

Abstract
The type and quantity of dietary fat ingested contributes to the onset and progression of chronic diseases, like diabetes and atherosclerosis. The liver plays a central role in whole body lipid metabolism and responds rapidly to changes in dietary fat composition. Polyunsaturated fatty acids (PUFA) play a key role in membrane composition and function, metabolism and the control of gene expression. Certain PUFA, like the n-3 PUFA, enhance hepatic fatty acid oxidation and inhibit fatty acid synthesis and VLDL secretion, in part, by regulating gene expression. Our studies have established that key transcription factors, like PPARα, SREBP-1, ChREBP and MLX, are regulated by n-3 PUFA, which in turn control levels of proteins involved in lipid and carbohydrate metabolism. Of the n-3 PUFA, 22:6,n-3 has recently been established as a key controller of hepatic lipid synthesis. 22:6,n-3 controls the 26S proteasomal degradation of the nuclear form of SREBP-1. SREBP-1 is a major transcription factor that controls the expression of multiple genes involved fatty acid synthesis and desaturation. 22:6,n-3 suppresses nuclear SREBP-1 which, in turn suppresses lipogenesis. This mechanism is achieved, in part, through control of the phosphorylation status of protein kinases. This review will examine both the general features of PUFA-regulated hepatic gene transcription and highlight the unique mechanisms by which 22:6,n-3 impacts gene expression. The outcome of this analysis will reveal that changes in hepatic 22:6,n-3 content has a major impact on hepatic lipid and carbohydrate metabolism. Moreover, the mechanisms involve 22:6,n-3 control of several well-known signaling pathways, such as Akt, Erk1/2, Gsk3β and PKC (novel or atypical). 22:6,n-3 control of these same signaling pathways in non-hepatic tissues may help explain the diverse actions of n-3 PUFA on such complex physiological processes as visual acuity and learning.
Keywords: docosahexaenoic acid (22:6,n-3); PPARα; SREBP-1; ChREBP; MLX; gene transcription; hepatic fatty acid metabolism
Introduction
Dietary fat is an important macronutrient for growth and development of all animals. Excessive levels of dietary fat or an imbalance of saturated fat versus unsaturated fat or n6 versus n3 polyunsaturated fat (PUFA) have been implicated in the onset and progression of several chronic diseases, including coronary artery disease and atherosclerosis (Harris, 2003; Kris-Etherton et al., 2002), diabetes and obesity (Kelley, 2002; Wang et al., 2002), cancer (Cho, 2003; Kushi, 2002), Parkinson’s disease, major depressive disorders and schizophrenia (Ellis, 2005; Fedorova, 2006; Horrobin, 2003; Horrobin and Bennett, 1999; Johnson, 2006; Julien, 2006; Peet, 2002; Youdim, 2000; Youdim, 2006). As such, considerable clinical and basic science research has been directed at understanding the biochemical and molecular basis of fatty acid effects on complex physiological systems impacting human health.
Our understanding of the role dietary fat plays in these chronic diseases is complicated by the fact that fat has many physiological roles. Dietary fat is a substrate for energy metabolism, membrane formation and signaling molecules; dietary fat also regulates gene expression. The focus of this discussion is on how dietary fat control of gene expression with an emphasis on docosahexaenoic acid, 22:6,n-3 (DHA). The outcome of this discussion will reveal that while DHA shares many of the actions on gene expression as other n-3 and n-6 PUFA. However, DHA has unique effects on gene expression. The mechanistic basis for the general effects of PUFA and the unique action of DHA on gene expression will be discussed.
Since the original description of dietary fat as a regulator of gene expression over a decade ago, many transcription factors have been identified as prospective targets for fatty acid regulation, including several nuclear receptors like the peroxisome proliferator activated receptor family (PPAR) [α, β, γ1 & γ2], retinoid X receptor α (RXRα), liver X receptor α (LXRα) and hepatic nuclear factor α & γ (HNF4α & γ) as well as several basic helix-loop-helix leucine-zipper transcription factors (bHLH-LZ) like sterol regulatory element binding protein-1 (SREBP-1), carbohydrate regulatory element binding protein (ChREBP) and Max-Like Factor X (MLX) (Botolin, 2006; Dentin et al., 2005; Jump, 2004; Jump, 2005; Xu, 2006). Two general mechanisms characterize fatty acid control of these transcription factors. Fatty acids bind directly to the transcription factor and control transcription factor activity. In this fashion, fatty acids act like hydrophobic hormones regulating the function of nuclear receptors and their impact on transcriptional processes. Non-esterified fatty acids bind PPAR(α, β, γ1 & γ2) (Xu et al., 1999), HNF-4 (α & γ), (Dhe-Paganon et al., 2002; Wisely et al., 2002) RXRα (Mata de Urquiza, 2000) and LXRα (Ou et al., 2001). All of these proteins are members of the nuclear receptor superfamily of ligand-regulated transcription factors. Amongst these nuclear receptors, however, PPAR subtypes are the most widely accepted fatty acid-regulated transcription factors.
In the second mechanism, fatty acids control the nuclear abundance of key transcription factors, such as SREBP-1, NFκB, ChREBP and MLX (Dentin et al., 2005; Jump, 2004; Xu, 2006). The mechanisms controlling the nuclear abundance of these transcription factors is less clear, but likely does not involve direct binding of the fatty acid to the transcription factor. A goal of this chapter is to illustrate that while 22:6,n-3 acts like many PUFA to control gene expression by regulating transcription factor function, 22:6,n-3 also has unique regulatory effects on gene transcription. As such, these effects are not shared by other fatty acids. Accordingly, we will explore the underlying mechanisms that might account for these unique effects of 22:6,n-3 on gene transcription.
To gain an understanding of how dietary fat functions in a physiological context, we first discuss how PUFA control hepatic lipid metabolism through its impact on gene expression. In vivo and cell culture studies have established that dietary PUFA regulate multiple genes involved in hepatic carbohydrate and lipid metabolism, see Table 1 (Mater et al., 1999; Pan et al., 2000; Pawar, 2003; Ren et al., 1997; Wang, 2005; Worgall et al., 1998; Xu, 2006). We now know the key transcription factors controlling these genes. These are the aforementioned fatty acid-regulated transcription factors include PPARα, SREBP-1, ChREBP and MLX. These transcription factors play a major role in controlling hepatic carbohydrate and lipid synthesis and oxidation. Since the liver plays a central role in whole body carbohydrate and lipid metabolism, such regulatory schemes impact whole body metabolism and contribute to the onset and progression of several chronic diseases, including atherosclerosis, diabetes and obesity (Jump, 2004).
Table 1.
Involvement of Transcription Factors in Controlling Hepatic Glycolysis, Fatty Acid Synthesis and Oxidation.
| Protein | SREBP-1 | ChREBP/MLX | PPARα |
|---|---|---|---|
| Glycolysis and Gluconeogenesis | |||
| Glucose transporter-2 (Glut2) | No | Yes | No |
| Glucokinase (GK) | No | No | No |
| L-Pyruvate kinase (LPK) | No | Yes (+) | Yes (−) |
| Phosphoenolpyruvate carboxykinase (PepCk) | Yes (−) | No | Yes (+) |
| Glycose-6-Phosphatase (G6Pase) | No | No | Yes (+, weak) |
| Fatty Acyl Synthetases and Thioesterases | |||
| Fatty acyl synthetase 1 (ACS1) | No | No | Yes |
| Cytosolic Fatty Acyl Thioesterase (CTEI) | No | No | Yes (+) |
| Mitochondrial Fatty Acyl Thioesterase (MTEI) | No | No | Yes (+) |
| Peroxisomal Fatty Acyl Thioesterase (PTEI) | No | No | Yes (+) |
| Fatty Acid Synthesis, Desaturation and Elongation | |||
| ATP citrate lyase (ACL) | Yes (+) | No | No |
| Acetyl CoA carboxylase (ACC) | Yes (+) | Yes (+) | No |
| Fatty acid synthase (FAS) | Yes (+) | Yes (+) | No |
| Δ5-desaturase (Δ5D) | Yes (+, weak) | No | Yes (+, weak) |
| Δ6-desaturase (Δ6D) | Yes (+) | No | Yes (+, weak) |
| Stearoyl CoA desaturase (Δ9D) | Yes (+) | Yes (+) | Yes (+) |
| Fatty acid elongase-5 (Elovl5) | No | No | Yes (+) |
| Fatty acid elongase-6 (Elovl6) | Yes (+) | Yes (+) | Yes (+, weak) |
| Fatty Acid Oxidation | |||
| Mitochondrial HMG CoA synthase (mtHMGCoASyn) | No | No | Yes (+) |
| Peroxisomal Acyl CoA Oxidase (AOX) | No | No | Yes (+) |
| Microsomal cytochrome P450-4A (Cyp4A) | No | No | Yes (+) |
Overview of fatty acid effects on hepatic gene expression
An underlying assumption regarding fatty acid effects on gene expression has been that fatty acids must enter cells to control the activity or abundance of transcription factors (Fig. 1). Fatty acids, however, have been reported to regulate G-protein-coupled membrane receptors (Kostenis, 2004). Since downstream targets of G-protein signaling impact gene transcription, there is no strict requirement for fatty acids to enter cells. Analysis of the tissue distribution and ligand specificity of these membrane receptors shows that these receptors are not well expressed in liver. As such, this mechanism likely does not explain fatty acid effects on hepatic gene expression.
Figure 1. Summary of PUFA control of hepatic PPARα, SREBP-1 and MLX nuclear abundance.
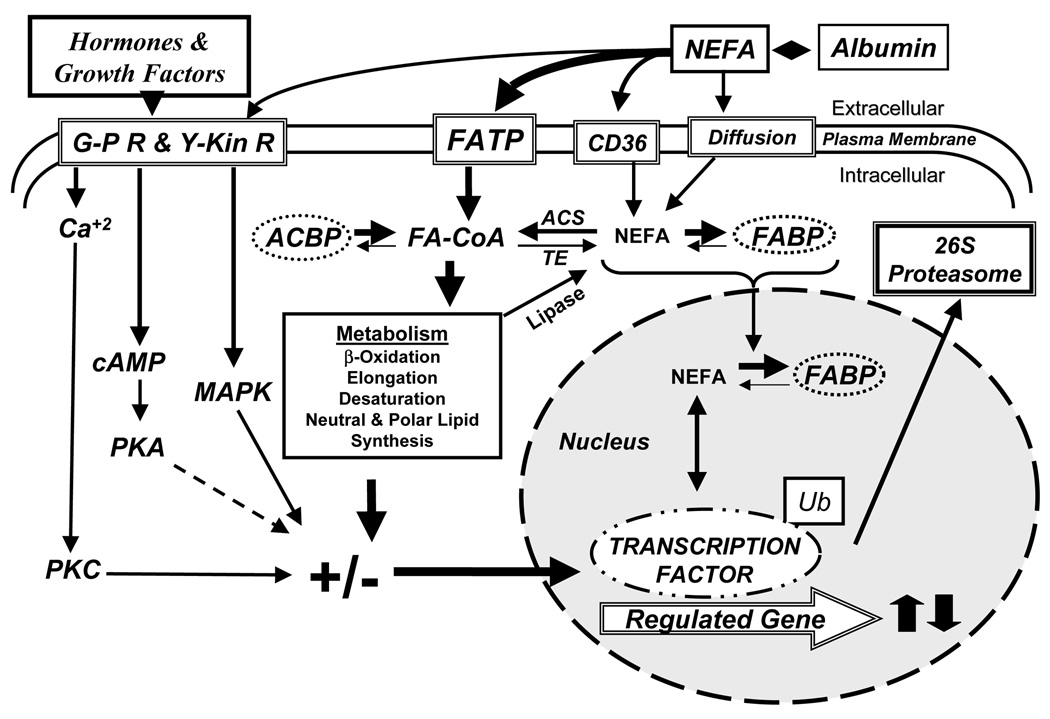
See text for explanation. G-PR, G-protein coupled receptors; Y-Kin R, tyrosine kinase-coupled receptors; FATP, fatty acid transport protein; CD36/FAT, a fatty acid transport protein; NEFA, non-esterified fatty acids; PKC, protein kinase C; PKA, protein kinase A; MAPK, mitogen activated protein kinase; ACBP, fatty acyl CoA binding protein; FA-CoA, fatty acyl CoA; ACS, acyl CoA synthetase; TK, fatty acyl thioesterase; FABP, fatty acid binding protein; Ub, ubiquitin.
