

Abstract
Objective
Osteoarthritis (OA) is associated with increased levels of reactive nitrogen and oxygen species and pro-inflammatory cytokines, such as interleukin-1 (IL-1). Nitric oxide (NO) can mediate a number of the catabolic effects of IL-1 in articular cartilage. The aims of this study were to determine if OA cartilage shows evidence of DNA damage, and if IL-1 could induce DNA damage in non-OA cartilage by increasing NO or superoxide.
Methods
Articular chondrocytes were isolated from porcine femoral condyles and embedded in 1.2% alginate. The effects of 24 hrs incubation with IL-1, the nitric oxide synthase 2 (NOS2) selective inhibitor, the free radical scavenger SOD, the NO donor NOC18, or the combined NO and peroxynitrite donor SIN-1 on DNA damage were tested, using the “comet” assay. NO production was measured using the Griess assay. The type of oxidative damage present was assessed using a modified comet assay.
Results
OA cartilage had significantly more DNA damage than non-OA cartilage (p < 0.001). IL-1 caused an increase in DNA damage (p < 0.01), which was associated with increased NO production (p < 0.01). Both oxidative DNA strand breaks and base modifications of purines and pyrimidines were observed. IL-1-induced DNA damage was inhibited by a NOS2 inhibitor or by superoxide dismutase (p < 0.01). Furthermore, NOC-18 or SIN-1 caused DNA damage (p < 0.001).
Conclusion
Our work shows chondrocytes in osteoarthritic cartilage exhibit DNA damage, and that IL-1 induces DNA damage and reactive oxygen and nitrogen species in non-OA chondrocytes in alginate.
Osteoarthritis (OA) is an age-associated joint disorder that affects the quality of life of over 20 million Americans1. One of the major symptoms of OA is pain associated with articular cartilage loss and degeneration. Articular cartilage is the tissue at the ends of diarthrodial joints, which function to allow a smooth, painless, low-frictional movement of synovial joints. This tissue contains a sparsely distributed population of highly specialized cells (chondrocytes) that are embedded within an extracellular matrix composed of collagens, proteoglycans, and noncollagenous proteins. Articular cartilage is avascular, aneural, and alymphatic, providing the tissue with a limited capacity to repair itself should it become damaged. At present, the sequence of events associated with cartilage degradation is not fully delineated.
Evidence suggests that abnormal loading of the joint and increased pro-inflammatory cytokines are risk factors for OA2,3. Pro-inflammatory mediators such as nitric oxide (NO) and other reactive nitrogen and oxygen species (RNS and ROS)4–6 are increased in OA. The increased levels of these reactive species have been correlated to increased levels of inflammatory cytokines, such as interleukin-1 (IL-1). IL-1 is implicated in the degeneration of cartilage due to its induction of proteoglycan loss and matrix degradation7. Elevated levels of IL-1 occur in the synovial fluid and cartilage tissue of patients with OA compared to healthy individuals8, implying a role in disease pathogenesis. IL-1 receptor antagonist, a natural competitor of IL-1, suppresses cartilage loss, further supporting the role of IL-1 in cartilage breakdown9.
Both IL-1 and mechanical loading of cartilage increase NO production10–12 by up-regulating nitric oxide synthase 2 (NOS2), which catalyzes the formation of NO and citrulline from arginine in the presence of molecular oxygen and NADPH. NOS2 inhibitors can inhibit the progression of OA in experimental animal models13, and osteoarthritic joint pathology is significantly inhibited in the collagen–induced arthritis model in NOS2 deficient mice14.
Some of the destructive effects of NO in articular cartilage are linked to the ability of NO to combine with superoxide anions (O2−) to generate peroxynitrite15,16. Endogenous antioxidants such as superoxide dismutase (SOD) can serve to protect against oxidative stress. Extracellular SOD (EC-SOD) is decreased in osteoarthritic cartilage17, indicating the potential importance of EC-SOD to articular cartilage. Gene expression profiling has identified decreased expression of antioxidant defense genes in human OA cartilage, a situation that may contribute to increased oxidative stress18. The effects of pro-inflammatory cytokines, ROS and RNS, on DNA integrity in articular cartilage have not previously been investigated. Oxidative stress can induce premature chondrocyte senescence via DNA damage in serially passaged chondrocytes19. DNA damage is noted in the synovial tissue of rheumatoid arthritis and OA patients. Microsatellite instability, a form of DNA mutation, is increased in the RA synovium compared with OA synovium20, correlating with increased DNA mismatch repair enzymes in synovial tissue of RA and OA patients compared with non-arthritic tissue21. Prior studies have not investigated the occurrence of microsatellite instability and DNA repair protein within articular cartilage.
Since articular cartilage in diseased states such as OA exhibit elevated levels of NO and IL-1, and IL-1 can induce chondrocytes to produce NO in vitro, we investigated the susceptibility of porcine articular chondrocytes to oxidative DNA damage. We sought (1) to determine if OA cartilage contains more DNA damage than non-OA cartilage, and (2) to determine the role of NO in IL-1-induced DNA damage in non-osteoarthritic chondrocytes embedded in alginate.
Materials and Methods
Macroscopic grading of articular cartilage
The femoral condyles of 2–3 year-old, female, ex-breeder pigs were obtained from the local slaughterhouse. Pigs can develop a spontaneous OA, and the incidence increases with age22,23. The articular cartilage on the medial condyle was graded macroscopically according to the Collins scale24. In summary, the principal distinguishing features of each grade in the Collins scale are as follows: Grade 0: Normal healthy joint with smooth cartilage; Grade I: Superficial flaking of cartilage in areas of pressure and movement; Grade II: More extensive destruction of cartilage not denuding bone; Grade III: Total loss of cartilage in one or more pressure areas and obvious marginal osteophytes; Grade IV: Complete loss of cartilage from large areas with eburnation of bone; prominent osteophytes. Grades III and IV were not used in these studies due to lack of chondrocytes.
