect of various MAP kinase or inhibitors on quercetin mediated down-regulation of survivin expression.
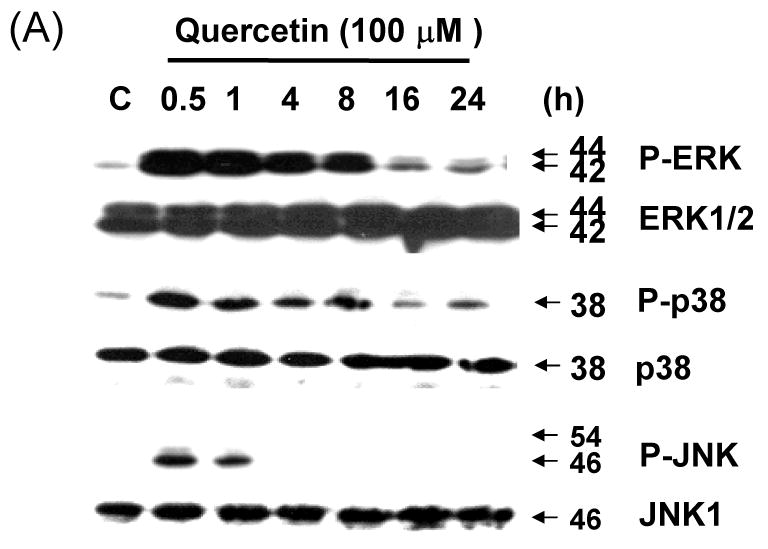
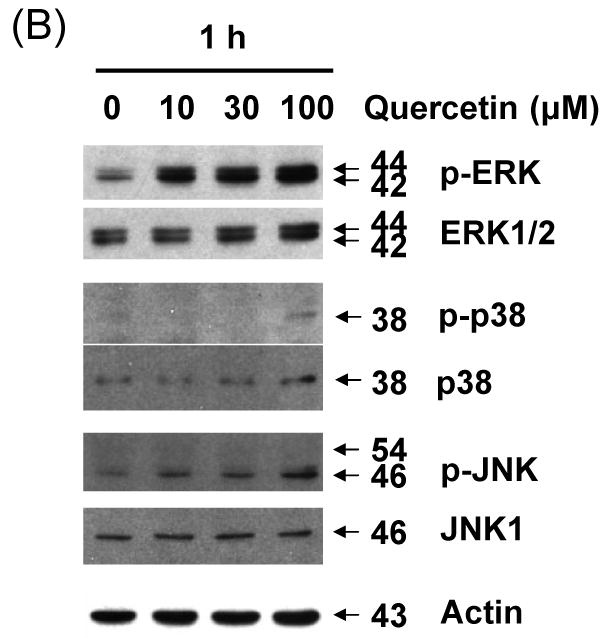
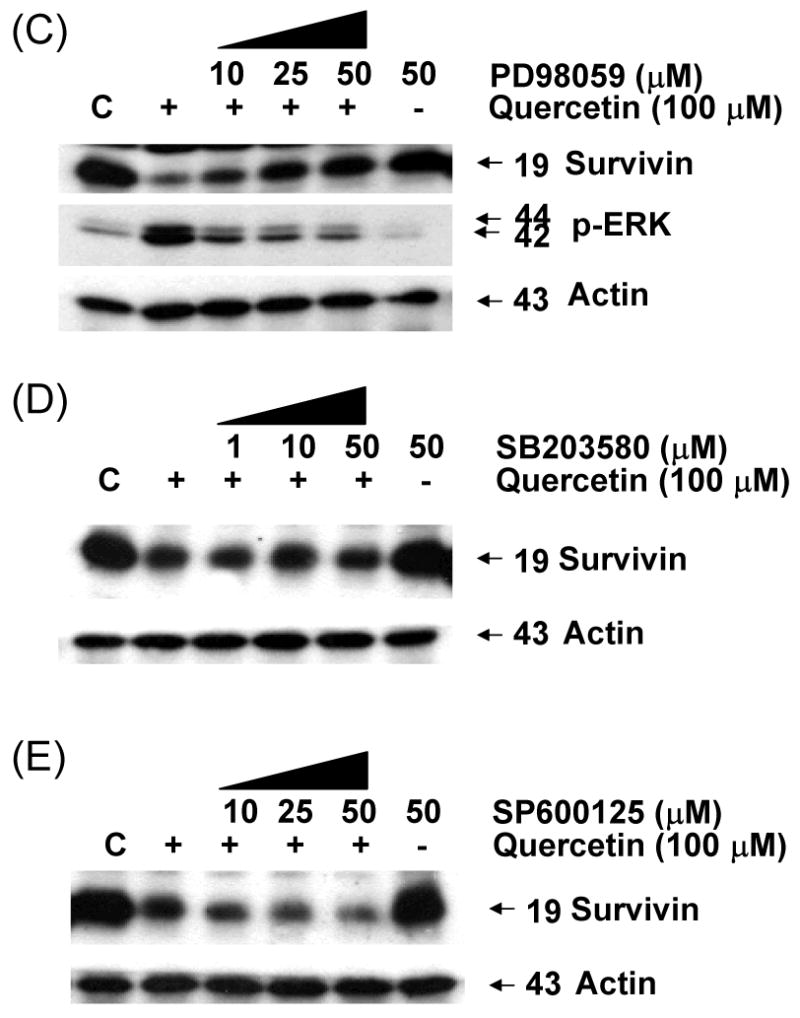

Quercetin affects the levels of phosphorylated mitogen-activated protein kinase. DU-145 cells were incubated with 100 μM of quercetin for indicated time (A) or with indicated amount of quercetin for 1 h (B) and the levels of phospho-ERK, -JNK, and –p38 were determined by western blotting with phosphor-specific antibodies. DU-145 cells were stimulated with quercetin (100 μM) in the presence or absence of several inhibitors: (C) PD98059 (a specific ERK inhibitor), (D) SB203580 (a specific p38 inhibitor), and (E) SP600125 (a specific JNK inhibitor) as indicated for 24 h and the levels of survivin were determined by western blotting. Protein expression levels of actin in cell lysates were used to confirm the amount of proteins loaded in each lane. (F) DU-145 cells were treated with quercetin (100 μM) and TRAIL (50 ng/ml) in the presence of indicated amount of PD98059 for 24 h. Cell survival was determined by the trypan blue exclusion assay. Error bars represent standard error from the mean (SEM) for three separate experiments.
3.7. Epigenetic regulation of histones by treatment with quercetin
Previous studies have shown that one of the main mechanisms involved in regulation of gene expression is the creation of an open (acetylated) or closed (deacetylated) chromatin structure which makes the promoter accessible or inaccessible to transcription factors, respectively [21]. To investigate the mechanism by which survivin expression is down-regulated by treatment with quercetin in prostate cancer cell, a possible epigenetic effect of