

Abstract
Alphaviruses are regarded as attractive systems for expression of heterologous genes and development of recombinant vaccines. Venezuelan equine encephalitis virus (VEE)-based vectors are particularly promising because of their specificity to lymphoid tissues and strong resistance to interferon. To improve understanding of the VEE genome packaging and optimize application of this virus as a vector, we analyzed in more detail the mechanism of packaging of the VEE-specific RNAs. The presence of the RNAs in the VEE particles during serial passaging in tissue culture was found to depend not only on the presence of packaging signal(s), but also on the ability of these RNAs to express in cis nsP1, nsP2 and nsP3 in the form of a P123 precursor. Packaging of VEE genomes into infectious virions was also found to be more efficient compared to that of Sindbis virus, in spite of lower levels of RNA replication and structural protein production.
Keywords: alphavirus, VEE, RNA replication, packaging, nonstructural proteins
INTRODUCTION
The alphavirus genus in the Togaviridae family contains almost 30 members, some of which are important human and animal pathogens (Griffin, 2001; Strauss and Strauss, 1994; Weaver and Barrett, 2004). Alphaviruses contain a single-stranded RNA genome of positive polarity of almost 12-kb in length (Strauss, Rice, and Strauss, 1984). The 5′ two-thirds of the genome encodes nonstructural proteins nsP1–4, forming together with cellular proteins the replicative enzyme complex RdRp. The nonstructural proteins are synthesized as 2 polyproteins, P123 and P1234, encoding nsP1–3 and nsP1–4, respectively. After partial cleavage of P1234, the released nsP4 and P123 precursor assemble into the primary RdRp that functions in the synthesis of minus-strand RNAs, forming replicative intermediates (Lemm and Rice, 1993; Shirako and Strauss, 1994). After complete processing of P123, nsP1–4 form mature RdRps that are active in synthesis of new positive strand viral genomes and transcription of the subgenomic 26S RNA (Lemm and Rice, 1993; Shirako and Strauss, 1994). The latter RNA is co-linear to the 3′ one-third of the viral genome and is transcribed from the subgenomic promoter located on the negative-strand replicative RNA intermediate (Rice and Strauss, 1981). The 26S RNA accumulates in the cells to higher concentration than the viral genome and is translated into the viral structural proteins: capsid, E2 and E1, forming infectious virions. These structural proteins are dispensable for replication and transcription of alphavirus-specific RNAs (Bredenbeek et al., 1993; Geigenmuller-Gnirke et al., 1991; Pushko et al., 1997). They can be deleted in viral genomes or replaced by heterologous genes, and, upon delivery into the cells, such RNAs (replicons) sustain self-replication and can efficiently express heterologous genes cloned under control of the subgenomic promoter.
The replicon-based gene expression systems were designed on the basis of Sindbis (SIN), Semliki Forest (SFV) and Venezuelan (VEE) and eastern (EEE) equine encephalitis virus genomes (Bredenbeek et al., 1993; Liljeström and Garoff, 1991; Petrakova et al., 2005; Pushko et al., 1997). These replicons can be delivered into the cell via transfection of the in vitro-synthesized replicon RNAs or by infection with replicon-containing viral particles. Packaging of replicons into these particles can be achieved by co-transfecting the cells by in vitro-synthesized replicon RNAs and so-called defective helper RNAs (DH RNAs). Transfected replicon RNAs supply nsP1–4 for their own replication and for replication of DH RNAs. These helper RNAs are incapable of producing a complete set of nsPs (if any nsPs at all), but can replicate if viral RdRp components are supplied in trans. Replicating DH RNAs drive the expression of the structural proteins, packaging replicons into infectious virions. As do defective interfering (DI) RNAs, DH RNAs contain virus-specific promoter elements at the 3′ and 5′ ends (cis-acting RNA elements). The cis-acting elements can be either the RNA fragments normally present at the very 3′ and 5′ ends of viral genomes, or some other sequences of cellular or viral origin. In addition, the DH RNAs also contain a subgenomic promoter that controls the transcription of the subgenomic RNA, encoding viral structural proteins.
Packaging of virus-specific RNAs (viral genome, replicon, DI RNA and DH RNAs) into viral particles was shown to depend on the presence of so-called packaging signals (PS) in their genomes (Frolova, Frolov, and Schlesinger, 1997; Weiss, Geigenmuller-Gnirke, and Schlesinger, 1994; White, Thomson, and Dimmock, 1998). These signals strongly enhance RNA encapsidation, and they were identified in SIN, SFV and Ross River virus (RRV) genomes, and the mechanism of their interaction with the capsid protein was intensively studied in a SIN model (Weiss, Geigenmuller-Gnirke, and Schlesinger, 1994; Weiss et al., 1989). The tRNAAsp sequence found in the naturally occurring SIN DI RNA was also shown to function not only as a promoter in RNA replication, but as a PS efficiently recognized by SIN structural proteins (Fayzulin et al., 2005; Frolova, Frolov, and Schlesinger, 1997).
In addition to being an important means of replicon packaging, alphavirus DH RNAs are useful tools for dissecting the mechanisms of viral RNA replication and packaging into infectious particles (Fayzulin et al., 2005; Frolova, Frolov, and Schlesinger, 1997). Helper RNAs do not need to encode functional replicative enzymes, and different RNA fragments of homologous and heterologous origin can be cloned into DH RNAs for testing their ability to function as packaging signals or promoters independently of their capability to express viral nsPs. The results of such studies not only improve our understanding of alphavirus replication, but they can be also directly applied in the subsequent development of alphavirus-based gene expression systems.
In the present work, we further investigated the packaging process of the alphaviruses using VEE as a model. The VEE-based replicons were packaged into virions by using one or two helpers, and we studied both the efficiency of the replicon packaging and the ability of helpers to self-package that is required for large-scale production of packaged replicons. We demonstrated that packaging of VEE-specific RNAs and their persistence in a viral population depend not only on the possession of packaging signal(s), but also on the ability of the helper RNAs to express functional nsP1–nsP3. The nsP1–nsP3 expression does not noticeably change the efficiency of packaging, but appears to have a strong positive impact on DH RNA replication (most likely on initiation of replication) that is critical for the ability of these defective RNAs to persist in a viral population. In addition, VEE demonstrated more efficient RNA packaging than that found for SIN, in spite of lower levels of RNA replication and translation of structural proteins. The data suggest that these specific features of VEE packaging could be critical determinants of the highly pathogenic phenotype of this virus. In addition, they may be useful in further development and application of VEE-based expression systems.
RESULTS
Packaging of VEE replicons using DH RNAs with deletion of all of the nonstructural genes
The VEE replicon used in our study was designed on the basis of the vaccine strain VEE TC-83 genome (Kinney et al., 1989). The only modification in the TC-83-specific sequence was a replacement of adenosine in the third position of the 5′ UTR by guanosine that is present in the wild type (wt) Trinidad donkey (TRD) strain of VEE. The presence of guanosine in this position was previously shown to be critical for the virus to exhibit an interferon-resistant phenotype and more efficient transcription of the subgenomic RNA (Petrakova et al., 2005; White et al., 2001). The GFP-encoding sequence was cloned under control of the subgenomic promoter, the second subgenomic promoter drove the expression of puromycin acetyltransferase (Pur), which makes cells resistant to puromycin. GFP expression was convenient for evaluating the titers of packaged replicons in infectious units (inf.u) (see Materials and Methods for details), Pac expression was used for generating stable PurR cell lines and measuring the titers in the colony-forming units (CFU). The previously designed SIN-based replicon SINrep/GFP (Gorchakov et al., 2004) demonstrated a cytopathic phenotype and contained a single subgenomic promoter that controlled transcription of GFP-encoding subgenomic RNA.
The initially constructed VEE DH RNA cassettes (Hvee/C+Gl, Hvee/C and Hvee/Gl) (Fig. 1A) had a design similar to that previously described for SIN, VEE and SFV helpers (Bredenbeek et al., 1993; Liljeström and Garoff, 1991; Pushko et al., 1997), in that the VEE genome fragment between nt 520 and 7290, encoding all of the nonstructural genes, was deleted. The subgenomic RNA of Hvee/C+Gl contained all of the VEE structural genes, and the Hvee/C and Hvee/Gl helpers were capable of expressing only capsid and glycoproteins, respectively. The subgenomic RNA in Hvee/Gl cassette encoded the VEE capsid with a deleted cluster of positively charged amino acids (see Materials and Methods for details). We considered it a very likely event that, as with the SIN capsid-coding sequence, the VEE capsid gene could contain a translational enhancer (Frolov and Schlesinger, 1996), which would increase the level of synthesis of viral structural proteins in the infected cells, in which the cellular protein synthesis would be significantly downregulated due to virus replication. Thus, the putative enhancer-containing sequence was left upstream of the glycoprotein genes. The deletion was aimed to make the capsid expressed from Hvee/Gl incapable of RNA packaging and to eliminate a possibility of infectious viral genome formation after recombination between the VEE replicon and Hvee/Gl helper. SIN-specific Hsin/C+Gl and tRNA/Hsin/C+Gl helpers were described in detail in our previous publication (Bredenbeek et al., 1993). They contained a 6914 nt-long deletion in the nonstructural genes and differed only in the 5′UTR. Hsin/C+Gl had a natural SIN 5′UTR, while tRNA/Hsin/C+Gl contained a tRNA structure (Monroe and Schlesinger, 1983) that increased the level of its replication and served as a packaging signal (Fayzulin et al., 2005; Frolova, Frolov, and Schlesinger, 1997).