Chondrocyte Cell Culture in Alginate Beads
Articular chondrocytes were enzymatically isolated, using pronase for 1 hour and collagenase type II for 2 hours, from site matched, full thickness slices of articular cartilage from the femoral condyles of skeletally mature 2–3 year old pigs and placed into alginate beads (1.2% alginate) at a concentration of 4 × 106 cells/ml. Alginate was used to maintain the chondrocytes in a rounded shape and ensure the collagen II expression associated with chondrocytes. If chondrocytes are grown as a monolayer, dedifferentiation to fibrochondrocytes occurs. Beads were cultured in high glucose DMEM (Gibco, Gaithersburg, MD) with 10% heat inactivated FBS (Hyclone), 0.1 mM non-essential amino acids (Gibco), 10 mM HEPES (Gibco), 100 U/ml penicillin and streptomycin (Gibco) 110 mg/L sodium pyruvate, 2 mM L-glutamine, at 37°C, 5% CO2, 95% air for either 24 or 72 hours prior to treatment
Treatment of Chondrocytes in Culture with Interleukin-1α
After 24 hrs in culture, chondrocytes encapsulated in alginate beads were treated for 24 hrs with 0 – 100 ng/ml recombinant porcine IL-α. The effects of the selective NOS2 inhibitor 1400W [N-(3-(Aminomethyl)benzyl)acetamidine, 2 mM, Alexis Chemical Co.] or superoxide dismutase (SOD, 50 µg/ml Sigma Chemical Co.) were tested. 1400W is a slow, tight binding inhibitor of NOS2, while SOD reduces production of peroxynitrite by breaking down superoxide.
We examined the effects of the pure NO donor NOC18 (DETA-NONOate, 0 – 500 µM) or the peroxynitrite generator SIN-1 (3-morpholinosydnonimine, 0 – 500 µM) by culturing with chondrocytes for 24 hrs. NOC18 is a stable NO-amine complex that spontaneously release NO, without cofactors, under physiological conditions. Unlike other NO donors such as nitroglycerin, nitroprusside, and S-nitroso-N-acetyl-L-penicillamine (SNAP), by-products of NO release do not interfere with cell activities. SIN-1, which uses molecular oxygen to generate superoxide and NO was chosen as it causes the spontaneous formation of peroxynitrite.
Comet Assay
The single cell gel electrophoresis assay, also known as a comet assay, allows DNA damage to be visualized and quantified at a single cell level. Specifically, the alkaline comet assay allows detection of single and double strand DNA breaks, as well as apurinic or apyrimidinic sites that are alkali labile and form breaks due to repair lesions produced during endogenous DNA repair (base excision or nucleotide excision)25. Chondrocytes were released from the alginate beads with calcium chelation in 55 mM sodium citrate and the alkaline comet assay (Trevigen, Gaithersburg, MD)26 was performed. Since changes in experimental procedures, such as electrophoresis times and lysis conditions may increase or decrease the sensitivity of the assay to detect DNA damage, our assays were carefully controlled by carrying out all groups from one experiment on the same slide.
The modified comet assay enables the detection of specific oxidative base lesions through the use of repair enzymes. This assay is based on the addition of specific glycosylases, which cleave the modified base from the DNA strand forming a single strand break27. The Fpg enzyme reveals oxidized purines by recognizing and binding duplex DNA containing oxidatively damaged bases, such as 8-oxo-2’-deoxyguanosine (8-oxoG), and formamidopyrimidines. EndoIII reveals oxidized pyrimidines. EndoIII enzyme recognizes and binds duplex DNA containing oxidatively damaged bases such as thymine and uracil glycol, thymine and cytosine hydrates and urea. Chondrocytes were cultured in alginate beads as above and exposed to 10 ng/ml IL-1 and processed as the comet assay. After the lysis step, the slides were washed three times for 5 min each in Buffer A (10 mM HEPES-KOH (pH 7.4), 10 mM EDTA (pH 8.0), and 0.1 M KCl) and tapped dry. The agarose-embedded cells were covered with either 1 µg/ml Fpg in Buffer A plus 100 µg/ml BSA, or 1 µg/ml Endo III in buffer A or buffer A alone and incubated in a moist atmosphere at 37°C for 1 h. The slides were then immersed in freshly prepared alkaline solution (pH > 13) and continued through the steps of the comet assay.
Slides were stained with 1x SYBR® Green I and imaged using a 20x objective on a confocal laser scanning microscope (LSM 510, Zeiss) followed by analysis using CASP™ software28. The Olive Tail Moment (OTM) was used to assess the amount of DNA damage29. The OTM is defined as the fraction of tail DNA multiplied by the distance between the profile centers of gravity for DNA in the head and tail. The distance the DNA moves is related to the size of free or relaxed pieces, while the intensity of the tail is a direct indication of the number of pieces that migrate. Since single- and double-stranded breaks causes DNA to become unwound and free to migrate towards the anode during electrophoresis (Fig. 1), nucleoids of comet-like structure are indicative of DNA damage. Over time, the comet tail length plateaus, but the amount of DNA entering the tail-like region increases. The OTM accounts for this feature. A total of 50 cells per group per joint were analyzed, avoiding edges and damaged areas of the gel, to give a representative result for the population of cells.
Figure 1. Comet assays of non-OA and OA porcine chondrocytes.
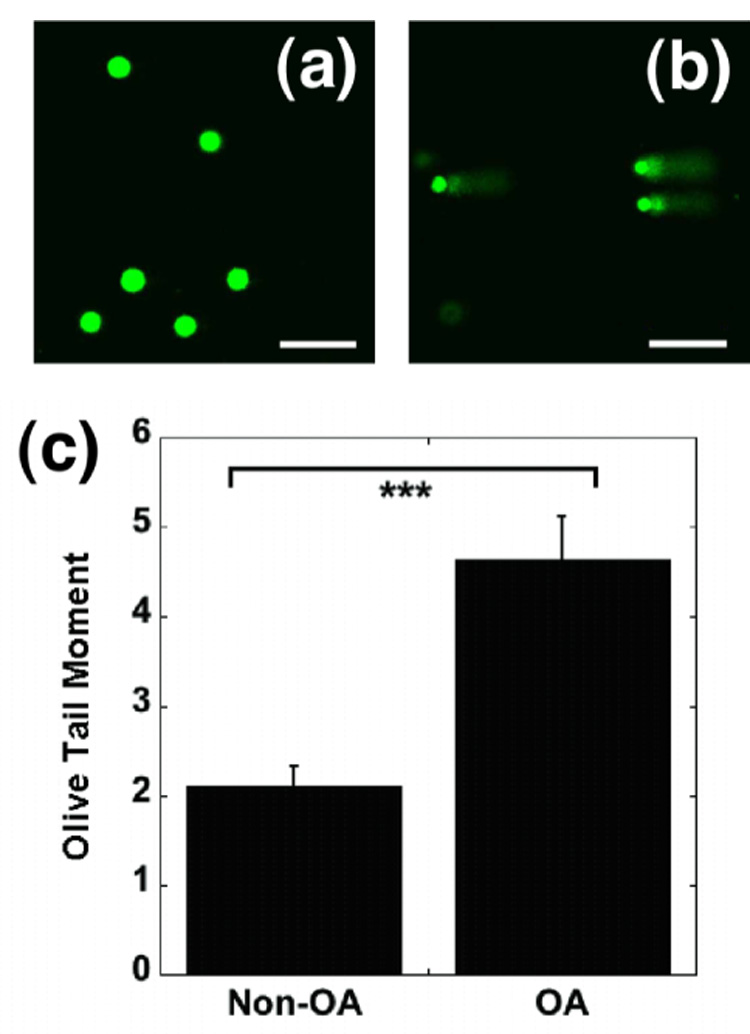
Chondrocytes from the medial femoral condyles of porcine cartilage were graded for OA using the Collins scale and analyzed for DNA damage using the comet assay, 24 hrs after cell isolation. In a typical nuclear profile of undamaged DNA (a), nuclear chromatin is tightly packed into a circular sphere. The nuclear profile of damaged DNA (b) reveals a comet-like appearance with highly fluorescent spherical heads and slightly less fluorescence in the tail, which is aligned in the direction of the anode. With higher levels of DNA damage, the further-fragmented DNA migrates in the electric field to give a longer tail containing more genetic material. Quantitation of DNA fragmentation using the OTM reveals significantly more fragmentation in OA cartilage (c) (Mean ± SEM ; N = 3 pigs with 50 observations per group; *** = p< 0.001. Scale bar represents 100 µm).