quercetin on histone acetylation was analyzed. In DU-145 cells, histone H3, but not histone H4, was significantly deacetylated by treatment with 100 μM quercetin for 24 h (Fig. 7A). Quercetin induced H3 deacetylation in a dose dependent manner in DU-145 (Fig. 7B) and PC-3 cells (Fig. 7C). H3 deacetylation also occurred in a time-dependent manner and it was quite evident after 8 h treatment with 100 μM of quercetin in DU-145 cells (Fig. 7D). But H2A and H2B deacetylation was not observed (data not shown), and H4 deacetylation occurred only slightly in both cell lines. We used MAPK chemical inhibitors to investigate whether these inhibitors affect quercetin-induced survivin down-regulation and H3 deacetylation in DU-145 cells (Fig 7E). Quercetin induced H3 deacetylation, which was suppressed by MEK inhibitor (PD98059). Other inhibitors (SB203580, SP600125) did not affect H3 deacetylation induced by quercetin. Our results indicated that quercetin induced H3 deacetylation was significantly prevented in the presence of the MEK inhibitor in DU-145 cells. We next analyzed the activity of HDAC enzymes during treatment with quercetin to determine how quercetin induces H3 deacetylation. We measured HDAC enzyme activity by absorbance at 405 nm (Fig 7F). These results show that quercetin increased HDAC enzyme activity. Our data also indicate that 30% increase in HDAC activity significantly decreases acetylation of H3 during long-term incubation (8-24 h), but not short-term incubation (0.5-4 h) with 100 μM quercetin. Taken together, these data suggest that quercetin induces down-regulation of survivin expression through H3 deacetylation by enhancement of HDAC enzyme activity.
Fig. 7. Effect of quercetin on acetylation of histone acetylation and HDAC activity.