In our view, fatty acid effects on hepatic gene expression require their entry into cells. Nonesterified fatty acids (NEFA) enter cells through transporters (FATP or FAT) or diffusion (Fig. 1). NEFA are rapidly converted to FACoA by FATP (DiRusso et al., 2005) or fatty acyl CoA synthetases (Coleman et al., 2002). When fatty acids enter hepatocytes as complex lipids, like chylomicron remnants, the complex lipids are hydrolyzed by lipases to form NEFA. In either case, intracellular NEFA is maintained at a very low level by quickly converting the NEFA to fatty acyl CoA. Fatty acyl CoAs are also kept at low intracellular levels. Both NEFA and FACoA are bound to FABP and ACBP, proteins that transport fatty acids to intracellular compartments for metabolism (Hertzel and Bernlohr, 2000) or to the nucleus to interact with transcription factors (Wolfrum, 2001). The bulk of FACoA is rapidly assimilated into complex lipids. Hepatocytes challenged with exogenous fatty acids rapidly assimilate the fatty acids into neutral and polar lipids, while a minor fraction will be β-oxidized. These metabolic pathways keep intracellular NEFA and FA-CoA very low. Intracellular NEFA, however, are not solely dependent on exogenous fatty acids; intracellular NEFA can also arise from the hydrolysis of complex lipids by lipases acting on phospholipids or triglycerides (Patton, 1994) or by the hydrolysis of FACoA by thioesterases (Hunt and Alexson, 2002). As such, these pathways may contribute to the pool of NEFA in cells. We view the intracellular NEFA fraction of cells as playing an important role in lipid mediated regulation of transcription factor function.
Studies with PPARα null mice and over expression of native and mutant forms of SREBP-1c, LXRα, ChREBP or MLX in primary hepatocytes have revealed several major metabolic pathways that are targeted by PUFA. Each pathway involves changes in gene expression. First, n-3 PUFA induction of mono- (microsomal) and β-oxidation (mitochondrial and peroxisomal) requires PPARα. Second, PUFA suppression of de novo lipogenesis (fatty acid synthase, FAS), fatty acid elongation (Elovl-6) and monounsaturated fatty acid synthesis (Δ9 desaturase [Δ9D], also known as stearoyl CoA desaturase, SCD1) involves three transcription factors, SREBP-1, ChREBP and MLX (Dentin et al., 2005; Ma et al., 2005; Wang, 2006; Xu, 2006). Third, PUFA suppression of the glycolytic enzyme, L-yruvate kinase, does not involve PPARα, SREBP-1 or LXRα (Pan et al., 2000; Pawar, 2003; Ren et al., 1997; Wang, 2005), but involves ChREBP and MLX heterodimer (Dentin et al., 2005; Wang, 2006; Xu, 2006). Fourth, PUFA suppression of PUFA synthesis lowers levels of fatty acid elongase-5 (Elovl-5), Δ5desaturase (Δ5D) and Δ6-desaturase (Δ6D). PUFA control of SREBP-1 nuclear abundance explains part of this mechanism (Wang, 2005; Wang, 2006; Xu, 2006). These studies indicate that PUFA, and in particular n-3 PUFA, function as feed-forward activators of fatty acid oxidation at the level of gene expression to control mitochondrial, peroxisomal and microsomal lipid metabolism. N-3 PUFA also function as feedback inhibitors of glycolysis, de novo lipogenesis, mono- and polyunsaturated fatty acid synthesis to control the production and cellular content of saturate, mono- and polyunsaturated fatty acids. These regulatory schemes not only reduce overall hepatic lipid content and VLDL secretion, but also eliminate excessive very long chain PUFA that may promote oxidant stress or impair membrane integrity.
Fatty Acid Regulation of Hepatic PPAR α
Of the multiple mechanisms by which fatty acids regulate gene expression, fatty acid control of PPAR represents the best understood. PPAR are fatty acid regulated nuclear receptors. PPAR form obligate heterodimer partners with RXR and bind regulatory elements in promoters of responsive genes. All PPAR subtypes (α, β/δ, γ1 and γ2) bind saturated and unsaturated fatty acids ranging in length from 16–20 carbons (Xu et al., 1999). Like all nuclear receptors, binding of the ligand, in this case the fatty acid, stimulates an exchange of co-activators for co-repressors on the chromatin-bound receptor and the recruitment of additional proteins involved in gene transcription such as RNA polymerase II. PPARα is the predominant PPAR subtype in rodent liver and it plays a dominant role in controlling genes involved in fatty acid oxidation, desaturation, elongation, transport, and binding. PPARα, however, is less active in human liver. Studies with PPARα null mice establish that PPARα is required for PUFA effects on genes encoding enzymes involved in fatty acid oxidation, desaturation and elongation, but not required for PUFA suppression of enzymes involved in glycolysis or lipogenesis (Pan et al., 2000; Ren et al., 1997).
Since PPARs are considered sensors of intracellular lipid and PPAR bind NEFA, we examined intracellular NEFA levels in primary rat hepatocytes before and after challenging cells with fatty acids (18:1, n-9 and 20:5,n-3) (Pawar, 2003). These studies revealed several important features as to how fatty acids control PPARα function. First, intracellular NEFA levels are maintained at very low levels representing <0.1% of the total lipid in the cell. Second, challenging cells with certain fatty acids, like 20:5,n-3, significantly change intracellular NEFA composition and promotes a robust response in PPARα target genes, like cytochrome P450-4A (CYP4A) and cytosolic fatty acyl thioesterase-1 (CTE1) (Pawar, 2003; Pawar, 2003). Other fatty acids, like 18:1,n-9, when added to cells are so rapidly assimilated as to not impact intracellular NEFA levels. As such, these fatty acids do not induce PPARα target genes. A factor that has a bearing on this response is the abundance of the fatty acid in the cell prior to challenge. In livers of chow fed animals or primary hepatocytes, 18:1,n-9 is a very abundant fatty acid in cell membrane phospholipids; it is also secreted as neutral lipids, in VLDL-triglycerides and VLDL-cholesterol esters. In contrast, 20:5,n-3 is not an abundant fatty acid in livers of chow fed animals. In fact, 20:5,n-3 levels in rat liver and plasma are <0.01% of 18:1,n-9. Feeding rats diets enriched in C20–22 n-3 PUFA increases 20:5,n-3 content in plasma and tissues, but it does not accumulate to the levels seen for 18:1,n-9. Nevertheless, the changes in intracellular 20:5,n-3 is recognized by PPAR and mechanisms are initiated to prevent excessive accumulation of this highly unsaturated fatty acid. Such studies support to the concept that PPARs are monitors of intracellular NEFA composition and respond accordingly to induce metabolic pathways that minimize damage brought on by excessive intracellular NEFA.
Is 22:6,n-3 (DHA) an effective PPARα ligand and activator of PPARα activity? Our studies have established that in primary rat hepatocytes, both 22:6,n-3 and 22:5,n-3 are weak activators of PPARα when compared to 20:5,n-3 (Pawar, 2003). Although co-crystals of 20:5,n-3 and PPAR β/δ have been described (Xu et al., 1999), we are unaware of similar studies assessing the binding of 22-carbon PUFA to any PPAR subtype. It is possible that the addition of the two additional carbon renders 22:5,n-3 and 22:6,n-3 a poor ligand for PPAR.
Another complicating factor in assessing effects of 22-carbon PUFA effects on PPAR activity is the fact that addition of 22:5,n-3 or 22:6,n-3 to hepatocytes leads to its retroconversion to 20:5,n-3. Retroconversion of very long chain fatty acids to shorter chain fatty acids is well described (Sprecher, 2000). Our studies established that not only does retroconversion occur in hepatocytes, it leads to an accumulation of 20:5,n-3 in esterified and non-esterified fatty acid fractions (Pawar, 2003). As such, retroconversion of 22-carbon PUFA generates a preferred PPARα ligand. Fig. 2 illustrates the conversion of dietary precursors (18:2,n-6 and 18:3,n-3) to the normal end products of PUFA synthesis, i.e., 20:4,n-6 and 22:6,n-3, respectively. The fatty acids exist as fatty acyl CoA during the elongation, desaturation and chain shortening reactions. When excessive 22:5,n-3 or 22:6,n-3 is added to cells, the bulk of this lipid is assimilated into triglycerides. 22-carbon PUFA are poor substrates for cholesterol ester formation (unpublished observation). A small fraction of the 22-carbon PUFA is chain shortened (retroconverted) to 20:5-CoA and this retroconverted 20:5,n-3 is assimilated into triglycerides. We speculate that the source of the accumulated non-esterified 20:5,n-3 in hepatocytes challenged with 22:5,n-3 or 22:6,n-3 is through the action of a lipase on triglycerides enriched in 22- carbon PUFA. Indirect support for this notion comes from our unpublished studies on fatty acyl thioesterases. We constructed a recombinant adenovirus expressing a peroxisomal fatty acyl thioesterase (PTE1). Since 22-carbon PUFAs are preferentially metabolized in the peroxisome (Reddy and Mannaerts, 1994), over expression of this peroxisomal fatty acyl thioesterase should affect intracellular NEFA and 22:6-mediated control of PPAR. Infection of rat primary hepatocytes with Ad- PTE1 leads to an increased hepatocyte thioesterase activity. This treatment, however, did not enhance the effect of 20:5,n-3 or 22:6,n-3 on PPARα-regulated genes4. The outcome of these studies implicates the action of lipases on complex lipids as a source of non-esterified 20:5,n-3. Lipase action on triglycerides or phospholipids enriched with 20:5,n-3 may contribute to the accumulation of nonesterified 20:5,n-3 following 22:6,n-3 treatment of primary hepatocytes.
Figure 2. Metabolic pathway for N-3 and N-6 PUFA synthesis.
