

Abstract
Alzheimer’s disease (AD) is associated with β-amyloid accumulation, oxidative stress and mitochondrial dysfunction. However, the effects of genetic mutation of AD on oxidative status and mitochondrial manganese superoxide dismutase (MnSOD) production during neuronal development are unclear. To investigate the consequences of genetic mutation of AD on oxidative damages and production of MnSOD during neuronal development, we used primary neurons from new born wild-type (WT/WT) and APP (NLh/NLh) and PS1 (P264L) knock-in mice (APP/PS1) which incorporated humanized mutations in the genome. Increasing levels of oxidative damages, including protein carbonyl, 4-hydroxynonenal (4-HNE) and 3-nitrotyrosine (3-NT), were accompanied by a reduction in mitochondrial membrane potential in both developing and mature APP/PS1 neurons compared to WT/WT neurons suggesting mitochondrial dysfunction under oxidative stress. Interestingly, developing APP/PS1 neurons were significantly more resistant to β-amyloid 1-42 treatment, whereas mature APP/PS1 neurons were more vulnerable than WT/WT neurons of the same age. Consistent with the protective function of MnSOD, developing APP/PS1 neurons have increased MnSOD protein and activity, indicating an adaptive response to oxidative stress in developing neurons. In contrast, mature APP/PS1 neurons exhibited lower MnSOD levels compared to mature WT/WT neurons indicating that mature APP/PS1 neurons lost the adaptive response. Moreover, mature APP/PS1 neurons had more co-localization of MnSOD with nitrotyrosine indicating a greater inhibition of MnSOD by nitrotyrosine. Overexpression of MnSOD or addition of MnTE-2-PyP5+ (SOD mimetic) protected against β-amyloid-induced neuronal death and improved mitochondrial respiratory function. Together, the results demonstrate that compensatory induction of MnSOD in response to an early increase in oxidative stress protects developing neurons against β-amyloid toxicity. However, continuing development of neurons under oxidative damage conditions may suppress the expression of MnSOD and enhance cell death in mature neurons.
Keywords: Alzheimer’s disease, APP/PS1, MnSOD, Oxidative stress, β-amyloid, MnTE-2-PyP5+ (SOD mimetic)
Introduction
Alzheimer’s disease (AD), a progressive neurodegenerative disorder, is the most common form of dementia among the elderly. The pathological hallmarks of AD are post-mortem β-amyloid accumulation and neurofibrillary tangles in the brain. Mutations of the amyloid precursor protein (APP) gene on chromosome 21 and the presenilin-1 (PS1) gene on chromosome 14 are thought to cause an early onset form of AD (Goate et al., 1991, Sherrington et al., 1995). β-amyloid peptides produced from APP processing cause neurodegeneration and disrupt cognitive function by several mechanisms including oxidative stress (Hardy and Selkoe, 2002, Walsh et al., 2002, Cleary et al., 2005, LaFerla et al., 2007). The oxidation of methionine 35 in β-amyloid peptides is thought to be an initiating step in the free radical chain reaction that causes oxidative damage to neurons in AD (Butterfield and Kanski, 2002, Butterfield and Boyd-Kimball, 2005).
Oxidative stress, the imbalance between antioxidants and reactive oxygen species (ROS), has been documented to be involved in AD (Markesbery, 1997), where macromolecules such as proteins, lipids, DNA and RNA are modified (Aksenov et al., 2001, Ding et al., 2005, Markesbery et al., 2005, Wang et al., 2006). Oxidative stress can be detected as early as the stage of mild cognitive impairment (MCI) (Pratico et al., 2002, Migliore et al., 2005, Butterfield et al., 2006), in which antioxidant capacity is low (Rinaldi et al., 2003, Guidi et al., 2006). The activity of copper-zinc superoxide dismutase (CuZnSOD), a cytoplasmic antioxidant enzyme, is reduced in several regions of the AD brain (Marcus et al., 1998). AD brains also have abnormal mitochondria (Hirai et al., 2001), but the status of the primary mitochondrial antioxidant enzyme, MnSOD, is unclear.
MnSOD is the only primary antioxidant enzyme that has been shown to be essential for the survival of all aerobic life. This enzyme rapidly converts superoxide radicals to molecular oxygen and H2O2, which can be further neutralized by glutathione peroxidase, peroxiredoxin reductase and catalase. Increasing evidence suggests that MnSOD is critical for neurons to survive oxidative damage. For example, MnSOD homozygous knockout mice die shortly after birth with severe pathology including neurodegeneration (Li et al., 1995, Lebovitz et al., 1996). Overexpression of MnSOD prevents oxidative insults to neurons from being injured (Gonzalez-Zulueta et al., 1998, Keller et al., 1998, Klivenyi et al., 1998). Recent studies suggest that MnSOD plays a protective role during AD development. For example, MnSOD deficiency increases β-amyloid levels and amyloid plaque burden, and accelerates the onset of behavioral alteration in APP transgenic mice (Li et al., 2004, Esposito et al., 2006). However, the fate of MnSOD during the development of AD is not known.
In this study, we used a homozygous knock-in APPNLh/NLh X PS-1P264L/P264L (APP/PS1) mouse that simulates the natural progression of β-amyloid pathology observed in AD patients (Reaume et al., 1996, Siman et al., 2000, Anantharaman et al., 2006) to investigate the fate of oxidative damages and MnSOD production during neuronal development. This is the first study to demonstrate the dynamics of MnSOD production under oxidative stress during neuronal development in an AD model.
We further employed a potent SOD mimetic, Mn(III) tetrakis (N-ethylpyridinium-2-yl) porphyrin, MnTE-2-PyP5+ (Batinic-Haberle et al., 1999). We have already shown that this porphyrin gets into mouse heart mitochondria at a 5.1 µM level after a single ip dose of 10 mg/kg (Spasojevic et al., 2007). In another study with submitochondrial particles we have shown that al levels > 3 µM, MnTE-2-PyP5+ is able to protect components of mitochondrial respiration against peroxynitrite damage (Ferrer-Sueta et al., 2006). Finally, in a mouse skin TPA carcinogenesis model we were able to show that MnTE-2-PyP5+ effectively substitutes for matrix enzyme, MnSOD (Zhao et al., 2005).
Experimental Procedures
APP-PS1 mouse and neuronal cultures
The humanized Alzheimer mice APPNLh/NLh x PS-1P264L/P264L used in this study were generated using Cre-lox© knock-in technology (Cephalon, Inc., West Chester, PA) (Reaume et al., 1996, Siman et al., 2000). The animal protocols were approved by the University of Kentucky Animal Care facility. Primary neuronal cell cultures were isolated from the cortexes of neonatal pups (P0–P1) from WT/WT and APP/PS1 mice (CD1 background). Briefly, brain meninges were removed and cortical slices were dissected under microscope in ice cold Hank’s balance salt solution and digested in 0.25% trypsin-EDTA in an incubator for 3 min at 37°C. The digestion was stopped by trypsin (soybean) inhibitor (50 mg/mL). DNA fibril was digested by DNAse-I (10 mg/mL). Isolated cell suspensions were filtered through a 0.44 micron nylon mesh (Falcon, Franklin Lake, NJ) and washed by centrifugation through neurobasal medium at 130 g for 5 min. The cells were seeded into poly-D-lysine coated plates and incubated in a cell culture incubator at 37°C for 1 h. The growth medium, neurobasal medium containing B27, glutamax, penicillin-streptomycin-neomycin and bFGF, replaced the pre-medium. Neuron cultures were maintained at 37°C in a humidified incubator containing 5% CO2. One-third of the original medium was changed at day 3 and then again every 2 days until day 10. In this study, day 3 neurons in culture were defined as the developing neurons and day 10 neurons were defined as mature neurons. These definitions were based on the results of a previous study, in which neuronal markers such as NeuN, synaptophysin, and synapsin IIa were present by day 10 but not before day 5 (Lesuisse and Martin, 2002).
Antibodies
All antibodies in the present study were purchased from commercial sources. Polyclonal anti-MnSOD and monoclonal anti-nitrotyrosine were obtained from Upstate (Lake Placid, NY). Monoclonal anti-beta actin was obtained from Sigma (St. Louis, MO). Polyclonal anti-3-nitrotyrosine, and polyclonal anti-hydroxynonenal were obtained from Chemicon (Temecula, CA). The protein oxidation detection kit was purchased from Intergen (Purchase, NY).
SOD mimetic
MnTE-2-PyP5+ was prepared as previously described (Batinic-Haberle et al., 1999).