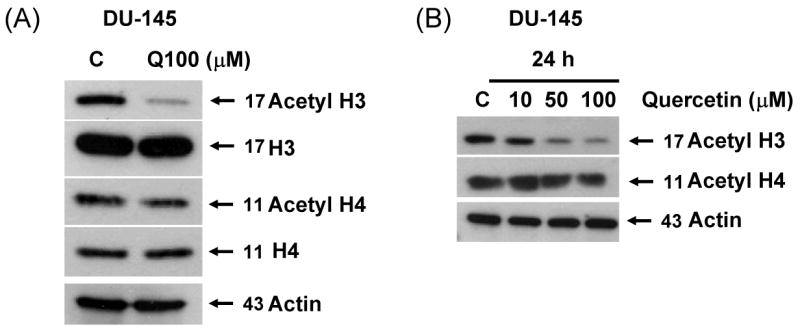
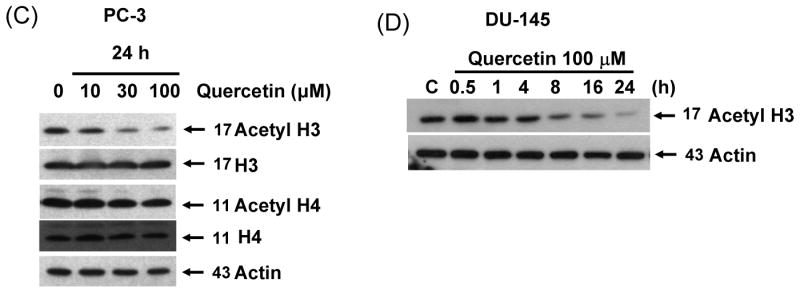
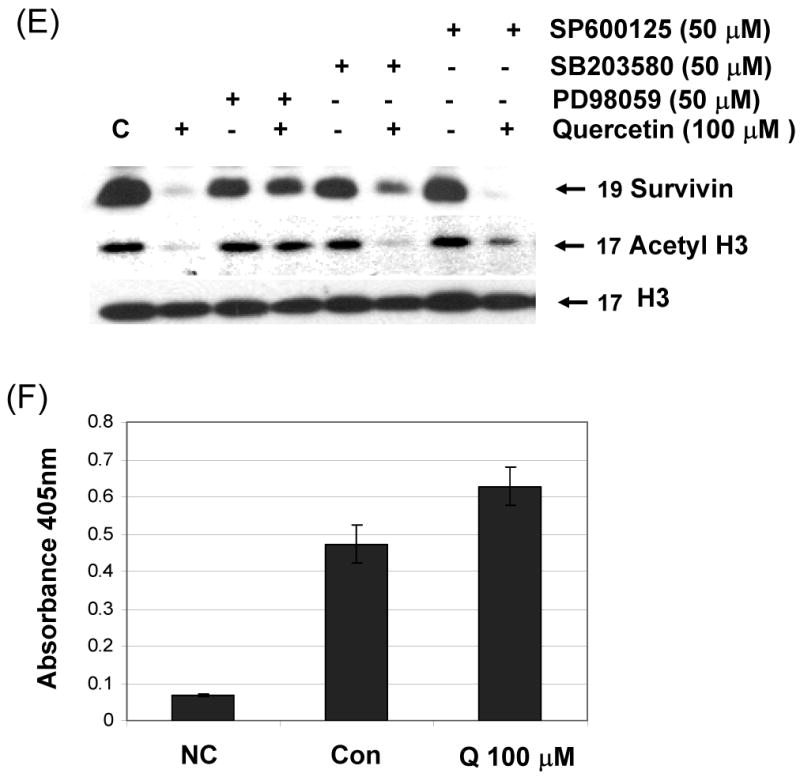
DU-145 cells were treated with 100 μM (A) or indicated amount (B) of quercetin for 24 h and total level and acetylation of histone H3 and H4 was analyzed. (C) PC-3 cells were treated with indicated amount of quercetin for 24 h and total level and acetylation of histone H3 and H4 was analyzed. (D) DU-145 cells were treated with 100 μM of quercetin for the indicated time and acetylation of histone H3 4 was analyzed. (E) DU-145 cells were treated with quercetin (100 μM) in the presence or absence of several inhibitors: PD98059 (a specific ERK inhibitor), SB203580 (a specific p38 inhibitor), and SP600125 (a specific JNK inhibitor) as indicated for 24 h and the levels of survivin and acetyl H3 were determined by western blotting. (F) HDAC enzyme activity was measured after 24 h exposure to 100 μM of quercetin. Representative results from three independent experiments carried out are shown. NC: negative control, Con: control.
3.8. Involvement of Sp1 in quercetin-induced survivin down-regulation
Survivin contains a zinc finger motif that shares a DNA-binding site with Sp1 [22]. To determine whether quercetin-reduced Sp1 expression plays a role in reduced survivin expression, we assessed the survivin expression levels in cells treated with quercetin. DU-145 cells were treated with 100 μM quercetin at the indicated times, and expression levels of survivin and Sp1 were determined by western blot analysis. As shown in Fig. 8A, after treatment with quercetin for 16 h, the intracellular level of Sp1 was gradually decreased. To examine the role of Sp1 in survivin expression, cells were transfected with the Sp1 expression vectors and treated with quercetin for 24 h. As expected, overexpression of Sp1 increased the expression of endogeneous survivin protein (Fig 8B), indicating that Sp1 expression positively recovered quercetin-mediated inhibition of survivin expression. To examine the functional influence of Sp1 on the survivin promoter, ChIP assays were performed with Sp1 specific antibody and PCR primers encompassing the seven putative Sp1-binding sequences present in the survivin promoter region (Fig. 8C). As shown in Figure 8C, Sp1 directly bound to survivin promoter region in the chromatin. Quercetin inhibited Sp1 binding activity. Taken together, these data suggest that transcription factor Sp1 plays an important role in survivin gene expression and quercetin down-regulates survivin gene expression by inhibiting Sp1 binding activity.