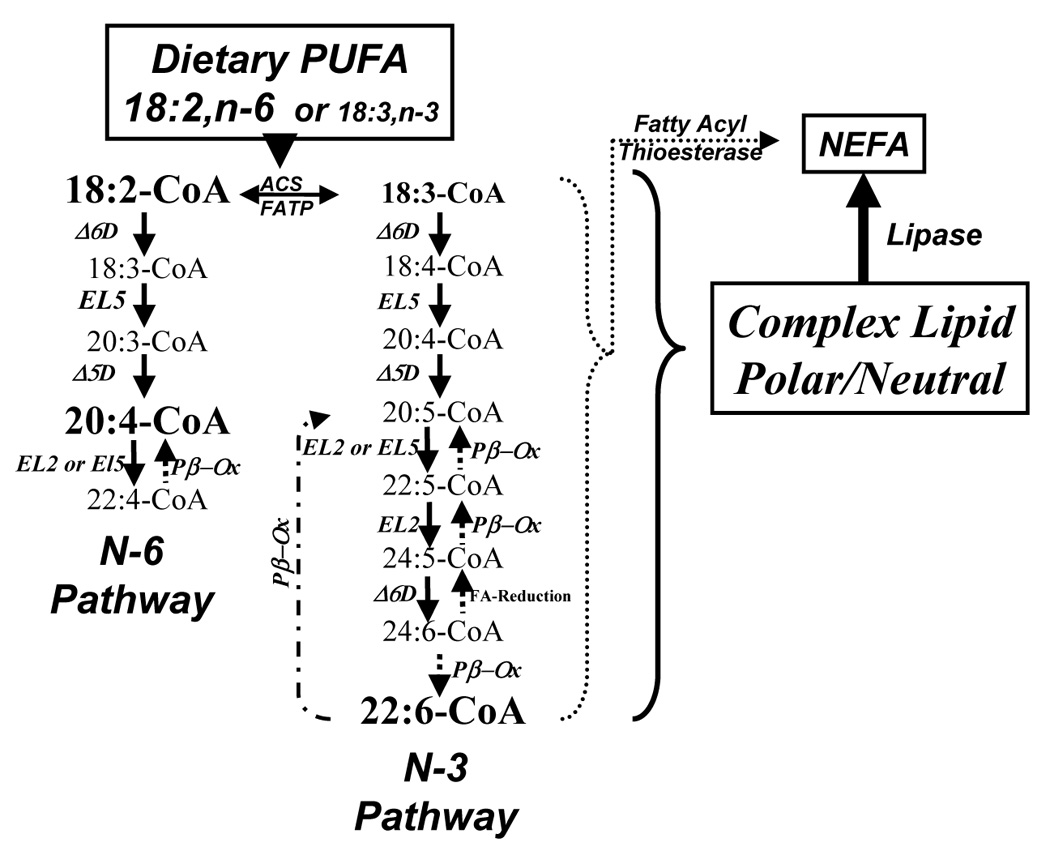
The diagram illustrates the metabolic pathway for n-3 and n-6 PUFA synthesis (solid line/arrows), retroconversion of 22:6,n-3 to 20:5,n-3 (stippled line/arrows) and the release of fatty acids from fatty acyl CoA or complex lipids by the action of fatty acyl thioesterases or lipases (dotted line/arrows), respectively. 18:2,n-6, 20:4,n-6 and 22:6,n-3 are the predominant fatty acid accumulating in tissues as neutral and phospholipids. See text for additional explanation. Δ5D, Δ5-desaturase; Δ6D, Δ6-desaturase; EL2, fatty acid elongase-2; EL5, fatty acid elongase-5; pβ-OX, peroxisomal β-oxidation.
To summarize, PPAR are nuclear receptors that bind fatty acids. When compared to the pharmacologic ligands like the fibrate drugs (Lopid or Trocor) or WY14,643, fatty acids are weak PPAR agonist. Nevertheless, fatty acid binding sets in motion a sequence of events that leads to activation of multiple genes involved in fatty acid transport, binding and metabolism (Table 1). Certain fatty acids, however, are better than others at activating PPAR. Structural studies have established that 20:5,n-3 is both a ligand (Xu et al., 1999) and a robust activator of PPAR (Pawar, 2003). 22:6,n-3 and 22:5,n-3, however, are weak PPARα activators. In our view, this is because they bind poorly to PPAR. Any stimulation of PPAR activity by 22-carbon PUFA likely requires prior retroconversion of the 22-carbon PUFA to the 20-carbon PUFA. In support of this concept, we attenuated the effect of 20:5,n-3 on PPAR-regulated genes by stimulating the conversion of 20:5 to 22:5,n-3. This was accomplished by elevating the activity of a specific fatty acid elongase, Elovl-2, in primary hepatocytes using a recombinant adenovirus4. These studies also highlight the fact that intracellular metabolism of 20-carbon PUFA is an important determinant in the control of PPAR activity and its target genes.
Hepatic Glycolysis and Lipogenesis: Targets for Dietary PUFA Control
Glycolysis and de novo lipogenesis (DNL) are two metabolic pathways for glucose utilization for anaerobic and aerobic respiration, as well as fuel storage in the form of glycogen and triglycerides. These pathways are subject to complex hormonal and nutrient control. Insulin, thyroid hormones, glucocorticoids and glucose stimulate glycolysis and DNL, while glucagon, epinephrine and PUFA suppress at least one glycolytic enzyme, i.e., L-pyruvate kinase, and several enzymes involved in DNL, i.e., ACL, ACC, 13 FAS, SCD1, Elovl-6 (Iizuka et al., 2004; Jump, 2004; Towle, 2005; Wang, 2005; Wang, 2006; Xu, 2006).
Key transcription factors controlling expression of proteins involved in these pathways include SREBP-1 and the ChREBP/MLX heterodimer (Table 1). Insulin induces the nuclear abundance of SREBP-1 and ChREBP/MLX. The mechanism by which insulin controls the nuclear abundance of these transcription factors, however, is different. Insulin induces SREBP-1 nuclear abundance through several mechanisms, including increased SREBP-1 gene transcription, stabilization of its RNA, proteolytic conversion of the precursor of SREBP-1 to its nuclear (mature) form and inhibition of 26S proteasomal degradation of the nuclear form (Jump, 2005). Insulin-stimulated elevation of ChREBP nuclear abundance, in contrast, requires insulin-regulated glucose metabolism (Cha and Repa, 2007; Towle, 2005; Uyeda, 2006). Below, we describe the recent advances in control of ChREBP/MLX and SREBP-1 nuclear abundance by insulin, glucose and fatty acids.
ChREBP and MLX are Targets for Fatty Acid Control in Hepatic Glycolysis and Lipid Synthesis
It is well established that glucose effects on hepatic carbohydrate and lipid metabolism go beyond glucose control of insulin secretion (Towle, 2005; Uyeda, 2006). Glucose stimulates the accumulation of ChREBP, a bHLH-LZ transcription factor, in hepatocyte nuclei. ChREBP binds carbohydrate regulatory elements (ChoRE, E-boxes) in promoters of responsive genes, like L-PK, ACC, FAS, and SCD1 (Table 1). Binding of ChREBP to these regulatory elements, however, requires MLX. Neither insulin, nor glucose regulate nuclear MLX content; nuclear MLX content appears constitutive(Stoeckman et al., 2004; Towle, 2005). Hormones that elevate cAMP or metabolic states that activate AMP-kinase (AMPK) induce phosphorylation of ChREBP. Phospho-ChREBP does not accumulate in nuclei or bind well to DNA (ChoRE).
When glucose is in excess, hepatic glucose metabolism through the pentose phosphate pathway increases, leading to elevated levels of xylulose-5-phosphate (X5P); X5P is an activator of protein phosphatases 2A (PP2A). PP2A dephosphorylates ChREBP, allowing ChREBP to accumulate in nuclei (Kabashima et al., 2003). The influx of ChREBP into nuclei likely triggers binding of ChREBP to its obligate heterodimer partner, Max-like factor-X (MLX) resulting in enhanced occupancy of ChoRE with nascent ChREBP/MLX. This heterodimer binds carbohydrate regulatory elements in promoters of target genes (Stoeckman et al., 2004; Xu, 2006). Binding of ChREBP/MLX to ChoRE in chromatin promotes the recruitment of RNA polymerase II to the promoter and the acetylation of histones (H3 and H4) on the promoter of ChREBP/MLX target genes (Stoeckman et al., 2004; Xu, 2006).
PUFA interfere with glucose activation of L-type pyruvate kinase (L-PK) gene transcription by targeting the ChoRE/HNF-4α region of the L-PK promoter (Jump et al., 1994; Liimatta et al., 1994; Pan et al., 2000). The discovery that ChREBP and MLX played a role in glucose-mediated control of gene expression made it possible to determine if these proteins were targets for PUFA regulation. Two recent reports examined this issue (Dentin et al., 2005; Xu, 2006). These studies indicated that PUFA suppression of L-PK was due to suppression of ChREBP (Dentin et al., 2005) or MLX (Xu, 2006) nuclear abundance. Both reports also indicated that the effect of PUFA on ChREBP or MLX nuclear abundance could not be explained by fatty acid induction of AMPK. Xu, et al 2006 reported that there was no differential effect in potency of 20:4,n-6, 20:5,n-3 or 22:6,n-3 on the control of L-PK transcription, or ChREBP or MLX nuclear abundance. Unlike PPARα (see above) and SREBP-1 (see below), PUFA regulation of ChREBP & MLX nuclear abundance is less responsive to the type of PUFA; ChREBP & MLX nuclear abundance responds more to the amount of PUFA in cells (Xu, 2006). The outcome of these studies provided a key missing link in our understanding of how PUFA 15 control glycolysis, in particular, L-pyruvate kinase. The molecular basis for PUFA control of MLX or ChREBP nuclear abundance, however, remains under investigation.
SREBP-1c is a Target for Fatty Acid Control in Hepatic Lipid Synthesis
SREBP-1c is one of 3 bHLH-LZ transcription factors (SREBP-1a, SREBP-1c & SREBP-2) that control of hepatic and whole body cholesterol and fatty acid synthesis. While SREBP-2 plays a major role in the regulation of cholesterol synthesis and uptake, SREBP-1 functions to regulate multiple facets of fatty acid synthesis and VLDL assembly (Horton et al., 2002; Horton, 2003).
SREBP effects on gene transcription are determined, in large part, by factors that control the nuclear abundance of the transcription factor (nSREBP). Two post-translational mechanisms control nSREBP, proteolytic processing (Goldstein et al., 2006; Horton et al., 2002) and proteasomal degradation (Sundqvist, 2005). All SREBPs are synthesized as precursors (pSREBP, ~125 kd) and tethered to the endoplasmic reticulum (ER). Precursor SREBPs are escorted from the ER to the Golgi complex by SREBP-cleavage activating protein (SCAP) for proteolytic processing. Proteases in the Golgi, i.e., site 1 and site 2 protease, cleave the SREBP precursor to generate the mature-nuclear form of SREBP (nSREBP)(Goldstein et al., 2006). nSREBP is transported to the nucleus as a dimer, via importin- β (Nagoshi et al., 1999). In nuclei, SREBP binds sterol regulatory elements (SRE) as dimers in promoters of target genes. Once bound, SREBPs recruit co-activators to the promoter and stimulate gene transcription (Bennett, 2004; Horton et al., 2002).