NOx Assay
NO production was assessed by measuring the concentration of total nitrate and nitrite (termed “NOx”) in the media as described previously30. This method first converts nitrate to nitrite using nitrate reductase, and then total nitrite is measured using the Griess reagent. NOx levels were normalized to the DNA content of the chondrocytes. Media were removed from beads, and NOx was determined. Chondrocytes in alginate beads were digested in 125 µg/ml papain solution at 60°C for 24 hrs. DNA content of the alginate beads was determined using the fluorescent picoGreen dsDNA quantification assay (Molecular Probes, Eugene, OR) using the diluted bead-papain digest solution.
Cell Viability
Cell viability was determined using the fluorescence-based viability assay (Live/Dead Assay, Molecular Probes). At the end of all culture conditions, the viability of the chondrocytes was not significantly different from control chondrocytes directly after isolation. Cell viability was found to be 95–99% in control or treated cells.
Statistical Analysis
Statistical analysis was performed using Student’s t-test for comparison of DNA damage in OA verses non-OA chondrocytes or analysis of variance with Duncan's post hoc comparison with significance reported at the 95% confidence level for all other comparisons.
Results
DNA damage in articular cartilage
Articular cartilage was harvested from the medial condyles of macroscopically “normal” tissue (Collins Grade O) and osteoarthritic cartilage (Collins Grades I and II). Levels of DNA damage in these cells were determined using the Comet assay. Nucleoids appeared as tightly supercoiled, spheroid structures (Fig. 1a), indicating absence of DNA damage. The nucleoids which showed tail-like structures (“comets”) (Fig. 1b) indicating presence of DNA damage. A significant increase in DNA damage was observed in the OA chondrocytes compared with the non-OA chondrocytes (Fig. 1c). Importantly, a small proportion of the chondrocytes analyzed from OA joints had no evidence of DNA damage as assessed by the comet assay.
Effects of IL-1α on DNA damage in articular cartilage
OA articular cartilage is characterized by an increase in the level of the pro-inflammatory cytokine IL-1. Therefore we investigated the effects of IL-1 on DNA damage in non-OA cartilage. IL-1α caused a concentration-dependent increase in comet tail length. Exposure to 1 ng/ml ≥ IL-1 over a 24h period caused a significant increase in DNA damage as measured by the OTM, compared to untreated non-OA chondrocytes (Fig. 2). There was a significant, concentration-dependent increase in NO production in response to IL-1 compared with control cells.
Figure 2. IL-1 induction of NO chondrocyte IL-1 production and DNA damage.
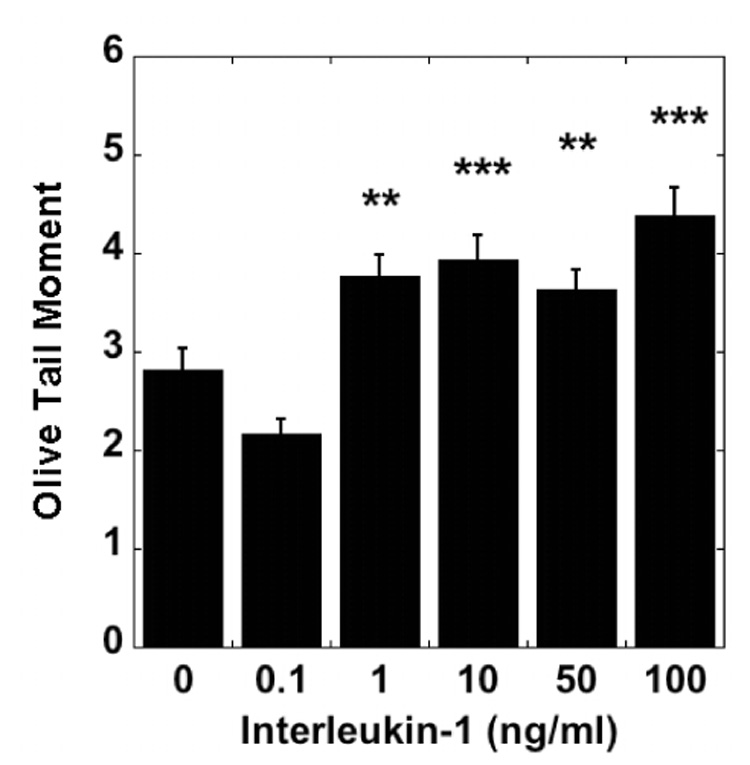
Porcine articular chondrocytes cultured in vitro in the alginate bead system were exposed to 0 - 100 ng/ml IL-1α. DNA damage is expressed as the OTM, (N = 3 pigs, with 50 observations per group). (Mean ± SEM; ** = p < 0.01; *** = p < 0.001).