Measurement of protein carbonyls
Protein carbonyl levels were measured from day 3 and day 10 neuronal cultures from WT/WT and APP/PS1 mice. Neuronal cells were collected in PBS and kept in −80°C for further analysis. Protein carbonyl levels were determined as adducts of 2,4-dinitrophenylhydrazine (Oliver et al., 1987). The sample (5 µL) was incubated for 20 min at room temperature (22°C) with 5 µL of 12% sodium dodecyl sulfate (SDS) and 10 µL of 2,4-dinitrophenylhydrazine that was diluted 10 times with water from a 200 mM stock. The samples were neutralized with 7.5 µL of neutralization solution (2 M Tris in 30% glycerol). The resulting sample was loaded 250 ng per well onto a nitrocellulose membrane in a slot-blot apparatus under vacuum pressure. The membrane was blocked with 3% bovine serum albumin (BSA) in PBS containing 0.2% (v/v) Tween 20 for 1 h and incubated with a 1:100 dilution of anti-dinitrophenylhydrazine (anti-DNP) polyclonal antibody for 1 h. Following completion of the primary antibody incubation, the membranes were washed three times for 5 min each. The membranes were incubated in an anti-rabbit IgG alkaline phosphatase secondary antibody (1:8000) for 1 h, washed three times for 5 min each and developed using Sigmafast tablets (BCIP/NBT substrate). Blots were dried, scanned with Adobe Photoshop (San Jose, CA), and quantified with Scion Image software (PC version of Macintosh-compatible NIH Image; National Institutes of Health, Bethesda, MD).
Measurement of 4-hydroxy-2- trans-nonenal (4-HNE)
Levels of HNE, which reflect lipid peroxidation, were quantified by slot-blot analysis (Lauderback et al., 2001). Anti-HNE antibody raised in rabbit was used as the primary antibody (1:200 dilution). The membranes were developed using Sigmafast tablets. Blots were dried, scanned with Adobe Photoshop, and quantifieded with Scion Image software (PC version of Macintosh-compatible NIH Image).
Measurement of 3-nitrotyrosine (3-NT)
The sample (10 µL) was incubated with 10 µL of buffer containing 0.125 M Tris base, pH 6.8, 4% (v/v) SDS, and 20% (v/v) glycerol. The resulting sample (250 ng) was loaded per well in the slot-blot apparatus. Samples were loaded onto a nitrocellulose membrane under vacuum pressure. The membrane was blocked with 3% (w/v) BSA in wash blot for 1 h and incubated with a 1:2000 dilution of 3-NT polyclonal antibody for 90 min. The remainder of the procedure was identical to that described above for HNE.
Western Blot
Neuronal cells were cultured and collected at days 3 and 10. The same amounts of crude extract were loaded into 12.5% SDS-polyacrylamide gel electrophoresis and transferred to nitrocellulose membranes. The membranes were then blocked with 5% nonfat dried milk in TBS-T buffer (pH 7.6) for at least 1 h at room temperature. Membranes were incubated in the primary antibody at room temperature for 2 h or at 4°C overnight. The primary antibody was diluted in TBS-T buffer containing 5% nonfat dried milk. The membranes were washed three times for 5 min each with TBS-T, and incubated with secondary antibody for 2 h. The membranes were then washed three times with TBS-T for 5 min and once with TBS for 5 min. Protein bands were detected using the enhanced chemiluminescence detection system (ECL®, Amersham Biosciences). Densitometry analysis was performed by using the Quantity One® Image analyzer software program (Bio-Rad).
Manganese superoxide dismutase (MnSOD) activity assay
MnSOD activity was determined by a modified nitroblue tetrazolium (NBT) assay (Spitz and Oberley, 1989). The assay is based on the competition reaction between SOD and the indicator molecule, NBT, for superoxide radicals generated by xanthine/xanthine oxidase. The rate of increase of 560 nm over a 3-min time period indicates the reduction of NBT by superoxide. Briefly, 20 µL of different amounts of total protein from neuronal cell homogenate were added to the 160 µL of reaction buffer (50 mM potassium phosphate, pH 7.8, 1 mM diethylenetriamine pentaacetic acid [DETAPAC], 10 U/mL catalase, 56 µM NBT, 0.1 mM xanthine) with 0.33 M NaCN and incubated for 30 min to inhibit CuZnSOD. 20 µL of properly diluted xanthine oxidase is added to initiate the reaction. The rate of reaction was followed for 3 min at 560 nm. The amount of protein needed for 50% inhibition is defined as one unit of the enzyme.
Confocal laser scanning microscopy
Neuronal cells were fixed with 4% paraformaldehyde at room temperature for 20 min. The cells were washed with PBS 3 times for 5 min each and permeabilized with 0.2% triton X100 for 10 min. After incubation, triton X100 was removed and the cells were blocked with 3% normal donkey serum for 30 min. To detect co-localization of MnSOD and nitrotyrosine, neurons were incubated with polyclonal anti-MnSOD and monoclonal anti-nitrotyrosine as primary antibodies. Primary antibodies were prepared in 3% normal donkey serum using 1:200 dilution. The cells were incubated with antibodies at room temperature for 4 h or at 4°C overnight. Primary antibody was decanted and the cells were washed 3 times with PBS for 5 min each. The cells were incubated with 1:200 dilution of secondary antibody at room temperature for 1 h. The cells were incubated with secondary donkey anti-rabbit conjugated with cy2 and secondary donkey anti-mouse conjugated with cy3 to detect MnSOD and nitrotyrosine, respectively. The cells were washed 3 times with PBS for 5 min each. Immunoreactive cells were captured in at least five random fields using 100X from a Leica microscope. Negative fluorescence controls for secondary antibodies were tested.
Neuronal cell survival
Cortical neurons at day 3 and day 10 were treated with 5 µM β-amyloid1-42 (Anaspec, Sanjoes, CA) for 24 h. Before treatment, neuronal pictures in triplicate were captured for 4 areas of one treatment. Twenty-four hours after treatment, the same area of neuronal cultures was captured again. The neurons remaining in the same area after treatment were counted and calculated as survival cells. Percent of cell survival was quantified from three sets of independent cell culture experiments. Neuronal cell survival was also determined in an experiment where neurons were pretreated with SOD mimetic (100 to 1000 pg/mL) before being treated with β-amyloid1-42.
Mitochondrial membrane potential
Mitochondrial membrane potential was measured by using 5,5′, 6,6′-Tetrachloro-1,1′,3,3′-tetraethylbenzimidazolocarbocyanine iodide or JC-1 (Sigma, St. Louis, MO). Briefly, cells were incubated with 5µM JC-1 in neurobasal medium for 20 min. Then cells were washed twice with PBS. Fluorescence signal was measured by spectrofluorometry with 485 nm excitation, and 525 nm and 590 nm for emission of green (monomer form) and red (aggregate form) fluorescence, respectively. The color of the dye changes reversibly from green to red as the mitochondrial membrane becomes more polarized. The ratio of red/green was used as relative increase in mitochondrial membrane potential. Cells with 1µM Carbonyl cyanide p-trifluoromethoxyphenylhydrazone (FCCP) were used as positive control samples.
Isolation of Mitochondria
Mitochondria were isolated from WT/WT (CD1 background), WT and MnSOD-overexpressing mice TgH (CB57BL/6 background). The expression and characterization of MnSOD transgenic mice have been previously reported (Yen et al., 1996, Daosukho et al., 2005). Briefly, the brain was rapidly removed and the forebrain was chopped and placed in glass homogenizer with 5 mL of isolation buffer (0.225 mol/L D-mannitol, 0.075 mol/L sucrose, 20 mmol/L HEPES, 1 mmol/L EGTA, and 1% bovine serum albumin, pH 7.2). Tissue was homogenized and centrifuged at 1,800 g at 4°C for 5 min. Supernatants were collected and centrifuged at 1,800 g at 4°C for 5 min. The supernatants were collected and centrifuged at 8,000 g at 4°C for 15 min and then the pellets were resuspended in 3% percoll solution and layered on a surface containing 6% percoll solution in isolation buffer. This density gradient was centrifuged at 8,000 g at 4°C for 30 min to separate the unintact mitochondria from the intact mitochondria in the pellets. The pellets were washed again in isolation buffer by centrifugation at 8,000 g at 4°C for 5 min and resuspended in mitochondrial respiration buffer (0.25 mol/L sucrose, 50 mmol/L HEPES, 2 mmol/L MgCl2, 1 mmol/L EGTA, 10 mmol/L KH2PO4, and 0.5% bovine serum albumin, pH 7.4).
Mitochondrial Respiration assay
After protein determination by Bradford assay, 250 µg protein were used to measure oxygen consumption using a Clark-type electrode oxygraph (Hansatech Inc., Norfolk, UK) with 10 mmol/L pyruvate and 5 mmol/L malate as substrates in the absence of exogenous ADP (state II) and after addition of 300 mmol/L ADP (state III). Mitochondria treated with SOD mimetic (1000 pg/mL) and mitochondria not treated were measured for function after one min. The ATP synthase inhibitor oligomycin (100 µg/mL) was then added to inhibit mitochondrial respiration (state IV). The respiratory control ratio (RCR) was the ratio between the rate of oxygen consumption/min of state III and state IV.
