

Abstract
2-Chloro-5′ -N-methylcarboxamidoadenosine analogues containing the (N)-methanocarba (bicyclo[3.1.0]hexane) ring system as a ribose substitute display increased selectivity as agonists of the human A3 adenosine receptor (AR). However, the selectivity in mouse was greatly reduced due to an increased tolerance of this ring system at the mouse A1AR. Therefore, we varied substituents at the N6 and C2 positions in search of compounds that have improved A3AR selectivity and are species independent. An N6-methyl analogue was balanced in affinity at mouse A1/A3ARs, with high selectivity in comparison to the A2AAR. Substitution of the 2-chloro atom with larger and more hydrophobic substituents, such as iodo and alkynyl groups, tended to increase the A3AR selectivity (up to 430-fold) in mouse and preserve it in human. Extended and chemically functionalized alkynyl chains attached at the C2 position of the purine moiety preserved A3AR selectivity more effectively than similar chains attached at the 3 position of the N6–benzyl group.
Keywords: nucleoside, G protein-coupled receptor, mouse, adenosine receptor, radioligand binding
Adenosine is a protective mediator that has been described as a general endogenous signal for tissue protection and regeneration and even “the signal of life”.1,2 Adenosine activates four different receptor subtypes - A1, A2A, A2B, and A3 - which are widely but differentially distributed throughout the body.3 The A3 adenosine receptor (AR) is located in some neurons, astrocytes, various immune cell populations (neutrophils, eosinophils, mast cells) and potentially muscle cells and endothelial cells.4-7 The A3AR is coupled to inhibition of adenylate cyclase and also activates Akt and calcium mobilization.8,9 Two potent and selective agonists of the A3AR, IB-MECA 1a and Cl-IB-MECA 1b, are currently in Phase II clinical trials for the treatment of rheumatoid arthritis, several other autoimmune inflammatory diseases, and cancer.10-12 Protective mechanisms in the envisioned disease applications of A3AR agonists appear to be a downregulation of the NF-κB system, common to both arthritis and cancer treatments,12 and a widespread correction of gene dysregulation induced in an inflammatory bowel model.13 Cardioprotection by selective A3AR agonists has been extensively explored in various species.14-16
Other selective A3AR agonists have recently been reported, based on introduction of large substituents at the N6 and C2 positions and modification of the ribose ring, particularly at the 4′ and 5′ positions.16-19 For example, the 4′ -thio analogue 2 is among the most selective known A3AR agonists,17 and 3′ -amino substitution is possible.å16 Carbocyclic nucleosides have also been developed as A3AR agonists.20-22 Conformationally constrained methanocarba (bicyclo[3.1.0]hexane) nucleoside analogues were used to determine that the biologically active conformation of the ribose ring, i.e. that required in order to bind to and activate the receptor, corresponds to a North (N) conformation. Furthermore, the addition of a 5′ -N-methyl or ethyl uronamide group assures that the efficacy of the nucleoside to activate the A3AR is maintained in combination with various structural changes at the N6 and C2 positions, which modulate the AR selectivity profile and might otherwise reduce the A3AR efficacy. The conformational flexibility and H-bonding in the region of the 5′ -uronamide are critical factors affecting the efficacy at the A3AR, as deduced from structure activity relationship (SAR) studies and from rhodopsin-based dynamic molecular modeling and ligand docking.23