The effects of the NOS2 inhibitor 1400W, or the free radical scavenger SOD on IL-1-mediated DNA damage in chondrocytes were investigated. A significant reduction in the OTM was seen in chondrocytes incubated with IL-1 and 1400W compared with chondrocytes treated with IL-1 alone. SOD also inhibited IL-1-induced DNA damage (Fig. 3). There was no significant change in levels of DNA damage when chondrocytes were incubated with 2 mM 1400W or 50 µM SOD (Data not shown).
Figure 3. Effects of IL-1, NOS2 inhibitor, and SOD on DNA damage in chondrocytes.
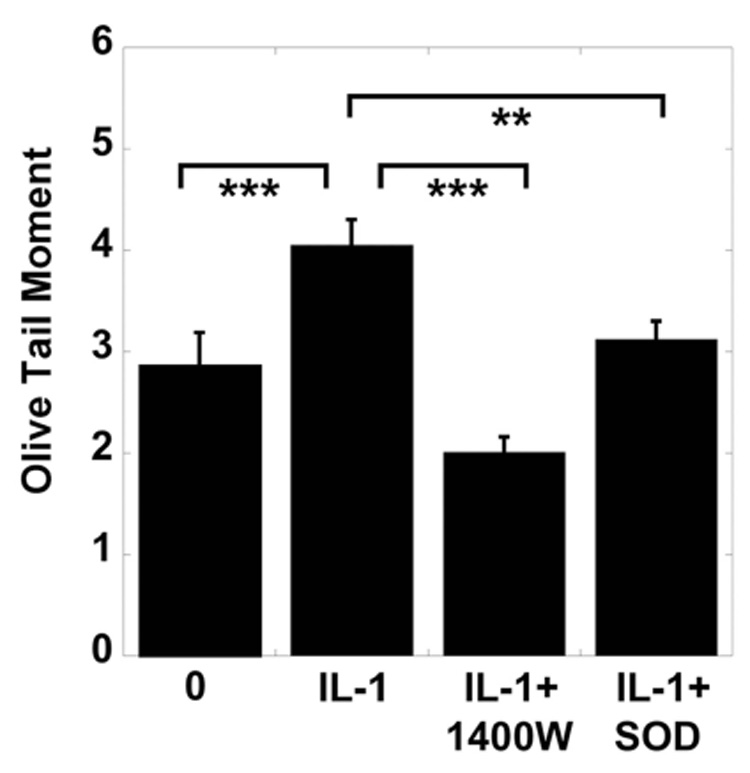
Porcine articular chondrocytes cultured in vitro in alginate bead system were exposed to 0 and 10 ng/ml IL-1, 10 ng/ml IL-1 with 2 mM 1400W or 10 ng/ml IL-1 with 50 µg/ml SOD (N = 3, with 50 observations). DNA damage is expressed as the OTM. (Data represent Mean ± SEM; ** = p < 0.01; *** = p < 0.001).
Effects of NO donors on DNA damage
To further confirm that NO or superoxide could induce DNA damage in non-OA chondrocytes, we tested the effects of NO donors on DNA damage. NOC18, a pure NO donor, caused a concentration-dependent increase in DNA damage (Fig. 4a) and, as predicted more NO was noted in the cultures (Fig. 4b). SIN-1, which releases both NO and superoxide and generates peroxynitrite, had similar effects (Fig. 4c & 4d).
Figure 4. Effects of an NO donor (NOC18) and peroxynitrite generator (SIN-1) on DNA damage in chondrocytes.
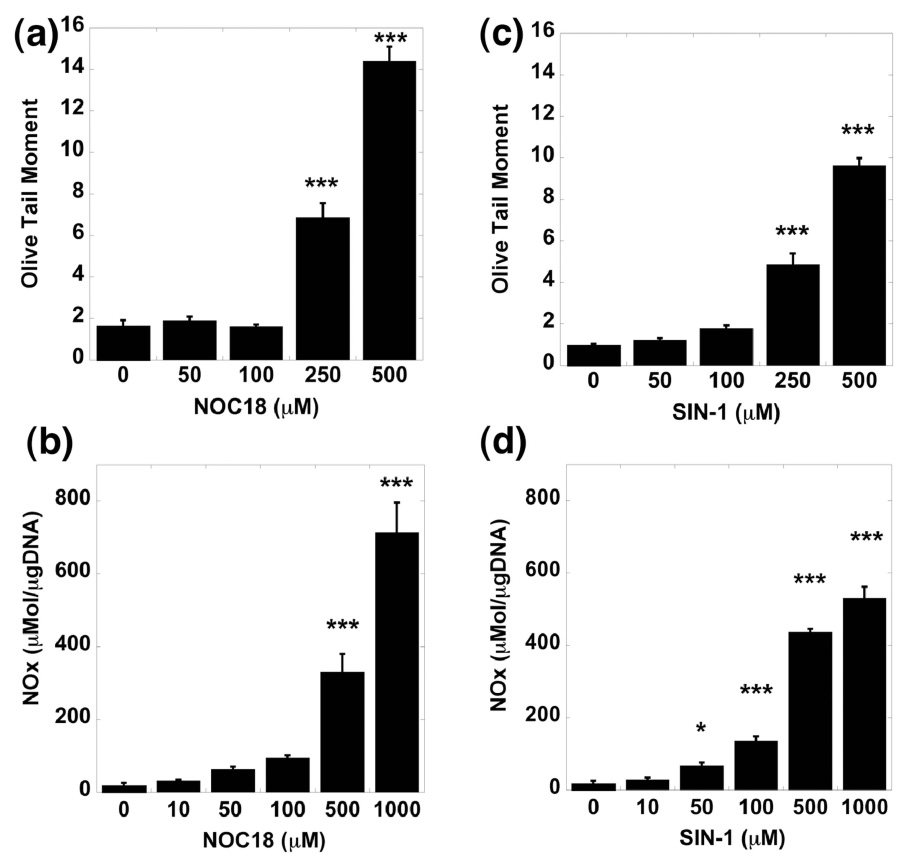
Porcine articular chondrocytes cultured in vitro in an alginate bead system were exposed to 0 to 1000 µM NOC18. (a) DNA damage (OTM) with culture with the NO donor NOC18. (b) NO elaboration after culture with NOC18. Porcine articular chondrocytes cultured in vitro in alginate bead system were exposed to 0 to 1000 µM SIN-1. (c) DNA damage (OTM) with culture with the peroxynitrite generator SIN-1. (d) NO elaboration after culture with SIN-1. (Data represents Mean ± SEM, *= p < 0.05; ** = p < 0.01; *** = p < 0.001; N = 3 pigs, with 50 observations per group).