Sterols regulate nSREBP levels by controlling the proteolytic processing step at the level of the endoplasmic reticulum and Golgi, not the proteasomal degradation of the nuclear form of SREBP. Sterols induce the ER-resident proteins, Insig-1 or Insig-2 to bind SCAP; the Insig-SCAP-SREBP complex is retained in the ER preventing its movement to the Golgi for cleavage and maturation to nSREBP (Adams et al., 2004). In this fashion cholesterol functions as a feedback inhibitor of cholesterol synthesis by preventing SREBP-2 from accumulating in nuclei and inducing expression of key genes involved in cholesterol synthesis.
While SREBP-1 and SREBP-2 are structurally similar, their regulation in the liver by nutrients and hormones is quite different. Both SREBP-1a and -1c are expressed in the liver, but SREBP-1c is the predominant subtype based on its transcript abundance. In contrast to SREBP-2, SREBP-1c nuclear abundance is controlled by several mechanisms (Jump, 2005). Insulin and oxysterols (LXR agonists) induce SREBP-1c gene transcription; LXR/RXR heterodimers bind the SREBP-1c promoter. Insulin also stabilizes mRNASREBP-1c, and inhibit 26S proteasomal degradation of nSREBP-1, but not SREBP- 2 (Botolin, 2006). The outcome of these effects is to elevate the nuclear abundance of SREBP-1, but not SREBP-2, so as to selectively induce the transcription of genes involved in de novo lipogenesis and mono-unsaturated fatty acid synthesis (Jump, 2005). Insulin regulates SREBP-1c gene transcription through PI3 kinase and Akt (Ribaux and Iynedjian, 2003), changes in Insig-1 and -2 expression (Yabe et al., 2003), and inhibition of 26S proteasomal degradation (Botolin, 2006; Sundqvist, 2005). In fact, removal of insulin from primary hepatocytes leads to a prompt decline in nSREBP-1 (T1/2~10 hrs). This loss of nSREBP-1 following insulin removal is blocked by inhibitors of 26S proteasomal degradation (Botolin, 2006).
N-3 and n-6 PUFA are well-established suppressors of SREBP-1, but not SREBP-2, nuclear abundance (Mater et al., 1999; Worgall et al., 1998; Xu et al., 1999; Xu et al., 2001). PUFA suppression of SREBP-1 accounts for much of the well-known suppression of de novo lipogenesis by dietary PUFA (Jump and Clarke, 1999; Mater et al., 1999; Yahagi et al., 1999). We and others examined the effects of various fatty acids on nSREBP-1; fatty acids inhibit SREBP-1 gene transcription, induce mRNASREBP-1 instability and inhibit SREBP processing, for review see (Hannah et al., 2001; Jump, 2005; Mater et al., 1999; Ou et al., 2001; Xu et al., 1999; Xu et al., 2001). Many of these studies used high levels of fatty acids. More recently, we re-examined PUFA control of SREBP- 1 nuclear abundance and found that 22:6,n-3 was the most potent suppressor of SREBP-1 nuclear abundance (ED50 <100 µM). 20:4,n-6 and 22:6,n-3 are the end products of PUFA synthesis and the predominant n-6 and n-3 PUFA accumulating in livers of animals fed essential fatty acid sufficient diets (Botolin, 2006). At a 100 µM dose, 20:4,n-6, 20:5,n-3 and 22:6,n-3 had equivalent, but minor, effects on SREBP-1c promoter activity, mRNA SREBP-1c abundance and SREBP-1 precursor levels. Of these 3 fatty acids, only 22:6,n-3 significantly (>75%) suppressed nSREBP-1 abundance. These studies indicated that 22:6,n-3 was likely the major regulator of nSREBP-1 abundance in vivo. A major component of this control was directed at a post-translational level (Botolin, 2006). This is interesting because the mass of 20:4,n-6 exceeds the mass of 22:6,n-3 in the liver. While both 20:4,n-6 and 22:6,n-3 are end products of PUFA synthesis (Figure 2), it is 22:6,n-3, and not 20:4,n-6, that is the major regulator of nuclear SREBP-1 abundance and its target genes.
Post-translational control of SREBP-1 nuclear abundance also occurs in vivo, particularly during post-natal development when lipogenesis and nuclear SREBP-1 are induced at weaning (Botolin and Jump, 2003). Since expression of the 2 desaturases (Δ5 and Δ6 desaturase, Table 1) involved in PUFA synthesis are controlled by SREBP-1 (Matsuzaka et al., 2002; Wang, 2006), this mechanism likely plays a key role in the management of hepatic and whole body 22:6,n-3 levels.
Post-translational mechanisms controlling SREBP-1 include proteolytic conversion of the precursor to the mature (nuclear) form and 26S proteasomal degradation. 22:6,n-3 was found to have little effect on SCAP expression and 22:6,n-3 modestly induced Insig-1 expression. Like insulin, 22:6,n-3 strongly suppressed Insig-2 expression. These studies provided little support for 22:6,n-3 regulation of SCAP or Insig expression as the mechanism by which 22:6,n-3 controls SREBP-1 nuclear abundance (Botolin, 2006).
Since a compelling case for 22:6,n-3 control of SREBP-1 proteolytic processing could not be made, we turned out attention to the 26S proteasomal degradation of nuclear SREBP-1 as a mechanism regulating SREBP-1 nuclear abundance. SREBPs are phosphoproteins and their phosphorylation status controls their capacity to transactivate genes as well as their nuclear abundance. Protein kinase A (PKA), extracellular receptor kinase-1/2 (Erk1/2) and glycogen synthase kinase-3β (Gsk-3β) phosphorylate SREBPs. cAMP-activated PKA phosphorylates SREBPs within the DNA binding domain of the protein and inhibits SREBP binding to sterol regulatory elements (Lu, 2006). Active Erk (pErk1/2) phosphorylates SREBP-1a at Ser117; this event impairs insulin-stimulated regulation of the LDLR promoter by SREBP-1a (Roth et al., 2000). Ser117corresponds to Ser92 in SREBP-1c. We have identified other Erk sites in SREBP-1c at Ser39, Ser73, and Thr395 by using a computer assisted approach (http://scansite.mit.edu/). SREBPs are also phosphorylated by the active (dephosphorylated) form of Gsk-3β (Punga, 2006; Sundqvist, 2005). The phosphorylation sites in SREBP-2 are LM431SPPA435SD; in SREBP-1a, the sites are TL425TTPPP430SD; and in SREBP-1c the sites are TL395TPPP399SD. Phosphorylation at these sites promotes binding of the ubiquitin ligase SCFFbw7 and ubiquitination of SREBP; these events stimulate 26S proteasome degradation of SREBPs (Punga, 2006; Sundqvist, 2005). Thus, Gsk-3β appears to be an important mediator of SREBP proteasomal degradation.
The phosphorylation status of Gsk-3β is regulated by insulin (Srivastava, 1998; Welsh, 1996). Since 22:6,n-3 controlled SREBP-1 nuclear abundance through 26S proteasomal degradation, we considered Gsk-3β as a target of PUFA regulation. Gsk-3β activity is inhibited by Akt-mediated phosphorylation of Ser-9 of Gsk3β. Akt is a down-stream target of insulin action and required for insulin control of SREBP-1 nuclear abundance. Our studies established that removal of insulin from primary hepatocytes leads to a decline in nuclear SREBP-1 with a T1/2~10 hrs; inhibitors of 26S proteasomal degradation blocked this decline (Botolin, 2006). This observation supported the role of the 26S proteasome in insulin-mediated control of SREBP nuclear abundance (Sundqvist, 2005). Treatment of primary hepatocytes with 22:6,n-3 accelerates the loss of nSREBP-1 from a T1/2 of 10 hours when insulin was omitted to a T1/2~4 hrs when cell were treated with 22:6,n-3 in the absence of insulin. The 22:6,n-3-mediated loss of nSREBP-1 was completely blocked by 26S proteasome inhibitors, like MG132 and lactacystin. Of all the fatty acids tested, only 22:6,n-3 controlled nuclear SREBP-1 abundance through a 26S-proteasomal dependent pathway; inhibitors of 26S proteasomal completely blocked the 22:6,n-3 suppression of nSREBP-1. Since no other PUFA tested regulated SREBP-1 at the level of 26S proteasomal degradation, the outcome of these studies revealed a unique role for 22:6,n-3 in the control SREBP-1 nuclear abundance; through a 26S proteasome-dependent pathway.
To determine if the PUFA-regulated 26S proteasomal regulation of SREBP-1 nuclear abundance is relevant in vivo, we used our suckling rat model (Botolin and Jump, 2003). In suckling animals, blood levels of insulin are low (≤20% of adult levels (0.5 ng/ml plasma) (Girard et al., 1994). Moreover, enzymes involved in PUFA synthesis (Δ5 and Δ6 desaturases and the fatty acid elongase, Elovl-5) are induced immediately after birth (Wang, 2005). Thus, enhanced PUFA synthesis coupled with low circulating insulin may contribute to the absence of hepatic nuclear SREBP-1 in the neonate. As stated above, SREBP-1 mRNA is very abundant in neonatal liver. The mechanism for suppression of SREBP-1 nuclear abundance can not be explained by pre-translational mechanisms (transcription & RNA stability) since the mRNA encoding both SREBP-1a and SREBP-1c are abundance in livers of pre-weaned animals. To gain some insight in to the post-translational mechanisms controlling SREBP-1 in the neonate, we injected neonatal animals with sufficient insulin (5 U/100 gbw, ip) to restore insulin levels to adult values; we also administered the 26S proteasomal inhibitor (MG132, 10 µg/100 gbw, ip). While SREBP-2 nuclear abundance and microsomal levels of SREBP-1 and -2 were unaffected by these treatments, nuclear SREBP-1 (but not SREBP-2) was transiently (within 90 mins) increased by insulin and MG132 (not shown). These studies suggest that in vivo, insulin rapidly controls hepatic nuclear SREBP-1 content through a 26S proteasomal pathway. Our original report on developmental regulation of hepatic SREBP-1 and SREBP-2 nuclear abundance (Botolin and Jump, 2003) attributed differences in SREBP-1 and SREBP-2 nuclear abundance in the neonate to selective proteolytic cleavage. Based on these more recent studies (Botolin, 2006), we now believe this difference is due to selective 26 S proteasomal degradation of SREBP-1, a process regulated by insulin and 22:6,n-3.
How does 22:6,n-3 control SREBP-1 26S proteasomal degradation? Gsk3β-mediated phosphorylation of SREBP is clearly involved in the ubiquitination and 26S proteasomal degradation of SREBP (Sundqvist, 2005). Insulin suppresses Gsk-3β activity by inducing its phosphorylation through an Akt-dependent pathway. We found that both insulin and 22:6,n-3 induced Gsk-3β phosphorylation. In fact, all 20–22 carbon PUFA tested induced Gsk-3β phosphorylation4. Moreover, 22:6,n-3 transiently suppressed insulin-induced Akt-phosphorylation; phospho-Akt is active. Over expression of a constitutively active Akt failed to abolish the 22:6,n-3 suppression of nSREBP-1 (Botolin, 2006). Taken together, the outcome of these studies indicated that Akt and Gsk-3β are not involved in the 22:6,n-3 mediated suppression of SREBP-1 nuclear abundance.