In this study, we report that certain known A3AR agonists are much less selective at the A3AR in the mouse than in other species, such as human, and are therefore unsuitable for use alone as definitive pharmacological probes in mouse. Thus, additional A3AR agonists displaying high affinity and selectivity that are independent of species are desired. We previously explored the SAR surrounding both N6 and C2 positions of 5′ -N-methylcarboxamido adenosine analogues that contained an (N)-methanocarba ring system as a ribose substitute, with the objective of designing potent, and selective A3AR agonists.22 There is considerable evidence that appropriate modification of the N6 and C2 positions of nucleoside agonists is tolerated at the human A3AR, while SAR at the mouse ARs has not been systematically explored.3,16-19,23-25
Table 1 lists the structures of the adenosine derivatives that were assayed for binding affinity at ARs from several species. Compounds 3 – 12, previously synthesized and evaluated at human and rat ARs,21,22 were evaluated at the mouse ARs. Based on indications that the 2-iodo derivative 12 maintained selectivity at the mouse A3AR, compounds 13 – 19 and 25 – 27 were synthesized. Synthetic routes to the novel derivatives are shown in Schemes 1 and 2. In the synthetic route to each of the new A3AR agonists, after substitution of the chlorine at C6 in the purine ring of 20 and 21 with a corresponding primary amine R1NH2, the 5′ -ester group was aminolyzed with methylamine solution. Resultant compounds of type 22 or 23 were either hydrolyzed to afford agonists 3-10, 12, 13, 19, or underwent Sonogashira coupling26 with corresponding alkynes followed by hydrolysis to afford agonists 15 – 17. Agonist 14 was prepared from agonist 15 through desilylation with tetrabutylammnium fluoride. An N6-methoxy derivative 19, based on a recent report by Volpini et al. showing high potency at the human A3AR of similar derivatives in the ribose series,18 was prepared by substitution of the 6-Cl of 20 with O-methylhydroxylamine followed by deprotection of the 2′ and 3′ hydroxyl groups.
Table 1.
Affinity of a series of (N)-methanocarba adenosine derivatives at three subtypes of ARs in various species. Compounds 1a and 1b are 9-riboside derivatives.
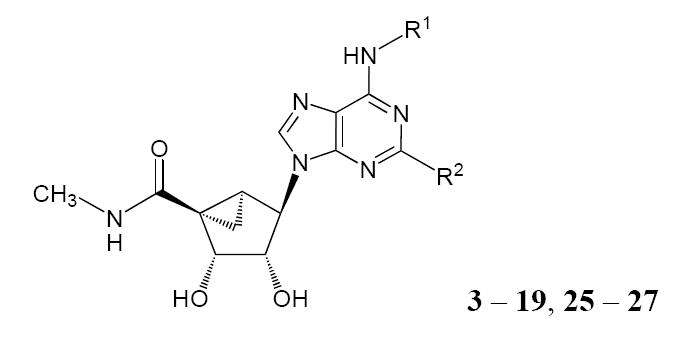
| Affinity | Selectivity | ||||||
|---|---|---|---|---|---|---|---|
| Compound | R1 | R2 | Species | A1a Ki, nM | A2Aa Ki, nM (or % inhib.) | A3a Ki, nM | A1/A3 |
| 1a | Mb,c | 5.9 | ~1000 | 0.087 | 68 | ||
| H | 49.3±3.7 | 93.1±4.2 | 1.74±0.36 | 28d | |||
| Re | 54 | 56 | 1.1 | 49 | |||
| 1b | Mb,c | 35 | ~10,000 | 0.18 | 190 | ||
| Hd | 220 | 5400 | 1.4 | 160 | |||
| Re | 820 | 470 | 0.33 | 2500 | |||
| 3 | CH3 | Cl | M | 55.3±6.0 | 20,400±3,200 | 49.0±3.9 | 1.1 |
