

Abstract
The oxidative treatment of vinyl tris(trimethylsilyl)silanes with hydrogen peroxide in aqueous sodium hydroxide in tetrahydrofuran generates reactive silanol or siloxane species that undergo Pd-catalyzed cross-couplings with aryl, heterocyclic and alkenyl halides in the presence of Pd(PPh3)4 and tetrabutylammonium fluoride. Hydrogen peroxide and base are necessary for the coupling to occur while activation of the silanes with fluoride is not required. The conjugated and unconjugated tris(trimethylsilyl)silanes serve as good cross-coupling substrates. The (E)-silanes undergo coupling with retention of stereochemistry while coupling of (Z)-silanes occurred with lower stereoselectivity to produce an E/Z mixture of products.
Keywords: Cross-couplings, Organosilanes, Pd-catalyzed reactions, Tris(trimethylsilyl)silanes
Introduction
Pd-catalyzed cross-coupling reactions are a powerful method for the formation of carbon-carbon bonds under conditions that are compatible with a broad range of functional groups.1 Among these methods, couplings between organometallics derived from Group 14 metals and various electrophiles are well developed. Thus, the Stille reaction has been widely applied in modern synthetic organic chemistry.2 Furthermore, the Hiyama coupling has received increased attention due to the lower toxicity of organosilicon compounds.3 Procedures for coupling with organogermanes have also been reported.4,5
Hiyama and Hatanaka reported on the positive effect that fluoride ion imparts on the nucleophilicity of certain substituents bonded to trialkylsilanes and attributed this enhancement to the formation of pentacoordinated silicon species.6 This strategy has opened up the door for the wide application of structurally diverse silanes including: (i) heteroatom functionalized precursors such as halosilanes, oxysilanes, silanols and polysiloxanes and (ii) all-carbon substituted silicon species such as phenyl-, benzyl-, 2-thienyl-, 2-pyridyldimethylsilanes.3
Denmark et al. developed Pd-catalyzed coupling reactions employing siletanes (alkenylsilacyclobutanes) in the presence of TBAF and Pd(0) such as Pd2(dba)3.7a The siletanes were found to be converted in situ into reactive alkenyl(propyl)silanol and disiloxane species via the ring opening.7b When the alkenylsilanols were synthesized independently, they were also found to undergo coupling.8 Hiyama, Mori and coworkers independently found that silver(I) oxide is an excellent activator for the coupling of alkenylsilanols with aryl iodides.9 Generally, the organosilanols can couple by two mechanistically distinct processes. One is operative under basic conditions and requires only formation of the silanolates while the second pathway involves the use of a fluoride activator that can form an active fluorosiliconate.10
Recent developments in Si-cross-coupling include the application of triallyl(aryl)silanes for the synthesis of biaryls,11a a palladium-phosphinous acid-catalyzed and NaOH-promoted coupling of arylsiloxanes in water,11b application of alkenyl- and aryl[2-(hydroxymethyl)phenyl]dimethylsilanes as a reusable silicon based precursor for the fluoride-free couplings via intramolecular activation,11c vinylation of aryl bromides using an inexpensive vinylpolysiloxane,11d application of (2-pyridyl)allyldimethylsilanes as novel pyridyl transfer,11e and fluoride-free methodologies.11f,g Herein, we report application of various tris(trimethylsilyl)silanes as “masked” substrates for the cross-coupling reactions with aryl halides and alkenyl bromides which occurred under oxidative conditions in the presence of aqueous sodium hydroxide with or without tetrabutylammonium fluoride.
Results and Discussion
The (E)-vinyl tris(trimethylsilyl)silanes (TTMS-silanes) 2a,c-f were prepared by stereoselective radical-mediated silyldesulfonylations5a of the corresponding (E)-vinyl sulfones 1 with (TMS)3SiH (Scheme 1). The cyclohexylidene silane 2g was similarly prepared. The (Z)-vinyl silanes 4a-d were synthesized in 80-92% yield by the radical hydrosilylation12 of the corresponding alkynes 3 with (TMS)3SiH. Attempted hydrosilylation of 1-alkynes 3a-c with (TMS)3SiH in the presence of Rh(COD)2BF4/PPh3/NaI or RhCl(PPh3)3/NaI catalyst systems13 produced E isomers 2a-c in high yields but complete control of stereochemistry was generally difficult since Z isomers 4a-c were also formed (~5-20%). Thus, hydrosilylation of 3b gave 2b/4b (E/Z, ~9:1) mixture, which was purified to afford 2b (82%). Isomerization (20 h, 60 °C) of the (Z)-silanes 4a and 4c with 0.5 equivalent of (TMS)3SiH in the presence of Rh catalyst13 also afforded the (E)-silanes 2a (78%) and 2c (89%). Prolonged heating (54 h) of 4b in the presence of (TMS)3SiH/RhCl(PPh3)3/NaI effected quantitative conversion of 4b into 2b.
Scheme 1. Stereoselective synthesis of (E)- and (Z)-vinyl tris(trimethylsilyl)silanes (2 and 4)a.