Fig. 1.
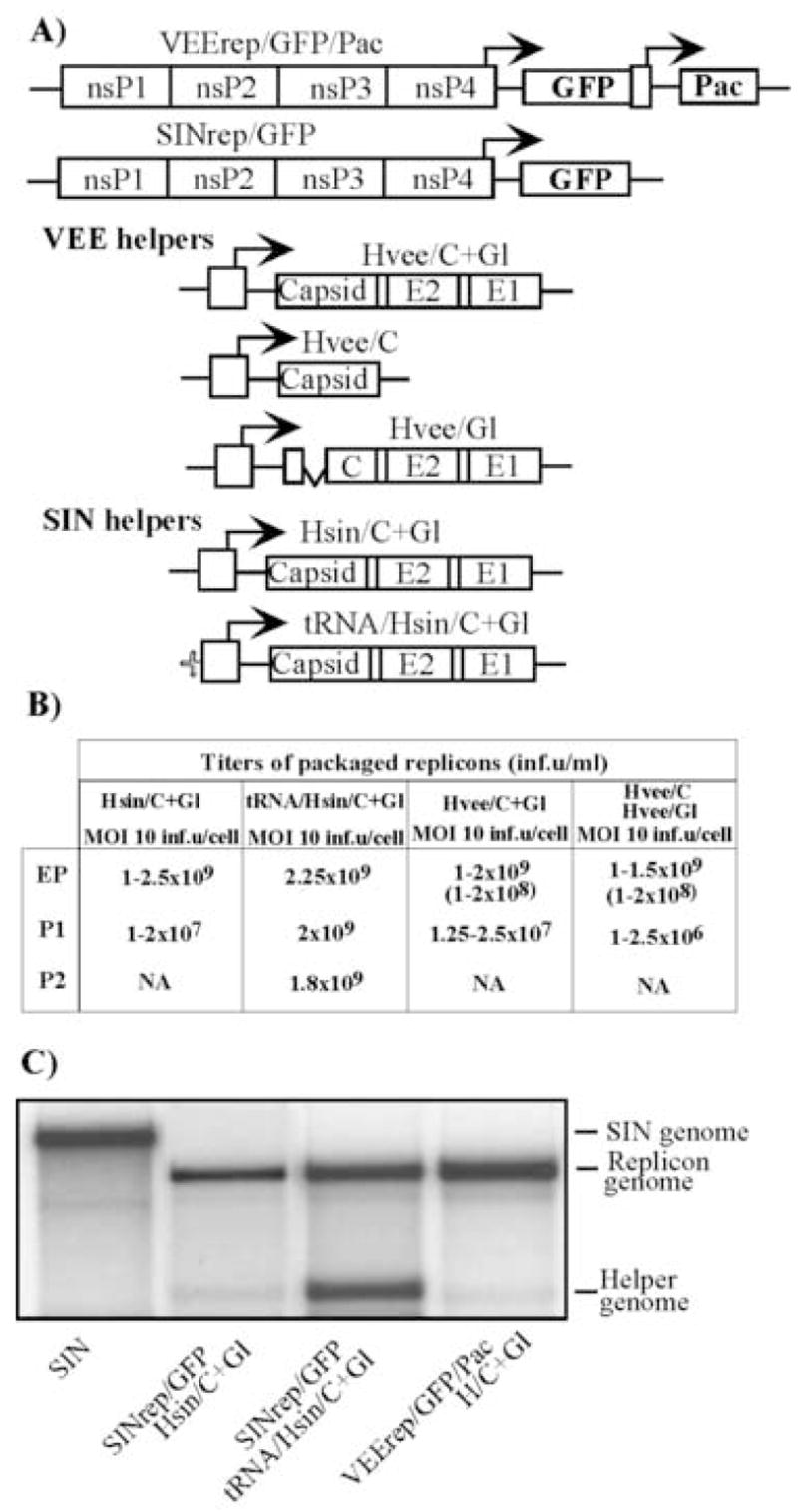
Packaging of VEE and SIN replicons in BHK-21 cells using helpers with deletions of nsP1-4-coding genes. (A) Schematic representation of SIN and VEE replicons and helper genomes. All of the VEE helpers, Hvee/C+Gl, Hvee/C and Hvee/Gl, had natural 5′UTR, derived from VEE TC-83 virus genome, and deletion of nt 520–7290. Hsin/C+Gl and tRNA/Hsin/C+Gl had natural SIN 5′UTR and the 5′ tRNAAsp sequence derived from the naturally occurring SIN DI RNA, respectively. SIN helpers contained a deletion of nt 421–7334 of SIN genome. (B) Titers of packaged VEE and SIN replicons after electroporation and further passaging of the samples. BHK-21 cells were co-transfected with VEErep/GFP/Pac or SINrep/GFP replicons and indicated helper RNAs. Harvested viral particles were used for the next round of infection of naïve BHK-21 cells at the indicated MOIs (measured in inf.u/cell). Viruses were harvested after development of CPE and used for further passaging. Titers refer to unconcentrated virus-containing media. Numbers in the brackets indicate titers in colony-forming units (CFU/ml). All of the experiments were performed multiple times and generated very reproducible titers. NA indicates “not applicable,” because concentration of the packaged replicons after passage 1 was insufficient for infecting cells on the next passage at an MOI of 10 inf.u/cell. (C) Packaging of replicon and helper RNAs into viral particles. 32P-labeled RNAs were isolated from the viral particles released from the cells co-transfected with the indicated replicon and helper RNAs (see Materials and Methods for details). RNA isolated from SIN virus was used as a positive control.
SIN and VEE replicons were transfected into BHK-21 cells together with the homologous helpers, indicated in Fig. 1A, and particles were harvested 24 h post electroporation, after development of profound cytopathic effect (CPE). VEE replicons were packaged very efficiently both by single helper Hvee/C+Gl and by two helpers, Hvee/C and Hvee/Gl, (Fig. 2B). In multiple experiments, titers of the replicon-containing particles reproducibly approached 1–2 × 109 inf.u/ml. Packaged VEE replicons were capable of establishing persistent replication (see titers in the brackets), and BHK-21 cells infected with replicons confirmed a PurR phenotype and expressed GFP. The titers in CFU/ml were noticeably lower than titers in infectious units, but this was an expected phenomenon. During the acute, early phase of replication, wt VEE replicons demonstrate some level of cytopathogenicity (Petrakova et al., 2005).
Fig. 2.
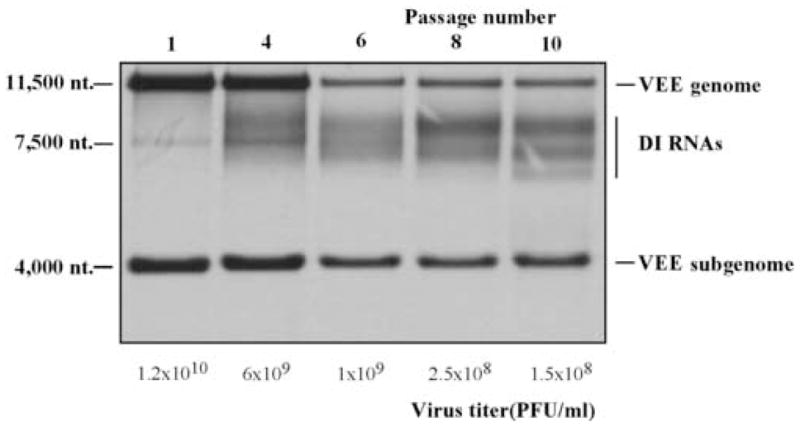
Accumulation of the DI RNAs in the samples of VEE TC-83 passaged in BHK-21 cells at high MOI. Passaging was performed as described in Materials and Methods. RNA labeling in the presence of ActD and analysis were performed for the samples harvested after passage 1, 4, 6, 8 and 10 as described in Materials and Methods. Virus-specific RNAs and viral titers are indicated.