Despite this outcome, these studies revealed a novel target of fatty acids, i.e., Gsk3β. Gsk-3β phosphorylation is mediated by both Akt and PKC (Goode et al., 1992; Lavoie et al., 1999). We and others have reported that PUFA inhibit insulin-induced Akt phosphorylation (Botolin, 2006; Talukdar et al., 2005). This outcome implicated PKC as a likely target for PUFA control. Defining the role PUFA plays in controlling Gsk-3β activity has high significance because this signaling pathway plays a central role in many cellular events, e.g., glycogen synthesis (glycogen synthase), protein synthesis (eIF2B), tubulin polymerization (Tau), cell cycle (cyclin D) and Wnt signaling (β-catenin)(Srivastava, 1998; Welsh, 1996). Moreover, Gsk-3 has attracted considerable interest as a drug target for neurological disorders (Chang et al., 2006; Kozlovsky et al., 2006; Li et al., 2006; Terstappen et al., 2006), heart disease (Palfi et al., 2006), inflammation (Yan and Tai, 2006), and cancer (De Toni et al., 2006; Ougolkov and Billadeau, 2006; Samanta et al., 2006). Gsk-3β is likely the key target for lithium, a well know treatment of certain neurological disorders. N-3 polyunsaturated fat (PUFA) are essential fatty acids (Spector, 1999). N-3 PUFA deficiency leads to problems with visual acuity and learning (Greiner et al., 2003; Rojas et al., 2002; Salem et al., 2001) as well as metabolic problems associated with lipid and carbohydrate metabolism [reviewed (Jump, 2002; Jump, 2002; Jump, 2004)]. Defining the mechanism for n-3 PUFA control of hepatic SREBP-1 and Gsk-3β may provide important clues to explain how n-3 PUFA operate in other tissues. Understanding the impact of n-3 PUFA on physiological process is an important human health issue. Its importance increases because of the increased availability of n-3 PUFA in various processed foods, e.g., baby formula (Infamile- Lipil/Mead-Johnson) and Promise and Smart Balance margarines/butter spreads, as well as the availability of omega-3 capsules in groceries and health food stores.
Protein Kinase C and Erk1/2 are likely involved in the 22:6,n-3 control of SREBP-1 nuclear abundance
The phosphorylation of Gsk-3β at Ser9 determines it activity; when not phosphorylated the kinase is active; phosphorylation at Ser9 inactivates the kinase. Ser9 on Gsk3β is phosphorylated by at least two signaling pathways, Akt or PKC (Goode et al., 1992; Srivastava, 1998; Welsh, 1996). Akt subtypes are activated by insulin and inhibited by 22:6,n-3 (Botolin, 2006; Scheid, 2001; Talukdar et al., 2005). Atypical PKC subtypes are targets of insulin action (Farese et al., 2005; Standaert et al., 2004). Moreover, PKC subtypes are well established target of fatty acid regulation (Hanks, 1988; Newton, 1997; Nishizuka, 1992). Since PUFA suppress the insulin-induced phosphorylation of Akt and Akt activity (Botolin, 2006; Talukdar et al., 2005), Akt is likely not the mediator of 22:6-mediated phosphorylation of Gsk-3β. As such, we considered PKC as a possible mediator of PUFA control of Gsk-3β. Three families of PKC have been defined, each with multiple subtypes: conventional (α, β (I & II), γ), novel (δ, ε, η, θ, μ) and atypical (ζ, λ, τ). Of these, PKC α, β, γ & ζ have been reported to phosphorylate Gsk-3β (Desbois-Mouthon et al., 2002; Goode et al., 1992; Kirshenboim et al., 2004). To determine if PKC might be involved in PUFA control of Gsk-3β, primary rat hepatocytes were treated with 22:6,n-3, in the absence and presence of two PKC inhibitors; calphostin C (a pan inhibitor of all PKC subtypes) and Gö6976 (an inhibitor of PKC α, β (I & II), γ). The 22:6,n-3 induction of Gsk-3β phosphorylation was blocked by calphostin C, but not Gö6976. The 22:6,n-3 suppression of nuclear SREBP-1 (a 26S proteasome-mediated process) was also blocked by calphostin C, but not Gö6976. These results suggest that either novel or atypical PKCs are involved in the 22:6-control of both Gsk-3β phosphorylation and SREBP-1 nuclear abundance.
Fatty acids also regulate other protein kinases cascades; including the mitogen-activated protein kinase (MAPK) pathways (P38, JNK & Erk). The Salati lab has identified P38 as a target for PUFA control and the regulation of hepatic glucose-6-phosphate dehydrogenase mRNA splicing (Talukdar et al., 2005). Our studies, however, ruled out the P38 and JNK pathways as being involved in PUFA control of SREBP-1 nuclear abundance. Instead, our studies implicated the Erk pathway in the 22:6,n- 6 control of SREBP-1 (Botolin, 2006). The ERK pathway is a well established target of insulin action. Insulin rapidly, but transiently, induces ERK phosphorylation in rat primary hepatocytes (Botolin, 2006; Xu et al., 2005). Only C22 PUFA induced ERK phosphorylation and regulate SREBP-1 26S proteasomal degradation (Botolin, 2006). 22:6,n-3 treatment of primary hepatocytes enhanced Erk-phosphorylation; elevated Erk-phosphorylation correlated with suppressed nSREBP-1 in primary hepatocytes and in livers of suckling rats. ERK1/2 is activated through phosphorylation by MEK. MEK inhibitors (PD98059 & U0126) rapidly (within 30 mins) attenuated 22:6, n-3-mediated suppression of nuclear SREBP-1 (Botolin, 2006). Based on these results, the MEK/ERK pathway controls SREBP-1 nuclear abundance.
While such studies connect ERK phosphorylation to the control of SREBP-1 nuclear abundance, the molecular details are missing. Particularly intriguing is the fact that PUFA control is directed exclusively at SREBP-1, while SREBP-2 nuclear abundance remains unaffected by these manipulations. The mechanism by which 22:6,n-3 controls ERK phosphorylation and SREBP-1 nuclear abundance remain two important unanswered questions. There are, however, some hints as to how this mechanism might work. First, Busik and colleagues have focused on the impact of PUFA, and in particular 22:6,n-3, on ameliorating the deterioration of the retina associated with diabetic retinopathy (Chen et al., 2003; Chen, 2005; Chen, 2007). The key observations are that treating human retinal endothelial cells with 20:4,n-6 promotes cytokine production, while 22:6,n-3 inhibits this process. Activation of cytokine production is linked to NFκB activation and its migration to the nucleus; 22:6,n-3 inhibits NFκB migration to the nucleus (Chen, 2005). In their most recent studies, Busik et al (Chen et al., 2007) reported that treating human retinal endothelial cells with 22:6,n-3 enriched membranes with 22:6,n-3. Membrane enrichment with 22:6,n-3 depleted membranes of cholesterol from caveolae/lipid rafts, but not bulk membranes. A key event associated with these changes in membrane composition was the loss of the Src family kinases, Fyn and c-Yes, from lipid rafts. These kinases are thought to be crucial for activation of NFκB signaling.
Lipid rafts are regions within the exoplasmic leaflet of the plasma membrane that are enriched in cholesterol and sphingolipids (Ikonen, 2001). Rafts selectively incorporate proteins and govern protein-protein and protein-lipid interactions. These membrane microdomains contribute to the structure and function of caveolae, plasma membrane invaginations, signal transduction, endocytosis, transcytosis, cholesterol trafficking as well as tyrosine kinase and sphingolipid cell signaling. Proteins acylated with saturated fatty acids (14:0 or 16:0) partition into the cytoplasmic membrane lipid leaflet with high affinity for rafts. A number of proteins involved in cell signaling are found in lipid rafts, including G-proteins (αs, αq), members of the Src-kinase family, caveolin, Gap43 (Moffett, 2000).
The notion that PUFA impact lipid rafts composition and cell signaling is not new. A well-defined model for studying PUFA effects on the function of rafts is T-cell activation (Fan et al., 2004; Fan et al., 2003; Ma et al., 2004; Pike et al., 2002; Pohl et al., 2004; Stulnig et al., 2001). The two Src family kinases, Lck and Fyn, as well as the transmembrane adaptor protein LAT (linker for activation of T cells) are concentrated on the cytoplasmic side of lipid rafts. Stimulation of the TCR triggers the activation of Lck and Fyn that leads to an increase in Ca+2-signaling, an activation of ERK, a MAP kinase pathway, and other downstream signaling events (Stulnig et al., 2001; Switzer et al., 2004). Prior treatment of Jurkat T-cells with 20:4,n-6 or 20:5,n-3 leads to a reduction in Lck and Fyn in lipid rafts, a reduction in calcium signaling and a decline in ERK activation. Both n3 and n6 PUFA affect raft composition and function through an eicosanoid-independent mechanism.
In the context of 22:6,n-3 effects on ERK phosphorylation status and the control of SREBP-1 nuclear abundance, a key issue may be the location of ERK in the cell. It is well established that activated (phospho-) ERK moves to the nucleus to phosphorylate nuclear proteins. ERK also interacts with membranes and this interaction is determined by membrane cholesterol content (Wang, 2003). Changes in membrane cholesterol content alter this association and impact the spectrum of proteins phosphorylated by ERK. As such, changes in membrane cholesterol content induced by 22:6,n-3 introduction into membrane phospholipids might impacts ERK signalling. Whether changes in cholesterol content in membranes is mediated by physical repulsion by 22:6,n-3 or by regulating an membrane-associated enzyme that affects membrane cholesterol content e.g., sphingomyelinase (SanGiovanni, 2005; Wu, 2005) remains to be tested.
Key future experiments to define the molecular basis for 22:6,n-3 control of 26S proteasomal degradation of nuclear SREBP-1 must focus on those mechanisms at the membrane and nuclear level that alter signaling pathways controlling selective degradation of nuclear SREBP-1. An important first step will be to determine which phosphorylated amino acids in SREBP-1 are required for 22:6- mediated control of SREBP-1 nuclear abundance.
Does 22:6,n-3 antagonize insulin action?
The preceding discussion has pointed out that many of the biochemical events regulated by 22:6,n-3 are also regulated by insulin (Table 2). In some cases, 22:6,n-3 antagonizes insulin action. For example, 22:6,n-3 inhibits the inductive effect of insulin on de novo lipogenesis, PUFA synthesis, L-pyruvate kinase gene transcription. 22:6,n-3 suppresses the nuclear abundance of the key transcription factors (ChREBP, MLX and SREBP-1) controlling the expression of glycolytic (L-pyruvate kinase) and lipogenic (ACC, FAS, SCD1, Elovl6) genes. Of the key signaling pathways regulated by insulin, e.g., Akt, Gsk3β, Erk1/2 and PKC, only the insulin regulation of the Akt pathway is antagonized by 22:6,n-3. For the others, 22:6,n-3 is insulinmimetic. These observations suggest that 22:6,n-3 likely does not interfere with early events in insulin signaling, but impacts key downstream events. The underlying mechanisms for these effects are not fully defined. These observations are consistent with the finding that feeding rodents or human diets supplemented with n-3 PUFA does not promote insulin resistance (Lombardo and Chicco, 2006; Sirtori et al., 1997; Steerenberg et al., 2002; Winzell et al., 2006).