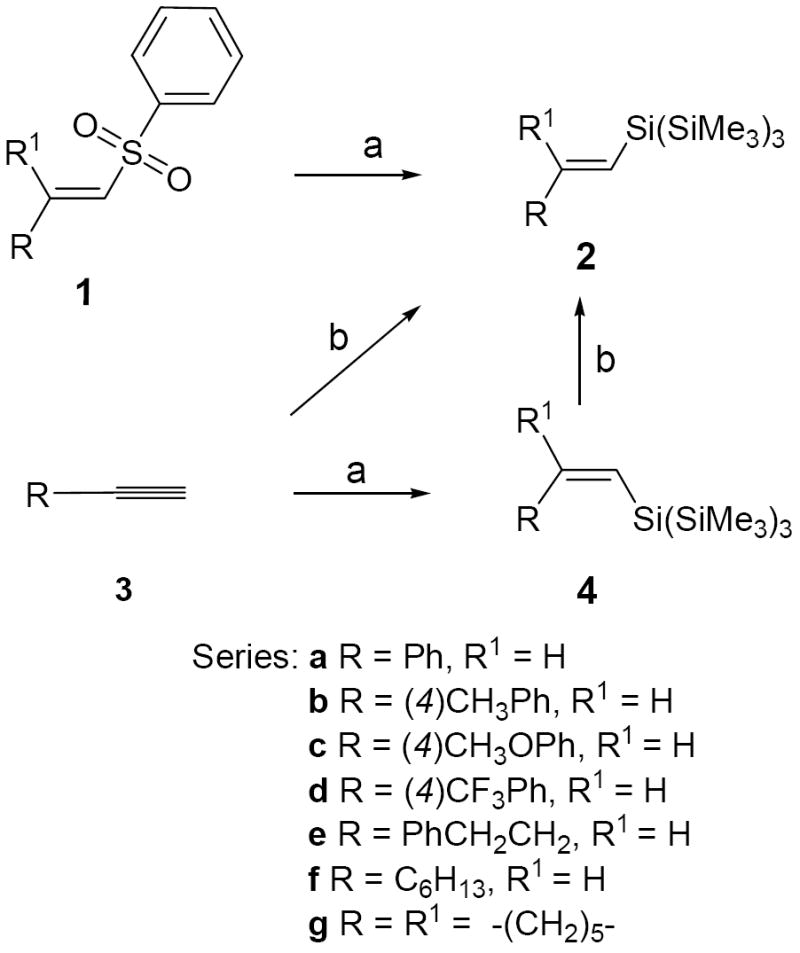
a Reagents and conditions: (a) (Me3Si)3SiH/AIBN/toluene or benzene (85 °C; oil-bath); (b) (Me3Si)3SiH/RhCl(PPh3)3 or Rh(COD)2BF4/PPh3/NaI/neat/Δ.
We started by examining different coupling conditions. Thus, attempted coupling of (Z)-vinyl silane 4b with iodobenzene under typical coupling conditions employed for organosilanes6 [e.g., TBAF/Pd(0)/THF] yielded the desired product 5b in less than 5% (Table 1, entry 1). However, we found that fluoride-free treatment of 4b with H2O2/NaOH5a in aqueous THF for 15 min. followed by addition of iodobenzene and Pd(PPh3)4 provided (E/Z)-5b in 61% yield (entry 2). When NaOH was replaced with KOSiMe3 base14 the yield did not improve, although the reaction mixture became homogeneous with the combination of KOSiMe3/H2O2 (entry 3). TBAF was found to be an excellent activator under aqueous (H2O2/NaOH) and “anhydrous” (H2O2/KOSiMe3) oxidative conditions furnishing coupling product 5b in 90% and 81% yield, respectively (entries 4 and 5). Both peroxide and base were necessary since only 18% conversion to 5b was obtained without NaOH or KOSiMe3 (entry 6) and almost no product was formed without peroxide (entry 7). Other fluoride sources such as NaF and CsF did not show improvement over TBAF (entries 8 and 9). Anhydrous oxidative conditions [t-BuOOH/KH/Pd(0)] with or without TBAF did not give satisfactory results (entry 10), although such conditions worked well for the coupling of analogous TTMS-germanes.5b These results indicate that the peroxide and base are necessary for the coupling of TTMS-silanes to occur while fluoride facilitates this conversion (vide infra Table 2 and 3 vs Table 4).
Table 1.
Effect of Reaction Parameters on the Cross-Coupling of TTMS-Silanes
| entry | peroxide | base | fluoride | yielda (%) | E/Zb |
|---|---|---|---|---|---|
| 1 | none | none | TBAF | <5c | 5/95 |
| 2 | H2O2 | NaOH | none | 61d,e,f | 15/85 |
| 3 | H2O2 | KOSiMe3 | none | 60 | 25/75 |
| 4 | H2O2 | NaOH | TBAF | 90 | 3/97 |
| 5 | H2O2 | KOSiMe3 | TBAF | 81 | 2/98 |
| 6 | H2O2 | none | TBAF | 18c | 67/33 |
| 7 | none | KOSiMe3g | TBAF | <2c | n/a |
| 8 | H2O2 | NaOH | NaF | 60 | 40/60 |
| 9 | H2O2 | NaOH | CsF | 12c | 60/40 |
| 10 | t-BuOOH | KH | noneh | 15c | 75/25 |
Isolated yields (combined for both isomers of 5b). Couplings were performed on 0.1 mmol scale of silane (0.03 mM).
Determined by GC-MS [with internal standard of (E)- and (Z)- stilbenes] and/or 1H NMR of the crude reaction mixture.
Based on GC-MS.
Pd2(dba)3 also gave 5b (52%; E/Z, 25:75).
Attempted couplings with H2O2 (without NaOH) or with NaOH (without H2O2) failed to give 5b.
Coupling with bromobenzene instead of iodobenzene gave 5b (40%; E/Z, 20:80).
Reaction with NaOH instead of KOSiMe3 was also unsuccessful.
Coupling in the presence of TBAF gavec 5b in ~2% yield.
Table 2.
The Coupling of Vinyl (Z)-TTMS-Silanes
| entry | silane | R1X | product | yielda (%) | E/Zb |
|---|---|---|---|---|---|
| 1 | 4a | PhBr | 5a | 82 | 40/60 |
| 2 | 4a | 2-iodopyridine | 9a | 60 | 2/98 |
| 3 | 4b | PhI | 5b | 90 | 3/97 |
| 4 | 4b | PhBr | 5b | 86 | 30/70 |
| 5 | 4b | PhCl | 5b | <5c | n/a |
| 6 | 4b | PhOTf | 5b | <5c | n/a |
| 7 | 4b | PhCH=CHBrd | 6b | 72 | 44/56e |
| 8 | 4b | (CH3)2C=CHBr | 7b | 87 | 4/96 |
| 9 | 4b | iodothiophenef | 8b | 75 | 15/85 |
| 10 | 4b | (4)BuPhI | 10b | 61 | 24/76 |
| 11 | 4b | 1-iodonaphthaleneg | 11b | 73 | 15/85 |
| 12 | 4c | PhBr | 5c | 97 | 9/91 |
| 13 | 4d | PhBr | 5d | 70 | 55/45 |
Isolated yields (combined for the E/Z isomers). Couplings were performed on 0.1-1.0 mmol scale of silanes (0.03 mM). Pd(PPh3)4 (10% mol).
Determined by GC-MS and/or 1H NMR of the crude reaction mixture.
GC-MS.
E/Z = 88:12.
1E ,3E/1Z ,3E.
2-Iodo- 5-methylthiophene.
Coupling with 1-bromonaphthalene gave 11b (51%, E/Z = 27:73).
Table 3.