| H | 2100±1700 | (6%)d,f | 2.2±0.6 | 950 | |||
| R | 805g | >10,000g | 160±30d | 5.0 | |||
| 4h | 3-Cl-Bn | Cl | M | 15.3±5.8 | 10,400±1,700 | 1.49±0.46 | 10.3 |
| Hc,d | 260 | 2300 | 0.29 | 900 | |||
| Rd | ND | ND | 1.0 | ||||
| 5 | 3-Br-Bn | Cl | M | 8.79±0.12 | 6,390±870 | 0.90±0.22 | 9.8 |
| Hc,d | 270 | 1300 | 0.38 | 710 | |||
| Rc | ND | ND | 0.76 | ||||
| 6h | 3-I-Bn | Cl | M | 7.32±1.5 | 5,350±860 | 0.80±0.14 | 9.2 |
| Hc | 136 | 784 | 1.5 | 91 | |||
| R | 83.9g | 1660g | 1.1 | 76 | |||
| 7 | 3-(C≡C- CH2OH)-Bn | Cl | M | 111±22 | (11%)i | 1.94±1.1 | 57.2 |
| Hd | 2600±300 | (56%)f | 2.9±0.7 | 900 | |||
| Rd | ND | ND | 1.6±0.6 | ||||
| 8 | 2,5-(OCH3)2Bn | Cl | M | 29.0±3.3 | 44,700±5,700 | 1.72±0.04 | 17 |
| Hc,d | 1600 | ~10,000 | 1.4 | 1100 | |||
| Rd | ND | ND | 0.87 | ||||
| 9 | CH2CH(Ph)2 | Cl | M | 6.83±1.5 | 1,810±581 | 1.67±0.09 | 4.1 |
| Hc,d | 1300±100 | 1600±100 | 0.69±0.02 | 1900 | |||
| Rd | ND | ND | 10±4 | ||||
| 10 | c-Pr-Ph | Cl | M | 6.60±1 .3 | 38,200±5,300 | 2.79±0.89 | 2.4 |
| Hc,d | 770±50 | 4800±200 | 0.78±0.06 | 990 | |||
| 11 | 3-Cl-Bn | SCH3 | M | 98.9±18.8 | (32%)i | 1.19±0.09 | 83 |
| Hd | 610 | ~10,000 | 1.5 | 410 | |||
| 12h | 3-Cl-Bn | I | M | 210±34.4 | (40%)i | 1.18±0.11 | 178 |
| Hd | 2200 | >10,000 | 3.6 | 610 | |||
| Rd | ND | ND | 3.9 | ||||
| 13h | 2,5-(OCH3)2Bn | I | M | 293±29 | (14%)i | 1.51±0.36 | 194 |
| H | 3070±820 | (35%)f | 1.30±0.27 | 2360 | |||
| 14 | 3-Cl-Bn | C≡CH | M | 45.6±7.9 | (41%)i | 0.85±0.08 | 53.6 |
| H | 174±23 | (48%)f | 1.30±0.38 | 134 | |||
| 15 | 3-Cl-Bn | C≡C- Si(CH3)3 | M | 159±22 | (20%)i | 4.46±0.57 | 35.6 |
| H | 160±40 | (52%)f | 0.98±0.14 | 160 | |||
| 16 | 3-Cl-Bn | C≡C(CH2)2- CH3 | M | 1390±430 | (42%)i | 6.06±1.21 | 229 |
| H | 1040±83 | (80%)f | 0.82±0.20 | 1300 | |||
| 17 | 3-Cl-Bn | C≡C(CH2)3- COOCH3 | M | 1340±330 | (50%)i | 4.65±0.53 | 288 |
| H | 482±23 | (49%)f | 1.17±0.27 | 412 | |||
| 18ah | 3-Cl-Bn | C≡ C(CH2)3- COOH | M | 10,500±1900 | (8%)i | 24.4±3.1 | 431 |
| H | 14,900±3500 | (43%)f | 2.38±0.56 | 6260 | |||
| 18bh | 3-Cl-Bn | C≡ C(CH2)3- CONH- | M | 546±62 | (31%)i | 8.60±1.02 | 64 |
| H | 454±44 | (81%)f | 2.17±0.51 | 209 | |||
| (CH2)2NH2 | |||||||
| 19 | OCH3 | Cl | M | 1,160±130 | (2%)f | 877±149 | 1.3 |
| H | 265±45 | (2%)f | 149±15 | 1.8 | |||
| 25 | 3-(C≡ C(CH2)3- COOH)-Bn | Cl | M | 703±71 | (5%)i | 14.4±2.5 | 49 |
| H | 320±31 | (14%)f | 17.1±1.2 | 19 | |||
| 26 | 3-(C≡ C(CH2)3- CON(CH2)2- NH2)-Bn | Cl | M | 151±18 | (39%)i | 11.9±2.4 | 13 |
| H | 271±23 | (58%)f | 5.21±0.91 | 52 | |||
| 27 | 3-(C≡ C(CH2)3- CONH- (CH2)2NH- COCH3)-Bn | Cl | M | 45.4±3.4 | (68%)i | 4.65±0.22 | 9.8 |
| H | 181±22 | (80%)f | 2.88±0.54 | 63 | |||
Competition radioligand binding assays using [125I]N6-(4-amino-3-iodobenzyl)adenosine-5′ -N-methyl-uronamide (A1 and A3ARs) and [3H]2-[p-(2-carboxyethyl)phenyl-ethylamino]-5′ -N-ethylcarboxamidoadenosine (A2AAR) were conducted with membranes prepared from HEK293 cells expressing recombinant mouse A1, A2A, or A3ARs. At rat and human ARs, the A1 radioligand was either [3H]R-phenylisopropyladenosine or [3H]2-chloro-N6-cyclopentyladenosine. Values are expressed as the mean ± SEM. ND, not determined.