Further passaging of VEE replicon-containing samples, aimed at testing the possibility of generating larger stocks, was inefficient. Hvee/C+Gl helper genomes were present in released particles at low concentrations (Fig. 1C). After infecting naïve cells by samples packaged with one or two helpers at an MOI of 10 inf.u/cell, we detected the release of replicons only to the titer of 1–2 × 107 and 2.5 × 106 inf.u/ml, respectively. These titers were 2–3 orders of magnitude below the titers obtained after electroporation. There was no apparent difference when the the infections were performed at higher MOIs (data not shown). The titers described here for VEE helpers were consistent with those obtained for SINrep/GFP packaged with Hsin/C+Gl. The latter helper contained natural SIN 5′UTR, and it packaged a homologous SIN replicon very efficiently, but was incapable of self-packaging (Fig. 1C). Consequently, the samples could not be serially passaged at MOI of 10 inf.u/cell (Fig. 1B) or higher (data not shown) to produce titers exceeding 107 inf.u/ml. Consistently with our previous work, a SIN helper with the 5′UTR derived from the naturally occurring DI RNA (tRNA/Hsin/C+Gl) packaged not only the SIN replicon to high titers, but also packaged itself (Fig. 1B and C), making viral samples capable of being serially passaged without reductions in titers. Based on these data, we assumed that concentrations of helper-containing particles in the samples of VEE replicons packaged with either Hvee/C+Gl or Hvee/Gl and Hvee/C were insufficient for delivery of helper RNAs into the cells at concentrations adequate for supporting next rounds of productive infection.
VEE DI RNAs
Alphavirus defective interfering RNAs are composed of i) cis-acting RNA elements required for their replication by replicative enzymes produced by replicating helper virus, and ii) the sequences that promote DI RNA packaging into viral particles, PSs (Strauss and Strauss, 1994). Structure of the DI RNAs was intensively studied for SFV and SIN (Lehtovaara et al., 1982; Monroe and Schlesinger, 1983; Monroe and Schlesinger, 1984; White, Thomson, and Dimmock, 1998), but there was no available information about the replication and sequences of VEE-specific DI RNAs. Thus, we generated VEE DI RNAs by serial passaging of cDNA-derived VEE TC-83 in BHK-21 cells at high MOIs. The first passages were performed at an MOI of close to 10,000 PFU/cell. At the late passages, the MOI became lower (close to 100 PFU/cell) due to accumulation of the DI RNAs (Fig. 2). These RNAs were readily detectable by the metabolic labeling with [3H]uridine, followed by electrophoresis in denaturing conditions. After 10 passages, there was no accumulation of clearly visible dominating DI RNA species, and the DI RNAs genomes were distributed on the gels between 6,000 and 8,000 nt, albeit 1–3 diffuse RNA bands could be detected (Fig. 2). Based on the length of the VEE DI RNA genomes, we hypothesized that as has been demonstrated for SIN and SFV, these DI RNAs contained a deletion of the viral structural genes, but, most likely, retained a major segment of RdRp-coding sequence. To test this hypothesis, we performed an RT-PCR analysis of VEE DI RNA genomes using primers specific to 3′ UTR and nsP3 gene (nt 5312–5333). The PCR product was a diffuse band, containing DNA fragments ranging between 450 and 850 nt. After this material was cloned, we selected at random 6 clones that were revealed to contain the end of the VEE nsP3 gene and fragments of nsP4 and 3′ UTR of different lengths (data not shown). Thus, a significant fraction of the RNAs had a common feature: deletions of the structural genes and the nsP4-coding sequence. This was an indication that either i) the genome sequence between nt 520 and the end of nsP3 gene (deleted in Hvee helpers) could contain an efficient packaging signal(s) utilized by VEE capsid, or ii) the nonstructural protein(s) had to be expressed in cis from the DI RNA genomes to support the efficient persistence of the latter RNAs in the viral population, or iii) both the presence of PS and expression of nsPs cis could have synergistic positive effects on RNA packaging and/or replication.
Replication of VEE helper RNAs encoding nsP1–nsP3
To further investigate the effect of nsP1-nsP3-coding sequences on RNA packaging and the presence of virus-specific RNAs in viral populations, we designed a set of recombinant helpers that were either capable of expressing nsP1–3 (H123/C+Gl, H123/Gl and H123/C) (see Materials and Methods for details) or contained essentially the same nsP1-nsP3-coding nucleotide sequence except for an insertion of 4 nt at the 1620 position (Hstop123/C+Gl). These few extra nucleotides destroyed the open reading in the nsP1-coding sequence, making this helper incapable of nsPs production. Helpers H123/C+Gl encoded all of the VEE structural genes under control of the subgenomic promoter, and helpers H123/C and H123/Gl contained VEE capsid- and glycoprotein-coding cassettes, respectively, that were the same as those described above for Hvee/Gl and Hvee/C. Upon transfection into the cells, H123/C+Gl efficiently packaged replicons to the titers exceeding 109 inf.u/ml. Samples harvested after electroporation were passaged at an MOI of 10 and 1 inf.u/cell, and replicons were reproducibly packaged to a concentration of 1–2.5 × 109 inf.u/ml, indicating that helpers were present in the harvested samples at levels sufficient for supporting productive viral replication during serial passaging. In the case of the two-helper packaging system (H123/C and H123/Gl helpers), titers of packaged replicons were detectably lower, but during passaging at MOIs 10 and 1 inf.u/cell, remained at the levels exceeding 108 inf.u/ml, which suggested that replicons and both helpers were efficiently packaged into viral particles. Harvested samples of replicons packaged with H123/C+Gl or both H123/C and H123/Gl helpers demonstrated plaque-forming activity, indicating the presence of the bi- and tri-partite genome particles (or aggregates of the particles) containing both replicon and helper genomes in the virions (Fayzulin et al., 2005; Geigenmuller-Gnirke et al., 1991). However, the measured titers were 3 and 5 orders of magnitude lower for H123/C+Gl or H123/C + H123/Gl, respectively, than were titers determined in infectious units (data not shown). Thus, replicon and helper RNAs were predominantly packaged into separate virions. Compared to VEE TC-83, the tri-component genome virus with VEErep/GFP/Pac + H123/C + H123/Gl genome demonstrated slower growth kinetics (Fig. 3C), but to comparable final titers.
Fig. 3.
Packaging of VEE replicons in BHK-21 cells using helpers with deletions of nsP4-coding gene. (A) Schematic representation of VEE replicons and helper genomes. All of the helpers were derived from the VEE TC-83 genome, in which nt 5702–7500 encoding almost the entire nsP4 was deleted. The TGA codon was inserted after nt 5701. Hstop123/C+Gl helper contained an additional insertion of 4 nt sequence after nt 1620. (B) Titers of packaged VEE replicons after electroporation and further passaging of the samples. BHK-21 cells were co-transfected with VEErep/GFP/Pac replicon and indicated helper RNAs. Harvested viral particles were used for the next round of infection of naïve BHK-21 cells at the indicated MOIs (measured in inf.u/cell). Viruses were harvested after the development of CPE and used for further passaging. Titers refer to unconcentrated harvested virus-containing media. NA indicates “not applicable,” because concentration of the packaged replicons after the previous passage was insufficient for infecting cells at an MOI of 10 inf.u/cell. (C) Growth curves of the tri-component genome VEE (VEErep/GFP/Pac + H123/C + H123/Gl) and VEE TC-83. BHK-21 cells were infected at an MOI of 10 inf.u/cell or PFU/cell with tri-component virus and VEE TC-83, respectively. At the indicated times, media were replaced and titers of packaged replicons and virus were determined as described in the Materials and Methods. (D) Packaging of replicon and helper RNAs into viral particles. 32P-labeled RNAs were isolated from the viral particles released from the cells co-transfected with VEErep/GFP/Pac and indicated helper RNAs (see Materials and Methods for details). RNA isolated from VEE TC-83 virus was used as a positive control.
The Hstop123/C+Gl helper was also efficient in replicon RNA packaging, but further passaging of the viral samples generated by electroporation was unsuccessful. To additionally investigate if Hstop123/C+Gl helper RNAs were packaged into virions, we metabolically labeled RNAs with 32P and analyzed the RNA content of purified viral particles. After electroporation, Hstop123/C+Gl helper genomes were packaged as efficiently as H123/C+Gl RNAs (Fig. 3D). This result indicated that the nsP1-nsP3-coding sequence likely contained a packaging signal(s), that made both H123/C+Gl and Hstop123/C+Gl genomes more efficiently packaged into virions, when compared to the encapsidation of the Hvee/C+Gl helper RNA that had the same 5′ and 3′ cis-acting RNA elements. These helpers demonstrated very similar levels of intracellular replication after co-transfection with replicons into the cells (data not shown). Nevertheless, at the next passage, the Hstop123/C+Gl-containing viral populations were unable to establish productive replication, even at an MOI of 10 inf.u/cell, and replicon-containing infectious virions were released into the media to 3 orders of magnitude lower titers.