Table 2.
A Comparison of Insulin and 22:6,n-3 Effects on Hepatic Signaling Mechanisms and Fatty Acid Synthesis.
| Insulin | 22:6,n-3 | |
|---|---|---|
| De novo lipogenesis | Induces | Suppresses |
| PUFA synthesis | Increases | Suppresses |
| Glucokinase Gene transcription | Increases | No Effect |
| L-Pyruvate Kinase Gene transcription | Increases | Decreases |
| ChREBP/MLX Nuclear abundance | Increases | Decreases |
| SREBP-1 Nuclear abundance | Increases | Decreases |
| PKB/Akt Phosphorylation (Activates) | Decreases | Increases |
| Gsk-3β Phosphorylation (Inactivates) | Increases | Increases |
| Erk1/2 Phosphorylation (Activates) | Increases | Increases |
| Transiently | Sustained | |
| Protein kinase C Activity | Activates | Activates |
| Subtypes | Atypical PKC | Atypical and/or novel PKCs |
Summary
As a polyunsaturated fatty acid, 22:6,n-3 has many of the same effects on gene expression as other PUFA. 22:6,n-3 activates PPARα, but suppresses the nuclear abundance of SREBP-1, ChREBP and MLX. In doing so, 22:6,n-3 promotes fatty acid oxidation and inhibits fatty acid synthesis. 22:6,n-3, however, differs from other PUFA in having different potencies on these various regulatory systems. 22:6,n-3 is a weak activator of PPARα; this process likely requires retroconversion of 22:6,n-3 to 20:5,n-3. 20:5,n-3, but not 22:6,n-3, is a robust activator of PPARα. In contrast to PPARα, the effect of 22:6,n-3 on the control of glycolytic gene expression and ChREBP & MLX nuclear abundance can not be distinguished from other PUFA. Where 22:6,n-3 truly distinguishes itself as a regulator of gene expression is its effect on 26S proteasomal degradation of SREBP-1. This unique event is seen only with 22:6,n-3; no other PUFA controls SREBP-1 nuclear abundance through this mechanism. Insulin inhibits the 26S proteasomal degradation of nuclear SREBP-1. Elevated nuclear content of SREBP-1 is one of the key factors involved in the positive effect of insulin on de novo lipogenesis. 22:6,n-3 antagonizes the insulin control of this process through the Erk1/2 and PKC (novel or atypical) pathways. Precisely how this is achieved is under investigation.
These studies have also uncovered effects of 22:6,n-3 on signaling pathways (Erk1/2, Gsk3β and PKC [novel or atypical]) that have broad effects on multiple regulatory processes in many tissues. Unknown is whether 22:6,n-3 regulates these signaling pathways in non-hepatic tissues. If such regulation is operative in other tissues, e.g., the central nervous or visual systems, then control of these pathways may explain some of the actions of 22:6,n-3 on such complex processes as learning and visual acuity.
Footnotes
This project was supported by the National Institutes of Health (DK43220) and the National Research Initiative of the USDA Cooperative State Research, Education and Extension Service (2003-5200-3400).
Abbreviations: ChREBP, carbohydrate regulatory element binding protein; DHA, docosahexaenoic acid (22:6,n-3); FABP, fatty acid binding protein; FACoA, fatty acyl CoA; FAT, fatty acid transporter (CD36); FATP, fatty acid transport protein; CTEI, cytosolic thioesterase I; CYP4A, cytochrome P450- 4A; basic helix-loop-helix-leucine-zipper transcription factor, bHLH-LZ; HNF-4, hepatic nuclear factor-4; LXR, liver X-receptor; NEFA, non-esterified fatty acid; MLX, Max-like factor X; PPAR, peroxisome proliferator activated receptor; PPRE, peroxisome proliferator regulatory element; peroxisomal thioesterase, PTE1; PUFA, polyunsaturated fatty acid; RXR, retinoid X receptor; SREBP, sterol regulatory element binding protein.
Unpublished observation.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
LITERATURE CITED
- Adams CM, et al. Cholesterol and 25-hydroxycholesterol inhibit activation of SREBPs by different mechanisms, both involving SCAP and Insigs. J Biol Chem. 2004;279(50):52772–52780. doi: 10.1074/jbc.M410302200. [DOI] [PubMed] [Google Scholar]
- Bennett M, Toth JI, Osborne TF. Selective association of sterol regulatory element binding protein isoforms with target promoters in vivo. J Biol Chem. 2004;279:37360–37367. doi: 10.1074/jbc.M404693200. [DOI] [PubMed] [Google Scholar]
- Botolin D, Jump DB. Selective proteolytic processing of rat hepatic sterol regulatory element binding protein-1 (SREBP-1) and SREBP-2 during postnatal development. J Biol Chem. 2003;278(9):6959–6962. doi: 10.1074/jbc.M212846200. [DOI] [PubMed] [Google Scholar]
- Botolin D, Wang Y, Christian B, Jump DB. Docosahexaneoic acid [22: 6, n-3] stimulates rat hepatic sterol regulatory element binding protein-1c (SREBP-1c) degradation by an Erk-and 26S proteasome-dependent pathway. J Lipid Res. 2006;47:181–192. doi: 10.1194/jlr.M500365-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cha JY, Repa JJ. The liver X receptor (LXR) and hepatic lipogenesis. The carbohydrate-response element-binding protein is a target gene of LXR. J Biol Chem. 2007;282(1):743–751. doi: 10.1074/jbc.M605023200. [DOI] [PubMed] [Google Scholar]
- Chang KA, et al. Phosphorylation of amyloid precursor protein (APP) at Thr668 regulates the nuclear translocation of the APP intracellular domain and induces neurodegeneration. Mol Cell Biol. 2006;26(11):4327–4338. doi: 10.1128/MCB.02393-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Jump DB, Esselman WJ, Busik JV. Inhibition of cytokine signaling in human retinal endothelial cells through modification of caveolae/lipid rafts by docosahexaenoic acid. Invest Ophthalmol Vis Sci. 2007;48(1):18–26. doi: 10.1167/iovs.06-0619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Jump DB, Grant MB, Esselman WJ, Busik JV. Dyslipidemia, but not hyperglycemia, induces inflammatory adhesion molecules in human retinal vascular endothelial cells. Invest Ophthalmol Vis Sci. 2003;44(11):5016–5022. doi: 10.1167/iovs.03-0418. [DOI] [PubMed] [Google Scholar]
- Chen W, Jump DB, Esselman WJ, Busik JV. Anti-inflammatory effect of docosahexaenoic acid on cytokine-induced adhesion molecule expression in human retinal endothelial cells. Invest Ophthalmol Vis Sci. 2005;46:4342–4347. doi: 10.1167/iovs.05-0601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Jump DB, Esselman WJ, Busik JV. Inhibition of cytokine signaling in human retinal endothelial cells through modification of caveaolae/lipid rafts by docosahexaenoic acid. Invest Ophthalmol Vis Sci. 2007;48:18–26. doi: 10.1167/iovs.06-0619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho E, Spiegelman D, Hunter DJ, Chen WY, Stampfer MJ, Colditz GA, Willett WC. Premenopausal fat intake and risk of breast cancer. J. Natl. Cancer Inst. 2003;95:1079–1085. doi: 10.1093/jnci/95.14.1079. [DOI] [PubMed] [Google Scholar]
- Coleman RA, Lewin TM, Van Horn CG, Gonzalez-Baro MR. Do long-chain acyl-CoA synthetases regulate fatty acid entry into synthetic versus degradative pathways? J Nutr. 2002;132(8):2123–2126. doi: 10.1093/jn/132.8.2123. [DOI] [PubMed] [Google Scholar]
- De Toni F, et al. A crosstalk between the Wnt and the adhesion-dependent signaling pathways governs the chemosensitivity of acute myeloid leukemia. Oncogene. 2006;25(22):3113–3122. doi: 10.1038/sj.onc.1209346. [DOI] [PubMed] [Google Scholar]
- Dentin R, et al. Polyunsaturated fatty acids suppress glycolytic and lipogenic genes through the inhibition of ChREBP nuclear protein translocation. J Clin Invest. 2005;115(10):2843–2854. doi: 10.1172/JCI25256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desbois-Mouthon C, et al. Dysregulation of glycogen synthase kinase-3beta signaling in hepatocellular carcinoma cells. Hepatology. 2002;36(6):1528–1536. doi: 10.1053/jhep.2002.37192. [DOI] [PubMed] [Google Scholar]
- Dhe-Paganon S, Duda K, Iwamoto M, Chi YI, Shoelson SE. Crystal structure of the HNF4 alpha ligand binding domain in complex with endogenous fatty acid ligand. J Biol Chem. 2002;277(41):37973–37976. doi: 10.1074/jbc.C200420200. [DOI] [PubMed] [Google Scholar]
- DiRusso CC, et al. Comparative biochemical studies of the murine fatty acid transport proteins (FATP) expressed in yeast. J Biol Chem. 2005;280(17):16829–16837. doi: 10.1074/jbc.M409598200. [DOI] [PubMed] [Google Scholar]
- Ellis CE, Murphy EJ, Mitchell DC, Golovko MY, Scaglia F, Barcelo-Coblijn GC, Nussbaum RL. Mitochondiral lipid abnormality and electron transport chain impairment in mice lacking alpha-synuclein. Mol Cell Biol. 2005;25:10190–10201. doi: 10.1128/MCB.25.22.10190-10201.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan YY, Ly LH, Barhoumi R, McMurray DN, Chapkin RS. Dietary docosahexaenoic acid suppresses T cell protein kinase C theta lipid raft recruitment and IL-2 production. J Immunol. 2004;173(10):6151–6160. doi: 10.4049/jimmunol.173.10.6151. [DOI] [PubMed] [Google Scholar]
- Fan YY, McMurray DN, Ly LH, Chapkin RS. Dietary (n-3) polyunsaturated fatty acids remodel mouse T-cell lipid rafts. J Nutr. 2003;133(6):1913–1920. doi: 10.1093/jn/133.6.1913. [DOI] [PubMed] [Google Scholar]
- Farese RV, Sajan MP, Standaert ML. Atypical protein kinase C in insulin action and insulin resistance. Biochem Soc Trans. 2005;33(Pt 2):350–353. doi: 10.1042/BST0330350. [DOI] [PubMed] [Google Scholar]