Data from Ge et al.15
Data from Tchilibon et al.22
Data from Kim et al.24
Percent inhibition at 10 μM.
Data from Lee et al.21
Percent inhibition at 100 μM.
4, MRS3558; 6, MRS1898; 12, MRS3609; 13, MRS5128; 18a, MRS5151; 18b, MRS5166.
Some of the 2-alkynyl derivatives contained chemically functionalized, extended alkynyl chains to serve as functionalized congeners for conjugation to other biologically active moieties or to carriers.27-29 These included carboxylic acids 18a and 25, amines 18b and 26, and an acetylamino derivative 27. The Sonogoshira reaction sequence using methyl hexynoate in combination with an iodo-derivatized nucleoside, either a 2-iodo (Scheme 1) or N6-(3-iodobenzyl) (Scheme 2) derivative, provided a mixture of the product methyl ester and corresponding carboxylic acid, which were separated by silica gel column chromatography. The primary amine congeners 18b and 26 were prepared by treatment of the appropriate methyl ester with excess ethylenediamine.
Scheme 2.
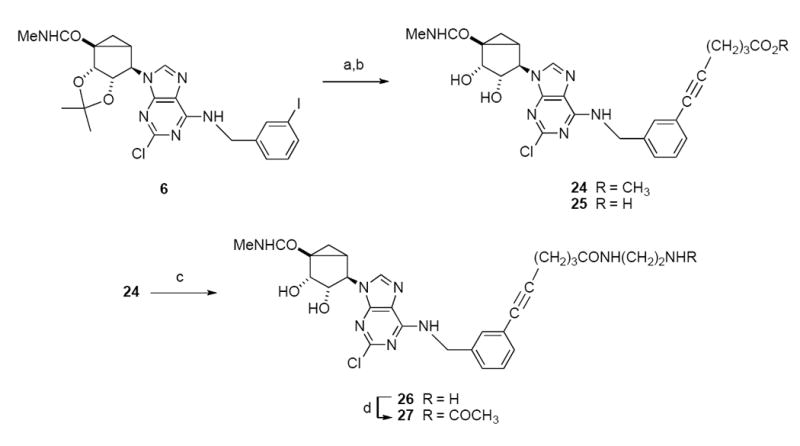
a Synthesis of novel (N)-methanocarba A3AR agonists containing functionalized alkynyl chains attached at the N6-benzyl 3 position. Note that 24 and 25 were both isolated chromatographically from the same reaction.
a Reagents & conditions: a) HC≡C(CH2)3COOCH3, PdCl2(PPh3)2, CuI, DMF, Et3N; b) TFA, H2O, MeOH, Δ; c) ethylenediamine; d) acetic anhydride.