Taken together, the results indicated that possession of the packaging signal(s) and cis-acting promoter RNA elements are insufficient by themselves for the persistence of VEE-specific RNAs in a viral population. The ability of the RNAs to serve as a template for nsP1–nsP3 translation, in addition to being competent in utilizing the RdRp by the promoter sequences, appeared to play a critical role in replication and following packaging of defective VEE genomes.
Expression of nsP1, nsP2 and nsP3 in cis is required for persistence of VEE-specific RNAs in viral populations
To further investigate the phenomenon that synthesis of the nonstructural proteins in cis is beneficial for the persistence of VEE-specific RNAs in a viral population during serial passaging, we tested whether synthesis of the entire P123 was required or if particular nsPs could be sufficient. DH RNA capable of the production of only nsP1 could be easily designed (H1stop/C+Gl), but the constructs capable of expressing only nsP2 or nsP3 had to have major changes in the DH genome sequence that would make interpretation of the data impossible. Therefore, we designed new helpers as three nearly identical cassettes (Fig. 4A): H123/C+Gl, H1stop/C+Gl and H12stop/C+Gl had only 3-nt differences in the nsP-coding region. Thus, all of these DH constructs were expected to contain the same packaging signal(s). As described above, H123/C+Gl encoded the intact P123; H1stop/C+Gl had an insertion of a stop codon immediately downstream of the nsP1-coding sequence. Similarly, a stop codon was inserted into the nsP2/nsP3 cleavage site of H12stop/C+Gl, making this construct capable of P12, nsP1 and nsP2, but not nsP3, production. After co-electroporation of the replicon and newly designed helper RNAs into the cells, VEErep/GFP/Pac was efficiently packaged into infectious viral particles (Fig. 4B and C), and, for reasons that are not clear yet, the DH H1stop/C+Gl was the most efficient in replicon-containing particle production. All of the three helpers were found efficiently packaged into viral particles after co-electroporation with replicons (Fig. 4B). However, after the next passage at an MOI of 10 inf.u/cell, titers of packaged replicons were 2–3 orders of magnitude lower in H1stop/C+Gl- and H12stop/C+Gl-derived viral samples (Fig. 4C). In contrast, H123/C+Gl was capable of supporting serial passaging without a decrease in titers (Fig. 4C). These data indicated that nsP1–nsP3 expressed in cis (most likely in their unprocessed form P123) were critical for helper functioning in terms of its presence in the samples during serial passaging of packaged replicons and helpers in tissue culture. We cannot completely rule out a possibility that the expression of nsP3 alone from the helper could be sufficient for supporting passaging. However, formation of the alphavirus replicative complexes is a sophisticated process, and it is difficult to expect proper nsP3 functioning when it is synthesized not in P123 context.
Fig. 4.
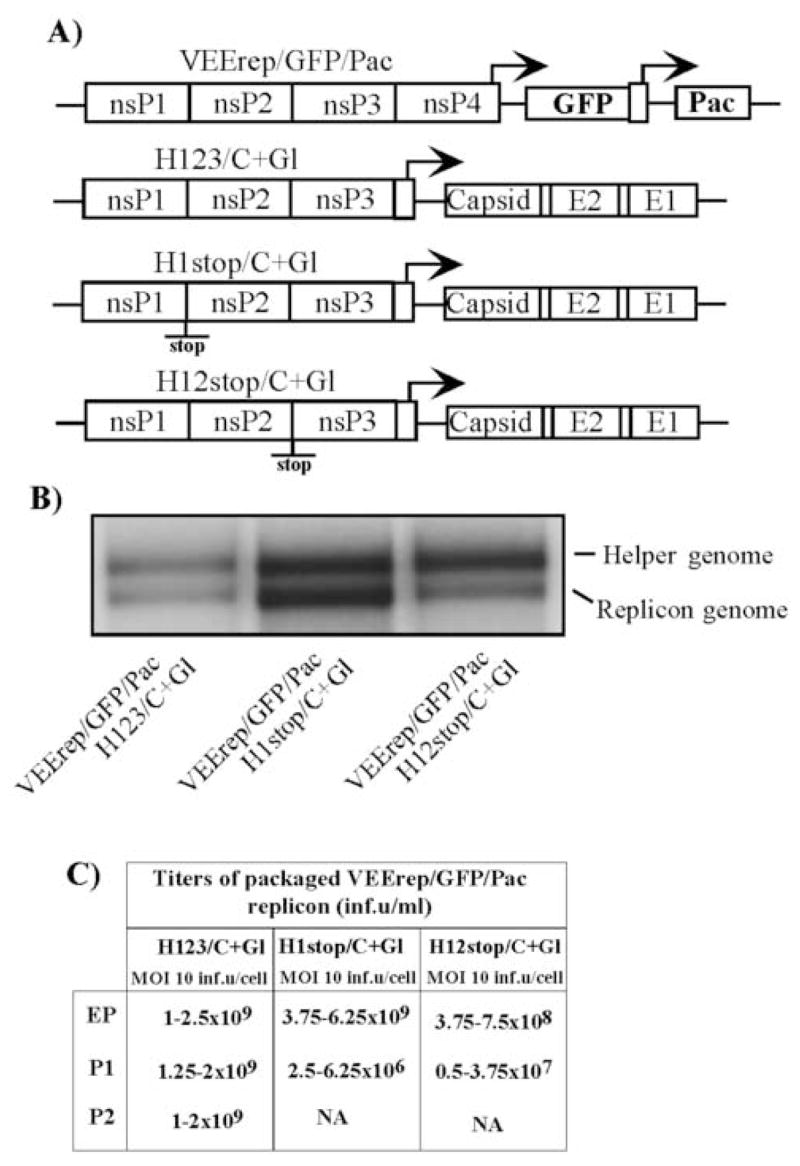
Packaging of VEE replicons in BHK-21 cells using helpers with deletions of nsP4-coding gene. (A) Schematic representation of VEE replicons and helper genomes. All of the helpers were derived from the VEE TC-83 genome, in which nt 5702–7500 were deleted. In all of them, TGA codons were inserted after the last amino acid of nsP3. The TGA codon was also inserted after the last amino acid of nsP1 and nsP2 in H1stop/C+Gl and H12stop/C+Gl, respectively. (B) Packaging of replicon and helper RNAs into viral particles. 32P-labeled RNAs were isolated from the viral particles released from the cells co-transfected with the indicated replicon and helper RNAs (see Materials and Methods for details). (C) Titers of packaged VEE replicons after electroporation and further passaging of the samples. BHK-21 cells were co-transfected with VEErep/GFP/Pac replicon and indicated helper RNAs. Harvested viral particles were used for the next round of infection of naïve BHK-21 cells at the indicated MOIs (measured in inf.u/cell). Viruses were harvested after development of CPE. Titers refer to unconcentrated harvested media. NA indicates “not applicable,” because concentration of the packaged replicons after the previous passage was insufficient for infecting cells at an MOI of 10 inf.u/cell.
Interestingly, the requirement of nsP1–3 expression in cis for persistence of virus-specific RNAs in viral populations during serial passaging is a requisite of VEE, but not of SIN infection. We designed a set of SIN helpers, Hsin123/C+Gl, Hsin123/C and Hsin123/Gl, having essentially the same genome strategy as VEE-specific H123 constructs encoding nsP1–3 (Fig. 5A). After co-electroporation of SIN replicon and these helpers into BHK-21 cells, they were efficiently packaged into viral particles (Fig. 5B) and replicated in the transfected cells (Fig. 5C upper panel). However, at the next passage, their replication in the infected cells was below detectable levels, and only a control helper tRNA/Hsin/C+Gl was replicating (Fig. 5C lower panel). As a result, titers of packaged replicons became 3 orders of magnitude lower than those after electroporation.
Fig. 5.
Packaging of SIN replicons in BHK-21 cells using helpers with deletions of the nsP4-coding gene. (A) Schematic representation of SIN replicons and helper genomes. The detailed information is presented in Materials and Methods. (B) Packaging of replicon and helper RNAs into viral particles. 32P-labeled RNAs were isolated from the viral particles released from the cells co-transfected with the indicated replicon and helper RNAs (see Materials and Methods for details). RNA isolated from SIN virus was used as a positive control. (C) Replication of SIN-specific RNAs in BHK-21 cells after electroporation (upper panel) and during passage 1 performed at an MOI 10 inf.u/cell. At 3 h post transfection or post infection, media were replaced by the same media supplemented with dactinomycin (1 μg/ml) and [3H]uridine (20 μCi/ml). After 4 h of incubation at 37°C, RNAs were isolated and analyzed as described in Materials and Methods. SINrep/GFP and Hsin123/C plus Hsin123/Gl helpers, lanes 1; SINrep/GFP and Hsin123/C+Gl helper, lanes 2; SINrep/GFP and Hsin/C plus Hsin/Gl helpers, lanes 3; SINrep/GFP and Hsin/C+Gl helper, lanes 4; SINrep/GFP and tRNA/Hsin/C+Gl helper, lanes 5. (D) Titers of packaged VEE replicons after electroporation and further passaging of the samples. BHK-21 cells were co-transfected with SINrep/GFP replicon and indicated helper RNAs. Harvested viral particles were used for the next round of infection of naïve BHK-21 cells at the indicated MOIs. Viruses were harvested after development of CPE. Titers refer to unconcentrated harvested media. NA indicates “not applicable,” because concentration of the packaged replicons after the previous passage was insufficient for infecting cells at an MOI of 10 inf.u/cell.
