

Abstract
6-Gingerol, a natural product of ginger, has been known to possess anti-tumorigenic and pro-apoptotic activities. However, the mechanisms by which it prevents cancer are not well understood in human colorectal cancer. Cyclin D1 is a proto-oncogene that is overexpressed in many cancers and plays a role in cell proliferation through activation by β-catenin signaling. Nonsteroidal anti-inflammatory drug (NSAID)-activated gene-1 (NAG-1) is a cytokine associated with pro-apoptotic and anti-tumorigenic properties. In the present study, we examined whether 6-gingerol influences cyclin D1 and NAG-1 expression and determined the mechanisms by which 6-gingerol affects the growth of human colorectal cancer cells in vitro. 6-Gingerol treatment suppressed cell proliferation and induced apoptosis and G1 cell cycle arrest. Subsequently, 6-gingerol suppressed cyclin D1 expression and induced NAG-1 expression. Cyclin D1 suppression was related to inhibition of β-catenin translocation and cyclin D1 proteolysis. Furthermore, experiments using inhibitors and siRNA transfection confirm the involvement of the PKCε and glycogen synthase kinase (GSK)-3β pathways in 6-gingerol-induced NAG-1 expression. The results suggest that 6-gingerol stimulates apoptosis through upregulation of NAG-1 and G1 cell cycle arrest through downregulation of cyclin D1. Multiple mechanisms appear to be involved in 6-gingerol action, including protein degradation as well as β-catenin, PKCε, and GSK-3β pathways.
Keywords: 6-gingerol, NAG-1, cyclin D1, β-catenin, PKC, GSK-3β
INTRODUCTION
Colorectal carcinoma (CRC), the third leading cause of cancer mortality in the United States, is a highly preventable cancer [1]. Of several strategies that can reduce colon cancer risk, dietary phytochemicals are emerging as a promising chemoprevention approach [2]. Of these phytochemicals, gingerols are a group of structurally related polyphenolic compounds isolated from ginger [3], and 6- gingerol has been known as a novel component for prevention of increased risk of cancer due to the compound’s chemo-protective properties in several tissues of experimental animals. In fact, topical application of 6-gingerol onto mice inhibits 7,12- dimethylbenz[a]anthracene-induced skin cancer [4], and 6-gingerol inhibits TPA-induced cyclooxygenase- 2 expression in mouse skin [5]. In addition, dietary 6-gingerol inhibits pulmonary metastasis in mice implanted with melanoma cells [6]. Furthermore, in vitro studies have shown that 6-gingerol induces apoptosis in cultured leukemia cells [7], inhibits EGF-induced cell transformation and AP-1 activation in mouse epidermal cells [8], and inhibits angiogenesis in human endothelial cells [9]. Although 6-gingerol shows anti-cancer activity in several types of cancer, it has not been determined whether 6-gingerol inhibits tumorigenic activity in human colorectal cancer.
Many signaling pathways play a pivotal role in colorectal tumorigenesis. Specifically, the β-catenin signaling pathway is important in development and tumorigenesis [10,11]. The β-catenin protein interacts with adenomatous polyposis coli (APC), glycogen synthase kinase (GSK)-3β, casein kinase 1α, axin, or the closely related factor conductin/Axil [12]. The β-catenin protein also binds to the members of the T cell factor (TCF) family and transactivates several target genes. Among the target genes of the β-catenin/ TCF complex, several cell growth related genes have been identified including cyclin D1 [13,14], c-myc [15], and PPAR-δ [16]. Thus, activation of β-catenin signaling leads to the induction of cyclin D1 at the transcriptional level and further results in the progression of the G1/S phase and the increase of proliferation. Another important pathway in colorectal tumorigenesis is the protein kinase C (PKC) pathway. PKC is a family of the serine/threonine kinase involved in intracellular signal transduction pathways that regulate gene transcription, differentiation, cell cycle, cytoskeletal functions, apoptosis, and growth factor response. The PKC superfamily contains to date 11 isoforms encoded by 10 genes [17]. In general, PKC family members are broad-specificity kinases, which phosphorylate many proteins. However, in most cases, isotype specificity is poorly defined, and even contrary functions for a single PKC have been reported mostly because appropriate molecular and genetic tools were missing to specifically assess the contribution of single PKC isoforms in vivo. For example, PKCε has been proposed to act both as tumor promoter and a tumor suppressor via multiple signaling pathways [18–23].
The nonsteroidal anti-inflammatory drug (NSAID)- activated gene (NAG-1), one of the transforming growth factor-beta (TGF-β) superfamily genes, has been reported as a pro-apoptotic and anti-tumorigenic gene [24]. NAG-1 expression is increased in mature intestinal epithelial cells, whereas it is reduced in human CRC and neoplastic intestinal polyps of Min mice [25]. Our recent data show that transgenic mice overexpressing NAG-1 (NAG-1-Tg) are resistant to azoxymethane-induced aberrant crypt foci, and NAG-1-Tg-Min mice showed less tumor load in the small intestine compared with littermate Min control mice [26]. NAG-1 expression is induced not only by NSAIDs, but also by several anti-tumorigenic compounds, including PPARγ ligands [27,28] and dietary compounds such as conjugated linoleic acid [29], indole-3-carbinol [30], resveratrol [31], genistein [32], catechins [33], and anti-inflammatory plant extracts [34]. These results indicate thatNAG-1 acts as a tumor suppressor gene and a target protein of several chemopreventive compounds [35].
The current study was performed to elucidate whether 6-gingerol affects human colorectal tumorigenesis. To our knowledge, we report for the first time that 6-gingerol increases cell cycle arrest and apoptosis in human colorectal cancer cells. 6-Gingerol affects the PKC and β-catenin pathways, resulting in the induction of NAG-1 and the reduction of cyclin D1 expression. These results imply that the anti-cancer activity of 6-gingerol is mediated by multiple mechanisms in human colorectal cancer cells.
MATERIALS AND METHODS
Materials
Human CRC cells, HCT-116, SW480, HT-29, LoVo, and Caco-2 were purchased from American Type Culture Collection (Manassas, VA). Antibodies for p53, PKCε, cyclin D3, p21, cdk-4, p-Rb (Ser780), and actin and PKCε small interfering RNA (siRNA) were purchased from Santa Cruz (Santa Cruz, CA), and antibodies for cyclin D1 and GSK-3β, and GSK-3α/β siRNA were purchased from Cell Signaling (Beverly, MA). Antibody for β-catenin was purchased from BD Biosciences (San Jose, CA), and control siRNA was purchase from Ambion (Austin, TX). The antibodies for hemagglutinin (HA) and p27 were purchased from Covance (Berkeley, CA) and NeoMarkers (Fremont, CA), respectively. Cyclin D1 promoter constructs [29] and antibody for NAG-1 [24] were previously described. 6-Gingerol was purchased from BIOMOL (Plymouth Meeting, PA), and 4′,6-diamidino- 2-phenylindole (DAPI) was purchased from Roche (Indianapolis, IN). Lithium chloride was purchased from Sigma (St. Louis, MO), and RO-31- 8220, Rottlerin, Gö6983, MG-132, and cycloheximide (CHX) were purchased from Calbiochem (San Diego, CA). Rhodamine conjugated Goat anti-mouse IgG was purchased from Southern Biotechnology Associates (Birmingham, AL). Cell culture media and Alexa Fluor 488 goat anti-rabbit IgG antibody were purchased from Invitrogen (Carlsbad, CA). All chemicals were purchased from Fisher Scientific, unless otherwise specified.