- Fedorova Ias N., Jr Omega-3 fatty acids and rodent behavior. Prostaglandins Leukot Essent Fatty Acids. 2006;75:271–289. doi: 10.1016/j.plefa.2006.07.006. [DOI] [PubMed] [Google Scholar]
- Girard J, Perdereau D, Foufelle F, Prip-Buus C, Ferre P. Regulation of lipogenic enzyme gene expression by nutrients and hormones. Faseb J. 1994;8(1):36–42. doi: 10.1096/fasebj.8.1.7905448. [DOI] [PubMed] [Google Scholar]
- Goldstein JL, DeBose-Boyd RA, Brown MS. Protein sensors for membrane sterols. Cell. 2006;124(1):35–46. doi: 10.1016/j.cell.2005.12.022. [DOI] [PubMed] [Google Scholar]
- Goode N, Hughes K, Woodgett JR, Parker PJ. Differential regulation of glycogen synthase kinase-3 beta by protein kinase C isotypes. J Biol Chem. 1992;267(24):16878–16882. [PubMed] [Google Scholar]
- Greiner RS, Catalan JN, Moriguchi T, Salem N., Jr Docosapentaenoic acid does not completely replace DHA in n-3 FA-deficient rats during early development. Lipids. 2003;38(4):431–435. doi: 10.1007/s11745-003-1080-2. [DOI] [PubMed] [Google Scholar]
- Hanks SK, Quinn AM, Hunter T. The protein kinase family: conserved features and deduced phylogeny of the catalytic domains. Science. 1988;241:42–52. doi: 10.1126/science.3291115. [DOI] [PubMed] [Google Scholar]
- Hannah VC, Ou J, Luong A, Goldstein JL, Brown MS. Unsaturated fatty acids down-regulate srebp isoforms 1a and 1c by two mechanisms in HEK-293 cells. J Biol Chem. 2001;276(6):4365–4372. doi: 10.1074/jbc.M007273200. [DOI] [PubMed] [Google Scholar]
- Harris WS. n-3 Long-chain polyunsaturated fatty acids reduce risk of coronary heart disease death: extending the evidence to the elderly. Am J Clin Nutr. 2003;77(2):279–280. doi: 10.1093/ajcn/77.2.279. [DOI] [PubMed] [Google Scholar]
- Hertzel AV, Bernlohr DA. The mammalian fatty acid-binding protein multigene family: molecular and genetic insights into function. Trends Endocrinol Metab. 2000;11(5):175–180. doi: 10.1016/s1043-2760(00)00257-5. [DOI] [PubMed] [Google Scholar]
- Horrobin DF. Omega-3 fatty acids for schizophrenia. Am. J. Psychiatry. 2003;160:188–189. doi: 10.1176/appi.ajp.160.1.188. [DOI] [PubMed] [Google Scholar]
- Horrobin DF, Bennett CN. Depression and bipolar disorder: relationships to impaired fatty acid and phospholipid metabolism and to diabetes, cardiovascular disease, immunological abnormalities, cancer, ageing and osteoporosis. Possible candidate genes. Prostaglandins Leukot Essent Fatty Acids. 1999;60(4):217–234. doi: 10.1054/plef.1999.0037. [DOI] [PubMed] [Google Scholar]
- Horton JD, Goldstein JL, Brown MS. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest. 2002;109(9):1125–1131. doi: 10.1172/JCI15593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton JD, Shah NA, Warrington JA, Anderson NN, Park SW, Brown MS, Goldstein JL. Combined analysis of oligonucleotide microarray data from transgenic and knockout mice identifies direct SREBP target genes. Proc Natl Acad Sci U S A. 2003;100:12027–12032. doi: 10.1073/pnas.1534923100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt MC, Alexson SE. The role Acyl-CoA thioesterases play in mediating intracellular lipid metabolism. Prog Lipid Res. 2002;41(2):99–130. doi: 10.1016/s0163-7827(01)00017-0. [DOI] [PubMed] [Google Scholar]
- Iizuka K, Bruick RK, Liang G, Horton JD, Uyeda K. Deficiency of carbohydrate response element-binding protein (ChREBP) reduces lipogenesis as well as glycolysis. Proc Natl Acad Sci U S A. 2004;101(19):7281–7286. doi: 10.1073/pnas.0401516101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikonen E. Role of lipid rafts in membrane transport. Current Opinions in Cell Biology. 2001;13:470–477. doi: 10.1016/s0955-0674(00)00238-6. [DOI] [PubMed] [Google Scholar]
- Johnson EJ, Schaefer EJ. Potential role of dietary n-3 fatty acids in the prevention of dementia and macular degeneration. Am J Clin Nutr. 2006;83:14945–14985. doi: 10.1093/ajcn/83.6.1494S. [DOI] [PubMed] [Google Scholar]
- Julien C, Berthiaume L, Hadj-Tahar A, Rujput AH, Bedard PJ, Di Paolo T, Julien P, Calon F. Postmortem brain fatty acid profile of levodopa-treated Parkinson disease patients and parkinsonian monkeys. Neurochem Int. 2006;48:404–414. doi: 10.1016/j.neuint.2005.12.002. [DOI] [PubMed] [Google Scholar]
- Jump DB. The biochemistry of n-3 polyunsaturated fatty acids. J Biol Chem. 2002;277(11):8755–8788. doi: 10.1074/jbc.R100062200. [DOI] [PubMed] [Google Scholar]
- Jump DB. Dietary polyunsaturated fatty acids and regulation of gene transcription. Curr Opin Lipidol. 2002;13(2):155–164. doi: 10.1097/00041433-200204000-00007. [DOI] [PubMed] [Google Scholar]
- Jump DB. Fatty acid regulation of gene transcription. Cric. Rev. Clin. Lab Sci. 2004;41:41–78. doi: 10.1080/10408360490278341. [DOI] [PubMed] [Google Scholar]
- Jump DB, Botolin D, Wang Y, Xu J, Christian B, Demeure O. Fatty acid regulation of gene transcription. J. Nutr. 2005;135:2503–2506. doi: 10.1093/jn/135.11.2503. [DOI] [PubMed] [Google Scholar]
- Jump DB, Clarke SD. Regulation of gene expression by dietary fat. Annu Rev Nutr. 1999;19:63–90. doi: 10.1146/annurev.nutr.19.1.63. [DOI] [PubMed] [Google Scholar]
- Jump DB, Clarke SD, Thelen A, Liimatta M. Coordinate regulation of glycolytic and lipogenic gene expression by polyunsaturated fatty acids. J Lipid Res. 1994;35(6):1076–1084. [PubMed] [Google Scholar]
- Kabashima T, Kawaguchi T, Wadzinski BE, Uyeda K. Xylulose 5-phosphate mediates glucose-induced lipogenesis by xylulose 5-phosphate-activated protein phosphatase in rat liver. Proc Natl Acad Sci U S A. 2003;100(9):5107–5112. doi: 10.1073/pnas.0730817100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley DE, Goodpaster BH, Storlien L. Muscle triglyceride and insulin resistance. Annu. Rev. Nutr. 2002;22:325–346. doi: 10.1146/annurev.nutr.22.010402.102912. [DOI] [PubMed] [Google Scholar]
- Kirshenboim N, Plotkin B, Shlomo SB, Kaidanovich-Beilin O, Eldar-Finkelman H. Lithium-mediated phosphorylation of glycogen synthase kinase-3beta involves PI3 kinase-dependent activation of protein kinase C-alpha. J Mol Neurosci. 2004;24(2):237–245. doi: 10.1385/JMN:24:2:237. [DOI] [PubMed] [Google Scholar]
- Kostenis E. A glance at G-protein-coupled receptors for lipid mediators: a growing receptor family with remarkably diverse ligands. Pharmacol Ther. 2004;102(3):243–257. doi: 10.1016/j.pharmthera.2004.04.005. [DOI] [PubMed] [Google Scholar]
- Kozlovsky N, Amar S, Belmaker RH, Agam G. Psychotropic drugs affect Ser9-phosphorylated GSK-3 beta protein levels in rodent frontal cortex. Int J Neuropsychopharmacol. 2006;9(3):337–342. doi: 10.1017/S1461145705006097. [DOI] [PubMed] [Google Scholar]
- Kris-Etherton PM, Harris WS, Appel LJ. Fish consumption, fish oil, omega-3 fatty acids, and cardiovascular disease. Circulation. 2002;106(21):2747–2757. doi: 10.1161/01.cir.0000038493.65177.94. [DOI] [PubMed] [Google Scholar]
- Kushi LaG E. Dietary fat and cancer. Am. J. Med. 2002;113:63S–70S. doi: 10.1016/s0002-9343(01)00994-9. [DOI] [PubMed] [Google Scholar]
- Lavoie L, Band CJ, Kong M, Bergeron JJ, Posner BI. Regulation of glycogen synthase in rat hepatocytes. Evidence for multiple signaling pathways. J Biol Chem. 1999;274(40):28279–28285. doi: 10.1074/jbc.274.40.28279. [DOI] [PubMed] [Google Scholar]
- Li F, Chong ZZ, Maiese K. Microglial integrity is maintained by erythropoietin through integration of Akt and its substrates of glycogen synthase kinase-3beta, beta-catenin, and nuclear factor-kappaB. Curr Neurovasc Res. 2006;3(3):187–201. doi: 10.2174/156720206778018758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liimatta M, Towle HC, Clarke S, Jump DB. Dietary polyunsaturated fatty acids interfere with the insulin/glucose activation of L-type pyruvate kinase gene transcription. Mol Endocrinol. 1994;8(9):1147–1153. doi: 10.1210/mend.8.9.7838147. [DOI] [PubMed] [Google Scholar]
- Lombardo YB, Chicco AG. Effects of dietary polyunsaturated n-3 fatty acids on dyslipidemia and insulin resistance in rodents and humans. A review. J Nutr Biochem. 2006;17(1):1–13. doi: 10.1016/j.jnutbio.2005.08.002. [DOI] [PubMed] [Google Scholar]
- Lu M, Shyy JY-J. Sterol regulatory element binding protein 1 is negatively modulated by PKA phosphorylation. Am J Physiol Cell Physiol. 2006;290:C1477–C1486. doi: 10.1152/ajpcell.00374.2005. [DOI] [PubMed] [Google Scholar]
- Ma DW, et al. n-3 PUFA and membrane microdomains: a new frontier in bioactive lipid research. J Nutr Biochem. 2004;15(11):700–706. doi: 10.1016/j.jnutbio.2004.08.002. [DOI] [PubMed] [Google Scholar]
- Ma L, Tsatsos NG, Towle HC. Direct role of ChREBP.Mlx in regulating hepatic glucose-responsive genes. J Biol Chem. 2005;280(12):12019–12027. doi: 10.1074/jbc.M413063200. [DOI] [PubMed] [Google Scholar]
- Mata de Urquiza A, Liu S, Sjoberg M, Zetterstrom RH, Criffith W, Sjovall J, Perlmann T. Docosahexeanoic acid, a ligand for retinoic X receptor in mouse brain. Science. 2000;290:2140–2144. doi: 10.1126/science.290.5499.2140. [DOI] [PubMed] [Google Scholar]
- Mater MK, Thelen AP, Pan DA, Jump DB. Sterol regulatory element-binding protein 1c (SREBP1c) is involved in the polyunsaturated fatty acid suppression of hepatic S14 gene transcription. J Biol Chem. 1999;274(46):32725–32732. doi: 10.1074/jbc.274.46.32725. [DOI] [PubMed] [Google Scholar]