Binding assays were carried out using standard radioligands in Chinese hamster ovary (CHO) cells expressing the human A1 or A3ARs and in HEK293 cells expressing the human A2AAR.22 Also, the mouse ARs were expressed in HEK293 cells for binding assays.15 A functional assay in guanine nucleotide binding ([35S]GTPγS)20,31 in membranes of CHO cells expressing the human A3AR showed that 18a is a full agonist. The pEC50 value of 18a was 7.85 ± 0.15, in comparison to the pEC50 value of NECA of 6.46 ± 0.13.
The binding affinity at the mouse and rat A3ARs of the N6-methyl derivative 3 was considerably weaker than at the human A3AR (Table 1). Thus, this compound was balanced in affinity at mouse A1/A3ARs, with high selectivity in comparison to the mouse A2AAR. Other agonists having this mixed AR selectivity were explored for their cardioprotective properties.29
N6-Benzyl derivatives of adenosine have previously been shown to favor selectivity at the A3AR.24 This observation led to the design of 1a and 1b. Although these two 9-riboside derivatives maintain selectivity for the mouse A3AR, the selectivity in mouse of similar N6-substituted benzyl (N)-methanocarba derivatives 4 – 6 was greatly reduced due to increased tolerance of this bicyclic ring system at the mouse A1AR. Therefore, we varied substituents at the N6 and C2 positions in an effort to reduce affinity at the mouse A1AR. Extension of the 3-benzyl group as an alkyne in 7 reduced mouse A1 and A2AAR affinity without greatly reducing affinity at the mouse, rat, or human A3ARs, resulting in a 57- fold selectivity for the mouse A3AR. A 2,5-dimethoxy substitution in 8 showed only a slight enhancement of mouse A3AR selectivity in comparison to 6, the (N)-methanocarba equivalent of Cl-IB-MECA. Modified N6-phenylethyl analogues 9 and 10, although both very potent at the mouse A3AR, were essentially nonselective in comparison to the mouse A1AR.
Modification at the adenine C2 position was generally more beneficial than structural changes of the N6 substituent, with respect to mouse A3AR selectivity. 2-Cl was replaced with small hydrophobic groups,22 which greatly increased the selectivity for the mouse A3AR. For example, a 2-iodo analogue 12 was 178-fold and >180,000-fold selective in binding to the mouse A3AR in comparison to mouse A1 and A2AAR, respectively, with a Ki value of 1.18 nM. The corresponding 2-iodo-N6-(2,5-dimethoxybenzyl) analogue 13 was similarly selective. Thus, the 2-iodo modification resulted in increased selectivity for the mouse A3AR; i.e., the selectivity ratio of 2-iodo compounds (12 and 13) was increased over that of the corresponding 2-chloro analogues (4 and 8, respectively) by 11 – 17-fold. Other hydrophobic substituents at the C2 position, such as an ethynyl group in 14 and its trimethylsilyl adduct 15, provided moderate mouse A3AR selectivity. Flexible, extended ethynyl chains in 16 and 17 increased the ratio of A3AR selectivity. 2-Alkynyl groups were previously reported to enhance affinity of adenosine derivatives at the rat and human A3ARs.25,30 Compound 18a, a long chain carboxylic acid congener, and its corresponding methyl ester 17 were highly selective for the mouse A3AR in comparison to the A1AR by 431- and 288-fold, respectively. Both compounds had greater A3AR selectivity than Cl-IB-MECA 1b, although they were less potent. A primary amino congener 18b displayed moderate A3AR selectivity, with a Ki value of 8.6 nM. Functionalized congeners in which a functionalized ethynyl chain was positioned on the N6-benzyl moiety were only moderately (a carboxylic acid 25) or weakly (a primary amine 26 and its acetyl analogue 27) selective for the mouse A3AR. Thus, the attachment of alkynyl chains at the 3 position of the N6-benzyl moiety did not preserve A3AR selectivity as well as the placement of similar chains at the adenine C2 position.
The use of an alkynyl substitutent at the 3-position of an N6-benzyl group, to serve as a covalent linking site for conjugation, was shown previously to maintain A3AR affinity in the series of 9-ribosides.28,29 In the present study of the (N)-methanacarba series, a variety of adenosine derivatives bearing a 2-alkynyl group have been shown to bind potently and selectively to the A3AR.