Cell Culture and Treatment
HCT-116 cells and HT-29 cells were maintained in McCoy’s 5A medium. SW480 cells, LoVo, and Caco-2 cells were maintained in RPMI1640, Ham’s F-12, and DMEM medium, respectively. All media were supplemented with 10% fetal bovine serum (FBS) and 10 μg/mL of gentamicin, respectively. Cells were grown at 37°C under a humidified atmosphere of 5% CO2. The cells were plated in a 12-well or 60-mm culture dish and incubated until cells were 70–80% confluent. The cells were then treated with different concentrations of 6-gingerol at different time points as indicated in figure legends.
Cell Proliferation and Flow Cytometric Detection of Apoptotic Cells
Cell proliferation assay was performed using the CellTiter 96 Aqueous One Solution Cell Proliferation Assay (Promega, Madison, WI). Briefly, cells were seeded at a concentration of 1000 cells/well in 96- well tissue culture plates in four replicates and maintained overnight. The cells were then treated with 0, 50, 100, 150, and 200 μM of 6-gingerol for 72 h. Twenty microliter of CellTiter96®Aqueous One solution was added to each well, and the plate was incubated for 1 h at 37°C. Absorbance at 490 nm was recorded in an ELISA plate reader (Bio-Tek Instruments, Inc., Winooski, VT). Apoptosis was measured using the Annexin V-FITC apoptosis detection kit (BD Biosciences). Briefly, cells were plated in 6-well tissue culture dishes and incubated with 0, 25, 50, 100, and 200 μMof 6-gingerol for 72 h. The attached and floating cells were harvested together, washed with PBS, stained with Annexin V-FITC, and the early and late apoptosis was quantified according to the manufacturer’s instructions. For the cell cycle analysis, the cells were treated as described above and stained with propidium iodide, and cell cycle distribution was analyzed. Apoptosis and cell cycle distribution were analyzed using Beckman Coulter Epixs XL flow cytometer equipped with EXPO32 ADC and ModFit LT software, respectively.
Immunofluorescence
The cells were seeded on eight-well glass slides (Lab-Tek Chamber slide system, Nalge Nunc International, Rochester, NY) and grown in culture media. The cells were then washed twice with PBS and treated with 200 μM of 6-gingerol for 24 h. After treatment, the cells were washed twice with PBS, fixed in an ice-cold acetone for 15 min, and washed in PBS three times for 5 min. The nonspecific binding of the antibodies was blocked by universal blocking solution (Biogenex, San Ramon, CA) for 30 min at room temperature, and the cells were incubated with a mixture of specific antibodies for cyclin D1 (1:250), NAG-1 (1:250), and β-catenin (1:250) overnight at 4°C. The cells were washed with PBS containing 0.1% Tween 20 (PBS-T) three times for 5 min and incubated with secondary antibodies goat antimouse rhodamine conjugate (1:250) and goat anti-rabbit Alexa Fluor 488 (1:300) for 1 h at room temperature in darkness. The cells were stained with 0.5 mg/mL of DAPI for 2 min to counter stain the nucleus, and then washed three times for 5 min with PBST. The excess PBST was decanted and the slide was mounted with a few drops of an aqueous mounting medium. The expression of proteins was detected using a fluorescence microscope Nikon Eclipse E600 (Melville, NY). The TIF images were captured using QCapture software version 2.66.4 with 400× or 600× magnification.
Transient Transfections
Transient transfections were performed using the Lipofectamine (Invitrogen) according to the manufacturer’s instruction. The cells were plated in 12- well plates at the concentration of 2 × 105 cells/well. After growth overnight, the cells were transfected with plasmid containing β-catenin/TCF-LEF-responsive (TOP-FLASH) and mutant (FOP-FLASH) promoters or cyclin D1 promoters for 5 h as described previously [29]. Then, the cells were harvested in 1× luciferase lysis buffer, and luciferase activity was determined and normalized to the pRL-null luciferase activity using a dual luciferase assay kit (Promega). The expression vector for wild type (WT) and dominant negative (DN) PKCε was transfected as described previously [36]. For RNA interference of GSK-3 and PKCε, cells were transfected with the GSK- 3α/β siRNA, PKCε siRNA, or control siRNA at a concentration of 100 nM, using Trans-IT-TKO transfection reagent (Mirus, Madison, WI), as described previously [29]. After 24 h transfection, the cells were treated with vehicle or 6-gingerol for 24 h.
Western Blotting
Cells were washed with PBS, and cell lysates were isolated in RIPA buffer (50 mM Tris-HCl pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, 1% sodium deoxycholate, 0.1% SDS) supplemented with protease inhibitors (1mMPMSF, 1 μg/mL aprotinin, 1 μg/mL leupeptin) and phosphatase inhibitors (10 mM NaF, 0.1 mM Na3VO4,) and centrifuged at 12 000 rpm for 5 min at 4°C. Protein concentration was determined by the BCA protein assay (Pierce, Rockford, IL), using BSA as the standard. The proteins were separated on SDS–PAGE and transferred to nitrocellulose membranes (Osmonics, Minnetonka, MN). The membranes were incubated with a specific primary antiserum in TBS containing 0.05% Tween 20 (TSB-T) and 5% nonfat dry milk at 4°C overnight. After four washes with TBS-T, the blots were incubated with peroxidase-conjugated IgG for 1 h at room temperature and visualized using ECL (Amersham Biosciences, Piscataway, NJ).
Preparation of Nuclear Extracts
Nuclear extracts were isolated from the cells treated with vehicle or 200 μof 6-gingerol for 24 h, using the Nuclear Extract Kit (Active Motif, Carlsbad, CA), according to the manufacturer’s protocol.
Isolation and Analysis of RNA
Total RNA was prepared using an RNA isolation kit (Eppendorf, Hamburg, Germany), according to the manufacturer’s instructions. Ten microgram of total RNA were fractionated on 1.0% agarose-2.2M formaldehyde gels and transferred to a nylon membrane (Micron Separations, Inc., Westborough, MA). The cDNA probe for NAG-1 was synthesized using the Biotin random prime kit (Pierce). The probes used were full-length NAG-1 fragments [24]. Hybridization and chemiluminescent signal detection were performed using the North2South chemiluminescent detection kit (Pierce), according to the manufacturer’s instructions. For semi-quantitative RTPCR, RNA was treated with DNase I (Invitrogen) prior to cDNA synthesis. Total RNA (2 μg) was reverse-transcribed with an iScript cDNA kit (BioRad, Hercules, CA), according to the manufacturer’s instruction. PCR was carried out using Ready- Mix Taq polymerase (Sigma) with primers for human cyclin D1 and GAPDH as follows: cyclin D1; forward 5′-atggaacaccagctcctgtgctgc-3′, and reverse 5′-tcagatgtccacgtcccgcacgt-3′, GAPDH; forward 5′-gggctgcttttaactctggt-3′, and reverse 5′-tggcaggtttttctagacgg-3′.
Statistical Analysis
Statistical analysis was performed with the Student’s unpaired t-test, with statistical significance set at *P<0.05; **P<0.01; ***P<0.001.