The Coupling of Vinyl (E)-TTMS-Silanes

| entry | silane | R2X | producta (E) | yieldb (%) |
|---|---|---|---|---|
| 1 | 2a | PhI | 5a | 83 |
| 2 | 2a | PhBr | 5a | 67 |
| 3 | 2a | (4)CH3OPhI | 5c | 79 |
| 4 | 2a | (4)CF3PhI | 5d | 90 |
| 5 | 2a | (CH3)2C=CHBr | 7a | 85 |
| 6 | 2a | iodothiophenec | 8a | 58 |
| 7 | 2a | 2-iodopyridine | 9a | 70 |
| 8 | 2b | PhI | 5b | 72 |
| 9 | 2b | iodothiophenec | 8b | 66 |
| 10 | 2b | (4)BuPhI | 10b | 59 |
| 11 | 2b | 1-bromonaphthalene | 11b | 48 |
| 12 | 2b | 1-iodonaphthalene | 11b | 70 |
| 13 | 2c | PhBr | 5c | 63 |
| 14 | 2c | PhCH=CHBrd | 6ce | 60 |
| 15 | 2d | PhBr | 5d | 86 |
| 16 | 2e | PhI | 5e | 85 |
| 17 | 2e | PhBr | 5e | 72 |
| 18 | 2f | PhI | 5f | 70 |
| 19 | 2f | PhBr | 5f | 78 |
| 20 | 2g | PhI | 5g | 84 |
| 21 | 2g | PhBr | 5g | 65 |
| 22 | 2g | (CH3)2C=CHBr | 7g | 50 |
| 23 | 2g | iodothiophenec | 8g | 83 |
Only E-isomers were detected (1H NMR, GC-MS) except for the g series where stereochemistry is not relevant. Couplings were performed on 0.1-0.5 mmol scale of silanes (0.03 mM). Pd(PPh3)4 (10% mol).
Isolated yields.
2-Iodo-5-methylthiophene.
E/Z = 88:12.
1E/3E.
Table 4.
Fluoride-Free Coupling of the Vinyl TTMS-Silanes
| entry | silane | R1X | product | Yield a (%) | E/Zbb |
|---|---|---|---|---|---|
| 1 | 2a | PhI | 5a | 75c | 100/0 |
| 2 | 2a | PhBr | 5a | 50 | 100/0 |
| 3 | 2b | iodothiophened | 8b | 52 | 100/0 |
| 4 | 2b | 1-iodonaphthalene | 11b | 46 | 100/0 |
| 5 | 2c | PhCH=CHBre | 6c | 45 | 100/0f |
| 6 | 2e | PhI | 5e | 56 | 100/0 |
| 7 | 2g | PhI | 5g | 65 | n/a |
| 8 | 2g | (CH3)2C=CHBr | 7g | 42 | n/a |
| 9 | 4a | PhBr | 5a | 55 | 25/75 |
| 10 | 4b | PhI | 5b | 61g | 15/85 |
| 11 | 4b | PhBr | 5b | 40h | 20/80 |
| 12 | 4b | 1-bromonaphthalene | 11b | 30 | 17/83 |
Isolated yields. Couplings were performed on 0.1 mmol scale of silanes (0.03 mM). Pd(PPh3)4 (10% mol).
Determined by 1H NMR and/or GC-MS of the crude reaction mixture.
With KOSiMe3 instead of NaOH yield was 60% (E/Z, 100:0).
2-Iodo-5-methylthiophene.
E/Z, 88:12.
Only E,E isomer was observed.
With KOSiMe3 instead of NaOH yield was 60% (E/Z, 25:75).
With KOSiMe3 instead of NaOH yield was 48% (E/Z, 10:90).
The protocols established above (Table 1, entries 2-5) have proven to be general for the coupling of a range of alkenyl, aryl and heterocyclic iodides and bromides with an array of vinyl TTMS-silanes. Thus, treatment of the conjugated silane (Z)-4a with H2O2/H2O (30%, 3 equiv.) and NaOH (3 equiv.)/H2O in THF followed by addition of bromobenzene, Pd(PPh3)4 and TBAF gave stilbene 5a (82%, Table 2, entry 1). Analogously, (Z)-4b coupled with iodobenzene and bromobenzene to give p-methylstilbene 5b in 90% and 86% yield, respectively (entries 3 and 4). Less reactive electrophiles such as chlorobenzene and aryl triflate15 failed to give the desired coupling products (entries 5 and 6). The substituent on the phenyl ring in (Z)-silanes 4a-d (p-MeO, p-Me, H, p-CF3) effects coupling reactions with bromobenzene, increasing both the yields (from 70% to 97%) and steroselectivity outcome (E/Z from 55:45 to 9:91) as the substituent changed from an electron-withdrawing group to an electron-donating group (entries 1, 4, 12 and 13). TTMS-silanes also coupled with both π-deficient (entry 2) and electron-rich heterocyclic halides (entry 9) as well as with iodonaphthalene (entry 11).
Not only aryl halides but also vinyl halides can be coupled with vinyl TTMS-silanes. Thus, oxidative treatment of (Z)-4b with β-bromostyrene produced the diene 6b (72%, entry 7). The aliphatic 1-bromo-2-methyl-1-propene coupled with (Z)-4b yielding 7b in high yield and high stereoselectivity (entry 8).
The (E)-TTMS-silanes 2a-f underwent coupling with retention of stereochemistry. Thus, coupling of conjugated silanes 2a-d with aryl, heterocyclic, and aliphatic bromides or iodides (H2O2/NaOH/Pd(0)/TBAF) provided products stereoselectively in good to excellent yields (48-90%, Table 3, entries 1-15). The electron-deficient aryl iodides gave to some extent higher yields than the electron rich aryl iodides in the reactions with silane 2a (entries 1, 3 and 4).
The nonconjugated (E)-vinyl silanes 2e-f also undergo coupling with iodobenzene and bromobenzene under oxidative conditions in the presence of aqueous NaOH and TBAF to give the desired E alkenes 5e-f (70-85%; entries 16-19). The vinyl silane 2g, derived from cyclohexanone, successfully coupled with aryl (entries 20 and 21), alkenyl (entry 22) and heterocyclic halides (entry 23) to produce trisubstituted alkenes.