Results
Characterization of oxidative damages in APP/PS1 neurons
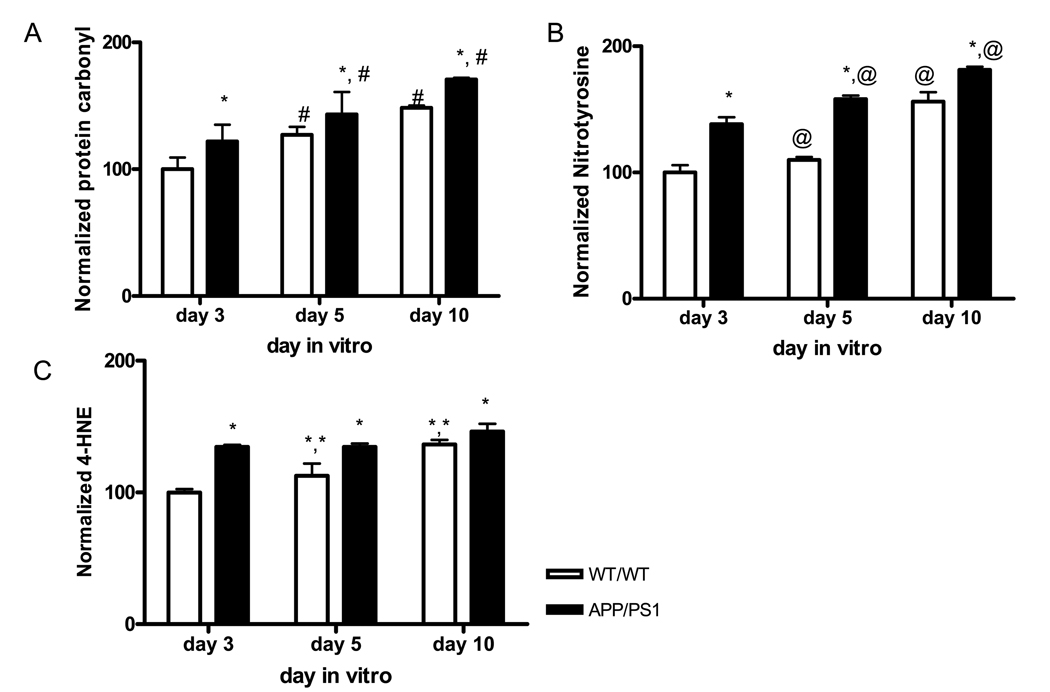
We have previously demonstrated that APP/PS1 mice generate β-amyloid plaques in the brain (Anantharaman et al., 2006). Our present study evaluated the oxidatively modified biochemical effects of the genetic mutation factor of AD during neuronal development. The levels of oxidative stress markers including protein carbonyl, 3-nitrotyrosine (3-NT), and 4-hydroxynonenal (4-HNE) in primary cortical neurons were measured in cell lysates from days 3, 5 and 10 of primary neuron cultures. All oxidative markers were increased in APP/PS1 neurons by day 3 (Fig. 1). The average increases of protein carbonyl, 3-NT and 4-HNE levels were 20%, 38% and 34%, respectively, at day 3 in APP/PS1 neurons compared to the corresponding WT/WT. Both protein carbonyl and 3-NT levels were significantly increased in an age-dependent manner in both genotypes with the levels in APP/PS1 neurons being higher than those in WT/WT neurons at all ages (# P = 0.00016 and @ P < 0.0001), (Fig. 1A and B). The levels of 4-HNE in WT/WT neurons increased with age during neuronal development (** P = 0.01). The levels of 4-HNE in APP/PS1 reached the levels of day 10 WT/WT neurons by day 3, and these discrepancies were sustained throughout the entire experimental period (Fig. 1C). These results clearly demonstrate that oxidatively modified proteins increase early during neuronal development in neuron models of AD and continue to accumulate with age in both normal and AD neuron models.
Figure. 1.
Mitochondrial membrane potential in APP/PS1 neurons
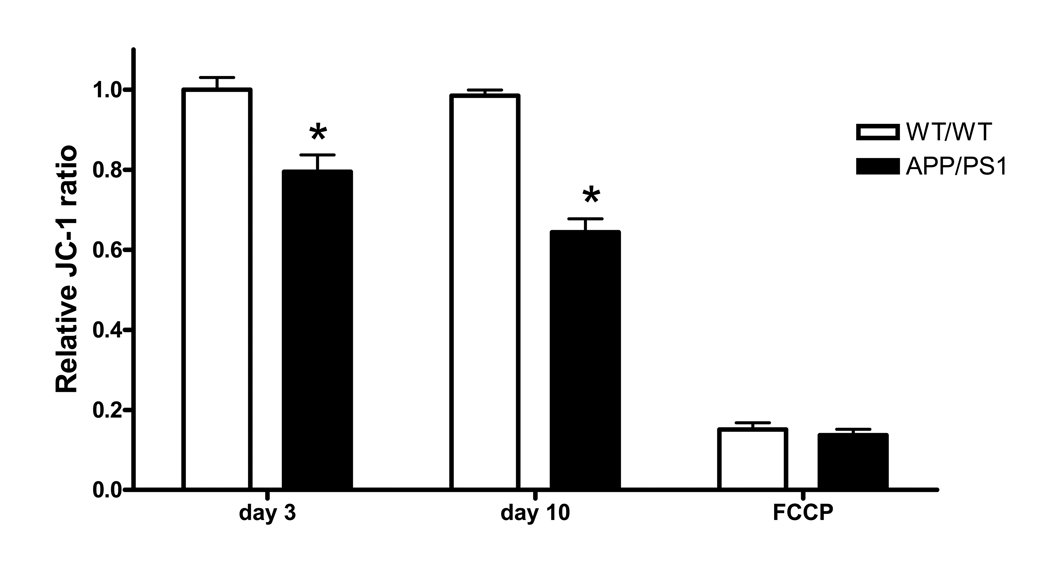
Mitochondrial membrane potential is an important indicator for cell death, the aging process and mitochondrial function. To measure mitochondrial membrane potential, we stained primary neurons with JC-1, the fluorescence-capable sensing dye. The relative changes in JC-1 ratio were 79% and 64% (* P < 0.0001, two-way ANOVA) in developing and mature APP/PS1 neurons, respectively, compared to WT/WT neurons of the same age (Fig. 2). The finding that oxidative damages increase in APP/PS1 neurons (Fig. 1) accompanied by a reduction of mitochondrial membrane potential suggests that oxidative stress may cause mitochondrial damage and dysfunction in APP/PS1 neurons.
Figure. 2.
Differential susceptibility of young and mature neurons to β-amyloid toxicity
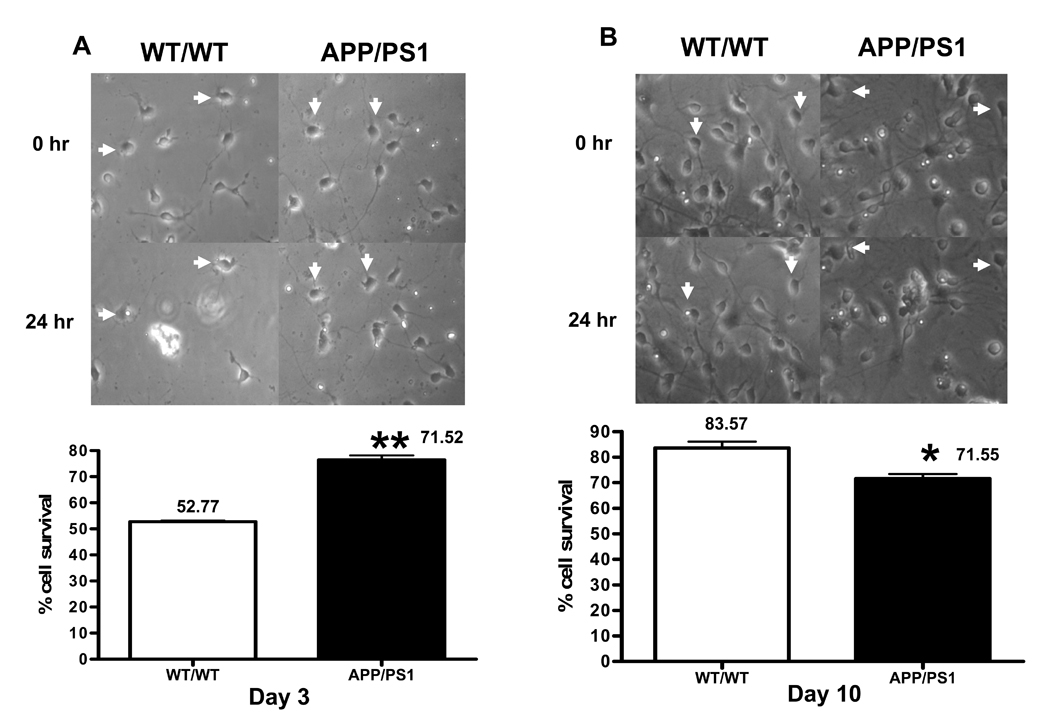
To test whether genetic predisposition of β-amyloid peptides alters susceptibility of neurons to additional oxidative stress, we determined cell survival after neurons were exposed to 5 µM β-amyloid 1-42 for 24 h. Interestingly, cell survival in day 3 APP/PS1 neurons was higher than WT/WT neurons, 71.1% and 52.77%, respectively (** P = 0.001) (Fig. 3A). We further investigated neuronal susceptibility in mature WT/WT and APP/PS1 neurons. A higher cell survival of neurons to β-amyloid toxicity was observed in WT/WT neurons than in APP/PSI neurons, 83.57% and 71.55%, respectively (* P = 0.0175) (Fig. 3B).
Figure. 3.