SIN and VEE differ in the efficiency of infectious virion formation
To further investigate the specific characteristics of VEE packaging, we infected BHK-21 cells with VEE TC-83 and compared virus release, synthesis of virus-specific RNAs and viral structural proteins with those in SIN Toto1101-infected cells. At any time post infection (at an MOI 10 PFU/cell), VEE-specific structural proteins were synthesized 5–6-fold less efficiently than were those SIN-specific proteins (Fig. 6A and B). VEE RNA replication and transcription of the subgenomic RNAs were also 7- to 8-fold lower compared to the levels found in SIN-infected cells (Fig. 6C and D). VEE TC-83 virus was used in these experiments for safety reasons, but it should be noted that we previously demonstrated that A3→G mutation does not change the level of genome RNA replication (Petrakova et al., 2005). However, in contrast to more efficient production of both RNA and protein components of the virions, infectious SIN virus particles were released to the media at almost 50-fold lower rates, starting from as early as 4 h post infection, and accumulated to a 10- to 20-fold lower final concentration.
Fig. 6.
Virus replication and synthesis of virus-specific RNAs in SIN Toto1101- and VEE TC-83-infected cells. (A and B) BHK-21 cells (5 × 105 cells in 35-mm dishes) were infected with SIN Toto1101 or VEE TC-83 at an MOI of 10 PFU/cell. At the indicated times, proteins were pulse-labeled with [35S]methionine as described in Materials and Methods and analyzed on sodium dodecyl sulfate-10% polyacrylamide gels. The gels were dried and autoradiographed (A) or analyzed on a Storm 860 PhoshorImager (B). The levels of synthesis of virus-specific proteins were determined by measuring radioactivity in the protein band corresponding to capsid and were normalized to the number of cysteins and methionines in these proteins. (C and D) BHK-21 cells (5 × 105 cells in 35-mm dishes) were infected with SIN Toto1101 or VEE TC-83 at an MOI of 10 PFU/cell. At the indicated times, media were replaced by the same media supplemented with dactinomycin (1 μg/ml) and [3H]uridine (20 μCi/ml). After 4 h of incubation at 37°C, RNAs were isolated and analyzed as described in Materials and Methods (C). The levels of viral genome replication were determined by excising the bands corresponding to 49S viral genome RNA from the gel shown on panel C, followed by measuring radioactivity by scintillation counting (D). (E) BHK-21 cells (5 × 105 cells in 35-mm dishes) were infected with SIN Toto1101 or VEE TC-83 at an MOI of 10 PFU/cell. At the indicated times, the media were replaced and virus titers were determined as described in Materials and Methods.
One of the possible explanations for such strong disagreement between infectious virus release and the rates of viral proteins and RNA synthesis could be due to the lower infectivity of SIN virions. To rule out this possibility, freshly prepared stocks of VEE TC-83 and SIN Toto1101 viruses were purified to homogeneity on sucrose gradients. VEE TC-83 samples demonstrated a specific infectivity of 2.8–4.2 × 109 PFU/μg of protein. SIN Toto1101 infectivity was found to be 3- to 4-fold lower, namely, 0.97–1.5 × 109 PFU/μg of protein. However, the observed 3-to 4-fold difference in the infectivities could not explain the strong discrepancy between lower protein and RNA synthesis in the VEE-infected cells and higher titers of the released infectious virus. Thus, VEE appears to use a more efficient mechanism(s) of virus particle formation that leads to higher levels of virus replication in spite of the less efficient synthesis of the structural proteins and virus-specific RNAs.
DISCUSSION
The mechanism of packaging of viral genomes into infectious viral particles was studied to different degrees for at least three members of the alphavirus genus, including SIN, SFV and RRV (Frolova, Frolov, and Schlesinger, 1997; Lehtovaara et al., 1981; Lehtovaara et al., 1982; Monroe and Schlesinger, 1983; Monroe and Schlesinger, 1984; Weiss, Geigenmuller-Gnirke, and Schlesinger, 1994; White, Thomson, and Dimmock, 1998). The main efforts were focused on an investigation of the specificity of RNA-capsid binding during nucleocapsid formation and interaction of the capsid with glycoproteins that determines the formation of the viral envelope (Lopez et al., 1994). The DI RNAs were the main tools in the identification of the packaging signals, whose presence in the DI RNAs was critical for their persistence in the viral samples during serial passaging. The sequences of naturally occurring DI RNA genomes were intensively analyzed, and these studies were complemented by reverse genetics experiments, in which the effect of deletions on RNA replication and packaging was assessed.
However, it should be noted that the presence of packaging signals usually increases the packaging of replicating RNA by a range of only 10- to 20-fold. Moreover, in recent years, a variety of viable alphavirus chimeras has been developed (Kuhn et al., 1996; Paessler et al., 2003; Schoepp, Smith, and Parker, 2002). They encoded replicative machinery and cis-acting RNA promoter elements derived from one alphavirus and structural proteins from a heterologous virus. The efficient replication of SIN/RRV, RRV/SIN, SIN/VEE, VEE/SIN and SIN/SFV chimeric viruses in vitro and in vivo supports the idea that packaging signals could not be the only determinants of RNA packaging. In other experiments, alphavirus replicons were also packaged into structural proteins of heterologous alphaviruses (Perri et al., 2003), exhibiting very low sequence identity in the structural and nonstructural genes that makes it virtually impossible for heterologous capsid to find an appropriate packaging signal. Taken together, the data indicated that some other mechanism(s), besides the specific interaction of the alphavirus capsid with packaging signals, might be involved in RNA packaging.
In the present study, we started to investigate the mechanism of packaging of the VEE-specific RNAs into viral particles. VEE is a particularly important representative of the alphavirus genus not only as a significant human and equine pathogen, but also as a promising vector for development of recombinant vaccines (Gupta et al., 2005; Hevey et al., 1998; Lee, Hadjipanayis, and Welkos, 2003). Despite the intense interest in VEE, our understanding of the biology of this virus, including the mechanisms of RNA replication and viral particle formation, is far from complete. We found that, as do other alphaviruses, the VEE genome contains packaging signal(s) capable of increasing the level of RNA packaging into viral particles. The DH H123stop/C+Gl and DH H123/C+Gl helpers were packaged more efficiently than Hvee/C+Gl that did not contain nsP2-, nsP3- and the major part of nsP1-coding sequence. The more efficient packaging that we detected was not due to higher replication levels of longer DHs. In the electroporated cells, we detected identical levels of H123/C+Gl and Hvee/C+Gl helper replication that were evaluated based on the subgenomic RNA synthesis (data not shown). (However, the difference in stability of the RNAs cannot be ruled out.) Therefore, it is reasonable to expect that nt 520–7290 of the VEE genome contains sequence(s) that have strong, positive impact on RNA encapsidation.
Nevertheless, the presence of packaged DH RNA genomes in a viral population generated by electroporation was insufficient for supporting their persistence in the samples during serial passaging. Helpers had to encode the ORF for nsP1–nsP3 (the components of the RdRp) to keep their presence in the samples at concentrations sufficient for productive replication. Accordingly, H123/C+Gl and H123stop/C+Gl helpers (that had only a 4-nt sequence difference) were packaged equally efficiently after co-transfection into the cells, but only H123/C+Gl produced a high concentration of both replicon- and DH RNA-containing viral particles during the subsequent passaging of samples in tissue culture. Moreover, the expression of only nsP1, or nsP1 and nsP2, by DHs was insufficient for passaging of viral samples. We speculate that synthesis of the entire P123, the precursor of nsP1, nsP2 and nsP3, in cis led to its binding to DH RNAs and formation of the pre-replicative complex, lacking only nsP4. This could make initiation of replication (that now required only nsP4 supplied in trans by a replicon) a more feasible event. Alternatively, P123, synthesized in cis, might make the DH RNA more resistant to degradation before initiation of replication and/or target the RNA-P123 complex to the appropriate cellular compartment. We strongly believe that the requirement of P123 synthesis in cis is beneficial for VEE replication, because this is an efficient selection system that might reduce the possibility of an accumulation of defective genomes in the viral population.