RESULTS
6-Gingerol Suppresses Cell Proliferation and Induces Apoptosis/G1 Cell Cycle Arrest in Human Colorectal Cancer Cells
6-Gingerol has been implicated in tumor suppression in skin [4–6,8], blood [7], and endothelial cells [9]. To investigate the effects of 6-gingerol on the growth of colorectal cancer cells, HCT-116 cells were incubated with 0, 50, 100, 150, and 200 μM of 6- gingerol for 72 h and cell proliferation was measured. As shown in Figure 1A, HCT-116 cells treated with 150 and 200 μMof 6-gingerol reduced the cell growth rate by 22% and 28%, respectively. The retarded cell growth was also observed in other human colorectal cancer cells including SW480, HT-29, LoVo, and Caco-2 cells treated with 200 μM of 6-gingerol for 72 h. Growth arrest by 200 μMof 6-gingerol was seen in HCT-116 (28%), SW480 (17%), HT-29 cells (28%), LoVo (13%), and Caco-2 cells (8%). It is notable that the doubling time of LoVo and Caco-2 cells are much longer than for other cells. To investigate the effects of 6-gingerol on apoptosis, HCT-116 cells were treated with different concentrations of 6-gingerol for 72 h, and apoptosis analysis was performed by flow cytometry. As shown in Figure 1B, the number of apoptotic cells (Annexin+/PI− and Annexin+/ PI+) was increased by 1.3-, 1.8-, and 2.6-fold in HCT- 116 cells treated with 50, 100, and 200 μM of 6- gingerol, respectively. Apoptotic cells were also significantly increased in SW480 and LoVo cells, but not in HT-29 and Caco-2 cells. Cell cycle analysis demonstrated that 6-gingerol treatment resulted in an increase of the G1 phase (Vehicle, 80.04 ± 1.81 vs. 6-gingerol, 87.45 ± 2.14), and a decrease of the S phase (Vehicle, 17.90 ± 1.35 vs. 6-gingerol, 11.52 ± 1.96) and the G2 phase (Vehicle, 2.06 ± 0.49 vs. 6-gingerol, 1.02 ± 0.23) of the cell cycle in HCT- 116 cells (Figure 1C). 6-Gingerol treatment also increased the G1 cell cycle arrest and decreased G2 phase in LoVo cells, while 6-gingerol did not affect G1 and S phase in SW480 cells.
Figure 1.

6-Gingerol suppresses cell proliferation, induces apoptosis, and arrests at G1 cell cycle in human colorectal cancer cells. (A) Human colorectal cancer cells were treated with indicated concentration of 6-gingerol for 72 h. Cell growth was measured using CellTiter96 Aqueous One Solution Cell Proliferation Assay. Values are expressed as mean ± SD of four replicates. (Left panel) HCT-116 cells were treated with the indicated concentration of 6-gingerol and cell growth rate was determined. (Right panel) Four different human colorectal cancer cells were treated with 6-gingerol and measured cell growth. The data represent % of reduction compared to vehicle treatment. (B) Human colorectal cancer cells were treated with 200 μM of 6-gingerol for 72 h. The cells were stained with Annexin V-FITC and propidium iodide (BD Biosciences). Apoptosis was quantified by flow cytometry as described in Materials and Methods section. Values are expressed as mean ± SD of three replicates. (C) HCT-116, SW480, and LoVo cells were treated with 200 μM of 6-gingerol for 72 h. Cell cycle distribution was analyzed by FACS analysis of propidium iodide-stained cells. Values are expressed as mean ± SD of three replicates. *P<0.05; **P<0.01; ***P<0.001 versus vehicle-treated cells.
6-Gingerol Suppresses Cyclin D1 Expression and Increases NAG-1 Expression in HCT-116 Cells
To investigate the possible molecular mechanism of 6-gingerol on apoptosis induction and G1 cell cycle arrest, HCT-116 cells were incubated with different concentrations of 6-gingerol, and several cell cycle (cyclin D1, cyclin D3, CDK4, p21, p27) and apoptosis-related genes (NAG-1, p53) were measured using Western blot analysis. As shown in Figure 2A, cyclin D1 protein was decreased in a dose-dependent manner, especially when treated with 100, 150, and 200 μM of 6-gingerol. We also observed a significant decrease of CDK4 in the cells treated with 150 and 200 μM of 6-gingerol. Cyclin D3, p21 and p27 were not significantly changed by 6-gingerol treatment. Cyclin D1 was further examined to determine its expression by 6-gingerol treatment. Cyclin D1 mRNA expression was decreased in a dose-dependent manner (Figure 2B), and the protein level was also decreased in a time-dependent manner in the presence of 200 μM of 6-gingerol. This decrease started after 12 h and was much more pronounced after 20 h of treatment (Figure 2C). Cyclin D1 repression by 6-gingerol was seen in other human colorectal cancer cells, including SW480 and Caco-2 cells, whereas HT-29 and LoVo cells expressed very low amounts of cyclin D1 at the basal level (Figure 2D).
Figure 2.
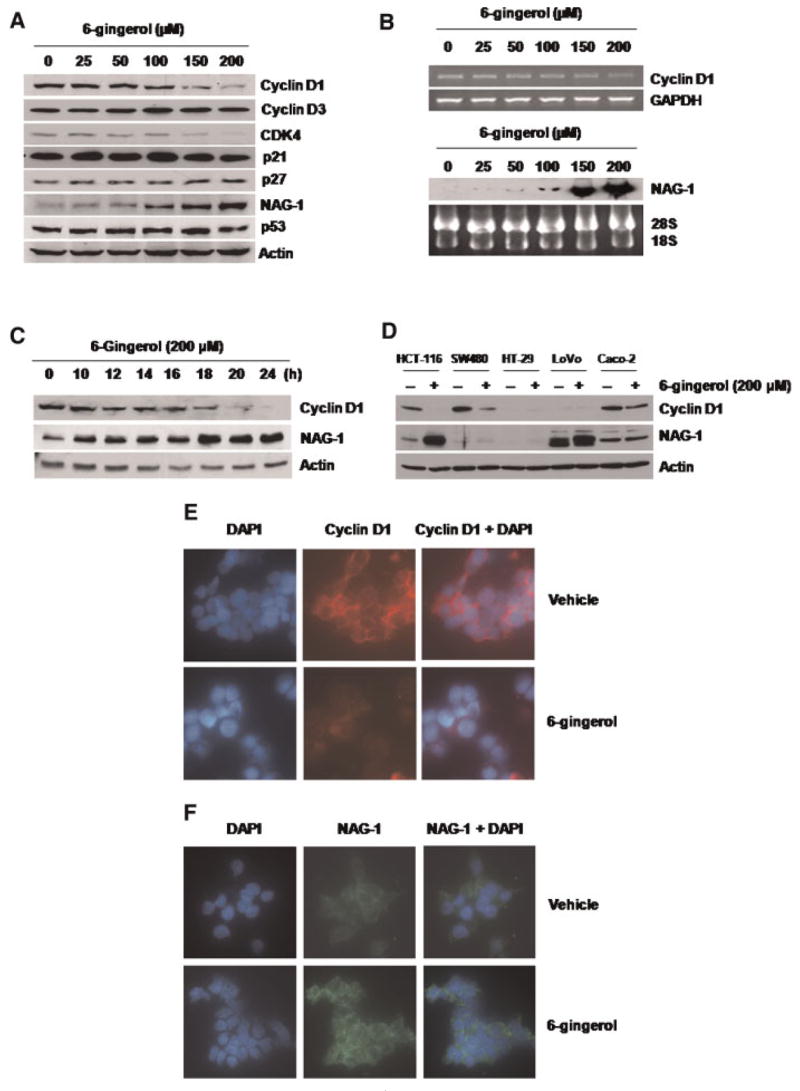
6-Gingerol suppresses cyclin D1 expression and increases NAG-1 expression in HCT-116 cells. (A) HCT-116 cells were treated with the indicated concentration of 6-gingerol for 24 h. Total cell lysates were harvested, and subsequently, 30 μg of total cell lysates were subjected to 14% SDS–PAGE. Cyclin D1, cyclin D3, CDK4, p21, p27, NAG-1, p53, and actin antibodies were probed. The results shown here are representative of three independent experiments. (B) HCT-116 cells were treated with the indicated concentrations of 6-gingerol for 24 h, and then RT-PCR (cyclin D1) or Northern blot analysis (NAG-1) was performed as described in Materials and Methods section. The GAPDH and 28S/18S shown represent a loading control. (C) HCT-116 cells were treated with 200 μM of 6-gingerol at indicated time points. Total cell lysates were harvested, and Western analysis was performed for cyclin D1, NAG- 1, and Actin antibodies. (D) HCT-116, SW480, HT-29, LoVo, and Caco-2 cells were grown as described in Materials and Methods section, and treated with 200 μM of 6-gingerol for 24 h. Western analysis was performed for cyclin D1, NAG-1, and Actin antibodies. (E and F) HCT-116 cells were grown in a glass slide chamber and then treated with 200 μM of 6-gingerol for 24 h. The cells were incubated with a specific antibody for cyclin D1 (1:250) or NAG-1 (1:250) overnight. Cyclin D1 and NAG-1 were detected by using rhodamine conjugate (red) and Alexa Fluor 488 conjugate (green), respectively, and visualized by fluorescence microscopy as described in Materials and Methods section. The DAPI staining (blue) was used to visualize the nuclei of the cells. Magnifications correspond to 400×. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
In contrast, treatment of the cells with 6-gingerol increased NAG-1 protein and mRNA levels at 100, 150, and 200 μM (Figure 2A and B), and the NAG-1 protein began to increase as early as the 10 h time point (Figure 2C). 6-Gingerol treatment increased NAG-1 expression in other colorectal cancer cells including LoVo cells (Figure 2D). Although the basal level of NAG-1 expression was low in HT-29 cells, we found that 6-gingerol also increased NAG-1 expression after longer exposure (data not shown). HCT- 116 cells were chosen for further study because 6- gingerol treatment altered both cyclin D1 and NAG- 1 expression.