Although TBAF promotes couplings of TTMS-silanes in the presence of H2O2/base, we found that fluoride activation of TTMS-silanes was not necessary for the cross-coupling to occur. Thus, oxidative treatment (H2O2/NaOH or KOSiMe3) of the conjugated silane (E)-2a with bromo- and iodobenzene also produced (E)- stilbene (entries 1 and 2, Table 4). Other conjugated and nonconjugated (E)- and (Z)-silanes coupled with aryl, heterocyclic and aliphatic alkenyl halides (entries 3-12). Again coupling of (Z)-silanes occurred with lower stereoselectivity to produce E/Z mixture (entries 9-12). It is noteworthy that TBAF containing reactions are, however, generally higher yielding and more stereoselective than the fluoride-free reactions.
The lack of stereoselectivity for the coupling of (Z)-silanes probably results from the isomerization of the vinyl siloxane intermediates derived from (Z)-TTMS-silanes under the coupling conditions. Isomerization16 of the products was excluded based on the following experiments: (i) no isomerization of the (Z)-stilbene 5a was observed when (Z)-5a was refluxed in THF in the presence of H2O2/NaOH or TMSOK with or without Pd(0) and/or TBAF, (ii) coupling of the (Z)-silane 4b under typical conditions [H2O2/NaOH/Pd(0)THF/with or without TBAF] with phenyl iodide or bromide in the presence of 0.25 or 1.0 equiv. of the (Z)-stilbene 5a produced product 5b as the E/Z mixture [see Table 2 (entries 3 and 4) and Table 4 (entries 10 and 11)], while isomerization of the (Z)-stilbene 5a into E isomer was not observed (GC/MS).
A “side-by-side” comparison of the coupling of (Z)-4b with 1-iodonaphthalene [1 h (30%, E/Z 0:100); 3 h (58%, E/Z 3:97)] and 1-bromonaphthalene [1 h (22%, E/Z 5:95); 3 h (35%, E/Z 13:87)] showed that product 11b is formed at different pace (Table 2, entry 11). It appears that coupling with the aryl iodides is faster and occurs with a higher degree of stereoretention than with the corresponding aryl bromides (see also Table 2, entry 3 vs. 4; Table 4, entry 10 vs. 11). Longer stirring of the silanes 2 and 4 with H2O2/NaOH (45 min. vs. 15 min.) prior to the addition of the aryl halide and the catalyst resulted in no improvement of yield and stereoselectivity.
Denmark and Tymonko have recently utilized substrates bearing two distinct silyl subunits [RSiMe2OH vs. RSiMe2Bn], which required complementary activations (TMSOK vs. TBAF), for the construction of unsymmetrical disubstituted 1,4-butadienes.17 TTMS-silanes can also serve as alternative organosilane substrates in Pd-catalyzed couplings. For example, TTMS-silane 2a was remained intact under typical conditions employed in the coupling of dimethylsilanols14,17 [TMSOK(2 equiv.)/Pd2(dba)3/dioxane/r.t./4 h] with more than 95% of 2a being recovered after 4 h and ~85% after 24 h. This experiment demonstrated that TTMS-silanes could act as masked silanols, which require hydrogen peroxide for initiation.
It is noteworthy that under the oxidative conditions required for coupling of TTMS-silanes, the reductive self-coupling of the halides has not been observed for the fluoride promoted reactions (Table 2 and 3) and was only sporadically observed for the fluoride-free reactions (Table 4, entries 2, 5 and 9; 1-3%, GC-MS). Moreover, byproducts resulting from the oxidative homocoupling18 of the vinyl silanes 2 and 4 have not been observed. Also, although the oxidative conditions employed for generation of the active organosilane species are similar to the ones used in Tamao-Kumada and Fleming oxidation of silanes to alcohols (including vinyl silanes to aldehydes and ketones), which involve cleavage of the C-Si bond,19 we did not observe conversion of the vinyl silanes 2 and 4 to the corresponding aldehydes. Apparently, Si-Si bond cleavage takes place chemoselectively with the C-Si bond tolerating the relatively mild oxidative conditions required for coupling.20
We have not yet had the opportunity to systematically investigate the mechanism(s) of the TTMS-silanes Pd-catalyzed coupling but it appears that hydrogen peroxide chemoselectively cleaves20b the Si–Si bond(s) to generate silanol species RSi(OH)n(SiMe3)3-n (n = 1, 2, or 3). Subsequently, the silanol(s) are converted by the base to a silanolate anion, which might follow the coupling mechanism suggested by Denmark et al. for the organosilanols.10 Alternatively, TTMS-silanes can be converted by hydrogen peroxide to siloxane species RSi(OSiMe3)n(SiMe3)3-n (n = 1, 2, or 3), that can be further transformed to the reactive pentacoordinate species (hypervalent silicate anion)3b,e,10 by fluoride or base.
In order to obtain additional mechanistic insights, we examined the coupling reaction of 2a with iodobenzene by 29Si NMR. Thus, treatment of 2a with hydrogen peroxide (THF-d8/NaOH/H2O) resulted in the appearance of new peaks at 9.82, 7.20 and 5.59 ppm, which are characteristic for the species having oxygen attached to silicon,7b,10a, 20a,b,21 with concurrent disappearance of the two distinctive peaks at -85.31 ppm (Si atom attached to sp2 carbon) and -14.37 pm (SiMe3) for the silicon atoms present in substrate 2a. Addition of Pd catalyst and phenyl iodide to the resulting mixture resulted in the formation of stilbene (E)-5a Moreover, when coupling of 4b with 1-iodonaphthalene under fluoride-free conditions was quenched after 2 h, the corresponding tris(trimethylsiloxy)silyl compound 12 [(4)CH3C6H4CH=CHSi(OSiMe3)3] was isolated in 7% yield in addition to product 11b (12%). The structure of 12 was assigned based on the HRMS and NMR spectra. Subjection of 12 to TBAF promoted coupling with 1-iodonaphthalene producing product 11b but in low yield (8%; GC/MS).