The (N)-methanacarba derivatives that were most potent (Ki 1 – 6 nM) and selective (180 – 290-fold) in binding to the mouse A3AR were, in order of decreasing affinity: 12, 13, 17, and 16. Compound 18a was the most selective novel agonist in this study at the mouse A3AR; however, the affinity was intermediate, with a Ki value of 24 nM. The selectivity for the human A3AR was >6000-fold. Thus, these C2 position-modified bicyclic nucleosides are good candidates for species-independent A3AR agonists.
In conclusion, the selectivity (but not affinity) of (N)-methanocarba-containing nucleosides as A3AR agonists was greatly reduced in the mouse due to increased tolerance of this ring system at the mouse A1AR. Several analogues having varied substitution at the N6 and C2 positions were balanced in affinity at mouse A1/A3ARs, with high selectivity in comparison to the A2AAR. Substitution of the 2-chloro atom with larger and more hydrophobic substituents, such as iodo and alkynyl groups, tended to increase the A3AR selectivity in mouse and preserve it in human. The carboxylic acid 18a and primary amino 18b derivatives are good candidates for use as functionalized congeners for covalent conjugation with retention of biological activity and receptor selectivity. Thus, we have identified novel (N)-methanocarba nucleosides that are A3AR- selective across several species and are especially suitable for pharmacological studies in the mouse.
Supplementary Material
Supplementary data (chemical synthesis, additional pharmacological procedures, and functional assay of 18a) associated with this article can be found, in the online version, at doi:.
Chart 1.
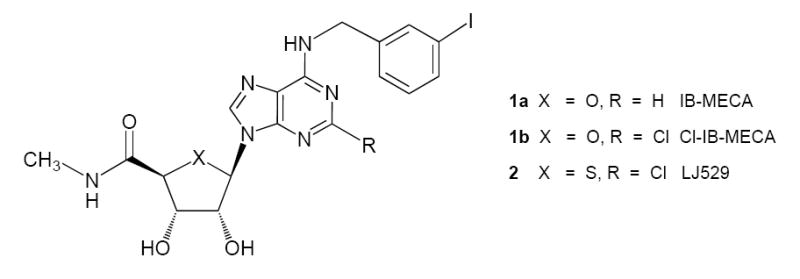
Prototypical selective A3AR agonists.
Scheme 1.

a Synthesis of novel (N)-methanocarba A3AR agonists with structural variation at the 2 and N6 positions. Note that 17 and 18a were both isolated chromatographically from the same reaction.
a Reagents & conditions: a) R1NH2; b) MeNH2, EtOH; c) RC≡CH, PdCl2(PPh3)2, CuI, DMF, Et3N; d) TFA, MeOH, H2O, Δ; e) TBAF, THF; f) ethylenediamine, MeOH.
Acknowledgments
This research was supported in part by the Intramural Research Program of the NIH, National Institute of Diabetes and Digestive and Kidney Diseases (KAJ) and by NIH R01 HL077707 (JAA). We thank Can-Fite Biopharma (Petah-Tikva, Israel) for financial support.