Identification of the type of DNA damage caused by IL-1
To determine the type of DNA damage occurring, the comet assay was performed in the presence of DNA repair enzymes. Fpg reveals oxidized purines, and Endo III reveals oxidized pyrimidines. The addition of the oxidative DNA damage lesion-specific enzymes Fpg (Fig. 5a) and Endo III (Fig. 5b) caused a significant increase in the OTM of control cells treated with 10 ng/ml IL-1. The mean values of Fpg-sensitive sites were significantly higher in chondrocytes cultured with IL-1 (10 ng/ml) for 24 hours compared to untreated chondrocytes. The mean values of EndoIII-sensitive sites were significantly higher in chondrocytes cultured with IL-1 (10 ng/ml) for 24 hours compared with untreated chondrocytes.
Figure 5. Determination of type of DNA damage induced by IL-1.
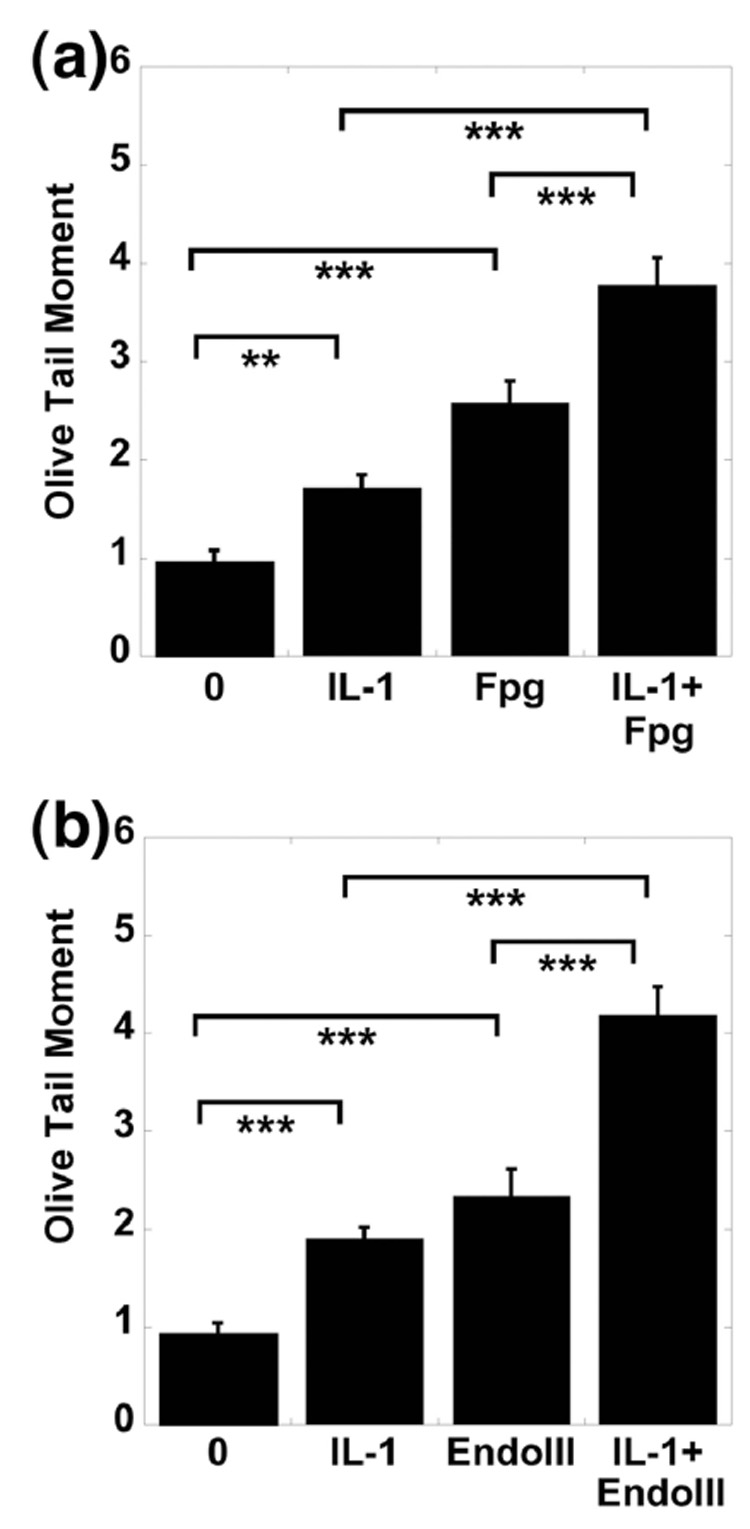
Porcine articular chondrocytes cultured in vitro in alginate bead system were exposed to 0 and 10 ng/ml IL-1, with and without the DNA glycosylase enzymes (a) Fpg or (b) Endo III (N = 3, with 50 observations). Fpg reveals oxidized purines, and Endo III reveals oxidized pyrimidines. DNA damage is expressed as the OTM. (Data represents Mean ± SEM; ** = p < 0.01; *** = p < 0.001. *= p < 0.05; ** = p < 0.01; *** = p < 0.001; N = 3 pigs, with 50 observations per group).
Discussion
Since OA is associated with increased reactive nitrogen and oxygen species and cartilage damage, we sought to determine if there was also cartilage DNA damage and if NO and superoxide might mediate DNA damage in chondrocytes. We found that chondrocytes in OA cartilage exhibit significantly more DNA damage than those in non-OA cartilage. Our in vitro studies showed that IL-1 causes a concentration-dependent increase in DNA damage in chondrocytes in alginate, and this damage is associated with increased NO production. We noted both oxidative DNA strand breaks and base modifications of purines and pyrimidines. IL-1-induced DNA damage is inhibited by a NOS2 inhibitor or by superoxide dismutase, indicating important contributing roles of NO and superoxide in the mechanism of DNA damage. Use of the pure NO donor (NOC-18) or peroxynitrite generator (SIN-1) causes DNA damage of chondrocytes in vitro. A small proportion of OA chondrocytes or non-OA chondrocytes treated with IL-1 or NO donors did not show evidence of DNA damage. Collectively, our data indicate that increased reactive nitrogen and oxygen associated with OA contribute to DNA damage in articular chondrocytes.