3.9. ERK mediates the phosphorylation of MSK1 by quercetin
We previously demonstrated that endogeneous survivin protein levels reduced by quercetin are severely affected in the presence of inhibitors of the ERK MAPK pathway. It is well known that mitogen- and stress-activated kinase 1 (MSK1), a kinase downstream of ERK, plays an important role in modification of histone H3 [23, 24]. We examined whether MSK1 is phosphorylated during treatment with quercetin. As shown in Fig. 9A, the kinase was phosphorylated rapidly, within 30 min, reaching maximal phosphorylation levels after 30 min of treatment. This phosphorylation declined thereafter, but remained considerably above basal levels during treatment with quercetin. Treatment with PD98059 resulted in significant decrease in quercetin-mediated phosphorylation of MSK1 (Fig. 9B). An inhibitor of MSK1, H-89, affected only the activity of the kinase but not phosphorylation levels of MSK1 and ERK (22; Figs. 9B and 9C). Nonetheless, both inhibitors of ERK and MSK1 significantly prevented quercetin-induced down-regulation of survivin (Fig. 9C). These results suggest that quercetin suppresses survivin expression by activating the ERK-MSK1 signal transduction pathway.
Fig. 9. Effect of quercetin on ERK-MSK1 signal pathway.
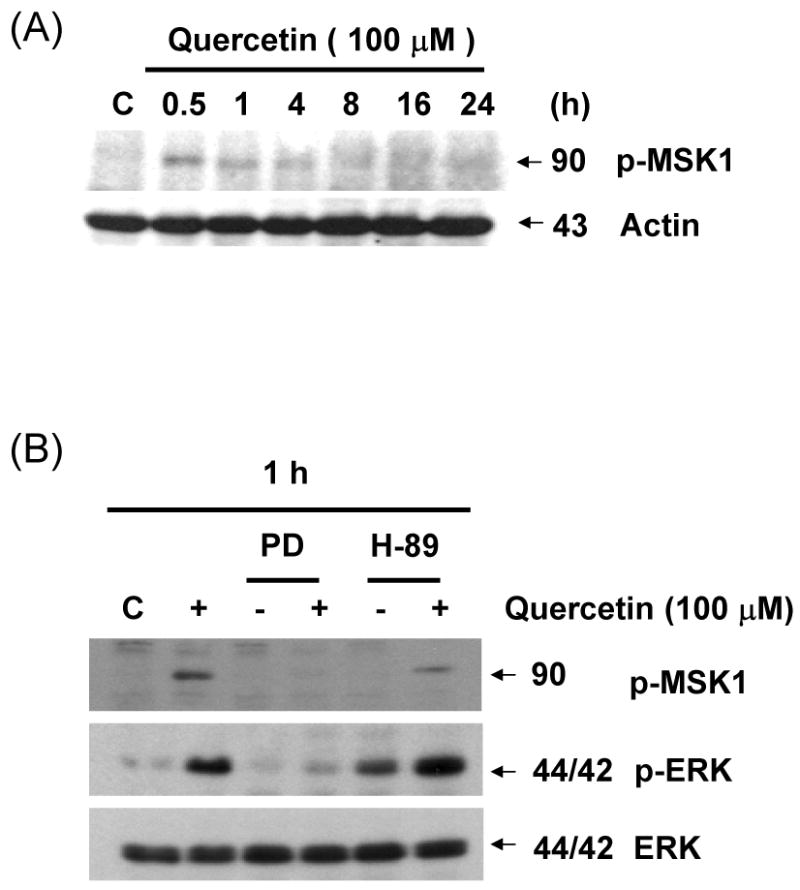
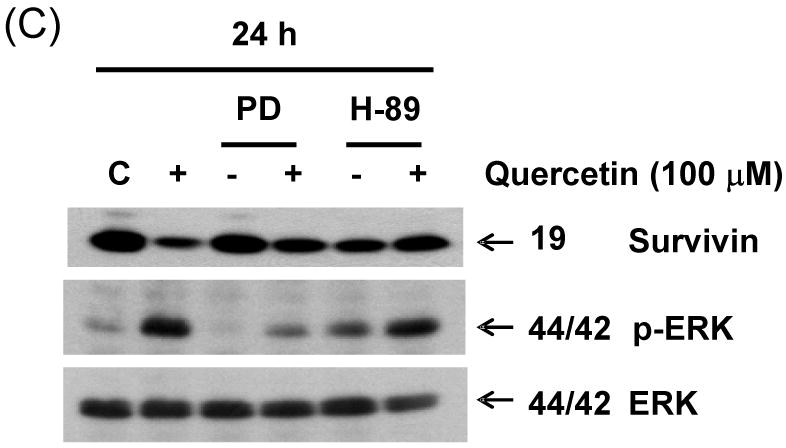
(A) DU-145 cells were incubated for the periods indicated in the presence or absence of quercetin. Intracellular levels of phospho-MSK1 were determined by western blot analysis. Cells were treated for 1 h (B) and 24 h (C) with 100 μM of quercetin in the absence or presence of the following inhibitors: PD98059 (50 μM), H-89 (20 μM). Immunoblotting was performed using an antibody against phospho-MSK1, phospho-ERK, survivin, or ERK.