Finally, we performed immunofluorescence analysis to detect relative abundance of cyclin D1 and NAG-1 after 6-gingerol treatment in HCT-116 cells. Cyclin D1 (red) and NAG-1 (green) immunofluorescence are shown with DAPI counterstain (blue) (Figure 2E and F). Immunofluorescence results revealed a decrease of cyclin D1 and an increase of NAG-1 expression. It is notable that NAG-1 was detected in vesicle, which is a typical phenomenon of secreted proteins, whereas 6-gingerol significantly decreased cyclin D1 expression in nuclei.
6-Gingerol Suppresses Cyclin D1 Expression at the Transcriptional and Translational Levels
To test whether 6-gingerol affects transcriptional regulation of the cyclin D1 gene, we measured promoter activity using three constructs (−1745/ +130, −963/+130, −163/+130) of cyclin D1 promoter. Each construct was transfected into HCT-116 cells and treated with 200 μM of 6-gingerol for 24 h. As shown in Figure 3A, 6-gingerol treatment resulted in the inhibition of cyclin D1 promoter activity in the three constructs tested (59%, 49%, and 58%, respectively), indicating that the −163/+130 region of the cyclin D1promoter is responsible for cyclinD1 inhibition by 6-gingerol. It has been reported that cyclin D1 and CDK4 leads to hyperphosphorylation of pRb, which then activates E2F family-induced cell proliferation [37]. Therefore, we investigated the phosphorylation of pRb from nuclear extract treated with 6-gingerol. The immunoblot analysis showed a decrease of p-pRb (Ser780), demonstrating that cyclin D1suppression by 6-gingerol results in the induction of hypophosphorylation of pRb (Figure 3B).
Figure 3.


6-Gingerol suppresses transactivation of the cyclin D1 gene through suppression of β-catenin activity and increases proteolysis of cyclin D1 protein. (A) HCT-116 cells were transfected with a reporter gene containing cyclin D1 promoter, and then the cells were treated with 200 μM of 6-gingerol for 24 h. Luciferase activity was measured as a ratio of firefly luciferase signal/renilla luciferase signal and is presented as a relative luciferase unit (RLU). The TCF binding site is shown as ■ [14], **P<0.01; ***P<0.001 versus vehicle-treated cells. (B) HCT-116 cells were treated with 200 μM of 6-gingerol for 24 h, and nuclear (N) and cytosol fractions (C) were isolated as described in Materials and Methods section. Western blot was performed for p-pRb (Ser780) and Actin antibodies. (C) HCT-116 cells were pretreated with vehicle or 200 μM of 6- gingerol for 1 h and then exposed to 10 μg/mL of CHX for the indicated time. Western analysis was performed for cyclin D1 and actin antibodies. (D) HCT-116 cells were pretreated with the indicated concentration of MG-132 for 30 min and then exposed to 200 μM of 6-gingerol for 24 h. Western analysis was performed for cyclin D1 and actin antibodies. (E) Human colorectal cancer cells were transfected with TOP-FLASH or FOP-FLASH constructs containing six copies of wild or mutated TCF binding sites. Then, the cells were treated with 200 μM of 6-gingerol for 24 h. Luciferase activity was measured as a ratio of TOP-FLASH over FOP-FLASH. *P<0.05; ***P<0.001 versus vehicle-treated cells. (F) SW480 cells were grown in a glass slide chamber and then treated with 200 μM of 6- gingerol for 24 h. The cells were incubated with a specific antibody for β-catenin (1:250) overnight, and β-catenin was detected by using rhodamine conjugate (red). The DAPI staining (blue) was used to visualize the nuclei of the cells (blue). Magnifications correspond to 600×. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
Several reports suggested that cyclin D1 is repressed through increased proteolysis by dietary compounds such as curcumin [38], retinoic acid [39], and troglitazone [40]. To determine whether 6- gingerol affects cyclin D1 expression via post-translational modification, we examined the effect of 6- gingerol on cyclin D1 in the presence of the protein synthesis inhibitor CHX. As shown in Figure 3C, addition of 6-gingerol rapidly degraded cyclin D1 in the presence of CHX. We also investigated whether MG-132, a proteasome inhibitor, blocks 6-gingerolinduced degradation of cyclin D1. The cells were pretreated with the indicated concentration of MG- 132 for 30 min and then exposed to 200 μM of 6- gingerol for 24 h. As shown in Figure 3D, cyclin D1 degradation by 6-gingerol was abolished in the presence of MG-132. These results clearly suggest that 6-gingerol represses cyclin D1 at both transcriptional and translational levels.
6-Gingerol Suppresses β-Catenin Signaling
Wnt signaling promotes the stabilization and accumulation of β-catenin, interacts with the TCF/ LEF family of transcription factors, and activates transcription of downstream genes including cyclin D1 [41]. To investigate whether 6-gingerol modulates β-catenin-dependent TCF transcriptional activity in human colorectal cancer cells, HCT-116, SW480, HT-29, LoVo, and Caco-2 cells were transiently transfected with the reporter plasmids TOP-FLASH or FOP-FLASH and pRL-null to normalize for transfection efficiency. 6-Gingerol was added to transfected cells and followed by assay for luciferase activity. As shown in Figure 3E, 6-gingerol treatment inhibits TCF activity by 30%, 39%, and 53% in HCT- 116, SW480, and Caco-2 cells, respectively, treated with 200 μM of 6-gingerol. However, HT-29 and LoVo cells showed lower TCF activity compared to other cells and no significant differences in the TCF activity after 6-gingerol treatment. This result is consistent with previous data, showing that cyclin D1 was decreased in HCT-116, SW480, and Caco-2 cells (Figure 2D).