Differential of MnSOD levels during neuronal development
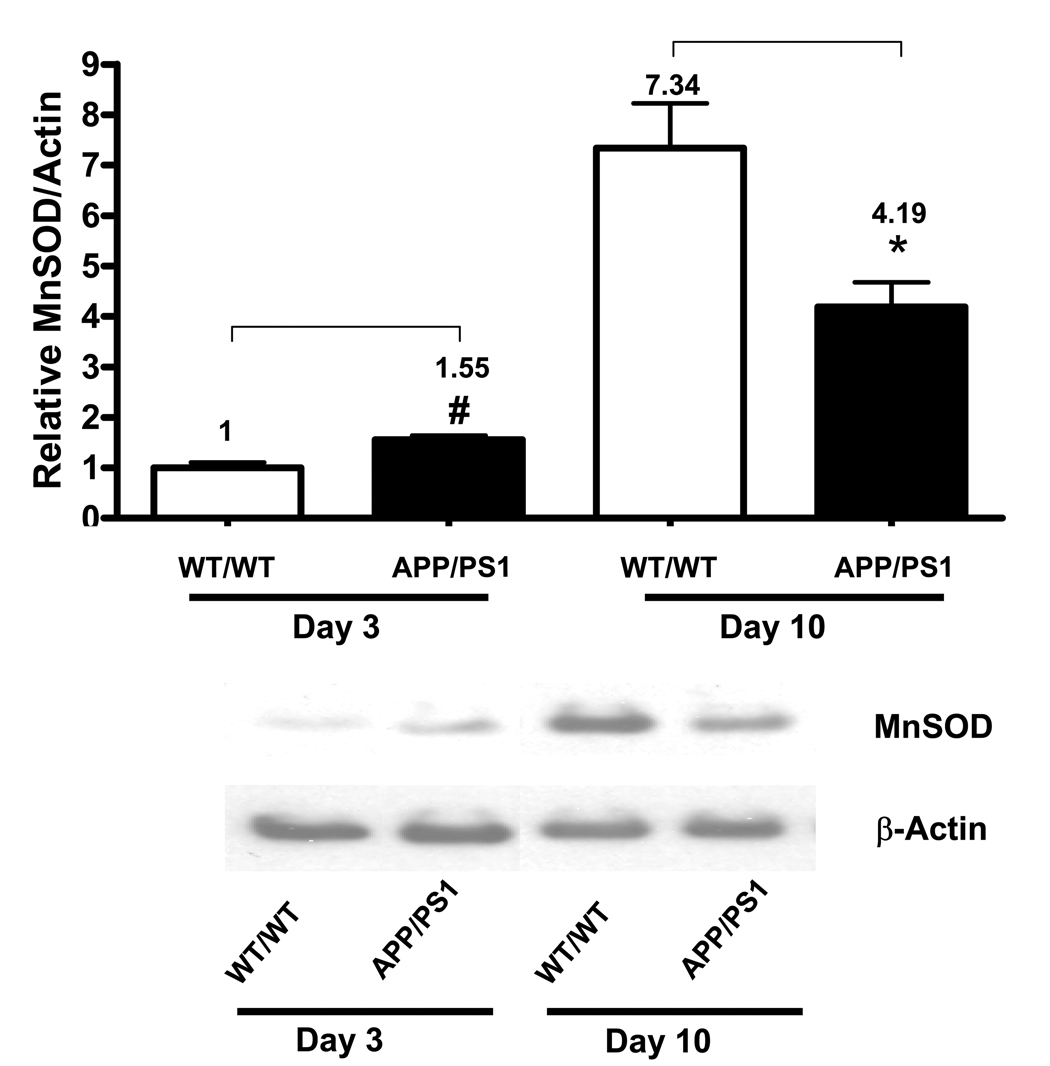
MnSOD, a primary mitochondrial antioxidant enzyme essential for survival under oxidative stress, is an inducible antioxidant enzyme which is highly responsive to agents that generate oxidative stress. Since oxidative stress occurs early in APP/PS1 neurons (Fig. 1) and developing APP/PS1 neurons can survive longer under additional oxidative stress (Fig. 3A), we investigated MnSOD levels in developing neurons isolated from WT/WT and APP/PS1 mice. Interestingly, MnSOD protein levels were 1.55-fold higher in day 3 APP/PS1 neurons compared to WT/WT of the same age (# P = 0.045, t-test) (Fig. 4), indicating that oxidative stress induces an antioxidant adaptive response in developing neurons. MnSOD protein levels increased with age in both genotypes. In day 10 cultures, WT/WT neurons had higher-fold increases of MnSOD than APP/PS1 neurons did, 7.34- and 4.19-fold (* P = 0.0359, t-test), respectively, compared to those in day 3 WT/WT (Fig. 4). However, the MnSOD levels in day 10 APP/PS1 neurons were only 2.7-fold of that in day 3 APP/PS1 neurons. These results suggest that day 10 APP/PS1 neurons lost the ability to overproduce MnSOD under sustained oxidative stress conditions.
Figure. 4.
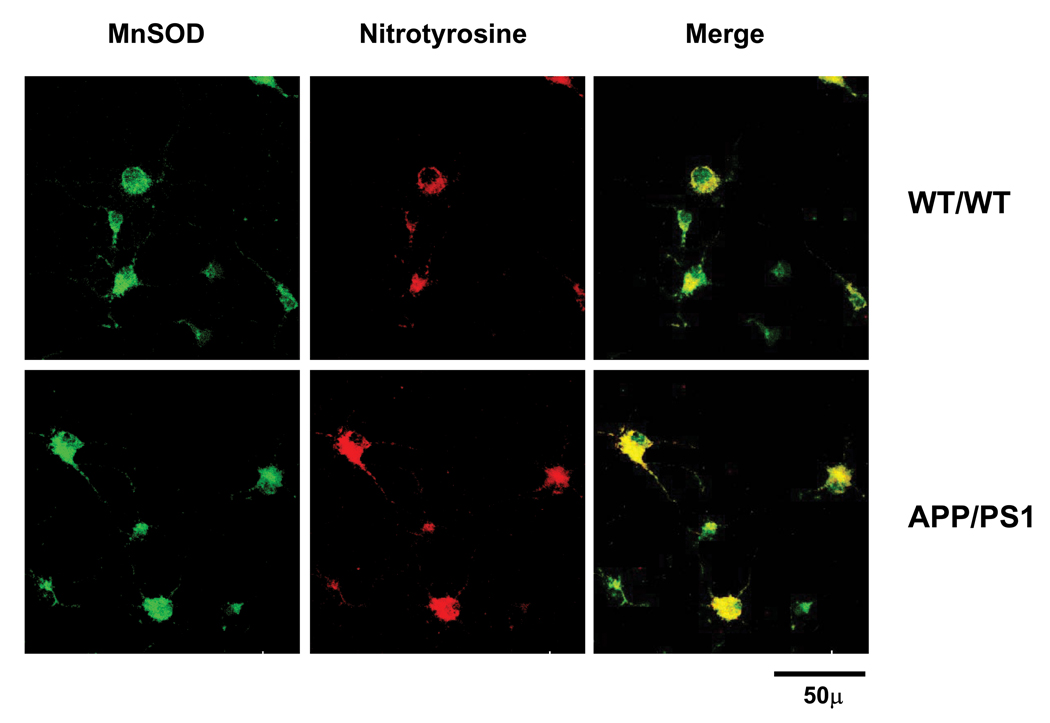
The 1.55-fold increase in MnSOD protein levels is consistent with the 1.53-fold increase in MnSOD activity in APP/PS1 day 3 neurons compared to WT/WT neurons of the same age. However, at day 10, there were 3.38- and 1.67-fold increases (* P = 0.006, t-test) in MnSOD activity in WT/WT and APP/PS1, respectively, compared to those in day 3 WT/WT neurons (Table 1). The changes in MnSOD protein levels in day 10 neurons from both genotypes were greater than the changes in enzyme activity (Fig. 4 and Table 1), suggesting that MnSOD was inactivated. The ratio between protein levels and activity were 2.17- and 2.51-fold in mature WT/WT and APP/PS1 neurons, respectively (Table 1). The greater increase in protein and activity ratio in day 10 APP/PS1 neurons suggests that inactivation of MnSOD was greater in APP/PS1 neurons. Since MnSOD can be inactivated by nitrotyrosine (MacMillanCrow et al., 1996, MacMillan-Crow et al., 1998), we further measured co-localization of MnSOD with nitrotyrosine in mature WT/WT and APP/PS1 neurons. The greater co-localization of MnSOD and nitrotyrosine indicates the greater inactivation of MnSOD by nitrotyrosine in mature APP/PS1 neurons (Fig. 5). These results demonstrate that oxidative stress affects both MnSOD production and activity during neuronal development.
Table. 1.
| MnSOD Activity (U/mg protein) | Fold change (Activity) | Fold change (Protein) | Protein Activity | ||
|---|---|---|---|---|---|
| Day 3 | WT/WT | 20.45 ± 12.54 | 1 | 1 | 1 |
| APP/PS1 | 31.31 ± 15.38 | 1.53 | 1.55 | 1.01 | |
| Day 10 | WT/WT | 69.12 ± 8.15 | 3.38 # | 7.34 | 2.17 |
| APP/PS1 | 34.22 ± 8.02 | 1.67 * | 4.19 | 2.51 |
Figure. 5.