In summary, we demonstrated that the conjugated and unconjugated vinyl tris(trimethylsilyl)silanes undergo Pd-catalyzed cross-coupling with aryl, heterocyclic and alkenyl iodides and bromides under aqueous oxidative conditions in the presence of sodium hydroxide with or without fluoride activation. Contrary to (E)-silanes, which undergo coupling with retention of stereochemistry, coupling of (Z)-silanes occurred with lower stereoselectivity giving an E/Z mixture of products. The best stereoselectivity was achieved when either aryl iodides or electron-rich TTMS-silanes were used. Under the oxidative coupling conditions neither reductive self-coupling of the halides nor oxidative homocoupling of the vinyl TTMS-silanes were observed. The tris(trimethylsilyl)silanes remained intact under typical basic conditions employed in the coupling of dimethylsilanols, thus making stable and readily accessible vinyl TTMS-silanes alternative substrates (“masked” silanols) for the Hiyama coupling. Hydrogen peroxide presumably chemoselectively cleaves Si–Si bond(s) generating active silanol/siloxane species that undergo coupling in the presence of base.
Experimental Section
1H (Me4Si) NMR spectra at 400 or 600 MHz, 13C (Me4Si) at 100.6 MHz, 19F (CCl3F) at 376.4 MHz and 29Si (Me4Si) at 119.2 MHz were determined with solutions in CDCl3 unless otherwise noted. Mass spectra (MS) were obtained by electron ionization (EI) technique and HRMS were acquired by AP-ESI technique. Reagent grade chemicals were used and solvents were dried by reflux over and distillation from CaH2 under an argon atmosphere. TLC was performed on Merck kieselgel 60-F254 and products were detected with 254 nm light or by development of color with I2. Merck kieselgel 60 (230-400 mesh) was used for column chromatography. Purity and identity of the products (crude and/or purified) were also established using a GC/MS (EI) system with a HP 5973 mass selective detector [capillary column HP-5MS (30 m × 0.25 mm × 25 μm)]. The vinyl sulfones 1a,5a 1c,22 1e,5a 1f,5a 1g5a and silanes 2e-g,5a 4a 12a were prepared as reported.
(E)-2-(4-Methoxyphenyl)-1-[tris(trimethylsilyl)silyl]ethene (2c). Procedure A
Argon was bubbled through a solution of 1c22 (E; 150 mg, 0.55 mmol) in anhydrous benzene (10 mL) for 15 min at ambient temperature. (Me3Si)3SiH (0.51 mL, 410 mg, 1.65 mmol) and AIBN (24 mg, 0.14 mmol) were added, and deoxygenation was continued for another 10 min. The resulting solution was refluxed at 85 °C for 4 h [additional AIBN (92 mg, 0.55 mmol) in degassed benzene (2 mL) was injected through a septum via an automatic syringe over the 4 h period or dropwise manually]. The volatiles were evaporated, and the residue was chromatographed in hexane to give 2c (134 mg, 64%) as a colorless oil: 1H NMR δ 0.25 (s, 27H), 3.84 (s, 3H), 6.29 (d, J = 18.8 Hz, 1H), 6.87 (d, J = 18.8 Hz, 1H), 6.89 (d, J = 8.8 Hz, 2H), 7.35 (d, J = 8.8 Hz, 2H); 13C NMR δ 1.0, 55.4, 114.0, 119.6, 127.2, 132.4, 145.0, 159.3; MS (EI) m/z 380 (15, M+), 174 (100). AP-ESI-HRMS Calcd for C18H36ONaSi4 (MNa+): 403.1735. Found: 403.1739.
Treatment of 4c (381 mg, 1 mmol) with (Me3Si)3SiH (0.15 mL, 124 mg, 0.5 mmol) in the presence of Rh catalyst, as described for 2a [GC-MS of the crude reaction mixture: 2c/4c (E/Z, 92:8; tR 22.02 min, Z, tR 22.34 min, E)], gave 2c (338 mg, 89%).
(Z)-2-(4-Methylphenyl)-1-[tris(trimethylsilyl)silyl]ethene (4b). Procedure B
(Me3Si)3SiH (0.31 mL, 248 mg, 1 mmol) was added in one portion via a syringe to a degassed solution of 3b (0.13 mL, 116 mg, 1 mmol) in dry benzene (3 mL) at ambient temperature under nitrogen atmosphere. AIBN (83.8 mg, 0.50 mmol) was then added and the resulting solution was heated (oil bath, 85 °C) for 3 h or until the alkyne was consumed (GC). The volatiles were evaporated in vacuo and the oily residue was flash chromatographed (hexane) on silica gel to give 4b (336 mg, 92%) as a colorless oil: 1H NMR δ 0.16 (s, 27H), 2.37 (s, 3H), 5.82 (d, J = 14.5 Hz, 1H), 7.16 (d, J = 7.8 Hz, 2H), 7.22 (d, J = 7.8 Hz, 2H), 7.40 (d, J = 14.5 Hz, 1H); 13C NMR δ 1.4, 21.3, 123.1, 128.1, 129.0, 137.1, 137.8, 146.6; 29Si NMR δ -88.33 [s, Si(SiMe3)3], -11.67 [s, Si(SiMe3)3]; GC-MS: (tR 22.12 min) m/z 364 (6, M+), 174 (100). HRMS Calcd for C18H36Si4 (M+): 364.1894. Found: 364.1896.
(E)-1,2-Diphenylethene (5a). Procedure C
Solution of NaOH (60 mg, 1.5 mmol) and H2O2 (30% solution, 0.15 mL, 1.5 mmol) in deionized H2O (1.5 mL) were added to a stirred solution of 2a (175 mg, 0.5 mmol) in THF (15 mL) at ambient temperature. After 15 minutes, iodobenzene (84 μL, 153 mg, 0.75 mmol), Pd(PPh3)4 (58 mg, 0.05 mmol) and tetrabutylammonium fluoride (1M/THF, 1.5 mL, 1.5 mmol) were added and the resulting brownish mixture was heated at 55 °C (oil bath) for 10 h. The volatiles were evaporated and the residue was partitioned (H2O/CHCl3). The aliquot of the organic layer was subjected to GC-MS and/or 1H NMR analysis in order to establish the overall stereochemistry. The organic layer was dried (MgSO4), evaporated and column chromatographed (hexane) to give (E)-5a (74 mg, 83%) with data identical to commercial sample: GC-MS (tR 17.9 min, E) m/z 180 (100, M+).
Treatment of 2a (35 mg, 0.10 mmol) with bromobenzene (16 μL, 23.6 mg, 0.15 mmol) by procedure C gave (E)-5a (12 mg, 67%).
Analogous treatment of 2a (35 mg, 0.10 mmol) with iodobenzene (17 μL, 31 mg, 0.15 mmol) by procedure C (without TBAF) gave (E)-5a (13.5 mg, 75%).