4. Discussion
In search for strategies to overcome resistance of tumor cells, we tested the antitumor activity of quercetin on human tumor cell lines. Here, we report that quercetin is a potent sensitizer for TRAIL-induced apoptosis in human prostate adenocarcinoma DU-145 and PC-3 cell line through reduction of survivin expression in the ERK-MSK1 signal pathway. Similar enhancement of TRAIL-induced apoptosis by down regulation of survivin has been reported for resveratrol, another naturally occurring chemotherapeutic agent [25]. It is possible that the cell cycle-specific regulation of survivin in tumor cells contributes to the regulation of apoptosis during cell proliferation. However, our data ruled out this possibility. Survivin, a member of the inhibitor of apoptosis gene family, is expressed in a cell cycle-dependent manner, increasing in the G2/M phase of the cell cycle followed by a rapid decline in the G1 phase [26, 27]. Recent studies have revealed that quercetin inhibits cell proliferation by causing cell cycle arrest in G2/M phase [28, 29]. Although quercetin causes cells to arrest in G2/M phase, expression of survivin is down-regulated by treatment with quercetin (Fig. 3B). These results suggest that quercetin-enhanced TRAIL cytotoxicity is not related to quercetin-induced cell cycle arrest.
Although there are reports on the interaction between survivin and caspases 3, 7, and 9, others have failed to demonstrate a direct effect on these proteases [30, 31]. One of the important features for TRAIL treatment to be effective is that the cancer cells must be sensitive to the effects of TRAIL to prevent emerging TRAIL resistance. This may be accomplished by the administration of quercetin. The molecular mechanism of sensitization for TRAIL induced apoptosis by quercetin is based on decreased survivin expression.
We observed that quercetin-enhanced TRAIL cytotoxicity is mediated through down-regulating survivin expression. Interestingly, quercetin treatment did not affect the levels of other IAPs (XIAP, cIAP1, cIAP2), emphasizing the specific effect of quercetin on survivin expression. Although the role of survivin in apoptosis inhibition has been well established in various systems [32], the question as to whether survivin functions in cytoprotection or in the regulation of cell death has been controversially discussed and may vary between various cell types.
Sp1 is known to play a role in the regulation of genes lacking a functional TATA box. The presence of numerous Sp1 binding sites in the promoter region of survivin suggested that Sp1 might be involved in the regulation of survivin activity. To test whether these Sp1 sites have any functional role in regulating survivin gene expression, Sp1 sites were simultaneously deleted (Figure 5B); the results suggested that Sp1 binding sites (at position -230 to -123) are important for the survivin promoter to attain promoter activity (Fig. 5C).
Moreover, quercetin induced phosphorylation of ERK, p38, and JNK MAP kinase in DU-145 cells. The specific ERK inhibitor, PD98059, was effective in protecting DU-145 cells from quercetin-mediated down-regulation of survivin expression and deacetylation of histone H3, but p38 and JNK inhibitors did not prevent these events. These results also show that quercetin regulated down-regulation of survivin by ERK activation through the deacetylation of histone H3. The acetylation of core histone tails is a dynamic process maintained by histone acetyltransferases (HAT) and histone deacetylases (HDAC). HDACs form complexes with transcriptional corepressors and are believed to repress transcription by removing the acetyl groups from the N-terminal tails of the core histones of chromatin [33]. Our data show that quercetin activated histone deacetylase enzyme activity, which reduced acetylation of histone H3.
MSKs (mitogen- and stress- activated kinase 1 and 2) are direct substrates of the MAPK family members ERK and p38, and the phosphorylation of MSKs by these kinases is required for MSK activity [34]. MSKs are constitutively localized to the nucleus suggesting that they may play a role in nuclear function [34]. It has been reported that MSKs are required for the mitogen or stress induced phosphorylation of the transcription factors CREB and ATF1, and the chromatin proteins histone H3 [35-37]. Our studies demonstrate that PD 98059, an inhibitor of MEK and consequently of ERK, largely suppresses the activation of endogeneous p-MSK1 by quercetin, while H-89, an inhibitor of MSK, does not suppresses this activation (Figure 9B). It has been shown that the inhibitor of MSK1, H-89, affects only the activity of the kinase but not its phosphorylation levels [30]. Consistent with these findings, MSK1 can be activated by ERK in vitro (Figure 9). Our data clearly demonstrate that MSK1 is activated by signals that trigger activation of the ERK cascade pathway. These results imply that MSK1 plays a role in integrating the effects of extracelluar signals.