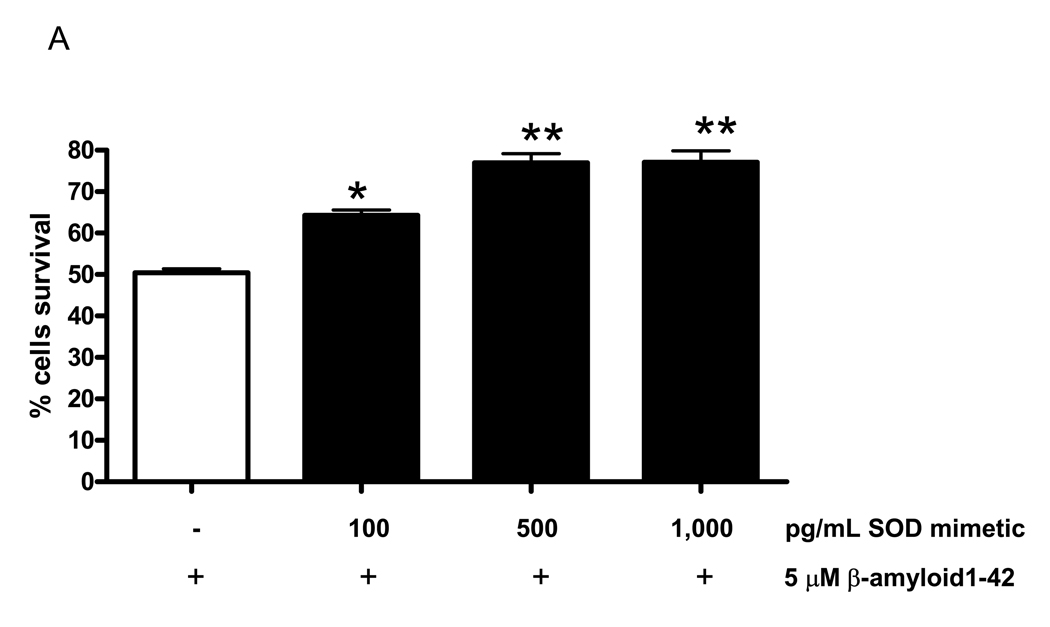
SOD mimetic protects neurons and improves mitochondrial function
To investigate the protective role of MnSOD against additional β-amyloid toxicity, we pretreated developing WT/WT neurons with SOD mimetic for 3h, followed by β-amyloid treatment for 24 h. Pretreatment with SOD mimetic increased neuronal survival against β-amyloid-induced cell death (* P < 0.05, ** P < 0.001, one-way ANOVA) (Fig. 6A). This result confirms the protective role of MnSOD and also establishes that induction of MnSOD is an adaptive event to protect developing APP/PS1 neurons against oxidative stress (Fig. 3A and Fig. 4).
Figure. 6.
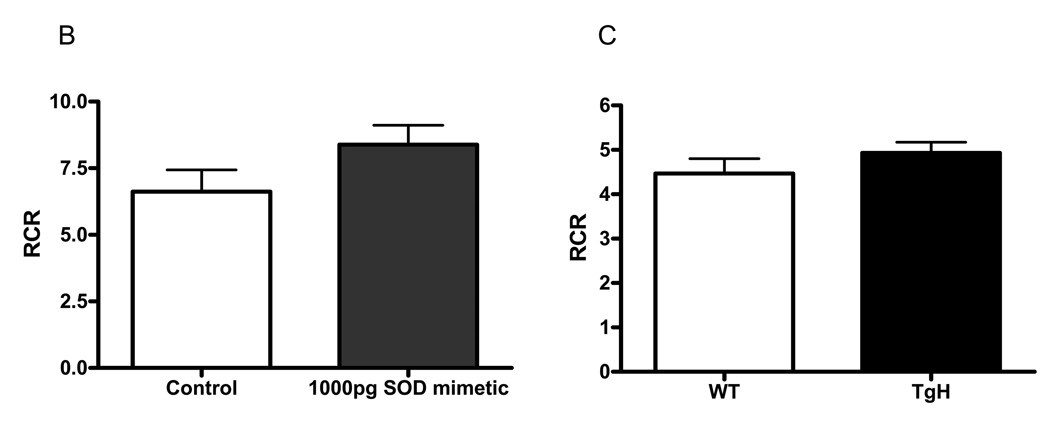
Mitochondria abnormalities have been reported in AD (Hirai et al., 2001). We have previously shown that mitochondria isolated from APP/PS1 mice have reduced respiratory function (Anantharaman et al., 2006). SOD mimetic was able to protect neurons from oxidative damage (Fig. 6A). To determine whether the protective role of SOD mimetic is related to its ability to protect mitochondrial function, we pretreated isolated mitochondria from adult WT/WT mice and measured mitochondrial respiration. Interestingly, SOD mimetic was not toxic when directly added to mitochondria but was able to enhance slightly the RCR value although it did not reach a significant level of 0.05. This result indicates that SOD mimetic is not toxic to mitochondria and may improve mitochondrial function (Fig 6B). To compare the effects of SOD mimetic and authentic MnSOD on mitochondrial function, we compared forebrain mitochondrial respiration from WT (CB57BL/6 background) with mitochondria isolated from transgenic mice overexpressing the human MnSOD gene, TgH. As observed with the SOD mimetic, there was no significant difference in RCR, but a trend of protection was observed. (Fig. 6C). These results suggest that SOD mimetic and authentic MnSOD exert a similar effect on the function of brain mitochondria.
Discussion
The present study demonstrates that mutations in the APP and PS genes increase oxidative stress in APP/PS1 neurons and result in increased MnSOD levels which act as an antioxidant adaptive response to oxidative stress. However, sustained exposure to high levels of oxidative stress is accompanied by a decline in MnSOD production and increased vulnerability to β-amyloid exposure.
Increasing the pro-oxidant side of pro-oxidant/antioxidant homeostasis or oxidative stress is thought to be a key factor in the pathogenesis of AD and MCI (Pratico et al., 2002, Rinaldi et al., 2003, Guidi et al., 2006). Several investigators have measured the levels of oxidatively modified biomacromolecules such as DNA, RNA, protein, and lipid in AD and MCI (Markesbery, 1997, Aksenov et al., 2001, Markesbery et al., 2005, Butterfield et al., 2006, Ding et al., 2006, Wang et al., 2006). Increasing incidences of oxidative damages in macromolecules strongly suggest that oxidative stress plays a role in AD; however, the precise pathogenesis mechanism of AD is still not completely known. In addition to other oxidatively modified biomacromolecules, protein carbonyl and hydroxynonenal (HNE) are toxic to the neuronal system (Picklo et al., 2002). Moreover, protein nitration affects the function of the protein and may contribute to neurodegeneration in AD (Smith et al., 1997). In the present study, we found early increases of protein carbonyl, 3-NT and 4-HNE in developing APP/PS1 neurons and sustained increases of oxidative damages during neuronal development (Fig. 1), which confirm a previous study on mature neurons (Abdul et al., 2006). Increasing levels of oxidative damages accompanied by reduced mitochondrial membrane potential in APP/PS1 neurons (Fig. 1 and Fig. 2) suggest a reduction of mitochondria function under oxidative stress conditions. These phenomena may prime neurons to aging and cell death processes since enhanced depolarization was found in mitochondria from old animals (Hagen et al., 1997). The finding that mature APP/PS1 neurons are more vulnerable to additional β-amyloid toxicity (as shown in Fig. 3B) supports this explanation. These results suggest that the genetic mutation risk factor of AD causes APP/PS1 neurons to be born with oxidative damages and that continuous exposure to higher levels of accumulated oxidative stress and mitochondrial dysfunction can eventually lead to the neuronal dysfunction and cell death observed in the AD brain.
MnSOD, a pro-survival mitochondrial antioxidant enzyme, has been shown to be essential for neuronal protection against oxidative damages (Gonzalez-Zulueta et al., 1998, Keller et al., 1998, Klivenyi et al., 1998). However, the fate of MnSOD during AD development is not completely known. The present study demonstrates the dynamics of MnSOD production during neuronal development. Interestingly, our results show an early increase in oxidative damages and accelerated increase in MnSOD levels in developing APP/PS1 neurons (Fig. 1 and Fig. 4). These results extend to demonstrate that developing neurons have the ability to cope with oxidative stress by induction of MnSOD. This result signifies the role of oxidative stress-induced MnSOD production. The neuroprotective role of SOD mimetic (Fig. 6A) confirms that an early increase in MnSOD may, in part, be an adaptive response that protects against β-amyloid-induced neuronal damage. Our results, which demonstrate that mature APP/PS1 neurons lose the ability to produce more of this enzyme than do WT/WT neurons of the same age (Fig. 4), suggest that sustained exposure to high levels of oxidative stress leads to a loss of adaptive response. Our results also suggest that sustained increase in MnSOD is essential for neuronal survival, and, thus, prevention of chronic oxidative stress-mediated MnSOD reduction may be a strategy to protect chronic neurodegenerative diseases such as AD.