Analogous treatment of 2a (35 mg, 0.10 mmol) with bromobenzene (16 μL, 23.6 mg, 0.15 mmol) by procedure C (without TBAF) gave (E)-5a (9 mg, 50%). Also, biphenyl (1%; e.g., 2% consumption of bromobenzene) was detected: GC-MS (tR 11.3 min) m/z 154 (100, M+).
Procedure D
KOSiMe3 (38.5 mg, 0.3 mmol) and H2O2 (30% solution, 31 μL, 0.30 mmol) were added to a stirred solution of 2a (35 mg, 0.10 mmol) in THF (3 mL) at ambient temperature. After 20 minutes, iodobenzene (17 μL, 31 mg, 0.15 mmol) and Pd(PPh3)4 (11 mg, 0.01 mmol) were added and the resulting mixture was heated at 55 °C (oil bath) for 10 h. Aqueous work-up and purification as described in procedure C gave (E)-5a (11 mg, 60%).
(E/Z)-1,2-Diphenylethene (5a)
Treatment of 4a12a (35 mg, 0.10 mmol) with bromobenzene (16 μL, 23.6 mg, 0.15 mmol) by procedure C gave 5a (E/Z, 40:60; 15 mg, 82%) with data identical to commercial sample: GC-MS (tR 15.1 min, Z; tR 17.9 min, E) m/z 180 (100, M+). HRMS Calcd for C14H13 (MH+): 181.1073. Found: 181.1079.
Analogous treatment of 4a (35 mg, 0.10 mmol) with bromobenzene (16 μL, 23.6 mg, 0.15 mmol) by procedure C (without TBAF) gave 5a (E/Z, 25:75; 10 mg, 55%). Also, biphenyl (3%; e.g. 6% consumption of bromobenzene) was detected (GC-MS).
(E)-1-(4-Methylphenyl)-2-phenylethene (5b)
Treatment of 2b (36.5 mg, 0.10 mmol) with iodobenzene (17 μL, 31 mg, 0.15 mmol) by procedure C gave (E)-5b (14 mg, 72%) with data as reported:23 1H NMR δ 2.22 (s, 3H), 6.92 (d, J = 18.1 Hz, 1H), 6.98 (d, J = 18.1 Hz, 1H), 7.03 (d, J = 7.9 Hz, 2H), 7.13 (t, J = 7.3 Hz, 1H), 7.22 (t, J = 7.4 Hz, 2H), 7.30 (d, J = 8.1 Hz, 2H), 7.39 (d, J = 7.9 Hz, 2H), GC-MS (tR 19.6 min) m/z 194 (100, M+).
(E/Z)-1-(4-Methylphenyl)-2-phenylethene (5b)
Treatment of 4b (364 mg, 1.0 mmol) with iodobenzene (0.17 mL, 306 mg, 1.5 mmol) by procedure C gave 5b23b (E/Z, 3:97; 175 mg, 90%): GC-MS (tR 16.8 min, Z; tR 19.6 min, E) m/z 194 (100, M+). HRMS Calcd for C15H15 (MH+): 195.1174. Found: 195.1179. (Z)-5b had: 1H NMR δ 2.20 (s, 3H), 6.43 (s, 2H), 6.92 (d, J = 7.9 Hz, 2H), 7.03 (d, J = 7.9 Hz, 2H), 7.11-7.20 (m, 5H).
Treatment of 4b (36.5 mg, 0.10 mmol) with bromobenzene (16 μL, 23.6 mg, 0.15 mmol) by procedure C gave 5b (E/Z, 30:70; 17 mg, 86%).
Analogous treatment of 4b (36.5 mg, 0.10 mmol) with iodobenzene (17 μL, 31 mg, 0.15 mmol) by procedure C (without TBAF) gave 5b (E/Z, 15:85; 12 mg, 61%). Identical coupling with bromobenzene (0.15 mmol) gave 5b (E/Z, 20:80; 8 mg, 40%).
Treatment of 4b (36.5 mg, 0.10 mmol) with iodobenzene (17 μL, 31 mg, 0.15 mmol) by procedure D gave 5b (E/Z, 25:75; 11.6 mg, 60%). Identical coupling with bromobenzene (0.15 mmol) gave 5b (E/Z, 10:90; 9 mg, 48%).
Analogous treatment of 4b (36.5 mg, 0.10 mmol) with iodobenzene (17 μL, 31 mg, 0.15 mmol) by procedure D [with addition of TBAF (0.3 mmol) as described in procedure C] gave 5b (E/Z, 2:98; 15.7 mg, 81%).
Analogous treatment of 4b (36.5 mg, 0.10 mmol) with iodobenzene by procedure C [using aqueous NaF (12.6 mg, 0.3 mmol) instead of TBAF] gave 5b (E/Z, 40:60; 11.6 mg, 60%).
Analogous treatment of 4b (36.5 mg, 0.10 mmol) with iodobenzene by procedure C [using Pd2(dba)3 (9.2 mg, 0.01 mmol) instead of Pd(PPh3)4 and without addition of TBAF] gave 5b (E/Z, 25:75; 10 mg, 52%).
(Z)-2-(4-Methylphenyl)-1-[tris(trimethylsiloxy)silyl]ethene (12)
Treatment of 4b (50 mg, 0.14 mmol) with 1-iodonaphtalene (22 μL, 35 mg, 0.14 mmol) by procedure C [without TBAF, 2 h, NaOH (5 equiv.)] and column chromatography (hexane) gave 12 (4 mg, 7%) and 11b (E/Z, 7:93; 4 mg, 12%). Compound 12 had: 1H NMR δ 0.07 (br s, 27H), 2.36 (s, 3H), 5.50 (d, J = 15.5 Hz, 1H), 7.12 (d, J = 7.9 Hz, 2H), 7.21 (d, J = 15.5 Hz, 1H) 7.44 (d, J = 8.0 Hz, 2H); GC-MS (tR 17.60 min) m/z 412 (6, M+), 175 (100); HRMS Calcd for C18H37O3Si4 (MH+): 413.1814. Found: 413.1823.