The ability of quercetin plus TRAIL as a combined agent to induce apoptosis was relatively low compared to its strong antiproliferative effect at low concentrations of TRAIL, indicating that the cytostatic activity of quercetin plus TRAIL is stronger than its cytotoxic activity. Importantly, the combination of quercetin as “sensitizer” together with TRAIL as “inducer” combined to trigger apoptosis. Thus, the potential of TRAIL for anticancer therapy may largely reside in its ability to sensitize tumor cells to death induction by its antiproliferative effect, that is, to render tumor cells more susceptible for death induction or even to overcome resistance. Clinically, resistance to apoptosis is a major cause of primary or acquired nonresponsiveness of cancer cells, leading to treatment failure. Thus, our results may have therapeutic implications in this aspect, which identifying the chemopreventive compound quercetin, previously considered to act predominantly as an antioxidant, to be a novel therapeutic to target survivin expression in cancer. This possibility supported by several studies that survivin targeting is a viable anticancer approach to potently trigger apoptosis in vitro and also in vivo [31].
The present study demonstrates that simultaneous administration of TRAIL and subtoxic doses of quercetin strongly potentiates the triggering of an apoptotic cascade in DU-145 and PC-3 cells. The detailed analysis of the mechanisms reveals that the increase in cell death is mediated by enhanced activation of the caspase cascade concomitant with down-regulation of the anti-apoptotic protein survivin in an ERK-MSK1 dependent pathway.
Acknowledgments
This work was supported by the following grants: NCI grant funds (CA95191, CA96989 and CA121395), DOD prostate program funds (PC020530 and PC040833), Susan G. Komen Breast Cancer Foundation fund (BCTR60306).
Abbreviations
- DTT
dithiothreitol
- FLICE
Fas-associated death domain-like interleukin-1 β-converting enzyme
- FLIP
FLICE inhibitory protein
- IAP
inhibitor of apoptosis
- PAGE
polyacrylamide gel electrophoresis
- PARP
poly (ADP-ribose) polymerase
- PBS
phosphate-buffered saline
- PDK-1
phosphoinositide-dependent kinase-1
- PI3K
phosphatidylinositol 3-kinase
- PP1
protein phosphatase 1
- SDS
sodium dodecyl sulfate
- TNF
tumor necrosis factor
- TRAIL
tumor necrosis factor-related apoptosis-inducing ligand
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kelley SK, Ashkenazi A. Targeting death receptors in cancer with Apo2L/TRAIL. Curr Opin Pharmacol. 2004;4:333–9. doi: 10.1016/j.coph.2004.02.006. [DOI] [PubMed] [Google Scholar]
- 2.Thomas LR, Henson A, Reed JC, Salsbury FR, Thorburn A. Direct binding of Fas-associated death domain (FADD) to the tumor necrosis factor-related apoptosis-inducing ligand receptor DR5 is regulated by the death effector domain of FADD. J Biol Chem. 2004 Jul 30;279:32780–5. doi: 10.1074/jbc.M401680200. [DOI] [PubMed] [Google Scholar]
- 3.Rowinsky EK. Targeted induction of apoptosis in cancer management: the emerging role of tumor necrosis factor-related apoptosis-inducing ligand receptor activating agents. J Clin Oncol. 2005;23:9394–407. doi: 10.1200/JCO.2005.02.2889. [DOI] [PubMed] [Google Scholar]
- 4.Block G, Patterson B, Subar A. Fruit, vegetables, and cancer prevention: a review of the epidemiological evidence. Nutr Cancer. 1992;18:1–29. doi: 10.1080/01635589209514201. [DOI] [PubMed] [Google Scholar]
- 5.Agullo G, Gamet-Payrastre L, Manenti S, Viala C, Remesy C, Chap H, Payrastre B. Relationship between flavonoid structure and inhibition of phosphatidylinositol 3-kinase: a comparison with tyrosine kinase and protein kinase C inhibition. Biochem Pharmacol. 1997;53:1649–57. doi: 10.1016/s0006-2952(97)82453-7. [DOI] [PubMed] [Google Scholar]