ABBREVIATIONS
- AR
adenosine receptor
- CGS21680
2-[p-(2-carboxyethyl)phenylethylamino]-5′ -N-ethylcarboxamido-adenosine
- CHO
Chinese hamster ovary
- Cl-IB-MECA
2-chloro-N6-(3-iodobenzyl)-5′ -N-methylcarboxamidoadenosine
- CPA
N6-cyclopentyladenosine
- DMEM
Dulbecco’s modified Eagle’s medium
- I-AB-MECA
N6-(4-amino-3-iodobenzyl)-5′ -N-methylcarboxamidoadenosine
- NECA
5′ -N-ethylcarboxamidoadenosine
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes
- 1.Linden J. Mol Pharmacol. 2005;67:1385. doi: 10.1124/mol.105.011783. [DOI] [PubMed] [Google Scholar]
- 2.Engler RL. Circulation. 1991;84:951. doi: 10.1161/01.cir.84.2.951. [DOI] [PubMed] [Google Scholar]
- 3.Jacobson KA, Gao ZG. Adenosine receptors as therapeutic targets. Nature Rev Drug Disc. 2006;5:247. doi: 10.1038/nrd1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yaar R, Jones MR, Chen JF, Ravid K. Animal models for the study of adenosine receptor function. J Cell Physiol. 2005;202:9. doi: 10.1002/jcp.20138. [DOI] [PubMed] [Google Scholar]
- 5.Gessi S, Varani K, Merighi S, Cattabriga E, Iannotta V, Leung E, Baraldi PG, Borea PA. Mol Pharmacol. 2002;61:415. doi: 10.1124/mol.61.2.415. [DOI] [PubMed] [Google Scholar]
- 6.Zheng J, Wang R, Zambraski E, Wu D, Jacobson KA, Liang BT. Am J Physiol Heart and Circ Physiol. 2007;293:3685. doi: 10.1152/ajpheart.00819.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Platts SH, Duling BR. Circ Res. 2004;94:77. doi: 10.1161/01.RES.0000108262.35847.60. [DOI] [PubMed] [Google Scholar]
- 8.Gao Z, Li BS, Day YJ, Linden J. Mol Pharmacol. 2001;59:76. doi: 10.1124/mol.59.1.76. [DOI] [PubMed] [Google Scholar]
- 9.Shneyvays V, Zinman T, Shainberg A. Cell Calcium. 2004;36:387. doi: 10.1016/j.ceca.2004.03.004. [DOI] [PubMed] [Google Scholar]
- 10.Madi L, Ochaion A, Rath-Wolfson L, Bar-Yehuda S, Erlanger A, Ohana G, Harish A, Merimski O, Barer F, Fishman P. Clin Cancer Res. 2004;10:4472. doi: 10.1158/1078-0432.CCR-03-0651. [DOI] [PubMed] [Google Scholar]
- 11.Bar Yehuda S, Silverman MH, Kerns WD, Ochaion A, Cohen S, Fishman P. Exp Opin Invest Drugs. 2007;16:1601. doi: 10.1517/13543784.16.10.1601. [DOI] [PubMed] [Google Scholar]
- 12.Fishman P, Jacobson KA, Ochaion A, Cohen S, Bar-Yehuda S. Immunology Endocrine and Metabolic Agents in Medicinal Chemistry. 2007;7:298. doi: 10.2174/187152207781369878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guzman J, Yu JG, Suntres Z, Bozarov A, Cooke H, Javed N, Auer H, Palatini J, Hassanain HH, Cardounel AJ, Javed A, Grants I, Wunderlich JE, Christofi FL. Inflamm Bowel Dis. 2006;12:766. doi: 10.1097/00054725-200608000-00014. [DOI] [PubMed] [Google Scholar]
- 14.Liang BT, Jacobson KA. Proc Natl Acad Sci USA. 1998;95:6995. doi: 10.1073/pnas.95.12.6995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ge ZD, Peart JN, Kreckler LM, Wan TC, Jacobson MA, Gross GJ, Auchampach JA. J Pharmacol Exp Ther. 2006;319:1200. doi: 10.1124/jpet.106.111351. [DOI] [PubMed] [Google Scholar]
- 16.a) DeNinno MP, Masamune H, Chenard LK, DiRico KJ, Eller C, Etienne JB, Tickner JE, Hill RJ, Kennedy SP, Knight DR, Kong J, Oleynek JJ, Tracey WR. J Med Chem. 2003;46:353. doi: 10.1021/jm0255724. [DOI] [PubMed] [Google Scholar]; b) DeNinno MP, Masamune H, Chenard LK, DiRico KJ, Eller C, Etienne JB, Tickner JE, Kennedy SP, Knight DR, Kong J, Oleynek JJ, Tracey WR, Hill RJ. Bioorg Med Chem Lett. 2006;16:2525. doi: 10.1016/j.bmcl.2006.01.088. [DOI] [PubMed] [Google Scholar]