Superoxide dismutase did not reduce the level of DNA damage as much as the NOS2-selective inhibitor 1400W. Our data agree with findings in other eukaryotic cell types in which NO can cause DNA damage31–33. Specifically, NO but not superoxide, caused DNA damage in rat islets of Langerhans and insulin-containing HIT-T15 cells treated with IL-1β (0.1 nM)31. However, in our system, superoxide was responsible for some of the IL-1-induced DNA damage. The advantage of 1400W over SOD in reducing IL-1-induced DNA damage in vitro in articular cartilage might be accounted for by differences in the rate constants of the NO/superoxide reaction compared with the superoxide/SOD reaction. The rate constant of the NO/superoxide reaction to form peroxynitrite is 6.7×109 M−1 s−1, a rate constant which is 3.5 times faster than that for the dismutation of superoxide by SOD34.
Although we found that IL-1 and NO damage chondrocyte DNA significantly in our system, no cell death was observed under these conditions. This finding is in agreement with previous studies showing neither IL-1, nor NOC 18 at these concentrations caused chondrocyte cell death15. SIN-1 can cause significant cell death in human chondrocytes cultured in serum free medium15, but SIN-1 did not cause cell death in our system using serum.
Our results demonstrate that Fpg-sensitive and EndoIII-sites in the DNA of chondrocytes are increased by incubation with IL-1α. This suggests NO might mediate formation of both oxidized purines and oxidized pyrimidines. In agreement with others, a significant number of Fpg and EndoIII sensitive sites were present even in the absence of IL-1 treatment35. This is likely related to spontaneous, basal oxidation that occurs normally in oxygen-containing atmospheres35. Oxidative DNA damage in articular chondrocytes has been reported in serially-passaged chondrocytes cultured at 21% O2 for 60–70 days19, but not in primary chondrocytes cultured in the alginate beads system that maintains the chondrocytic cellular phenotype in vitro. However, oxidative protein damage (3-nitrotyrosine formation) has been observed in articular cartilage explants and in articular chondrocytes cultured in alginate36,37 supporting the role of peroxynitrite in cell damage. In our studies, levels of NO are greater than 1 µM giving potential for reactive nitrogen species-mediated modification of proteins, as well as DNA38. Oxidation and nitration of bases are a more severe consequence of ROS and RNS. Many DNA base modifications can occur, but the oxidation of guanine to form 8-oxodG is one of the most common markers of base oxidation39.
The importance of the DNA damage observed in our studies in the pathogenesis of OA is unknown. Daughter cells inheriting oxidative- and nitrative-modified bases in their DNA might lead to mutagenic lesions due to base mis-incorporation opposite the lesion during mitosis. This would lead to base pair trans-versions, which in many tissues, such as gastric, liver, and colon tissue, can be carcinogenic39. However, in articular cartilage, after the completion of skeletal growth, little cell division occurs. Neuronal cells are another example of non-dividing cells. Large amounts of 8-oxoG are formed in the RNA in the neuronal tissues of some neurodegenerative diseases such as Parkinson’s disease, Alzheimer’s disease and Down’s syndrome40.
IL-1 and NO can cause catabolic effects on articular cartilage in vitro41. Free radical-induced DNA damage may modify transcription, causing transcription errors that result in erroneous protein formation40,42. The error rate of RNA transcription is estimated to be much greater for RNA transcription (10−5 per residues) than DNA replication (10−9 per residues). The DNA damage may alter binding of transcription factors43,44, or block gene transcription. A number of gene promoter elements contain a succession of guanine residues in their transcription factor recognition sequences, such as NFκB. Oxidative modification to 8-oxodG can alter the binding affinity of transcription factor NFκB45. Alternatively, the ROS and RNS could incapacitate DNA repair proteins, leading to further DNA damage over time46. Accumulation of damaged DNA, proteins, and lipids may be responsible for disrupting normal cell functions, which might explain the increased incidence of OA with age.
Some cell division may occur in late stage OA47, and the spontaneous replication of chondrocytes with impaired genetic material could result in chondrocyte apoptosis48. Since the development of OA is age-related, damage accumulation over time could be particularly significant when the chondrocyte does divide in the later stages of OA. Epigenetic changes, heritable changes in DNA without changes in the sequence, such as DNA methylation, are possibly important in the pathogenesis of OA49. Changes in DNA methylation and base modification could have a role in altering the chondrocyte phenotype in OA.
Collectively, our findings demonstrate that OA articular chondrocytes contain DNA damage, that high levels of IL-1 induced increases in NO result in DNA damage in non-OA articular chondrocytes in alginate through both strand breaks and base modifications. A NOS2 inhibitor and SOD reduce IL-1-mediated DNA damage. Although the full consequence(s) of chondrocyte DNA damage is not currently known, these results provide further evidence that agents that reduce reactive nitrogen and oxygen species in vivo might be beneficial for the treatment of OA.
Acknowledgements
This study was supported by NIH grants AR49790, AR50245, AG15768 and the VA Research Service.