- 6.Hagiwara M, Inoue S, Tanaka T, Nunoki K, Ito M, Hidaka H. Differential effects of flavonoids as inhibitors of tyrosine protein kinases and serine/threonine protein kinases. Biochem Pharmacol. 1988;37:2987–92. doi: 10.1016/0006-2952(88)90286-9. [DOI] [PubMed] [Google Scholar]
- 7.Aligiannis N, Mitaku S, Mitrocotsa D, Leclerc S. Flavonoids as cycline-dependent kinase inhibitors: inhibition of cdc 25 phosphatase activity by flavonoids belonging to the quercetin and kaempferol series. Planta Med. 2001;67:468–70. doi: 10.1055/s-2001-15807. [DOI] [PubMed] [Google Scholar]
- 8.Gamet-Payrastre L, Manenti S, Gratacap MP, Tulliez J, Chap H, Payrastre B. Flavonoids and the inhibition of PKC and PI 3-kinase. Gen Pharmacol. 1999;32:279–86. doi: 10.1016/s0306-3623(98)00220-1. [DOI] [PubMed] [Google Scholar]
- 9.Kim YH, Lee YJ. TRAIL apoptosis is enhanced by quercetin through Akt dephosphorylation. J Cell Biochem. 2007;100:998–1009. doi: 10.1002/jcb.21098. [DOI] [PubMed] [Google Scholar]
- 10.Constantinou A, Mehta R, Runyan C, Rao K, Vaughan A, Moon R. Flavonoids as DNA topoisomerase antagonists and poisons: structure-activity relationships. J Nat Prod. 1995;58:217–25. doi: 10.1021/np50116a009. [DOI] [PubMed] [Google Scholar]
- 11.Yoshizumi M, Tsuchiya K, Kirima K, Kyaw M, Suzaki Y, Tamaki T. Quercetin inhibits Shc- and phosphatidylinositol 3-kinase-mediated c-Jun N-terminal kinase activation by angiotensin II in cultured rat aortic smooth muscle cells. Mol Pharmacol. 2001;60:656–65. [PubMed] [Google Scholar]
- 12.Avila MA, Velasco JA, Cansado J, Notario V. Quercetin mediates the down-regulation of mutant p53 in the human breast cancer cell line MDA-MB468. Cancer Res. 1994;54:2424–8. [PubMed] [Google Scholar]
- 13.Xing N, Chen Y, Mitchell SH, Young CY. Quercetin inhibits the expression and function of the androgen receptor in LNCaP prostate cancer cells. Carcinogenesis. 2001;22:409–14. doi: 10.1093/carcin/22.3.409. [DOI] [PubMed] [Google Scholar]
- 14.Wei YQ, Zhao X, Kariya Y, Fukata H, Teshigawara K, Uchida A. Induction of apoptosis by quercetin: involvement of heat shock protein. Cancer Res. 1994;54:4952–7. [PubMed] [Google Scholar]
- 15.Ong CS, Tran E, Nguyen TT, Ong CK, Lee SK, Lee JJ, Ng CP, Leong C, Huynh H. Quercetin-induced growth inhibition and cell death in nasopharyngeal carcinoma cells are associated with increase in Bad and hypophosphorylated retinoblastoma expressions. Oncol Rep. 2004;11:727–33. [PubMed] [Google Scholar]
- 16.Gupta K, Panda D. Perturbation of microtubule polymerization by quercetin through tubulin binding: a novel mechanism of its antiproliferative activity. Biochemistry. 2002;41:13029–38. doi: 10.1021/bi025952r. [DOI] [PubMed] [Google Scholar]
- 17.Wang IK, Lin-Shiau SY, Lin JK. Induction of apoptosis by apigenin and related flavonoids through cytochrome c release and activation of caspase-9 and caspase-3 in leukaemia HL-60 cells. Eur J Cancer. 1999;35:1517–25. [PubMed] [Google Scholar]
- 18.Burow ME, Weldon CB, Tang Y, Navar GL, Krajewski S, Reed JC, Hammond TG, Clejan S, Beckman BS. Differences in susceptibility to tumor necrosis factor alpha-induced apoptosis among MCF-7 breast cancer cell variants. Cancer Res. 1998;58:4940–6. [PubMed] [Google Scholar]
- 19.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–5. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 20.Yuan H, Gong A, Young CY. Involvement of transcription factor Sp1 in quercetin-mediated inhibitory effect on the androgen receptor in human prostate cancer cells. Carcinogenesis. 2005;26:793–801. doi: 10.1093/carcin/bgi021. [DOI] [PubMed] [Google Scholar]
- 21.Jones PA, Baylin SB. The fundamental role of epigenetic events in cancer. Nat Rev Genet. 2002;3:415–28. doi: 10.1038/nrg816. [DOI] [PubMed] [Google Scholar]
- 22.Li F, Altieri DC. Transcriptional analysis of human survivin gene expression. Biochem J. 1999;344:305–11. doi: 10.1042/0264-6021:3440305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brami-Cherrier K, Lavaur J, Pages C, Arthur JS, Caboche J. Glutamate induces histone H3 phosphorylation but not acetylation in striatal neurons: role of mitogen- and stress-activated kinase-1. J Neurochem. 2007;101:697–708. doi: 10.1111/j.1471-4159.2006.04352.x. [DOI] [PubMed] [Google Scholar]