Many different groups reported that beta-amyloid is more toxic to mature neurons than young neurons (Liu et al., 2004). However, in the present study, we found that beta-amyloid is more toxic to developing neurons (WT/WT) compared to mature neurons (WT/WT). There are several potential reasons that may explain this apparent discrepancy. First, most reports, including Liu et al., use E18 neurons from rats. Our study used primary neurons from newborn mouse pups. The differences between neurons from rats and mice, as well as different conditions in uthero- and newborn- pups, could contribute, in part to the observed variations. An adaptive response may have developed in the mouse brain upon exposure to a higher level of oxygen after birth. Second, Liu et al. used A-beta 1-40 whereas we used A-beta 1-42. Treatment with β-amyloid1-40 in the Liu study might have primed young neurons to adapt against oxidative stress compared to treatment with the more toxic β-amyloid1-42 used in our present study. Abeta1-42 is more toxic than Abeta1-40. There are 2 possible reasons: (a) with two additional hydrophobic amino acids, Abeta1-42 could aggregate more quickly, forming toxic oligomers more readily than Abeta1-40. (b) The helix each forms when dissolved in the lipid bilayer as small oligomers has a dipole moment associated with the helix. The sulfuranyl free radical on Met-35 we propose would be more stabilized (i.e., live longer to then initate lipid peroxidation) in the longer peptide with the greater dipole moment. (Butterfield and Boyd-Kimball, 2005). Third, Liu et al. use MTT assay to determine cytotoxicity. Although this assay is the practical method to measure cell numbers, it represents the content of mitochondrial dehydrogenases. We use the disappearance of cells as a measure of cytotoxicity. Although this method is very tedious, we believe it directly indicates the amount of cell death.
MnSOD is a nuclear encoded mitochondrial enzyme (Wan et al., 1994). Induction of MnSOD is regulated by the combination of several transcription activators and repressors. Members of the NF-κB family, including p50, p65, and c-Rel, have been shown to induce MnSOD expression and protect neurons from oxidative damage (Mattson et al., 1997, Pizzi et al., 2005, Sompol et al., 2006). However, the molecular mechanism of MnSOD suppression in neurons remains unknown. A possible candidate for the suppression of MnSOD is p53. It has been shown that p53 level is increased in AD (Alves da Costa et al., 2006) and that expression of p53 suppresses MnSOD transcription in various cellular models (Drane et al., 2001, Dhar et al., 2006). Moreover, it is possible that mitochondrial injury observed in AD may suppress MnSOD expression since mitochondrial electron transport inhibitors have been shown to suppress MnSOD induction (Rogers et al., 2001). Mitochondrial injury has been shown to inhibit NF-κB and reduce p65, key transcription factors for MnSOD production, but to induce p53 expression (Schulze-Osthoff et al., 1993, Biswas et al., 1999, Behrend et al., 2005). In addition to transcriptional repression of MnSOD, it is also possible that the observed decrease of MnSOD may be due to oxidative stress-induced DNA damage and/or ribosomal oxidation, which have been observed in AD (Lovell et al., 2000, Ding et al., 2005, Wang et al., 2006). Additional studies will be needed to identify the exact cause for the loss of MnSOD induction upon sustained exposure to oxidative stress.
Our finding that MnSOD activity in mature APP/PS1 neurons declines more than do the levels of MnSOD protein (1.67-fold change in activity and 4.19-fold change in protein) is supportive evidence for MnSOD inactivation. It has been shown that MnSOD is particularly sensitive to oxidative stress-induced inactivation of enzyme activity by nitrotyrosine modification (MacMillanCrow et al., 1996, MacMillan-Crow et al., 1998). We have previously shown that MnSOD in APP/PS1 brain has a high level of nitrotyrosine modification (Anantharaman et al., 2006). The greater co-localization of MnSOD and nitrotyrosine in mature APP/PS1 neurons in the present study (Fig. 5) confirms those previous studies. These results suggest that AD is a neurodegenerative disease that accumulates oxidatively modified dysfunctional protein including MnSOD.
Oxidative stress is a cause of progressive aging and developing disease including neurodegeneration (Markesbery, 1997, Chan, 2006, Terman and Brunk, 2006). Our results demonstrate that neurons can adapt to the initial encounter with oxidative stress by increasing their antioxidant defense enzyme, MnSOD. Thus, preventing the decline in the antioxidant defense system may provide an effective means for intervention in the development of neurodegenerative disease and the aging process. The rate of brain development is highest within the first year of development (Dekaban, 1978). Our present study shows that oxidative stress increased during neuronal development but the young neuron with a higher level of MnSOD is protected. Thus, the most beneficial time at which to begin increasing the antioxidant system to protect the central nervous system from oxidative stress might be below the age of one year. Increasing the antioxidant system should continue from infancy through maturation and to the aging stages of the brain to reach full protection from the neurodegeneration caused by oxidative stress. Since our data indicate that genetically predisposed young neurons can adapt by increasing endogenous antioxidant, it would suggest that a genetically predisposed infant, even later in development, would benefit from supplementation of antioxidant to prevent , or at least slow down, the progress of neurodegeneration in AD.
Acknowledgments
This work was supported by NIH grants PO1AG 05119 and RO1 CA 49797 to Dr. Daret K. St. Clair and by the Royal Golden Jubilee Research Fellowship (The Thailand Research Fund) to Pradoldej Sompol. We are particularly grateful to Dr. Dorothy Flood, Cephalon Inc., for the critical review and valuable suggestions that improved the quality of this manuscript.
Abbreviations
- 3-NT
3-nitrotyrosine
- 4-HNE
4-hydroxynonenal
- AD
Alzheimer’s disease
- APP
amyloid precursor protein
- CuZnSOD
copper-zinc superoxide dismutase
- MCI
mild cognitive impairment
- MnSOD
manganese superoxide dismutase
- MnTE-2-PyP5+
SOD mimetic
- NF-κB
nuclear factor kappa B
- PS1
presenilin 1
- ROS
reactive oxygen species
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abdul HM, Sultana R, Keller JN, St Clair DK, Markesbery WR, Butterfield DA. Mutations in amyloid precursor protein and presenilin-1 genes increase the basal oxidative stress in murine neuronal cells and lead to increased sensitivity to oxidative stress mediated by amyloid beta-peptide (1-42), H2O2 and kainic acid: implications for Alzheimer's disease. J Neurochem. 2006;96:1322–1335. doi: 10.1111/j.1471-4159.2005.03647.x. [DOI] [PubMed] [Google Scholar]
- Aksenov MY, Aksenova MV, Butterfield DA, Geddes JW, Markesbery WR. Protein oxidation in the brain in Alzheimer's disease. Neuroscience. 2001;103:373–383. doi: 10.1016/s0306-4522(00)00580-7. [DOI] [PubMed] [Google Scholar]
- Alves da Costa C, Sunyach C, Pardossi-Piquard R, Sevalle J, Vincent B, Boyer N, Kawarai T, Girardot N, St George-Hyslop P, Checler F. Presenilin-dependent gamma-secretase-mediated control of p53-associated cell death in Alzheimer's disease. J Neurosci. 2006;26:6377–6385. doi: 10.1523/JNEUROSCI.0651-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anantharaman M, Tangpong J, Keller JN, Murphy MP, Markesbery WR, Kiningham KK, St Clair DK. beta-amyloid mediated nitration of manganese superoxide dismutase - Implication for oxidative stress in a APP(NLh/NLH) X PS-1(P264L/P264L) double knock-in mouse model of Alzheimer's disease. Am J Pathol. 2006;168:1608–1618. doi: 10.2353/ajpath.2006.051223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batinic-Haberle I, Spasojevic I, Hambright P, Benov L, Crumbliss AL, Fridovich I. Relationship among redox potentials, proton dissociation constants of pyrrolic nitrogens, and in vivo and in vitro superoxide dismutating activities of manganese(III) and iron(III) water-soluble porphyrins. Inorganic Chemistry. 1999;38:4011–4022. [Google Scholar]