To define whether inhibition of β-catenin-mediated transcription by 6-gingerol is mediated by βcatenin expression or nuclear localization, we performed immunofluorescence to compare β-catenin localization after 6-gingerol treatment using SW480 cells, which express no endogenous full-length APC protein. The result shows that 6-gingerol treatment decreased the accumulation of β-catenin (red) in the nuclei as shown with DAPI counterstain (blue) (Figure 3F).
6-Gingerol Activates NAG-1 Expression Via a PKCε-and GSK-3β-Dependent Pathway
Next, we investigated the signaling mechanisms responsible for NAG-1 induction by 6-gingerol. Since PKC has been implicated as an activator of NAG-1 [42], RO-31-8220, a PKC inhibitor, was used to assess the potential role of PKC in 6-gingerol-inducedNAG- 1 expression. Pretreatment of cells with 5 μM of RO- 31-8220 attenuated 6-gingerol-induced NAG-1 expression, whereas cyclin D1 was not affected by the RO-31-8220 compound (Figure 4A). We then examined the effects of different PKC inhibitors on the NAG-1 expression. Pretreatment with Rottlerin (a specific inhibitor for PKCδ) or Gö6983 (inhibitor for PKCα, β, γ, δ, ζ at 100 nM) did not attenuate NAG- 1 induction by 6-gingerol (Figure 4B). Because RO- 31-8220 also inhibits PKCα, β, γ, and ε, it is hypothesized that PKCε may play a role in the induction of NAG-1 by 6-gingerol. To confirm the role of PKCε in NAG-1 protein induction by 6- gingerol, the cells were transfected with WT, or DN form of HA-tagged PKCε expression vector and then treated with 6-gingerol for 24 h. As shown in Figure 4C, transfection of plasmid expressing DN PKCε attenuated NAG-1 induction by 6-gingerol.We also transfected siRNA of PKCε and treated the cells with vehicle or 6-gingerol for 24 h. As shown in Figure 4D, induction of NAG-1 by 6-gingerol was decreased in the cells transfected with siRNA of PKCε. Taken together, these results demonstrate a contributory role for PKCε in the positive regulation of NAG-1 by 6-gingerol.
Figure 4.
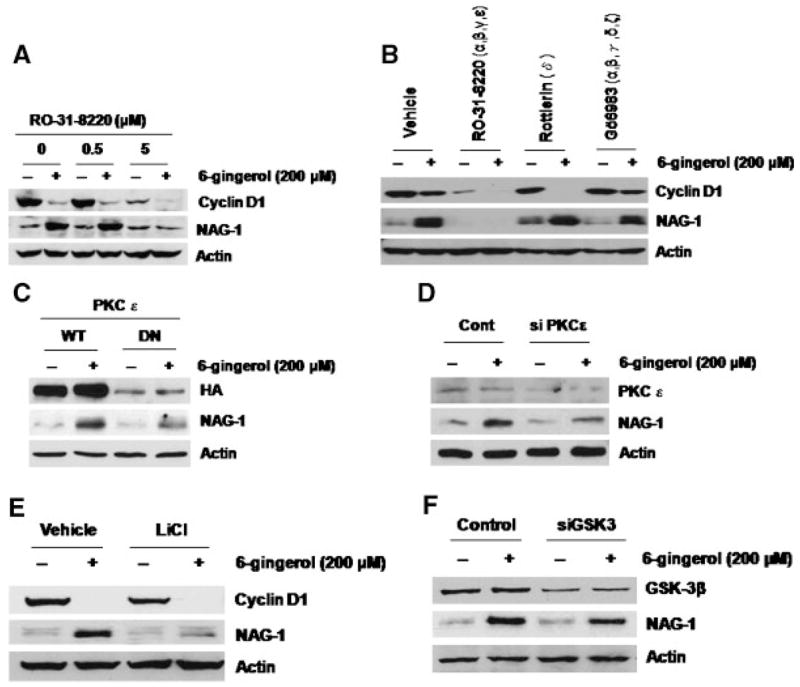
6-Gingerol activates NAG-1 expression via the PKCε and GSK-3β-dependent pathway. (A) HCT-116 cells were pretreated with the indicated concentration of RO-31-8220 for 30 min and then exposed to 200 μM of 6-gingerol for 24 h. Total cell lysates were harvested, and subsequently, 30 μg of total cell lysates were subjected to 14% SDS–PAGE. Cyclin D1, NAG-1, and Actin antibodies were probed. (B) HCT-116 cells were pretreated with 5 μM of RO-31-8220, 0.5 μM of Rottlerin, or 0.5 μM of Gö6983 for 30 min and then exposed to 200 μM of 6-gingerol for 24 h. Western analysis was performed for cyclin D1, NAG-1, and Actin antibodies. (C) HCT-116 cells were transfected with wild type (WT), or dominant negative PKCε expression vector (DN) as described previously [36]. The cells were then treated with 200 μM of 6-gingerol for 24 h. Western analysis was performed for hemagglutinin (HA), NAG-1, and Actin antibodies. (D) HCT-116 cells were transfected with control or PKCε siRNA as described in Materials and Methods section. Then, the cells were treated with 200 μM of 6-gingerol for 24 h. Western analysis was performed using 60 μg of total cell lyates for PKCε and 30 μg of total cell lysates for NAG-1 and Actin antibodies. (E) HCT-116 cells were pretreated with 20 mM of LiCl for 30 min and then exposed to 200 μM of 6-gingerol for 24 h. Western analysis was performed for cyclin D1, NAG-1, and actin antibodies. (F) HCT-116 cells were transfected with control or GSK-3 siRNA. Then, the cells were treated with 200 μM of 6-gingerol for 24 h. Western analysis was performed for GSK-3β, NAG-1, and actin antibodies.
We have reported that inhibition of PI3-kinase enhanced NAG-1 expression [43] and GSK-3β mediated conjugated linoleic acid-induced NAG-1 expression [29]. To investigate the possible regulatory effect of GSK-3 on NAG-1 induction, the cells were pretreated with LiCl, a potent GSK-3 inhibitor, or transfected with siRNA of GSK-3, followed by incubation with 6-gingerol for 24 h. Pretreatment with LiCl as well as transfection with GSK-3 siRNA attenuated NAG-1 induction, but not cyclin D1, by 6- gingerol (Figure 4E and F), indicating that GSK-3 plays an important role in 6-gingerol-induced NAG- 1, but not cyclin D1 expression.
DISCUSSION
Accumulating evidence demonstrates the inhibitory effects of 6-gingerol on the development of cancer [4–9]. However, underlying mechanisms by which 6-gingerol affects human CRC have not been determined. In this study, we demonstrated that 6- gingerol inducedG1 cell cycle arrest and apoptosis in human colorectal cancer cells. 6-Gingerol suppressed cyclin D1 expression by inhibiting the transcriptional regulation and by activating proteolysis of cyclin D1. In addition, 6-gingerol activated NAG-1 expression through the activation of the PKCε and GSK3 pathways. These results demonstrate that 6-gingerol is a pungent compound that affects many pathways involved in tumorigenesis.