- 24.Lee ER, McCool KW, Murdoch FE, Fritsch MK. Dynamic changes in histone H3 phosphoacetylation during early embryonic stem cell differentiation are directly mediated by mitogen- and stress-activated protein kinase 1 via activation of MAPK pathways. J Biol Chem. 2006;281:21162–72. doi: 10.1074/jbc.M602734200. [DOI] [PubMed] [Google Scholar]
- 25.Fulda S, Debatin KM. Sensitization for tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis by the chemopreventive agent resveratrol. Cancer Res. 2004;64:337–46. doi: 10.1158/0008-5472.can-03-1656. [DOI] [PubMed] [Google Scholar]
- 26.Chandele A, Prasad V, Jagtap JC, Shukla R, Shastry PR. Upregulation of survivin in G2/M cells and inhibition of caspase 9 activity enhances resistance in staurosporine-induced apoptosis. Neoplasia. 2004;6:29–40. doi: 10.1016/s1476-5586(04)80051-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Torres VA, Tapia JC, Rodríguez DA, Párraga M, Lisboa P, Montoya M, Leyton L, Quest AF. Caveolin-1 controls cell proliferation and cell death by suppressing expression of the inhibitor of apoptosis protein survivin. J Cell Sci. 2006;119:1812–23. doi: 10.1242/jcs.02894. [DOI] [PubMed] [Google Scholar]
- 28.Yang JH, Hsia TC, Kuo HM, Chao PD, Chou CC, Wei YH, Chung JG. Inhibition of lung cancer cell growth by quercetin glucuronides via G2/M arrest and induction of apoptosis. Drug Metab Dispos. 2006;34:296–304. doi: 10.1124/dmd.105.005280. [DOI] [PubMed] [Google Scholar]
- 29.Lee TJ, Kim OH, Kim YH, Lim JH, Kim S, Park JW, Kwon TK. Quercetin arrests G2/M phase and induces caspase-dependent cell death in U937 cells. Cancer Lett. 2006;240:234–42. doi: 10.1016/j.canlet.2005.09.013. [DOI] [PubMed] [Google Scholar]
- 30.Thomson S, Clayton AL, Hazzalin CA, Rose S, Barratt MJ, Mahadevan LC. The nucleosomal response associated with immediate-early gene induction is mediated via alternative MAP kinase cascades: MSK1 as a potential histone H3/HMG-14 kinase. EMBO J. 1999;18:4779–93. doi: 10.1093/emboj/18.17.4779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Altieri DC. Validating survivin as a cancer therapeutic target. Nat Rev Cancer. 2003;3:46–54. doi: 10.1038/nrc968. [DOI] [PubMed] [Google Scholar]
- 32.Zaffaroni N, Daidone MG. Survivin expression and resistance to anticancer treatments: perspectives for new therapeutic interventions. Drug Resist Updat. 2002;5:65–72. doi: 10.1016/s1368-7646(02)00049-3. [DOI] [PubMed] [Google Scholar]
- 33.Davie JR, Spencer VA. Control of histone modifications. J Cell Biochem. 1999;3233:141–8. doi: 10.1002/(sici)1097-4644(1999)75:32+<141::aid-jcb17>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 34.Deak M, Clifton AD, Lucocq LM, Alessi DR. Mitogen- and stress-activated protein kinase-1 (MSK1) is directly activated by MAPK and SAPK2/p38, and may mediate activation of CREB. EMBO J. 1998;17:4426–41. doi: 10.1093/emboj/17.15.4426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Arthur JS, Cohen P. MSK1 is required for CREB phosphorylation in response to mitogens in mouse embryonic stem cells. FEBS Lett. 2000;482:44–8. doi: 10.1016/s0014-5793(00)02031-7. [DOI] [PubMed] [Google Scholar]
- 36.Soloaga A, Thomson S, Wiggin GR, Rampersaud N, Dyson MH, Hazzalin CA, Mahadevan LC, Arthur JS. MSK2 and MSK1 mediate the mitogen- and stress-induced phosphorylation of histone H3 and HMG-14. EMBO J. 2003;22:2788–97. doi: 10.1093/emboj/cdg273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wiggin GR, Soloaga A, Foster JM, Murray-Tait V, Cohen P, Arthur JS. MSK1 and MSK2 are required for the mitogen- and stress-induced phosphorylation of CREB and ATF1 in fibroblasts. Mol Cell Biol. 2002;22:2871–81. doi: 10.1128/MCB.22.8.2871-2881.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]