The requirement of P123 translation in cis is not the only mechanism that differs in the replication processes of VEE and SIN. As we previously described (Paessler et al., 2003; Petrakova et al., 2005) and confirmed in this work (Fig. 6), compared to SIN, VEE-specific RNAs are reproducibly synthesized less efficiently, but cells release packaged VEE RNAs to the same concentration as did the cells transfected with similar SIN replicon and helper constructs. The higher rates of release of infectious VEE virions, in spite of a less efficient synthesis of viral structural proteins and RNAs, appeared to be the distinguishing feature of VEE replication (Fig. 6). These data suggested that VEE developed a more efficient mechanism of virion formation. A lower level of RNA replication could be a strong advantage for propagation of this virus in IFN-α/β-competent cells, both in vivo and in vitro. The number of replicative complexes is believed to determine the level of dsRNAs present in the infected cells, and this in turn may be one of critical determinants in the development of a virus-induced cell response (Sarkar et al., 2004; Sarkar and Sen, 2004; Sen, 2001). Therefore, a faster development of viremia, when the antiviral reaction is low, might be a critical factor in VEE pathogenesis. This possibility is indirectly supported by the evidence that VEE replication does not lead to changes in the intracellular environment to the same levels or rates as those found in SIN-infected cells. Replication of VEE-specific RNAs, inhibits cellular translation and, most likely, transcription of mRNAs to a lesser extent, and the VEE-based replicons are capable of establishing persistent replication more efficiently than similar SIN-based constructs. In some cell lines, the persistence occurs even without acquiring adaptive mutations in the nonstructural proteins (Petrakova et al., 2005).
Based on accumulated direct and indirect evidence, we suggest that packaging and persistence of previously studied SIN- and SFV-specific DI and DH RNAs in viral populations depends on at least two factors. i) DI or DH RNAs can be packaged at higher rates and to higher concentrations due to smaller sizes of the genome and the presence of additional homologous or heterologous packaging signals. One of the examples is SIN DI 25 RNA, in which the natural packaging signal is duplicated and a 5′ tRNAAsp sequence exhibits the packaging signal activity as well (Frolova, Frolov, and Schlesinger, 1997; Monroe et al., 1982; Monroe and Schlesinger, 1983). This more efficient packaging makes DI25 RNA genomes dominant in viral populations and leads to infection of the cells with numerous DI RNAs. ii) The defective genomes can attain cis-acting RNA elements that function more efficiently in RNA replication. Such 5′UTRs represented by tRNAAsp or 5′UTR of the 26S subgenomic RNA were found in SIN DI RNAs (Monroe and Schlesinger, 1983; Tsiang, Monroe, and Schlesinger, 1985). In our previous study, we have used the unique features of tRNAAsp to increase packaging and replication of SIN DH RNAs (Frolova, Frolov, and Schlesinger, 1997) and transformed SIN into bi- and tri-component genome viruses, in which the genome fragments encoding complementing functions were packaged into separate viral particles (Fayzulin et al., 2005).
However, the structure of the VEE-specific DI RNAs did not follow the same rules as did SIN DI RNAs. They were a few-fold longer than sequenced SIN and SFV DI RNAs, and a large fraction of them contained deletions of structural genes and nsP4. According to this finding, the persistence of VEE-specific DH RNAs in viral population during serial passaging was achieved by using an alternative approach. The designed VEE DH RNAs not only contained a large fragment of the viral genome with the packaging signal(s), but were capable of expressing the P123 that increased their ability to replicate (most likely, to initiate replication) in the infected cells in the presence of the replicon. Thus, VEE was transformed into bi- and tri-component genome viruses (Lazarowitz, 2001) that could be serially passaged in tissue culture. Unexpectedly, this P123-coding design was advantageous only for VEE, but not for SIN (Fig. 5). SIN-specific, P123-encoding helpers were efficiently packaged into viral particles after co-transfection with replicons, but did not replicate at the next passage at the level sufficient to support packaging. This result correlated with found fundamental differences between VEE-specific and previously published SIN-specific DI RNAs.
Based on the results of this study, it was not clear how electroporated Hvee/C+Gl, H1stop/C+Gl H12stop/C+Gl and H123stop/C+Gl helpers were capable of replicating and producing viral structural proteins at a level sufficient for packaging of replicons. These DH RNAs did not produce P123 and were inefficient in replication (most likely, initiation of replication) on the next passages regardless of having a packaging signal or not, but they functioned efficiently if delivered into the cells by transfetion. To understand this ambiguity, we compared VEE replication after electroporation of the in vitro-synthesized viral RNA genomes with replication of the same virus after infecting cells at different MOIs. The results indicated that electroporation of the RNAs at concentrations that we usually use for packaging of the replicons, is equivalent to infection at a very high MOI, ~100 PFU/cell (data not shown). Therefore, transfection of high amounts of DH and replicon RNAs into the cells made P123-negative helpers capable of using replicon-provided nsPs, replicating and producing viral structural proteins to the level sufficient for RNA packaging. However, at the next passage, the difference in replication became critical, when fewer DH RNA genomes were delivered into the cells, and the DH RNAs encoding no functional P123 could not supply structural proteins to the level required for productive infection.
In conclusion, we have shown that the packaging of VEE-specific RNAs during serial passaging of replicon- and helper-containing viral particles strongly depends on two conditions: the presence of packaging signal(s) in DH genomes, and their competence for efficient replication, which is determined by the ability to express nsP1, nsP2 and nsP3 in the form of P123 precursor. Moreover, VEE developed more efficient mechanism of infectious virions formation that allows this virus to replicate more efficiently than does SIN, using lower levels of the synthesized viral genome RNA and structural proteins. The found phenomena might be advantageous i) for supporting the integrity of the viral genome and developing high-titer viremia on the background of lower levels of RNA replication that are likely to lead to a less efficient antiviral response.
MATERIALS AND METHODS
Cell cultures
BHK-21 cells were kindly provided by Paul Olivo (Washington University, St. Louis, Mo). They were maintained at 37°C in alpha minimum essential medium (αMEM) supplemented with 10% fetal bovine serum (FBS) and vitamins.
Plasmid constructs
pVEErep/GFP/Pac and pSINrep/GFP plasmids encoding VEE replicon capable of expressing green fluorescent protein (GFP) and the puromycin acetyltransferase (Pac) gene, or the SIN replicon, encoding GFP, were described elsewhere (Gorchakov et al., 2004; Petrakova et al., 2005). pHsin/C+Gl, pHsin/C, pHsin/Gl and ptRNA/Hsin/C+Gl helper-coding plasmids were previously described (Bredenbeek et al., 1993; Fayzulin et al., 2005). They were formerly referred to as DH-BB(5′SIN), BB/C, BB/Gl and DH-BB, respectively. The terminology was changed to make their names consistent with those of other constructs developed in this work. These helpers encoded SIN genomes with either natural 5′UTR or tRNAAsp 5′UTR (derived from naturally occurring SIN DI RNA). The deletion of nt 421–7334 covered nsP2, nsP3, and most of the nsP1- and nsP4-coding regions. pHsin123/C+Gl plasmid encoded SIN genome with deletion of nt 5769–7334. This helper genome was capable of expressing nsP1, nsP2 and nsP3, and its subgenomic RNA encoded all of the SIN structural proteins. pHsin123/Gl and pHsin123/C helper-containing plasmids had the same deletion of the nsP4 gene as did pHsin123/C+Gl, but the subgenomic RNAs were derived from the previously described pDH-BB/C and pDH-BB/Gl (Fayzulin et al., 2005) and were capable of expressing only SIN capsid and SIN glycoproteins, respectively. pHvee/C+Gl, pHvee/C and pHvee/Gl encoded VEE helper RNAs, cloned under control of the SP6 promoter. They contained a deletion of nt 520–7290 covering almost the entire VEE P1234 polyprotein, encoding the RdRp. The Hvee/C+Gl helper contained the entire subgenomic RNA encoding all of the VEE TC-83 structural proteins. The subgenomic RNA of Hvee/C encoded only VEE capsid, because nt 8387–11326 were deleted. Hvee/Gl helper contained VEE subgenomic RNA, in which nt 7805–7897 (coding cluster of positively charged amino acids) was deleted to make the capsid incapable of RNA-binding. pH123/C+Gl encoded the helper genome containing a deletion of nt 5702–7500 (covering almost entire nsP4-coding sequence) with a TGA codon inserted downstream of the nsP3-coding sequence, at 5701 position. This helper contained the entire subgemomic RNA encoding all of the VEE TC-83 structural proteins. Subgenomic RNA of H123/C helper encoded only the VEE capsid, and nt 8387–11326 was deleted. H123/Gl helper contained VEE subgenomic RNA with the deletion of nt 7805–7897, making the capsid incapable of RNA binding. The Hstop123/C+Gl helper construct was essentially the same as H123/C+Gl, except for the insertion of a TCGA sequence after nt 1620. These 4 nucleotides were inserted to destroy the open reading frame (ORF). H1stop/C+Gl and H12stop/C+Gl helpers had the same genome structure as H123/C+Gl, except for insertion of TGA codons immediately after the nsP1- and nsP2-coding sequences, respectively. Standard recombinant DNA techniques were used for all plasmid constructions, and all of the required sequence modifications were carried out using PCR-based mutagenesis or by using existing, convenient restriction sites. Maps and sequences are available from the authors upon request.