- Behrend L, Mohr A, Dick T, Zwacka RM. Manganese superoxide dismutase induces p53-dependent senescence in colorectal cancer cells. Molecular and cellular biology. 2005;25:7758–7769. doi: 10.1128/MCB.25.17.7758-7769.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswas G, Adebanjo OA, Freedman BD, Anandatheerthavarada HK, Vijayasarathy C, Zaidi M, Kotlikoff M, Avadhani NG. Retrograde Ca2+ signaling in C2C12 skeletal myocytes in response to mitochondrial genetic and metabolic stress: a novel mode of inter-organelle crosstalk. The EMBO journal. 1999;18:522–533. doi: 10.1093/emboj/18.3.522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butterfield DA, Boyd-Kimball D. The critical role of methionine 35 in Alzheimer's amyloid beta-peptide (1-42)-induced oxidative stress and neurotoxicity. Biochim Biophys Acta. 2005;1703:149–156. doi: 10.1016/j.bbapap.2004.10.014. [DOI] [PubMed] [Google Scholar]
- Butterfield DA, Kanski J. Methionine residue 35 is critical for the oxidative stress and neurotoxic properties of Alzheimer's amyloid beta-peptide 1-42. Peptides. 2002;23:1299–1309. doi: 10.1016/s0196-9781(02)00066-9. [DOI] [PubMed] [Google Scholar]
- Butterfield DA, Poon HF, St Clair D, Keller JN, Pierce WM, Klein JB, Markesbery WR. Redox proteomics identification of oxidatively modified hippocampal proteins in mild cognitive impairment: insights into the development of Alzheimer's disease. Neurobiol Dis. 2006;22:223–232. doi: 10.1016/j.nbd.2005.11.002. [DOI] [PubMed] [Google Scholar]
- Chan DC. Mitochondria: Dynamic organelles in disease, aging, and development. Cell. 2006;125:1241–1252. doi: 10.1016/j.cell.2006.06.010. [DOI] [PubMed] [Google Scholar]
- Cleary JP, Walsh DM, Hofmeister JJ, Shankar GM, Kuskowski MA, Selkoe DJ, Ashe KH. Natural oligomers of the amyloid-beta protein specifically disrupt cognitive function. Nature neuroscience. 2005;8:79–84. doi: 10.1038/nn1372. [DOI] [PubMed] [Google Scholar]
- Daosukho C, Ittarat W, Lin SM, Sawyer DB, Kiningham K, Lien YC, St Clair DK. Induction of manganese superoxide dismutase (MnSOD) mediates cardioprotective effect of tamoxifen (TAM) J Mol Cell Cardiol. 2005;39:792–803. doi: 10.1016/j.yjmcc.2005.07.011. [DOI] [PubMed] [Google Scholar]
- Dekaban AS. Changes in brain weights during the span of human life: relation of brain weights to body heights and body weights. Ann Neurol. 1978;4:345–356. doi: 10.1002/ana.410040410. [DOI] [PubMed] [Google Scholar]
- Dhar SK, Xu Y, Chen Y, St Clair DK. Specificity protein 1-dependent p53-mediated suppression of human manganese superoxide dismutase gene expression. J Biol Chem. 2006;281:21698–21709. doi: 10.1074/jbc.M601083200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding QX, Markesbery WR, Cecarini V, Keller JN. Decreased RNA, and increased RNA oxidation, in ribosomes from early Alzheimer's disease. Neurochem Res. 2006;31:705–710. doi: 10.1007/s11064-006-9071-5. [DOI] [PubMed] [Google Scholar]
- Ding QX, Markesbery WR, Chen QH, Li F, Keller JN. Ribosome dysfunction is an early event in Alzheimer's disease. J Neurosci. 2005;25:9171–9175. doi: 10.1523/JNEUROSCI.3040-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drane P, Bravard A, Bouvard V, May E. Reciprocal down-regulation of p53 and SOD2 gene expression-implication in p53 mediated apoptosis. Oncogene. 2001;20:430–439. doi: 10.1038/sj.onc.1204101. [DOI] [PubMed] [Google Scholar]
- Esposito L, Raber J, Kekonius L, Yan FR, Yu GQ, Bien-Ly N, Puolivali J, Scearce-Levie K, Masliah E, Mucke L. Reduction in mitochondrial superoxide dismutase modulates Alzheimer's disease-like pathology and accelerates the onset of behavioral changes in human amyloid precursor protein transgenic mice. J Neurosci. 2006;26:5167–5179. doi: 10.1523/JNEUROSCI.0482-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer-Sueta G, Hannibal L, Batinic-Haberle I, Radi R. Reduction of manganese porphyrins by flavoenzymes and submitochondrial particles: a catalytic cycle for the reduction of peroxynitrite. Free Radic Biol Med. 2006;41:503–512. doi: 10.1016/j.freeradbiomed.2006.04.028. [DOI] [PubMed] [Google Scholar]
- Goate A, Chartierharlin MC, Mullan M, Brown J, Crawford F, Fidani L, Giuffra L, Haynes A, Irving N, James L, Mant R, Newton P, Rooke K, Roques P, Talbot C, Pericakvance M, Roses A, Williamson R, Rossor M, Owen M, Hardy J. Segregation of a Missense Mutation in the Amyloid Precursor Protein Gene with Familial Alzheimers-Disease. Nature. 1991;349:704–706. doi: 10.1038/349704a0. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Zulueta M, Ensz LM, Mukhina G, Lebovitz RM, Zwacka RM, Engelhardt JF, Oberley LW, Dawson VL, Dawson TM. Manganese superoxide dismutase protects nNOS neurons from NMDA and nitric oxide-mediated neurotoxicity. J Neurosci. 1998;18:2040–2055. doi: 10.1523/JNEUROSCI.18-06-02040.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guidi I, Galimberti D, Lonati S, Novembrino C, Bamonti F, Tiriticco M, Fenoglio C, Venturelli E, Baron P, Bresolin N, Scarpini E. Oxidative imbalance in patients with mild cognitive impairment and Alzheimer's disease. Neurobiol Aging. 2006;27:262–269. doi: 10.1016/j.neurobiolaging.2005.01.001. [DOI] [PubMed] [Google Scholar]
- Hagen TM, Yowe DL, Bartholomew JC, Wehr CM, Do KL, Park JY, Ames BN. Mitochondrial decay in hepatocytes from old rats: membrane potential declines, heterogeneity and oxidants increase. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:3064–3069. doi: 10.1073/pnas.94.7.3064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Hirai K, Aliev G, Nunomura A, Fujioka H, Russell RL, Atwood CS, Johnson AB, Kress Y, Vinters HV, Tabaton M, Shimohama S, Cash AD, Siedlak SL, Harris PLR, Jones PK, Petersen RB, Perry G, Smith MA. Mitochondrial abnormalities in Alzheimer's disease. J Neurosci. 2001;21:3017–3023. doi: 10.1523/JNEUROSCI.21-09-03017.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller JN, Kindy MS, Holtsberg FW, St Clair DK, Yen HC, Germeyer A, Steiner SM, Bruce-Keller AJ, Hutchins JB, Mattson MP. Mitochondrial manganese superoxide dismutase prevents neural apoptosis and reduces ischemic brain injury: Suppression of peroxynitrite production, lipid peroxidation, and mitochondrial dysfunction. J Neurosci. 1998;18:687–697. doi: 10.1523/JNEUROSCI.18-02-00687.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klivenyi P, St Clair D, Wermer M, Yen HC, Oberley T, Yang LC, Beal MF. Manganese superoxide dismutase overexpression attenuates MPTP toxicity. Neurobiol Dis. 1998;5:253–258. doi: 10.1006/nbdi.1998.0191. [DOI] [PubMed] [Google Scholar]
- LaFerla FM, Green KN, Oddo S. Intracellular amyloid-beta in Alzheimer's disease. Nat Rev Neurosci. 2007;8:499–509. doi: 10.1038/nrn2168. [DOI] [PubMed] [Google Scholar]
- Lauderback CM, Hackett JM, Huang FF, Keller JN, Szweda LI, Markesbery WR, Butterfield DA. The glial glutamate transporter, GLT-1, is oxidatively modified by 4-hydroxy-2-nonenal in the Alzheimer's disease brain: the role of Abeta1-42. Journal of neurochemistry. 2001;78:413–416. doi: 10.1046/j.1471-4159.2001.00451.x. [DOI] [PubMed] [Google Scholar]
- Lebovitz RM, Zhang HJ, Vogel H, Cartwright J, Dionne L, Lu NF, Huang S, Matzuk MM. Neurodegeneration, myocardial injury, and perinatal death in mitochondrial superoxide dismutase-deficient mice. Proc Natl Acad Sci USA. 1996;93:9782–9787. doi: 10.1073/pnas.93.18.9782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesuisse C, Martin LJ. Long-term culture of mouse cortical neurons as a model for neuronal development, aging, and death. J Neurobiol. 2002;51:9–23. doi: 10.1002/neu.10037. [DOI] [PubMed] [Google Scholar]
- Li F, Calingasan NY, Yu FM, Mauck WM, Toidze M, Almeida CG, Takahashi RH, Carlson GA, Beal MF, Lin MT, Gouras GK. Increased plaque burden in brains of APP mutant MnSOD heterozygous knockout mice. J Neurochem. 2004;89:1308–1312. doi: 10.1111/j.1471-4159.2004.02455.x. [DOI] [PubMed] [Google Scholar]