Grant Support: This study was supported by NIH grants AR49790, AR50245, AG15768 and the VA Research Service.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Buckwalter JA, Saltzman C, Brown T. The impact of osteoarthritis: implications for research. Clin Orthop Relat Res. 2004;427 Suppl:S6–S15. doi: 10.1097/01.blo.0000143938.30681.9d. [DOI] [PubMed] [Google Scholar]
- 2.Guilak F, Fermor B, Keefe FJ, Kraus VB, Olson SA, Pisetsky DS, et al. The role of biomechanics and inflammation in cartilage injury and repair. Clin Orthop Relat Res. 2004;423:17–26. doi: 10.1097/01.blo.0000131233.83640.91. [DOI] [PubMed] [Google Scholar]
- 3.Griffin TM, Guilak F. The role of mechanical loading in the onset and progression of osteoarthritis. Exerc Sport Sci Rev. 2005;33:195–200. doi: 10.1097/00003677-200510000-00008. [DOI] [PubMed] [Google Scholar]
- 4.Amin AR, Di Cesare PE, Vyas P, Attur M, Tzeng E, Billiar TR, et al. The expression and regulation of nitric oxide synthase in human osteoarthritis-affected chondrocytes: evidence for up-regulated neuronal nitric oxide synthase. J Exp Med. 1995;182:2097–2102. doi: 10.1084/jem.182.6.2097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Henrotin Y, Kurz B, Aigner T. Oxygen and reactive oxygen species in cartilage degradation: friends or foes? Osteoarthritis cartilage. 2005;13:643–654. doi: 10.1016/j.joca.2005.04.002. [DOI] [PubMed] [Google Scholar]
- 6.Yudoh K, Nguyen T, Nakamura H, Hongo-Masuko K, Kato T, Nishioka K. Potential involvement of oxidative stress in cartilage senescence and development of osteoarthritis: oxidative stress induces chondrocyte telomere instability and downregulation of chondrocyte function. Arthritis Res Ther. 2005;7:R380–R391. doi: 10.1186/ar1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kobayashi M, Squires GR, Mousa A, Tanzer M, Zukor DJ, Antoniou J, et al. Role of interleukin-1 and tumor necrosis factor alpha in matrix degradation of human osteoarthritic cartilage. Arthritis Rheum. 2005;52:128–135. doi: 10.1002/art.20776. [DOI] [PubMed] [Google Scholar]
- 8.Goldring SR, Goldring MB. The role of cytokines in cartilage matrix degeneration in osteoarthritis. Clin Orthop Relat Res. 2004:S27–S36. doi: 10.1097/01.blo.0000144854.66565.8f. [DOI] [PubMed] [Google Scholar]
- 9.Pelletier JP, Caron JP, Evans C, Robbins PD, Georgescu HI, Jovanovic D, et al. In vivo suppression of early experimental osteoarthritis by interleukin-1 receptor antagonist using gene therapy. Arthritis Rheum. 1997;40:1012–1019. doi: 10.1002/art.1780400604. [DOI] [PubMed] [Google Scholar]
- 10.Stadler J, Stefanovic-Racic M, Billiar TR, Curran RD, McIntyre LA, Georgescu HI, et al. Articular chondrocytes synthesize nitric oxide in response to cytokines and lipopolysaccharide. J Immunol. 1991;147:3915–3920. [PubMed] [Google Scholar]
- 11.Fermor B, Weinberg JB, Pisetsky DS, Misukonis MA, Banes AJ, Guilak F. The effects of static and intermittent compression on nitric oxide production in articular cartilage explants. J Orthop Res. 2001;19:729–737. doi: 10.1016/S0736-0266(00)00049-8. [DOI] [PubMed] [Google Scholar]
- 12.Agarwal S, Deschner J, Long P, Verma A, Hofman C, Evans CH, et al. Role of NFkB transciption factors in anti-inflammatory and proinflammatory actions of mechanical signals. Arthritis Rheum. 2004;50:3541–3548. doi: 10.1002/art.20601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pelletier JP, Jovanovic D, Fernandes JC, Manning P, Connor JR, Currie MG, et al. Reduced progression of experimental osteoarthritis in vivo by selective inhibition of inducible nitric oxide synthase. Arthritis Rheum. 1998;41:1275–1286. doi: 10.1002/1529-0131(199807)41:7<1275::AID-ART19>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 14.van den Berg WB, van de Loo F, Joosten LA, Arntz OJ. Animal models of arthritis in NOS2-deficient mice. Osteoarthritis cartilage. 1999;7:413–415. doi: 10.1053/joca.1999.0228. [DOI] [PubMed] [Google Scholar]
- 15.Del Carlo M, Jr., Loeser RF. Nitric oxide-mediated chondrocyte cell death requires the generation of additional reactive oxygen species. Arthritis Rheum. 2002;46:394–403. doi: 10.1002/art.10056. [DOI] [PubMed] [Google Scholar]
- 16.Pacher P, Beckman JS, Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiol Rev. 2007;87:315–424. doi: 10.1152/physrev.00029.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Regan E, Flannelly J, Bowler R, Tran K, Nicks M, Carbone BD, et al. Extracellular superoxide dismutase and oxidant damage in osteoarthritis. Arthritis Rheum. 2005;52:3479–3491. doi: 10.1002/art.21387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aigner T, Fundel K, Saas J, Gebhard PM, Haag J, Weiss T, et al. Large-scale gene expression profiling reveals major pathogenetic pathways of cartilage degeneration in osteoarthritis. Arthritis Rheum. 2006;54:3533–3544. doi: 10.1002/art.22174. [DOI] [PubMed] [Google Scholar]
- 19.Martin JA, Klingelhutz AJ, Moussavi-Harami F, Buckwalter JA. Effects of oxidative damage and telomerase activity on human articular cartilage chondrocyte senescence. J Gerontol A Biol Sci Med Sci. 2004;59:324–337. doi: 10.1093/gerona/59.4.b324. [DOI] [PubMed] [Google Scholar]
- 20.Lee SH, Chang DK, Goel A, Boland CR, Bugbee W, Boyle DL, et al. Microsatellite instability and suppressed DNA repair enzyme expression in rheumatoid arthritis. J Immunol. 2003;170:2214–2220. doi: 10.4049/jimmunol.170.4.2214. [DOI] [PubMed] [Google Scholar]
- 21.Simelyte E, Boyle DL, Firestein GS. DNA mismatch repair enzyme expression in synovial tissue. Ann Rheum Dis. 2004;63:1695–1699. doi: 10.1136/ard.2003.017210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reiland S. Pathology of so called leg weakness in the pig. Acta Radiologica - Supplementum. 1978;358:23–44. [PubMed] [Google Scholar]
- 23.Turner GV, Collett MG, Veary CM, Kruger C. Arthritis in slaughter pigs. Journal of the South African Veterinary association. 1991;62:107–109. [PubMed] [Google Scholar]