Colorectal tumorigenesis is affected by many signaling pathways including COX-2, β-catenin, p53, APC, and mismatch repair. 6-Gingerol uniformly inhibits cell proliferation in various human colorectal cancer models, including HCT-116, SW480, HT-29, LoVo, and Caco-2 cells, suggesting that suppression of cell growth by 6-gingerol is a common phenomenon in human CRC. However, the influence of 6-gingerol on apoptosis is inconsistent, inducing it in HCT-116, SW480, and LoVo cells, but not changing it in HT-29 and Caco-2 cells (Figure 1B). Because HT-29 and Caco-2 cells are COX- 2 expressing cells, while HCT-116, SW480, and LoVo cells are COX-2 negative cells [44], it is likely that 6- gingerol-induced apoptosis is affected by COX-2 expression. Along with this finding, we do not exclude the possibility that inflammatory status may affect 6-gingerol’s effects. Therefore, further studies are needed to elucidate the involvement of COX-2 in NAG-1-mediated apoptosis by 6-gingerol.
6-Gingerol induced G1 arrest in HCT-116 cells and LoVo cells, but not in SW480. It is notable that HCT- 116 and LoVo cells are p53-WT while SW480 is p53 mutant [45]. It has been known that inactivation of p53 results in chemoresistance by chemopreventive drugs in many types of cancer cells including colon cancer cell lines. Actually, colon tumor cell lines expressing wild-type p53 are more sensitive to 5-aza- CdR-mediated growth arrest [46], and p53 mutation decreased sensitivity to the G1/S interface of the cell cycle in prostate cancer cells [47]. Thus, it is assumed that effect of 6-gingerol on cell cycle might be dependent on p53 status.
The effect of 6-gingerol on β-catenin/TCF-dependent gene transcription can be important for the 6- gingerol induced anti-tumorigenesis. The importance of this effect is clearly indicated by the reduced expression of cyclin D1, a protein that plays an important role in cell-cycle transitions. 6-Gingerol led to a sustained suppression of cyclin D1 levels, a result consistent with the inhibition of G1 to S transitions and hypophosphorylation of p-Rb in HCT-116 cells. Our results indicate that there are multiple mechanisms by which 6-gingerol represses cyclin D1 expression. One of these mechanisms is through transcriptional regulation. The mRNA of cyclinD1 was decreased by 6-gingerol, and the cyclin D1 promoter assay, as determined by the reporter gene, indicated that the 6-gingerol response element is located between −163 and +130 of the 5′ region of the cyclin D1 promoter. Indeed, it has been reported that the TCF/LEF site is located in this region of the promoter and plays an important role in β-catenin-dependent transcriptional regulation of cyclin D1 [14]. In our studies, 6-gingerol decreased localization of β-catenin, which is followed by the inhibition of cyclin D1 expression at the transcriptional level. Another mechanism to suppress cyclin D1 expression is the activation of proteasome degradation. Our results are similar to those previously reported with curcumin [38], retinoic acid [39], and troglitazone [40], which downregulate cyclin D1 by proteolysis. This bidirectional downregulation of cyclin D1 supports previous reports that curcumin suppresses cyclin D1 transactivation as well as activates proteolysis [38].
GSK-3β is one of the primary target genes of the PI3K/AKT pathway and mediates apoptotic signals. In our previous study, transfection of specific GSK-3 siRNA blocked the induction of NAG-1 by the PI3K inhibitor [43], and GSK3 is the critical mediator of NAG-1 regulation by conjugated linoleic acid and LY294002 [29,43]. The current study supports that GSK-3β acts as an upstream target of NAG-1 expression and is responsible for NAG-1 induction by 6- gingerol. In addition, we found that PKCε is another mediator of NAG-1 induction by 6-gingerol. PKC constitutes a family of serine-threonine kinases, which are classified into three major groups based on their structure and activation mechanisms: conventional (α, βI, βII, γ), novel (δ, ε, η, θ, μ), and atypical (ζ, λ) [17]. Of the novel PKC isoforms, colon carcinoma cell lines express predominantly the PKCε and PKCδ isoforms [48]. Although PKCδ mediates NAG-1 expression in prostate cancer cells [42], our studies using PKC inhibitors and interferences of PKCε showed that PKCε at least partially mediates NAG-1 expression by 6-gingerol. PKCε’s effects on apoptosis and tumorigenesis are controversial. PKCε has been known to stimulate proliferation and inhibit apoptosis in prostate [18], glioma [19], and squamous carcinoma cells [20]. On the other hand, PKCε mediates the ability of vitamin D3 to prevent adenomas in colon tumors [21], and overexpression of PKCε sensitized LNCaP cells to induce apoptosis [22]. In addition, inactivation of PKCε using dominant- negative transfection has showed marked resistance to apoptosis in thyroid carcinoma cells [23]. Thus, the effect of PKCε on apoptosis may differ in tissues and cell types. In this study, we have shown that PKCε plays a pivotal role in 6-gingerol-induced NAG-1 expression, probably resulting in the induction of 6-gingerol-induced apoptosis.
The suppression of cyclin D1 and induction of NAG-1 by 6-gingerol were observed at 150 and 200 μM concentrations. This is in agreement with other observations that at least 300 μMof 6-gingerol was required to induce apoptosis [7,8]. It is believed that a higher concentration of 6-gingerol is required for inducing apoptosis in CRC and leukemia, but lower concentrations (30–50 μMof 6-gingerol) were effective to inhibit angiogenesis in endothelial cells [9] and skin carcinoma [5]. In addition, the large intestinal epithelia, including colon and rectum, could be highlighted as logical target tissue to further explore the relationship between 6-gingerol and colorectal cancer prevention. Investigation of ginger accumulation in the hindgut would serve to expand our understanding of the role of 6-gingerol in prevention of colorectal cancers. The efficacy of some metabolites of 6-gingerol will be particularly interesting because 6-gingerol in rats is biotransformed to various metabolites by gut flora and liver enzyme when orally administered [49]. In this regard, it is important that future studies expand beyond in vitro cell culture models to relevant in vivo models, addressing absorption and distribution of 6-gingerol and its metabolites.
In conclusion, the current study provides information on cellular events of pro-apoptotic and antitumorigenic activity by 6-gingerol. As depicted in Figure 5, 6-gingerol inhibits the transcription of cyclin D1 by suppression of β-catenin translocation into the nucleus and subsequent lowered β-catenin signaling in the nucleus. 6-Gingerol also leads to an increase of cyclin D1 proteolysis through proteosomal degradation. Downregulation of cyclin D1 results in suppression of Rb phosphorylation, resulting in cell growth arrest. In addition, 6-gingerol upregulates NAG-1 expression through GSK-3β and PKCε-mediated pathways. The resulting NAG-1 activation induces an apoptosis signaling pathway in colorectal cancer cells.
Figure 5.

Proposed mechanism by which 6-gingerol induces apoptosis and suppresses cell growth in human CRC. 6-Gingerol can activate GSK-3 and PKCε to increase NAG-1 expression and downregulate cyclin D1 by the inactivation of β-catenin signaling and increase of proteolysis. Upregulation of NAG-1 and downregulation of cyclin D1 by 6-gingerol results in the induction of apoptosis and cell cycle arrest in human colorectal cancer cells.
Acknowledgments
We thank Dr. Jae-Won Soh (Inha University, Korea) for providing the PKC expression vectors, Misty Bailey for her critical reading of manuscript, and Dianne Trent, Dr. Heon-Suk Lee and Dr. Guojun Zhao for their technical assistance.