RNA transcriptions
Plasmids were purified by centrifugation in CsCl gradients. Before the transcription reaction, the VEE replicon- and VEE helper-coding plasmids were linearized using an MluI restriction site located downstream of the poly(A) sequence. SIN replicon- and SIN helper-containing plasmids were linearized using an XhoI restriction enzyme. RNAs were synthesized by SP6 RNA polymerase in the presence of a cap analog using previously described conditions (Bredenbeek et al., 1993). The yield and integrity of transcripts were analyzed by gel electrophoresis under non-denaturing conditions. Aliquots of transcription reactions were used for electroporation without additional purification.
RNA transfections
BHK-21 cells were electroporated by previously described conditions (Liljeström et al., 1991). We always used 4 μg of in vitro-synthesized replicon RNA and 6 μg of each helper RNA for electroporation. Packaged replicons were harvested 24 to 28 h post electroporation. For measuring the titers of packaged replicons in infectious unit per ml (inf.u/ml), BHK-21 cells were seeded at a concentration of 5 × 105 cells/35-mm dish. After 4 h incubation at 37°C, monolayers were infected with 10-fold dilutions of the samples and incubated for 1 h at 37°C, and then 1 ml of complete medium was added to the wells. Incubation continued either at 37°C for 6–8 h or at 30°C for 16–18 h. The numbers of GFP-expressing cells were evaluated on an inverted fluorescent microscope. To measure the concentration of the packaged VEE replicon, VEErep/GFP/Pac, in colony-forming units (CFU/ml), cells infected with ten-fold dilutions of the packaged replicons were incubated in complete media, supplemented with 5 μg/ml of puromycin. Colonies of PurR cells were stained with crystal violet after 5–7 days of incubation at 37°C.
Viral replication analysis
BHK-21 cells were seeded at a concentration of 5 × 105 cells/35-mm dish. After 4 h incubation at 37°C, monolayers were infected with MOIs, as indicated in figure legends, and overlaid with 1 ml of complete medium. At indicated times post infection, media were replaced, and virus titers or titers of packaged replicons in the harvested samples were determined by a plaque assay on BHK-21 cells as previously described (Lemm et al., 1990) and by the above described method, respectively.
Generation of VEE TC-83 DI RNAs
2.5 × 106 BHK-21 cells were infected with 1 ml of the VEE vaccine strain TC-83 samples harvested after the previous passage. Viruses were harvested after development of profound cytopathic effect (CPE) that usually occurred within 30 h post infection. Titers were determined by a standard plaque assay on BHK-21 cells (Lemm et al., 1990). Accumulation of the DI RNAs was detected by infecting 5 × 105 BHK-21 cells with 200 μl of undiluted virus samples, followed by metabolic RNA labeling in 0.8 ml αMEM supplemented with 10% FBS, 1 μg/ml of ActD and 20 μCi/ml of [3H]uridine at 37°C between 3 and 7 h post infection. RNAs were isolated from the cells by TRIzol reagent, as recommended by the manufacturer (Invitrogen). They were denatured with glyoxal in dimethyl sulfoxide and analyzed by agarose gel electrophoresis using the previously described conditions (Bredenbeek et al., 1993).
RNA analysis
Cells were transfected with in vitro-synthesized RNAs or, in some experiments, infected with viral particles and seeded into 35-mm dishes at a concentration of 106 cells/dish. After incubation at 37°C for 16 h, cells were washed with phosphate-free Dulbecco’s Modified Eagle Medium (DMEM), and the incubation continued in 0.8 ml of the same phosphate-free DMEM supplemented with 1%FCS and 50 μCi/ml of [32P]phosphoric acid at 37°C for 8 h. Virus particles were harvested before CPE development and pelleted by centrifugation in the tabletop Optima ultracentrifuge (Beckman) in TLA-55 rotor at 52,000 rpm for 1 h at 4°C. RNAs were isolated from the pelleted viruses by TRIzol reagent, as recommended by the manufacturer (Invitrogen). They were denatured with glyoxal in dimethyl sulfoxide and analyzed by agarose gel electrophoresis using the previously described conditions (Bredenbeek et al., 1993). To test intracellular RNA replication, cells were transfected by electroporation or infected at the MOIs indicated in the figure legends with packaged replicons or viruses, and virus-specific RNAs were labeled with [3H]uridine as described in the figure legends. RNAs were isolated from the cells by TRIzol reagent, as recommended by the manufacturer (Gibco-BRL, Bethesda, MD), denatured with glyoxal in dimethyl sulfoxide and analyzed by agarose gel electrophoresis.
Analysis of protein synthesis
BHK-21 cells were seeded into 35-mm dishes at a concentration of 5 × 105 cells/well. After 4 h incubation at 37°C in 5% CO2 they were infected at an MOI of 10 PFU/cell with the viruses indicated in the figure legend, in 500 μl of alpha MEM supplemented with 1% FBS at room temperature for 1 h with continuous shaking. The medium was then replaced by corresponding complete medium, and incubation continued at 37°C. At the indicated times post infection, the cells were washed three times with phosphate-buffered saline (PBS) and then incubated for 30 minutes at 37°C in 0.8 ml of DMEM medium lacking methionine, supplemented with 0.1% FBS and 20 μCi/ml of [35S]methionine. After this incubation, cells were scraped into the media, pelleted by centrifugation and dissolved in 200 μl of standard protein loading buffer. Equal amounts of proteins were loaded onto sodium dodecyl sulfate-10% polyacrylamide gels. After electrophoresis, gels were dried, autoradiographed and analyzed on a Storm 860 PhosphorImager (Molecular Dynamics).
Acknowledgments
We wish to thank Dr. Scott Weaver for critical reading of the manuscript. This work was supported by Public Health Service grants AI053135.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bredenbeek PJ, Frolov I, Rice CM, Schlesinger S. Sindbis virus expression vectors: Packaging of RNA replicons by using defective helper RNAs. J Virol. 1993;67:6439–6446. doi: 10.1128/jvi.67.11.6439-6446.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fayzulin R, Gorchakov R, Petrakova O, Volkova E, Frolov I. Sindbis virus with a tricomponent genome. J Virol. 2005;79(1):637–43. doi: 10.1128/JVI.79.1.637-643.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frolov I, Schlesinger S. Translation of Sindbis virus mRNA: analysis of sequences downstream of the initiating AUG codon that enhance translation. J Virol. 1996;70(2):1182–90. doi: 10.1128/jvi.70.2.1182-1190.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frolova E, Frolov I, Schlesinger S. Packaging signals in alphaviruses. J Virol. 1997;71(1):248–58. doi: 10.1128/jvi.71.1.248-258.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geigenmuller-Gnirke U, Weiss B, Wright R, Schlesinger S. Complementation between Sindbis viral RNAs produces infectious particles with a bipartite genome. Proc Natl Acad Sci USA. 1991;88:3253–3257. doi: 10.1073/pnas.88.8.3253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorchakov R, Frolova E, Williams BR, Rice CM, Frolov I. PKR-dependent and -independent mechanisms are involved in translational shutoff during Sindbis virus infection. J Virol. 2004;78(16):8455–67. doi: 10.1128/JVI.78.16.8455-8467.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin DE. Alphaviruses. In: Knipe DM, Howley PM, editors. Fields’ Virology. 4. Lippincott, Williams and Wilkins; New York: 2001. pp. 917–962. [Google Scholar]
- Gupta S, Janani R, Bin Q, Luciw P, Greer C, Perri S, Legg H, Donnelly J, Barnett S, O’Hagan D, Polo JM, Vajdy M. Characterization of human immunodeficiency virus Gag-specific gamma interferon-expressing cells following protective mucosal immunization with alphavirus replicon particles. J Virol. 2005;79(11):7135–45. doi: 10.1128/JVI.79.11.7135-7145.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hevey M, Negley D, Pushko P, Smith J, Schmaljohn A. Marburg virus vaccines based upon alphavirus replicons protect guinea pigs and nonhuman primates. Virology. 1998;251(1):28–37. doi: 10.1006/viro.1998.9367. [DOI] [PubMed] [Google Scholar]