- 17.a) Jeong LS, Jin DZ, Kim HO, Shin DH, Moon HR, Gunaga P, Chun MW, Kim Y-C, Melman N, Gao Z-G, Jacobson KA. J Med Chem. 2003;46:3775. doi: 10.1021/jm034098e. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Jeong LS, Lee HW, Jacobson KA, Kim HO, Shin DH, Lee JA, Gao ZG, Lu C, Duong HT, Gunaga P, Lee SK, Jin DZ, Chun MW, Moon HR. J Med Chem. 2006;49:273. doi: 10.1021/jm050595e. [DOI] [PubMed] [Google Scholar]
- 18.Volpini R, Dal Ben D, Lambertucci C, Taffi S, Vittori S, Klotz KN, Cristalli GJ. J Med Chem. 2007;50:1222. doi: 10.1021/jm060963u. [DOI] [PubMed] [Google Scholar]
- 19.a) Elzein E, Palle V, Wu Y, Maa T, Zeng D, Zablocki J Med Chem. 2004;47:4766. doi: 10.1021/jm049682h. [DOI] [PubMed] [Google Scholar]; b) Cosyn L, Palaniappan KK, Kim SK, Duong HT, Gao ZG, Jacobson KA, Van Calenbergh S. J Med Chem. 2006;49:7373. doi: 10.1021/jm0608208. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Ohno M, Gao ZG, Van Rompaey P, Tchilibon S, Kim SK, Harris BA, Blaustein J, Gross AS, Duong HT, Van Calenbergh S, Jacobson KA. Bioorg Med Chem. 2004;12:2995. doi: 10.1016/j.bmc.2004.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jacobson KA, Ji X-d, Li AH, Melman N, Siddiqui MA, Shin KJ, Marquez VE, Ravi RG. J Med Chem. 2000;43:2196. doi: 10.1021/jm9905965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee K, Ravi RG, Ji X-d, Marquez VE, Jacobson KA. Bioorg Med Chem Lett. 2001;11:1333. doi: 10.1016/s0960-894x(01)00213-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tchilibon S, Joshi BV, Kim SK, Duong HT, Gao ZG, Jacobson KA. J Med Chem. 2005;48:1745. doi: 10.1021/jm049580r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim SK, Jacobson KA. J Chem Inf Model. 2007;47:1225. doi: 10.1021/ci600501z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim HO, Ji X-d, Siddiqi SM, Olah ME, Stiles GL, Jacobson KA. J Med Chem. 1994;37:3614. doi: 10.1021/jm00047a018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Costanzi S, Lambertucci C, Vittori S, Volpini R, Cristalli C. J Mol Graphics Model. 2003;21:253–262. doi: 10.1016/s1093-3263(02)00161-4. [DOI] [PubMed] [Google Scholar]
- 26.Chinchilla R, Nájera C. Chem Rev. 2007;107:874. doi: 10.1021/cr050992x. [DOI] [PubMed] [Google Scholar]
- 27.Jacobson KA, Kirk KL, Padgett WL, Daly JW. J Med Chem. 1985;28:1341. doi: 10.1021/jm00147a039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jacobson KA, Daly JW. Nucleosides Nucleotides. 1991;10:1029. [Google Scholar]
- 29.Jacobson KA, Xie R, Young L, Chang L, Liang BT. J Biol Chem. 2000;275:30272. doi: 10.1074/jbc.M001520200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Baraldi PG, Cacciari B, Pineda de las Infantas MJ, Romagnoli R, Spalluto G, Volpini R, Costanzi S, Vittori S, Cristalli G, Melman N, Park K-S, Ji X-d, Jacobson KA. J Med Chem. 1998;41:3174. doi: 10.1021/jm980147p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lorenzen A, Lang H, Schwabe U. Biochem Pharmacol. 1998;56:1287. doi: 10.1016/s0006-2952(98)00207-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary data (chemical synthesis, additional pharmacological procedures, and functional assay of 18a) associated with this article can be found, in the online version, at doi:.