Abbreviations
- CRC
colorectal carcinoma
- GSK
glycogen synthase kinase
- PKC
protein kinase C
- NSAIDs
nonsteroidal anti-inflammatory drugs
- NAG-1
NSAID-activated gene-1
- siRNA
small interfering RNA
- HA
hemagglutinin
- DAPI
4′,6-diamidino-2′-phenylindole dihydrochloride
- CHX
cycloheximide
- WT
wild type
References
- 1.Jemal A, Siegel R, Ward E, et al. Cancer statistics, 2006. CA Cancer J Clin. 2006;56:106–130. doi: 10.3322/canjclin.56.2.106. [DOI] [PubMed] [Google Scholar]
- 2.Surh YJ. Cancer chemoprevention with dietary phytochemicals. Nat Rev Cancer. 2003;3:768–780. doi: 10.1038/nrc1189. [DOI] [PubMed] [Google Scholar]
- 3.Dedov VN, Tran VH, Duke CC, et al. Gingerols: A novel class of vanilloid receptor (VR1) agonists. Br J Pharmacol. 2002;137:793–798. doi: 10.1038/sj.bjp.0704925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Park KK, Chun KS, Lee JM, Lee SS, Surh YJ. Inhibitory effects of [6]-gingerol, a major pungent principle of ginger, on phorbol ester-induced inflammation, epidermal ornithine decarboxylase activity and skin tumor promotion in ICR mice. Cancer Lett. 1998;129:139–144. doi: 10.1016/s0304-3835(98)00081-0. [DOI] [PubMed] [Google Scholar]
- 5.Kim SO, Kundu JK, Shin YK, et al. [6]-Gingerol inhibits COX-2 expression by blocking the activation of p38 MAP kinase and NF-kappaB in phorbol ester-stimulated mouse skin. Oncogene. 2005;24:2558–2567. doi: 10.1038/sj.onc.1208446. [DOI] [PubMed] [Google Scholar]
- 6.Suzuki F, Kobayashi M, Komatsu Y, Kato A, Pollard RB. Keishi-ka-kei-to, a traditional Chinese herbal medicine, inhibits pulmonary metastasis of B16 melanoma. Anticancer Res. 1997;17:873–878. [PubMed] [Google Scholar]
- 7.Lee E, Surh YJ. Induction of apoptosis in HL-60 cells by pungent vanilloids, [6]-gingerol and [6]-paradol. Cancer Lett. 1998;134:163–168. doi: 10.1016/s0304-3835(98)00253-5. [DOI] [PubMed] [Google Scholar]
- 8.Bode AM, Ma WY, Surh YJ, Dong Z. Inhibition of epidermal growth factor-induced cell transformation and activator protein 1 activation by [6]-gingerol. Cancer Res. 2001;61:850–853. [PubMed] [Google Scholar]
- 9.Kim EC, Min JK, Kim TY, et al. [6]-Gingerol, a pungent ingredient of ginger, inhibits angiogenesis in vitro and in vivo. Biochem Biophys Res Commun. 2005;335:300–308. doi: 10.1016/j.bbrc.2005.07.076. [DOI] [PubMed] [Google Scholar]
- 10.Widelitz RB, Jiang TX, Lu J, Chuong CM. Beta-catenin in epithelial morphogenesis: Conversion of part of avian foot scales into feather buds with a mutated beta-catenin. Dev Biol. 2000;219:98–114. doi: 10.1006/dbio.1999.9580. [DOI] [PubMed] [Google Scholar]
- 11.Polakis P. Wnt signaling and cancer. Genes Dev. 2000;14:1837–1851. [PubMed] [Google Scholar]
- 12.Bienz M. APC: The plot thickens. Curr Opin Genet Dev. 1999;9:595–603. doi: 10.1016/s0959-437x(99)00016-7. [DOI] [PubMed] [Google Scholar]
- 13.Shtutman M, Zhurinsky J, Simcha I, et al. The cyclin D1 gene is a target of the beta-catenin/LEF-1 pathway. Proc Natl Acad Sci USA. 1999;96:5522–5527. doi: 10.1073/pnas.96.10.5522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tetsu O, McCormick F. Beta-catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature. 1999;398:422–426. doi: 10.1038/18884. [DOI] [PubMed] [Google Scholar]
- 15.He TC, Sparks AB, Rago C, et al. Identification of c-MYC as a target of the APC pathway. Science. 1998;281:1509–1512. doi: 10.1126/science.281.5382.1509. [DOI] [PubMed] [Google Scholar]
- 16.He TC, Chan TA, Vogelstein B, Kinzler KW. PPARdelta is an APC-regulated target of nonsteroidal anti-inflammatory drugs. Cell. 1999;99:335–345. doi: 10.1016/s0092-8674(00)81664-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Soh JW, Lee YS, Weinstein IB. Effects of regulatory domains of specific isoforms of protein kinase C on growth control and apoptosis in MCF-7 breast cancer cells. J Exp Ther Oncol. 2003;3:115–126. doi: 10.1046/j.1359-4117.2003.01087.x. [DOI] [PubMed] [Google Scholar]
- 18.McJilton MA, Van Sikes C, Wescott GG, et al. Protein kinase Cepsilon interacts with Bax and promotes survival of human prostate cancer cells. Oncogene. 2003;22:7958–7968. doi: 10.1038/sj.onc.1206795. [DOI] [PubMed] [Google Scholar]
- 19.Okhrimenko H, Lu W, Xiang C, Hamburger N, Kazimirsky G, Brodie C. Protein kinase C-epsilon regulates the apoptosis and survival of glioma cells. Cancer Res. 2005;65:7301–7309. doi: 10.1158/0008-5472.CAN-05-1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Verma AK, Wheeler DL, Aziz MH, Manoharan H. Protein kinase Cepsilon and development of squamous cell carcinoma, the nonmelanoma human skin cancer. Mol Carcinog. 2006;45:381–388. doi: 10.1002/mc.20230. [DOI] [PubMed] [Google Scholar]
- 21.Wali RK, Bissonnette M, Khare S, et al. Protein kinase C isoforms in the chemopreventive effects of a novel vitamin D3 analogue in rat colonic tumorigenesis. Gastroenterology. 1996;111:118–126. doi: 10.1053/gast.1996.v111.pm8698190. [DOI] [PubMed] [Google Scholar]
- 22.Powell CT, Yin L. Overexpression of PKCepsilon sensitizes LNCaP human prostate cancer cells to induction of apoptosis by bryostatin 1. Int J Cancer. 2006;118:1572–1576. doi: 10.1002/ijc.21511. [DOI] [PubMed] [Google Scholar]
- 23.Knauf JA, Ouyang B, Croyle M, Kimura E, Fagin JA. Acute expression of RET/PTC induces isozyme-specific activation and subsequent downregulation of PKCepsilon in PCCL3 thyroid cells. Oncogene. 2003;22:6830–6838. doi: 10.1038/sj.onc.1206829. [DOI] [PubMed] [Google Scholar]
- 24.Baek SJ, Kim KS, Nixon JB, Wilson LC, Eling TE. Cyclooxygenase inhibitors regulate the expression of a TGF-beta superfamily member that has proapoptotic and antitumorigenic activities. Mol Pharmacol. 2001;59:901–908. [PubMed] [Google Scholar]
- 25.Kim KS, Baek SJ, Flake GP, Loftin CD, Calvo BF, Eling TE. Expression and regulation of nonsteroidal anti-inflammatory drug-activated gene (NAG-1) in human and mouse tissue. Gastroenterology. 2002;122:1388–1398. doi: 10.1053/gast.2002.32972. [DOI] [PubMed] [Google Scholar]