Supplementary Material
Experimental procedures and characterization data for compounds 1d, 2a, 2b, 2d, 4c, 4d, 5c-g, 6b, 6c, 7a, 7b, 7g, 8a, 8b, 8g, 9a, 10b, and 11b. This material will be published online alongside electronic version of the manuscript in Elsevier web products, including www.sciencedirect.com
Acknowledgments
We thank NIH/NIGMS program (S06GM08205) for supporting this research. LM and DD were sponsored by the MBRS RISE program (NIH/NIGMS; R25 GM61347). The support of US ARO (W911NF-04-1-0022) for the purchase of 600 MHz NMR spectrometer is deeply acknowledged. We thank Dr. Donald V. Eldred (Dow Corning Co., Midland, MI) and Dr. Vered Marks (FIU) for assistance with 29Si NMR and Myron Georgiadis from the Advanced Mass Spectrometry Facility at FIU for his assistance with GC/MS.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.(a) Diederich F, Stang PJ, editors. Metal-Catalyzed Cross-Coupling Reactions. Wiley-VCH; Weinheim: 1998. [Google Scholar]; (b) Miyaura N, editor. Top Curr Chem Cross coupling reactions. Springer-Verlag; Heidelberg: 2002. [Google Scholar]; (c) Meijere A, Diederich F, editors. Metal-Catalyzed Cross-Coupling Reactions. Second. Wiley-VCH; Weinhem, Germany: 2004. [Google Scholar]
- 2.Stille JK. Angew Chem Int Ed Engl. 1986;25:508–524. [Google Scholar]; (b) Fugami K, Kosugi M. Top Curr Chem. 2002;219:87–130. [Google Scholar]; (c) Mitchell TN. Chapter 3:125–164. in Ref. 1(c): [Google Scholar]; (d) Espinet P, Echavarren AM. Angew Chem Int Ed. 2004;43:4704–4734. doi: 10.1002/anie.200300638. [DOI] [PubMed] [Google Scholar]; (e) Powell DA, Maki T, Fu GC. J Am Chem Soc. 2005;127:510–511. doi: 10.1021/ja0436300. [DOI] [PubMed] [Google Scholar]; (f) Echavarren AM. Angew Chem Int Ed. 2005;44:3962–3965. doi: 10.1002/anie.200500918. [DOI] [PubMed] [Google Scholar]
- 3.(a) Hiyama T. Chapter 10:421–454. in Ref. 1(a): [Google Scholar]; (b) Hiyama T, Shirakawa E. Top Curr Chem. 2002;219:61–85. [Google Scholar]; (c) Denmark SE, Ober MH. Aldrichimica Acta. 2003;36:75–85. [Google Scholar]; (d) Denmark SE, Sweis RF. Chapter 4:163–216. in Ref. 1(c): [Google Scholar]; (e) Mowery ME, DeShong P. J Org Chem. 1999;64:1684–1688. doi: 10.1021/jo982463h. [DOI] [PubMed] [Google Scholar]; (f) Denmark SE, Neuville L, Christy MEL, Tymonko SA. J Org Chem. 2006;71:8500–8509. doi: 10.1021/jo061481t. [DOI] [PubMed] [Google Scholar]
- 4.(a) Kosugi M, Tanji T, Tanaka Y, Yoshida A, Fugami K, Kameyama M, Migita T. J Organometal Chem. 1996;508:255–257. [Google Scholar]; (b) Faller JW, Kultyshev RG. Organometallics. 2002;21:5911–5918. [Google Scholar]; (c) Nakamura T, Kinoshita H, Shinokubo H, Oshima K. Org Lett. 2002;4:3165–3167. doi: 10.1021/ol026613t. [DOI] [PubMed] [Google Scholar]; (d) Faller JW, Kultyshev RG, Parr J. Tetrahedron Lett. 2003;44:451–453. [Google Scholar]; (e) Enokido T, Fugami K, Endo M, Kameyama M, Kosugi M. Adv Synth Catal. 2004;346:1685–1688. [Google Scholar]
- 5.(a) Wnuk SF, Garcia PI, Jr, Wang Z. Org Lett. 2004;6:2047–2049. doi: 10.1021/ol049312n. [DOI] [PubMed] [Google Scholar]; (b) Wang Z, Wnuk SF. J Org Chem. 2005;70:3281–3284. doi: 10.1021/jo047773g. [DOI] [PubMed] [Google Scholar]; (c) Wang Z, Gonzalez A, Wnuk SF. Tetrahedron Lett. 2005;46:5313–5316. [Google Scholar]
- 6.(a) Hatanaka Y, Hiyama T. J Org Chem. 1988;53:920–923. [Google Scholar]; (b) Hiyama T, Hatanaka Y. Pure Appl Chem. 1994;66:1471–1478. [Google Scholar]
- 7.(a) Denmark SE, Choi JY. J Am Chem Soc. 1999;121:5821–5822. [Google Scholar]; (b) Denmark SE, Wehrli D, Choi JY. Org Lett. 2000;2:2491–2494. doi: 10.1021/ol006170y. [DOI] [PubMed] [Google Scholar]
- 8.(a) Denmark SE, Wehrli D. Org Lett. 2000;2:565–568. doi: 10.1021/ol005565e. [DOI] [PubMed] [Google Scholar]; (b) Denmark SE, Sweis RF. Org Lett. 2002;4:3771–3774. doi: 10.1021/ol026900x. [DOI] [PubMed] [Google Scholar]; (c) Denmark SE, Sweis RF. Acc Chem Res. 2002;35:835–846. doi: 10.1021/ar020001r. [DOI] [PubMed] [Google Scholar]
- 9.Hirabayashi K, Mori A, Kawashima J, Suguro M, Nishihara Y, Hiyama T. J Org Chem. 2000;65:5342–5349. doi: 10.1021/jo000679p. [DOI] [PubMed] [Google Scholar]
- 10.(a) Denmark SE, Sweis RF, Wehrli D. J Am Chem Soc. 2004;126:4865–4875. doi: 10.1021/ja037234d. [DOI] [PubMed] [Google Scholar]; (b) Denmark SE, Sweis RF. J Am Chem Soc. 2004;126:4876–4882. doi: 10.1021/ja0372356. [DOI] [PubMed] [Google Scholar]; (c) Denmark SE, Baird JD. Chem Eur J. 2006;12:4954–4963. doi: 10.1002/chem.200600034. [DOI] [PubMed] [Google Scholar]
- 11.(a) Sahoo AK, Oda T, Nakao Y, Hiyama T. Adv Synth Catal. 2004;346:1715–1727. [Google Scholar]; (b) Wolf C, Lerebours R. Org Lett. 2004;6:1147–1150. doi: 10.1021/ol049851s. [DOI] [PubMed] [Google Scholar]; (c) Nakao Y, Imanaka H, Sahoo AK, Yada A, Hiyama T. J Am Chem Soc. 2005;127:6952–6953. doi: 10.1021/ja051281j. [DOI] [PubMed] [Google Scholar]; (d) Denmark SE, Butler CR. Org Lett. 2006;8:63–66. doi: 10.1021/ol052517r. [DOI] [PubMed] [Google Scholar]; (e) Nokami T, Tomida Y, Kamei T, Itami K, Yoshida J-I. Org Lett. 2006;8:729–731. doi: 10.1021/ol052961u. [DOI] [PubMed] [Google Scholar]; (f) Alacid E, Najera C. Adv Synth Catal. 2006;348:945–952. [Google Scholar]; (g) Shi S, Zhang Y. J Org Chem. 2007;72:5927–5930. doi: 10.1021/jo070855v. [DOI] [PubMed] [Google Scholar]
- 12.Kopping B, Chatgilialoglu C, Zehnder M, Giese B. J Org Chem. 1992;57:3994–4000. For Lewis-acid controlled regioselective hydrosilylation of propiolate ester with (TMS)3SiH see: Liu Y, Yamazaki S, Yamabe S. J Org Chem. 2005;70:556–561. doi: 10.1021/jo048371b.