- Kinney RM, Johnson BJB, Welch JB, Tsuchiya KR, Trent DW. The full-length nucleotide sequences of the virulent Trinidad donkey strain of Venezuelan equine encephalitis virus and its attenuated vaccine derivative, strain TC-83. Virology. 1989;170:19–30. doi: 10.1016/0042-6822(89)90347-4. [DOI] [PubMed] [Google Scholar]
- Kuhn RJ, Griffin DE, Owen KE, Niesters HG, Strauss JH. Chimeric Sindbis-Ross River viruses to study interactions between alphavirus nonstructural and structural regions. J Virol. 1996;70(11):7900–9. doi: 10.1128/jvi.70.11.7900-7909.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarowitz SG. Plant Viruses. In: Knipe DM, editor. Fields Virology. Vol. 1. Lippincott Williams & Wilkins; Philadelphia: 2001. pp. 533–998. 2 vols. [Google Scholar]
- Lee JS, Hadjipanayis AG, Welkos SL. Venezuelan equine encephalitis virus-vectored vaccines protect mice against anthrax spore challenge. Infect Immun. 2003;71(3):1491–6. doi: 10.1128/IAI.71.3.1491-1496.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehtovaara P, Söderlund H, Keränen S, Pettersson RF, Kääriäinen L. 18S defective interfering RNA of Semliki Forest virus contains a triplicated linear repeat. Proc Natl Acad Sci USA. 1981;78:5353–5357. doi: 10.1073/pnas.78.9.5353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehtovaara P, Söderlund H, Keränen S, Pettersson RF, Kääriäinen L. Extreme ends of the genome are conserved and rearranged in the defective interfering RNAs of Semliki Forest virus. J Mol Biol. 1982;156:731–748. doi: 10.1016/0022-2836(82)90139-5. [DOI] [PubMed] [Google Scholar]
- Lemm JA, Durbin RK, Stollar V, Rice CM. Mutations which alter the level or structure of nsP4 can affect the efficiency of Sindbis virus replication in a host-dependent manner. J Virol. 1990;64:3001–3011. doi: 10.1128/jvi.64.6.3001-3011.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemm JA, Rice CM. Roles of nonstructural polyproteins and cleavage products in regulating Sindbis virus RNA replication and transcription. J Virol. 1993;67:1916–1926. doi: 10.1128/jvi.67.4.1916-1926.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liljeström P, Garoff H. A new generation of animal cell expression vectors based on the Semliki Forest virus replicon. BioTechnology. 1991;9:1356–1361. doi: 10.1038/nbt1291-1356. [DOI] [PubMed] [Google Scholar]
- Liljeström P, Lusa S, Huylebroeck D, Garoff H. In vitro mutagenesis of a full-length cDNA clone of Semliki Forest virus: the small 6,000-molecular-weight membrane protein modulates virus release. J Virol. 1991;65:4107–4113. doi: 10.1128/jvi.65.8.4107-4113.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez S, Yao JS, Kuhn RJ, Strauss EG, Strauss JH. Nucleocapsid-glycoprotein interactions required for assembly of alphaviruses. J Virol. 1994;68(3):1316–23. doi: 10.1128/jvi.68.3.1316-1323.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monroe SS, Ou JH, Rice CM, Schlesinger S, Strauss EG, Strauss JH. Sequence analysis of cDNAs derived from Sindbis virions and of defective interfering particles. J Virol. 1982;41:153–162. doi: 10.1128/jvi.41.1.153-162.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monroe SS, Schlesinger S. RNAs from two independently isolated defective interfering particles of Sindbis virus contain a cellular tRNA sequence at their 5′ ends. Proc Natl Acad Sci USA. 1983;80(11):3279–3283. doi: 10.1073/pnas.80.11.3279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monroe SS, Schlesinger S. Common and distinct regions of defective-interfering RNAs of Sindbis virus. J Virol. 1984;49:865–872. doi: 10.1128/jvi.49.3.865-872.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paessler S, Fayzulin RZ, Anishchenko M, Greene IP, Weaver SC, Frolov I. Recombinant sindbis/Venezuelan equine encephalitis virus is highly attenuated and immunogenic. J Virol. 2003;77(17):9278–86. doi: 10.1128/JVI.77.17.9278-9286.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perri S, Greer CE, Thudium K, Doe B, Legg H, Liu H, Romero RE, Tang Z, Bin Q, Dubensky TW, Jr, Vajdy M, Otten GR, Polo JM. An alphavirus replicon particle chimera derived from venezuelan equine encephalitis and sindbis viruses is a potent gene-based vaccine delivery vector. J Virol. 2003;77(19):10394–403. doi: 10.1128/JVI.77.19.10394-10403.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrakova O, Volkova E, Gorchakov R, Paessler S, Kinney RM, Frolov I. Noncytopathic replication of Venezuelan equine encephalitis virus and eastern equine encephalitis virus replicons in Mammalian cells. J Virol. 2005;79(12):7597–608. doi: 10.1128/JVI.79.12.7597-7608.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pushko P, Parker M, Ludwig GV, Davis NL, Johnston RE, Smith JF. Replicon-helper systems from attenuated Venezuelan equine encephalitis virus: expression of heterologous genes in vitro and immunization against heterologous pathogens in vivo. Virology. 1997;239(2):389–401. doi: 10.1006/viro.1997.8878. [DOI] [PubMed] [Google Scholar]
- Rice CM, Strauss JH. Nucleotide sequence of the 26S mRNA of Sindbis virus and deduced sequence of the encoded virus structural proteins. Proc Natl Acad Sci USA. 1981;78:2062–2066. doi: 10.1073/pnas.78.4.2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar SN, Peters KL, Elco CP, Sakamoto S, Pal S, Sen GC. Novel roles of TLR3 tyrosine phosphorylation and PI3 kinase in double-stranded RNA signaling. Nat Struct Mol Biol. 2004;11(11):1060–7. doi: 10.1038/nsmb847. [DOI] [PubMed] [Google Scholar]
- Sarkar SN, Sen GC. Novel functions of proteins encoded by viral stress-inducible genes. Pharmacol Ther. 2004;103(3):245–59. doi: 10.1016/j.pharmthera.2004.07.007. [DOI] [PubMed] [Google Scholar]
- Schoepp RJ, Smith JF, Parker MD. Recombinant chimeric western and eastern equine encephalitis viruses as potential vaccine candidates. Virology. 2002;302(2):299–309. doi: 10.1006/viro.2002.1677. [DOI] [PubMed] [Google Scholar]
- Sen GC. Viruses and interferons. Annu Rev Microbiol. 2001;55:255–81. doi: 10.1146/annurev.micro.55.1.255. [DOI] [PubMed] [Google Scholar]
- Shirako Y, Strauss JH. Regulation of Sindbis virus RNA replication: Uncleaved P123 and nsP4 function in minus strand RNA synthesis whereas cleaved products from P123 are required for efficient plus strand RNA synthesis. J Virol. 1994;185:1874–1885. doi: 10.1128/jvi.68.3.1874-1885.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strauss EG, Rice CM, Strauss JH. Complete nucleotide sequence of the genomic RNA of Sindbis virus. Virology. 1984;133:92–110. doi: 10.1016/0042-6822(84)90428-8. [DOI] [PubMed] [Google Scholar]
- Strauss JH, Strauss EG. The alphaviruses: gene expression, replication, evolution. Microbiol Rev. 1994;58:491–562. doi: 10.1128/mr.58.3.491-562.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsiang M, Monroe SS, Schlesinger S. Studies of defective interfering RNAs of Sindbis virus with and without tRNAAsp sequences at their 5′ termini. J Virol. 1985;54(4):38–44. doi: 10.1128/jvi.54.1.38-44.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver SC, Barrett AD. Transmission cycles, host range, evolution and emergence of arboviral disease. Nat Rev Microbiol. 2004;2(10):789–801. doi: 10.1038/nrmicro1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss B, Geigenmuller-Gnirke U, Schlesinger S. Interactions between Sindbis virus RNAs and a 68 amino acid derivative of the viral capsid protein further defines the capsid binding site. Nucleic Acids Res. 1994;22(5):780–6. doi: 10.1093/nar/22.5.780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss B, Nitschko H, Ghattas I, Wright R, Schlesinger S. Evidence for specificity in the encapsidation of Sindbis virus RNAs. J Virol. 1989;63:5310–5318. doi: 10.1128/jvi.63.12.5310-5318.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White CL, Thomson M, Dimmock NJ. Deletion analysis of a defective interfering Semliki Forest virus RNA genome defines a region in the nsP2 sequence that is required for efficient packaging of the genome into virus particles. J Virol. 1998;72(5):4320–6. doi: 10.1128/jvi.72.5.4320-4326.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White LJ, Wang JG, Davis NL, Johnston RE. Role of Alpha/Beta Interferon in Venezuelan Equine Encephalitis Virus Pathogenesis: Effect of an Attenuating Mutation in the 5′ Untranslated Region. J Virol. 2001;75(8):3706–18. doi: 10.1128/JVI.75.8.3706-3718.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]