- Li YB, Huang TT, Carlson EJ, Melov S, Ursell PC, Olson TL, Noble LJ, Yoshimura MP, Berger C, Chan PH, Wallace DC, Epstein CJ. Dilated Cardiomyopathy And Neonatal Lethality In Mutant Mice Lacking Manganese Superoxide-Dismutase. Nat Genet. 1995;11:376–381. doi: 10.1038/ng1295-376. [DOI] [PubMed] [Google Scholar]
- Liu T, Perry G, Chan HW, Verdile G, Martins RN, Smith MA, Atwood CS. Amyloid-beta-induced toxicity of primary neurons is dependent upon differentiation-associated increases in tau and cyclin-dependent kinase 5 expression. J Neurochem. 2004;88:554–563. doi: 10.1046/j.1471-4159.2003.02196.x. [DOI] [PubMed] [Google Scholar]
- Lovell MA, Xie CS, Markesbery WR. Decreased base excision repair and increased helicase activity in Alzheimer's disease brain. Brain Res. 2000;855:116–123. doi: 10.1016/s0006-8993(99)02335-5. [DOI] [PubMed] [Google Scholar]
- MacMillan-Crow LA, Crow JP, Thompson JA. Peroxynitrite-mediated inactivation of manganese superoxide dismutase involves nitration and oxidation of critical tyrosine residues. Biochemistry. 1998;37:1613–1622. doi: 10.1021/bi971894b. [DOI] [PubMed] [Google Scholar]
- MacMillanCrow LA, Crow JP, Kerby JD, Beckman JS, Thompson JA. Nitration and inactivation of manganese superoxide dismutase in chronic rejection of human renal allografts. Proc Natl Acad Sci USA. 1996;93:11853–11858. doi: 10.1073/pnas.93.21.11853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcus DL, Thomas C, Rodriguez C, Simberkoff K, Tsai JS, Strafaci JA, Freedman ML. Increased peroxidation and reduced antioxidant enzyme activity in Alzheimer's disease. Exp Neurol. 1998;150:40–44. doi: 10.1006/exnr.1997.6750. [DOI] [PubMed] [Google Scholar]
- Markesbery WR. Oxidative stress hypothesis in Alzheimer's disease. Free Radic Biol Med. 1997;23:134–147. doi: 10.1016/s0891-5849(96)00629-6. [DOI] [PubMed] [Google Scholar]
- Markesbery WR, Kryscio RJ, Lovell MA, Morrow JD. Lipid peroxidation is an early event in the brain in amnestic mild cognitive impairment. Ann Neurol. 2005;58:730–735. doi: 10.1002/ana.20629. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Goodman Y, Luo H, Fu WM, Furukawa K. Activation of NF-kappa B protects hippocampal neurons against oxidative stress-induced apoptosis: Evidence for induction of manganese superoxide dismutase and suppression of peroxynitrite production and protein tyrosine nitration. J Neurosci Res. 1997;49:681–697. doi: 10.1002/(SICI)1097-4547(19970915)49:6<681::AID-JNR3>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Migliore L, Fontana I, Trippi F, Colognato R, Coppede F, Tognoni G, Nucciarone B, Siciliano G. Oxidative DNA damage in peripheral leukocytes of mild cognitive impairment and AD patients. Neurobiol Aging. 2005;26:567–573. doi: 10.1016/j.neurobiolaging.2004.07.016. [DOI] [PubMed] [Google Scholar]
- Oliver CN, Ahn BW, Moerman EJ, Goldstein S, Stadtman ER. Age-related changes in oxidized proteins. The Journal of biological chemistry. 1987;262:5488–5491. [PubMed] [Google Scholar]
- Picklo MJ, Montine TJ, Amarnath V, Neely MD. Carbonyl toxicology and Alzheimer's disease. Toxicol Appl Pharmacol. 2002;184:187–197. doi: 10.1006/taap.2002.9506. [DOI] [PubMed] [Google Scholar]
- Pizzi M, Sarnico I, Boroni F, Benarese M, Steimberg N, Mazzoleni G, Dietz GP, Bahr M, Liou HC, Spano PF. NF-kappaB factor c-Rel mediates neuroprotection elicited by mGlu5 receptor agonists against amyloid beta-peptide toxicity. Cell Death Differ. 2005;12:761–772. doi: 10.1038/sj.cdd.4401598. [DOI] [PubMed] [Google Scholar]
- Pratico D, Clark CM, Liun F, Lee VYM, Trojanowski JQ. Increase of brain oxidative stress in mild cognitive impairment - A possible predictor of Alzheimer disease. Arch Neurol. 2002;59:972–976. doi: 10.1001/archneur.59.6.972. [DOI] [PubMed] [Google Scholar]
- Reaume AG, Howland DS, Trusko SP, Savage MJ, Lang DM, Greenberg BD, Siman R, Scott RW. Enhanced amyloidogenic processing of the beta-amyloid precursor protein in gene-targeted mice bearing the Swedish familial Alzheimer's disease mutations and a "humanized" A beta sequence. J Biol Chem. 1996;271:23380–23388. doi: 10.1074/jbc.271.38.23380. [DOI] [PubMed] [Google Scholar]
- Rinaldi P, Polidori MC, Metastasio A, Mariani E, Mattioli R, Cherubini A, Catani M, Cecchetti R, Senin U, Mecocci P. Plasma antioxidants are similarly depleted in mild cognitive impairment and in Alzheimer's disease. Neurobiol Aging. 2003;24:915–919. doi: 10.1016/s0197-4580(03)00031-9. [DOI] [PubMed] [Google Scholar]
- Rogers RJ, Monnier JM, Nick HS. Tumor necrosis factor-alpha selectively induces MnSOD expression via mitochondria-to-nucleus signaling, whereas interleukin-1beta utilizes an alternative pathway. The Journal of biological chemistry. 2001;276:20419–20427. doi: 10.1074/jbc.M008915200. [DOI] [PubMed] [Google Scholar]
- Schulze-Osthoff K, Beyaert R, Vandevoorde V, Haegeman G, Fiers W. Depletion of the mitochondrial electron transport abrogates the cytotoxic and gene-inductive effects of TNF. The EMBO journal. 1993;12:3095–3104. doi: 10.1002/j.1460-2075.1993.tb05978.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherrington R, Rogaev EI, Liang Y, Rogaeva EA, Levesque G, Ikeda M, Chi H, Lin C, Li G, Holman K, Tsuda T, Mar L, Foncin JF, Bruni AC, Montesi MP, Sorbi S, Rainero I, Pinessi L, Nee L, Chumakov I, Pollen D, Brookes A, Sanseau P, Polinsky RJ, Wasco W, Dasilva HAR, Haines JL, Pericakvance MA, Tanzi RE, Roses AD, Fraser PE, Rommens JM, Stgeorgehyslop PH. Cloning of a Gene Bearing Missense Mutations in Early-Onset Familial Alzheimers-Disease. Nature. 1995;375:754–760. doi: 10.1038/375754a0. [DOI] [PubMed] [Google Scholar]
- Siman R, Reaume AG, Savage MJ, Trusko S, Lin YG, Scott RW, Flood DG. Presenilin-1 P264L knock-in mutation: Differential effects on A beta production, amyloid deposition, and neuronal vulnerability. J Neurosci. 2000;20:8717–8726. doi: 10.1523/JNEUROSCI.20-23-08717.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MA, Richey Harris PL, Sayre LM, Beckman JS, Perry G. Widespread peroxynitrite-mediated damage in Alzheimer's disease. J Neurosci. 1997;17:2653–2657. doi: 10.1523/JNEUROSCI.17-08-02653.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sompol P, Xu Y, Ittarat W, Daosukho C, St Clair D. NF-kappaB-Associated MnSOD Induction Protects Against beta-Amyloid-Induced Neuronal Apoptosis. J Mol Neurosci. 2006;29:279–288. [PubMed] [Google Scholar]
- Spasojevic I, Chen Y, Noel TJ, Yu Y, Cole MP, Zhang L, Zhao Y, St Clair DK, Batinic-Haberle I. Mn porphyrin-based superoxide dismutase (SOD) mimic, MnIIITE-2-PyP5+, targets mouse heart mitochondria. Free Radic Biol Med. 2007;42:1193–1200. doi: 10.1016/j.freeradbiomed.2007.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitz DR, Oberley LW. An Assay for Superoxide-Dismutase Activity in Mammalian Tissue-Homogenates. Anal Biochem. 1989;179:8–18. doi: 10.1016/0003-2697(89)90192-9. [DOI] [PubMed] [Google Scholar]
- Terman A, Brunk UT. Oxidative stress, accumulation of biological 'garbage', and aging. Antioxid Redox Signal. 2006;8:197–204. doi: 10.1089/ars.2006.8.197. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416:535–539. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- Wan XS, Devalaraja MN, St Clair DK. Molecular structure and organization of the human manganese superoxide dismutase gene. DNA Cell Biol. 1994;13:1127–1136. doi: 10.1089/dna.1994.13.1127. [DOI] [PubMed] [Google Scholar]
- Wang J, Xiong S, Xie C, Markesbery WR, Lovell MA. Increased oxidative damage in nuclear and mitochondrial DNA in Alzheimer's disease. J Neurochem. 2006;93:953–962. doi: 10.1111/j.1471-4159.2005.03053.x. [DOI] [PubMed] [Google Scholar]
- Yen HC, Oberley TD, Vichitbandha S, Ho YS, St Clair DK. The protective role of manganese superoxide dismutase against adriamycin-induced acute cardiac toxicity in transgenic mice. J Clin Invest. 1996;98:1253–1260. doi: 10.1172/JCI118909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Chaiswing L, Oberley TD, Batinic-Haberle I, St Clair W, Epstein CJ, St Clair D. A mechanism-based antioxidant approach for the reduction of skin carcinogenesis. Cancer research. 2005;65:1401–1405. doi: 10.1158/0008-5472.CAN-04-3334. [DOI] [PubMed] [Google Scholar]