- 13.(a) Takeuchi R, Nitta S, Watanabe D. J Org Chem. 1995;60:3045–3051. [Google Scholar]; (b) Mori A, Takahisa E, Nishihara Y, Hiyama T. Can J Chem. 2001;79:1522–1524. [Google Scholar]; (c) Mori A, Takahisa E, Yamamura Y, Kato T, Mudalige AP, Kajiro H, Hirabayashi K, Nishihara Y, Hiyama T. Organometallics. 2004;23:1755–1765. [Google Scholar]
- 14.(a) Denmark SE, Sweis RF. J Am Chem Soc. 2001;123:6439–6440. doi: 10.1021/ja016021q. [DOI] [PubMed] [Google Scholar]; (b) Denmark SE, Tymonko SA. J Org Chem. 2003;68:9151–9154. doi: 10.1021/jo0351771. [DOI] [PubMed] [Google Scholar]
- 15.For a review on Pd-catalyzed coupling reactions of aryl chlorides, see: Littke AF, Fu GC. Angew Chem Int Ed Engl. 2002;41:4176–4211. doi: 10.1002/1521-3773(20021115)41:22<4176::AID-ANIE4176>3.0.CO;2-U.Faller, et al. reported4d that arylalkynyl oxagermatranes underwent coupling with aryl chlorides and triflates; substrates not normally successful under the conventional Sonogashira coupling reaction conditions
- 16.Methods for isomerization of cis alkenes into trans isomers usually involve radical or photochemical processes. For (Z)- to (E)-alkene isomerization in the presence of BzOOBz/NBS/AIBN see: Baag MM, Kar A, Argade NP. Tetrahedron. 2003;59:6489–6492. For the Pd(II)-catalyzed isomerization of cis-arylalkenes see: Yu J, Gaunt MJ, Spencer JB. J Org Chem. 2002;67:4627–4629. doi: 10.1021/jo015880u.
- 17.Denmark SE, Tymonko SA. J Am Chem Soc. 2005;127:8004–8005. doi: 10.1021/ja0518373. [DOI] [PubMed] [Google Scholar]
- 18.(a) Babudri F, Cicciomessere AR, Farinola GM, Fiandanese V, Marchese G, Musio R, Naso F, Sciacovelli O. J Org Chem. 1997;62:3291–3298. doi: 10.1021/jo9700437. [DOI] [PubMed] [Google Scholar]; (b) Yoshida H, Yamaryo Y, Ohshita J, Kunai A. Chem Commun. 2003:1510–1511. [Google Scholar]; (c) Itami K, Ushiogi Y, Nokami T, Ohashi Y, Yoshida J-I. Org Lett. 2004;6:3695–3698. doi: 10.1021/ol048620i. [DOI] [PubMed] [Google Scholar]
- 19.Joens GR, Landais Y. Tetrahedron. 1996;52:7599–7662. [Google Scholar]
- 20.For the cleavage of the Si-Si σ-bond which are known to have low oxidation potentials (with bond dissociation energies 332 kJ/mol vs. 394 kJ/mol for C-Si bond19d) see: Naka A, Yoshida K, Ishikawa M, Miyahara I, Hirotsu K, Cha S-H, Lee KK, Kwak Y-W. Organometallics. 2001;20:1204–1209. (with O2/AIBN or m-CPBA). Ackerhans C, Roesky HW, Labahan T, Magull J. Organometallics. 2002;21:3671–3674. (with H2O2/H2O). Mochida K, Shimizu H, Kugita T, Nanjo M. J Organomet Chem. 2003;673:84–94. (with tetracyanoethylene). Becerra R, Walsh R. In: The Chemistry of Organic Silicon Compounds. Part 1. Rappoport Z, Apeloig Y, editors. Chapter 4, Vol. 2. John Wiley; Chichester, U.K: 1998. pp. 153–180.
- 21.(a) Williams EA. In: The Chemistry of Organic Silicon Compounds. Patai S, Rappoport Z, editors. Chapter 8, Vol. 1. John Wiley; New York: 1989. pp. 511–544. [Google Scholar]; (b) Takeuchi Y, Takayama T. In: The Chemistry of Organic Silicon Compounds. Part 1. Rappoport Z, Apeloig Y, editors. Chapter 6, Vol. 2. John Wiley; Chichester, U.K.: 1998. pp. 267–354. [Google Scholar]
- 22.Lee JW, Oh DY. Synth Commun. 1989;19:2209–2212. [Google Scholar]
- 23.(a) Arvela R, Leadbeater N. J Org Chem. 2005;70:1786–1790. doi: 10.1021/jo048052k. [DOI] [PubMed] [Google Scholar]; (b) Lawrence NJ, Muhammad F. Tetrahedron. 1998;54:15361–15370. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Experimental procedures and characterization data for compounds 1d, 2a, 2b, 2d, 4c, 4d, 5c-g, 6b, 6c, 7a, 7b, 7g, 8a, 8b, 8g, 9a, 10b, and 11b. This material will be published online alongside electronic version of the manuscript in Elsevier web products, including www.sciencedirect.com