- 24.Collins DH, Mc ET. Sulphate (35SO4) uptake by chondrocytes in relation to histological changes in osteoarthritic human articular cartilage. Ann Rheum Dis. 1960;19:318–330. doi: 10.1136/ard.19.4.318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Collins AR, Dobson VL, Dusinska M, Kennedy G, Stetina R. The comet assay: what can it really tell us? Mutat Res. 1997;375:183–193. doi: 10.1016/s0027-5107(97)00013-4. [DOI] [PubMed] [Google Scholar]
- 26.Singh NP, McCoy MT, Tice RR, Schneider EL. A simple technique for quantitation of low levels of DNA damage in individual cells. Exp Cell Res. 1988;175:184–191. doi: 10.1016/0014-4827(88)90265-0. [DOI] [PubMed] [Google Scholar]
- 27.Collins AR, Duthie SJ, Dobson VL. Direct enzymic detection of endogenous oxidative base damage in human lymphocyte DNA. Carcinogenesis. 1993;14:1733–1735. doi: 10.1093/carcin/14.9.1733. [DOI] [PubMed] [Google Scholar]
- 28.Konca K, Lankoff A, Banasik A, Lisowska H, Kuszewski T, Gozdz S, et al. A cross-platform public domain PC image-analysis program for the comet assay. Mutat Res. 2003;534:15–20. doi: 10.1016/s1383-5718(02)00251-6. [DOI] [PubMed] [Google Scholar]
- 29.Olive PL, Banath JP, Durand RE. Heterogeneity in radiation-induced DNA damage and repair in tumor and normal cells measured using the "comet" assay. Radiat Res. 1990;122:86–94. [PubMed] [Google Scholar]
- 30.Granger DL, Anstey NM, Miller WC, Weinberg JB. Measuring nitric oxide production in human clinical studies. Methods Enzymol. 1999;301:49–61. doi: 10.1016/s0076-6879(99)01068-x. [DOI] [PubMed] [Google Scholar]
- 31.Delaney CA, Green MH, Lowe JE, Green IC. Endogenous nitric oxide induced by interleukin-1 beta in rat islets of Langerhans and HIT-T15 cells causes significant DNA damage as measured by the 'comet' assay. FEBS Lett. 1993;333:291–295. doi: 10.1016/0014-5793(93)80673-i. [DOI] [PubMed] [Google Scholar]
- 32.Jaiswal M, LaRusso NF, Burgart LJ, Gores GJ. Inflammatory cytokines induce DNA damage and inhibit DNA repair in cholangiocarcinoma cells by a nitric oxide-dependent mechanism. Cancer Res. 2000;60:184–190. [PubMed] [Google Scholar]
- 33.Li CQ, Wogan GN. Nitric oxide as a modulator of apoptosis. Cancer Lett. 2005;226:1–15. doi: 10.1016/j.canlet.2004.10.021. [DOI] [PubMed] [Google Scholar]
- 34.Burney S, Caulfield JL, Niles JC, Wishnok JS, Tannenbaum SR. The chemistry of DNA damage from nitric oxide and peroxynitrite. Mutat Res. 1999;424:37–49. doi: 10.1016/s0027-5107(99)00006-8. [DOI] [PubMed] [Google Scholar]
- 36.Collins AR, Dusinska M, Gedik CM, Stetina R. Oxidative damage to DNA: do we have a reliable biomarker? Environ Health Perspect. 1996;104 Suppl 3:465–469. doi: 10.1289/ehp.96104s3465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Loeser RF, Carlson CS, Del Carlo M, Cole A. Detection of nitrotyrosine in aging and osteoarthritic cartilage: Correlation of oxidative damage with the presence of interleukin-1 beta and with chondrocyte resistance to insulin-like growth factor 1. Arthritis Rheum. 2002;46:2349–2357. doi: 10.1002/art.10496. [DOI] [PubMed] [Google Scholar]
- 37.Del Carlo MD, Jr., Loeser RF. Increased oxidative stress with aging reduces chondrocyte survival: correlation with intracellular glutathione levels. Arthritis Rheum. 2003;48:3419–3430. doi: 10.1002/art.11338. [DOI] [PubMed] [Google Scholar]
- 38.Abramson SB, Amin AR, Clancy RM, Attur M. The role of nitric oxide in tissue destruction. Best Pract Res Clin Rheumatol. 2001;15:831–845. doi: 10.1053/berh.2001.0196. [DOI] [PubMed] [Google Scholar]
- 39.Kawanishi S, Hiraku Y. Oxidative and nitrative DNA damage as biomarker for carcinogenesis with special reference to inflammation. Antioxid Redox Signal. 2006;8:1047–1058. doi: 10.1089/ars.2006.8.1047. [DOI] [PubMed] [Google Scholar]
- 40.Ishibashi T, Hayakawa H, Ito R, Miyazawa M, Yamagata Y, Sekiguchi M. Mammalian enzymes for preventing transcriptional errors caused by oxidative damage. Nucleic Acids Res. 2005;33:3779–3784. doi: 10.1093/nar/gki682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Evans CH, Watkins SC, Stefanovic-Racic M. Nitric oxide and cartilage metabolism. Methods Enzymol. 1996;269:75–88. doi: 10.1016/s0076-6879(96)69011-9. [DOI] [PubMed] [Google Scholar]
- 42.Doetsch PW. Translesion synthesis by RNA polymerase:occurrence and biological implications for transcriptional mutagenesis. Mutation Research. 2002;510:131–140. doi: 10.1016/s0027-5107(02)00258-0. [DOI] [PubMed] [Google Scholar]
- 43.Ghosh R, Mitchell DL. Effect of oxidative DNA damage in promoter elements on transcription factor binding. Nucleic Acids Res. 1999;27:3213–3218. doi: 10.1093/nar/27.15.3213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Parsian AJ, Funk MC, Tao TY, Hunt CR. The effect of DNA damage on the formation of protein/DNA complexes. Mutat Res. 2002;501:105–113. doi: 10.1016/s0027-5107(02)00016-7. [DOI] [PubMed] [Google Scholar]
- 45.Hailer-Morrison MK, Kotler JM, Martin BD, Sugden KD. Oxidized guanine lesions as modulators of gene transcription. Altered p50 binding affinity and repair shielding by 7,8-dihydro-8-oxo-2'-deoxyguanosine lesions in the NF-kappaB promoter element. Biochemistry. 2003;42:9761–9770. doi: 10.1021/bi034546k. [DOI] [PubMed] [Google Scholar]
- 46.Jaiswal M, LaRusso NF, Shapiro RA, Billiar TR, Gores GJ. Nitric oxide-mediated inhibition of DNA repair potentiates oxidative DNA damage in cholangiocytes. Gastroenterology. 2001;120:190–199. doi: 10.1053/gast.2001.20875. [DOI] [PubMed] [Google Scholar]
- 47.Aigner T, Hemmel M, Neureiter D, Gebhard PM, Zeiler G, Kirchner T, et al. Apoptotic cell death is not a widespread phenomenon in normal aging and osteoarthritis human articular knee cartilage: a study of proliferation, programmed cell death (apoptosis), and viability of chondrocytes in normal and osteoarthritic human knee cartilage. Arthritis Rheum. 2001;44:1304–1312. doi: 10.1002/1529-0131(200106)44:6<1304::AID-ART222>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 48.D'Lima DD, Kuhn K, Lotz MK. Detection of apoptosis in cartilage in situ and in isolated chondrocytes. Methods Mol Med. 2004;100:275–290. doi: 10.1385/1-59259-810-2:275. [DOI] [PubMed] [Google Scholar]
- 49.Roach HI, Aigner T. DNA methylation in osteoarthritic chondrocytes: a new molecular target. Osteoarthritis cartilage. 2007;15:128–137. doi: 10.1016/j.joca.2006.07.002. [DOI] [PubMed] [Google Scholar]