- 26.Baek SJ, Okazaki R, Lee SH, et al. Nonsteroidal anti-inflammatory drug-activated gene-1 over expression in transgenic mice suppresses intestinal neoplasia. Gastroenterology. 2006;131:1553–1560. doi: 10.1053/j.gastro.2006.09.015. [DOI] [PubMed] [Google Scholar]
- 27.Baek SJ, Kim JS, Nixon JB, DiAugustine RP, Eling TE. Expression of NAG-1, a transforming growth factor-beta superfamily member, by troglitazone requires the early growth response gene EGR-1. J Biol Chem. 2004;279:6883–6892. doi: 10.1074/jbc.M305295200. [DOI] [PubMed] [Google Scholar]
- 28.Yamaguchi K, Lee SH, Eling TE, Baek SJ. A novel peroxisome proliferator-activated receptor gamma ligand, MCC-555, induces apoptosis via posttranscriptional regulation of NAG- 1 in colorectal cancer cells. Mol Cancer Ther. 2006;5:1352–1361. doi: 10.1158/1535-7163.MCT-05-0528. [DOI] [PubMed] [Google Scholar]
- 29.Lee SH, Yamaguchi K, Kim JS, et al. Conjugated linoleic acid stimulates an anti-tumorigenic protein NAG-1 in an isomer specific manner. Carcinogenesis. 2006;27:972–981. doi: 10.1093/carcin/bgi268. [DOI] [PubMed] [Google Scholar]
- 30.Lee SH, Kim JS, Yamaguchi K, Eling TE, Baek SJ. Indole-3- carbinol and 3,3’-diindolylmethane induce expression of NAG-1 in a p53-independent manner. Biochem Biophys Res Commun. 2005;328:63–69. doi: 10.1016/j.bbrc.2004.12.138. [DOI] [PubMed] [Google Scholar]
- 31.Baek SJ, Wilson LC, Eling TE. Resveratrol enhances the expression of non-steroidal anti-inflammatory drug-activated gene (NAG-1) by increasing the expression of p53. Carcinogenesis. 2002;23:425–434. doi: 10.1093/carcin/23.3.425. [DOI] [PubMed] [Google Scholar]
- 32.Wilson LC, Baek SJ, Call A, Eling TE. Nonsteroidal anti-inflammatory drug-activated gene (NAG-1) is induced by genistein through the expression of p53 in colorectal cancer cells. Int J Cancer. 2003;105:747–753. doi: 10.1002/ijc.11173. [DOI] [PubMed] [Google Scholar]
- 33.Baek SJ, Kim JS, Jackson FR, Eling TE, McEntee MF, Lee SH. Epicatechin gallate-induced expression of NAG-1 is associated with growth inhibition and apoptosis in colon cancer cells. Carcinogenesis. 2004;25:2425–2432. doi: 10.1093/carcin/bgh255. [DOI] [PubMed] [Google Scholar]
- 34.Yamaguchi K, Liggett JL, Kim NC, Baek SJ. Anti-proliferative effect of horehound leaf and wild cherry bark extracts on human colorectal cancer cells. Oncol Rep. 2006;15:275–281. [PMC free article] [PubMed] [Google Scholar]
- 35.Baek SJ, Eling TE. Changes in gene expression contribute to cancer prevention by COX inhibitors. Prog Lipid Res. 2006;45:1–16. doi: 10.1016/j.plipres.2005.10.001. [DOI] [PubMed] [Google Scholar]
- 36.Soh JW, Weinstein IB. Roles of specific isoforms of protein kinase C in the transcriptional control of cyclin D1 and related genes. J Biol Chem. 2003;278:34709–34716. doi: 10.1074/jbc.M302016200. [DOI] [PubMed] [Google Scholar]
- 37.Deshpande A, Sicinski P, Hinds PW. Cyclins and cdks in development and cancer: A perspective. Oncogene. 2005;24:2909–2915. doi: 10.1038/sj.onc.1208618. [DOI] [PubMed] [Google Scholar]
- 38.Mukhopadhyay A, Banerjee S, Stafford LJ, Xia C, Liu M, Aggarwal BB. Curcumin-induced suppression of cell proliferation correlates with down-regulation of cyclin D1 expression and CDK4-mediated retinoblastoma protein phosphorylation. Oncogene. 2002;21:8852–8861. doi: 10.1038/sj.onc.1206048. [DOI] [PubMed] [Google Scholar]
- 39.Spinella MJ, Freemantle SJ, Sekula D, Chang JH, Christie AJ, Dmitrovsky E. Retinoic acid promotes ubiquitination and proteolysis of cyclin D1 during induced tumor cell differentiation. J Biol Chem. 1999;274:22013–22018. doi: 10.1074/jbc.274.31.22013. [DOI] [PubMed] [Google Scholar]
- 40.Huang JW, Shiau CW, Yang YT, et al. Peroxisome proliferator-activated receptor gamma-independent ablation of cyclin D1 by thiazolidinediones and their derivatives in breast cancer cells. Mol Pharmacol. 2005;67:1342–1348. doi: 10.1124/mol.104.007732. [DOI] [PubMed] [Google Scholar]
- 41.Nelson WJ, Nusse R. Convergence of Wnt, beta-catenin, and cadherin pathways. Science. 2004;303:1483–1487. doi: 10.1126/science.1094291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shim M, Eling TE. Protein kinase C-dependent regulation of NAG-1/placental bone morphogenic protein/MIC-1 expression in LNCaP prostate carcinoma cells. J Biol Chem. 2005;280:18636–18642. doi: 10.1074/jbc.M414613200. [DOI] [PubMed] [Google Scholar]
- 43.Yamaguchi K, Lee SH, Eling TE, Baek SJ. Identification of nonsteroidal anti-inflammatory drug-activated gene (NAG- 1) as a novel downstream target of phosphatidylinositol 3- kinase/AKT/GSK-3beta pathway. J Biol Chem. 2004;279:49617–49623. doi: 10.1074/jbc.M408796200. [DOI] [PubMed] [Google Scholar]
- 44.Shao J, Sheng H, Inoue H, Morrow JD, DuBois RN. Regulation of constitutive cyclooxygenase-2 expression in colon carcinoma cells. J Biol Chem. 2000;275:33951–33956. doi: 10.1074/jbc.M002324200. [DOI] [PubMed] [Google Scholar]
- 45.Violette S, Poulain L, Dussaulx E, et al. Resistance of colon cancer cells to long-term 5-fluorouracil exposure is correlated to the relative level of Bcl-2 and Bcl-X(L) in addition to Bax and p53 status. Int J Cancer. 2002;98:498–504. doi: 10.1002/ijc.10146. [DOI] [PubMed] [Google Scholar]
- 46.Karpf AR, Moore BC, Ririe TO, Jones DA. Activation of the p53 DNA damage response pathway after inhibition of DNA methyltransferase by 5-aza-2’-deoxycytidine. Mol Pharmacol. 2001;59:751–757. [PubMed] [Google Scholar]
- 47.Sullivan GF, Yang JM, Vassil A, Yang J, Bash-Babula J, Hait WN. Regulation of expression of the multidrug resistance protein MRP1 by p53 in human prostate cancer cells. J Clin Invest. 2000;105:1261–1267. doi: 10.1172/JCI9290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Looby E, Long A, Kelleher D, Volkov Y. Bile acid deoxycholate induces differential subcellular localisation of the PKC isoenzymes beta 1, epsilon and delta in colonic epithelial cells in a sodium butyrate insensitive manner. Int J Cancer. 2005;114:887–895. doi: 10.1002/ijc.20803. [DOI] [PubMed] [Google Scholar]
- 49.Nakazawa T, Ohsawa K. Metabolism of [6]-gingerol in rats. Life Sci. 2002;70:2165–2175. doi: 10.1016/s0024-3205(01)01551-x. [DOI] [PubMed] [Google Scholar]