- Matsuzaka T, et al. Dual regulation of mouse Delta(5)- and Delta(6)-desaturase gene expression by SREBP-1 and PPARalpha. J Lipid Res. 2002;43(1):107–114. [PubMed] [Google Scholar]
- Moffett S, Brown DA, Linder ME. Lipid-dependent targeting of G-proteins into rafts. Journal of Biological Chemistry. 2000;275:2191–2198. doi: 10.1074/jbc.275.3.2191. [DOI] [PubMed] [Google Scholar]
- Nagoshi E, Imamoto N, Sato R, Yoneda Y. Nuclear import of sterol regulatory Nuclear import of sterol regulatory transcription factor, occurs through the direct interaction of importin beta with HLH-Zip. Mol Biol Cell. 1999;10(7):2221–2233. doi: 10.1091/mbc.10.7.2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton AC. Regulation of protein kinase C. Curr Opin Cell Biol. 1997;9:161–167. doi: 10.1016/s0955-0674(97)80058-0. [DOI] [PubMed] [Google Scholar]
- Nishizuka Y. Intracellular signaling by hydrolysis of phospholipids and activation of protein kinase C. Science. 1992;258:607–614. doi: 10.1126/science.1411571. [DOI] [PubMed] [Google Scholar]
- Ou J, et al. Unsaturated fatty acids inhibit transcription of the sterol regulatory element-binding protein-1c (SREBP-1c) gene by antagonizing ligand-dependent activation of the LXR. Proc Natl Acad Sci U S A. 2001;98(11):6027–6032. doi: 10.1073/pnas.111138698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ougolkov AV, Billadeau DD. Targeting GSK-3: a promising approach for cancer therapy? Future Oncol. 2006;2(1):91–100. doi: 10.2217/14796694.2.1.91. [DOI] [PubMed] [Google Scholar]
- Palfi A, et al. PARP inhibition prevents postinfarction myocardial remodeling and heart failure via the protein kinase C/glycogen synthase kinase-3beta pathway. J Mol Cell Cardiol. 2006;41(1):149–159. doi: 10.1016/j.yjmcc.2006.03.427. [DOI] [PubMed] [Google Scholar]
- Pan DA, et al. Evidence against the peroxisome proliferator-activated receptor alpha (PPARalpha) as the mediator for polyunsaturated fatty acid suppression of hepatic L-pyruvate kinase gene transcription. J Lipid Res. 2000;41(5):742–751. [PubMed] [Google Scholar]
- Patton GM, Fasulo JM, Robins SJ. Hepatic phosphatidylcholines: evidence for synthesis in the rat by extensive reutilization of endogenous acylglycerides. Journal of Lipid Research. 1994;35:1211–1221. [PubMed] [Google Scholar]
- Pawar A, Jump DB. Unsaturated fatty acid regulation of peroxisome proliferator activated receptor-alpha activity in primary rat hepatocytes. J Biol Chem. 2003;278:35931–35939. doi: 10.1074/jbc.M306238200. [DOI] [PubMed] [Google Scholar]
- Pawar A, Botolin D, Mangelsdorf DJ, Jump DB. The role of liver X receptor-alpha (LXR-alpha) in the fatty acid regulation of hepatic gene expression. J Biol Chem. 2003;278:40736–40743. doi: 10.1074/jbc.M307973200. [DOI] [PubMed] [Google Scholar]
- Peet M, Horrobin DF. A dose-ranging exploratory study of the effects of ethyl-eicosapentaenoate in patients with persistent schizophrenic symptoms. Journal of Psychiatric Research. 2002;36:7–18. doi: 10.1016/s0022-3956(01)00048-6. [DOI] [PubMed] [Google Scholar]
- Pike LJ, Han X, Chung KN, Gross RW. Lipid rafts are enriched in arachidonic acid and plasmenylethanolamine and their composition is independent of caveolin-1 expression: a quantitative electrospray ionization/mass spectrometric analysis. Biochemistry. 2002;41(6):2075–2088. doi: 10.1021/bi0156557. [DOI] [PubMed] [Google Scholar]
- Pohl J, et al. Long-chain fatty acid uptake into adipocytes depends on lipid raft function. Biochemistry. 2004;43(14):4179–4187. doi: 10.1021/bi035743m. [DOI] [PubMed] [Google Scholar]
- Punga T, Bengoechea-Alonso MT, Ericsson J. Phosphorylation and ubiquitination of the transcription factor sterol regulatory element-binding protein-1 in response to DNA binding. J Biol Chem. 2006:281. doi: 10.1074/jbc.M604983200. In Press. [DOI] [PubMed] [Google Scholar]
- Reddy JK, Mannaerts GP. Peroxisomal lipid metabolism. Annu Rev Nutr. 1994;14:343–370. doi: 10.1146/annurev.nu.14.070194.002015. [DOI] [PubMed] [Google Scholar]
- Ren B, Thelen AP, Peters JM, Gonzalez FJ, Jump DB. Polyunsaturated fatty acid suppression of hepatic fatty acid synthase and S14 gene expression does not require peroxisome proliferator-activated receptor alpha. J Biol Chem. 1997;272(43):26827–26832. doi: 10.1074/jbc.272.43.26827. [DOI] [PubMed] [Google Scholar]
- Ribaux PG, Iynedjian PB. Analysis of the role of protein kinase B (cAKT) in insulin-dependent induction of glucokinase and sterol regulatory element-binding protein 1 (SREBP1) mRNAs in hepatocytes. Biochem J. 2003;376(Pt 3):697–705. doi: 10.1042/BJ20031287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojas CV, et al. Long-term n-3 FA deficiency modifies peroxisome proliferator-activated receptor beta mRNA abundance in rat ocular tissues. Lipids. 2002;37(4):367–374. doi: 10.1007/s1145-002-0904-4. [DOI] [PubMed] [Google Scholar]
- Roth G, et al. MAP kinases Erk1/2 phosphorylate sterol regulatory element-binding protein (SREBP)-1a at serine 117 in vitro. Biol Chem. 2000;275(43):33302–33307. doi: 10.1074/jbc.M005425200. [DOI] [PubMed] [Google Scholar]
- Salem S., Jr Alterations in brain function after loss of docosahexaenoate due to dietary restriction of n-3 fatty acids. J Mol Neurosci. 2001;16(2–3):299–307. doi: 10.1385/JMN:16:2-3:299. discussion 317–21. [DOI] [PubMed] [Google Scholar]
- Samanta AK, Lin H, Sun T, Kantarjian H, Arlinghaus RB. Janus kinase 2: a critical target in chronic myelogenous leukemia. Cancer Res. 2006;66(13):6468–6472. doi: 10.1158/0008-5472.CAN-06-0025. [DOI] [PubMed] [Google Scholar]
- SanGiovanni JPaC EY. The role of omega 3-long chain PUFA in health and disease. Prog. Retin Eye Res. 2005;24:87–138. doi: 10.1016/j.preteyeres.2004.06.002. [DOI] [PubMed] [Google Scholar]
- Scheid MP, Woodgett JR. PkB/Akt: Functional insights from genetic models. Nature Reviews: Molecular Cell Biology. 2001;2:760–768. doi: 10.1038/35096067. [DOI] [PubMed] [Google Scholar]
- Sirtori CR, et al. N-3 fatty acids do not lead to an increased diabetic risk in patients with hyperlipidemia and abnormal glucose tolerance. Italian Fish Oil Multicenter Study. Am J Clin Nutr. 1997;65(6):1874–1881. doi: 10.1093/ajcn/65.6.1874. [DOI] [PubMed] [Google Scholar]
- Spector AA. Essentiality of fatty acids. Lipids. 1999;34:S1–S4. doi: 10.1007/BF02562220. [DOI] [PubMed] [Google Scholar]
- Sprecher H. Metabolism of highly unsaturated n-3 and n-6 fatty acids. Biochimica et Biophysica Acta. 2000;1486:219–231. doi: 10.1016/s1388-1981(00)00077-9. [DOI] [PubMed] [Google Scholar]
- Srivastava AKap SK. Potential mechanism(s) involved in the reglation of glycogen synthesis by insulin. Mol Cell Biochem. 1998;182:135–141. [PubMed] [Google Scholar]
- Standaert ML, et al. Insulin-induced activation of atypical protein kinase C, but not protein kinase B, is maintained in diabetic (ob/ob and Goto-Kakazaki) liver. Contrasting insulin signaling patterns in liver versus muscle define phenotypes of type 2 diabetic and high fati-nduced insulin-resistant states. J Biol Chem. 2004;279(24):24929–24934. doi: 10.1074/jbc.M402440200. [DOI] [PubMed] [Google Scholar]
- Steerenberg PA, et al. Long-term effect of fish oil diet on basal and stimulated plasma glucose and insulin levels in ob/ob mice. Diabetes Nutr Metab. 2002;15(4):205–214. [PubMed] [Google Scholar]
- Stoeckman AK, Ma L, Towle HC. Mlx is the functional heteromeric partner of the carbohydrate response element-binding protein in glucose regulation of lipogenic enzyme genes. J Biol Chem. 2004;279(15):15662–15669. doi: 10.1074/jbc.M311301200. [DOI] [PubMed] [Google Scholar]
- Stulnig TM, et al. Polyunsaturated eicosapentaenoic acid displaces proteins from membrane rafts by altering raft lipid composition. J Biol Chem. 2001;276(40):37335–373340. doi: 10.1074/jbc.M106193200. [DOI] [PubMed] [Google Scholar]
- Sundqvist A, Bengoechea-Alonso MT, Ye X, Lukiyanchuk Jin J, Harper JW, Ericsson J. Control of lipid metabolism by phosphorylation-dependent degradation of the SREBP family of transcription factors by SCGFBW7. Cell Metabolism. 2005;1:379–391. doi: 10.1016/j.cmet.2005.04.010. [DOI] [PubMed] [Google Scholar]
- Switzer KC, Fan YY, Wang N, McMurray DN, Chapkin RS. Dietary n-3 polyunsaturated fatty acids promote activation-induced cell death in Th1-polarized murine CD4+ T-cells. J Lipid Res. 2004;45(8):1482–1492. doi: 10.1194/jlr.M400028-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talukdar I, Szeszel-Fedorowicz W, Salati LM. Arachidonic acid inhibits the insulin induction of glucose-6-phosphate dehydrogenase via p38 MAP kinase. J Biol Chem. 2005;280(49):40660–40667. doi: 10.1074/jbc.M505531200. [DOI] [PubMed] [Google Scholar]
- Terstappen GC, Gaviraghi G, Caricasole A. The Wnt signaling pathway as a target for the treatment of neurodegenerative disorders. IDrugs. 2006;9(1):35–38. [PubMed] [Google Scholar]
- Towle HC. Glucose as a regulator of eukaryotic gene transcription. Trends Endocrinol Metab. 2005;16(10):489–494. doi: 10.1016/j.tem.2005.10.003. [DOI] [PubMed] [Google Scholar]
- Uyeda KaR JJ. Carbohydrate response element binding protein, ChREBP, a transcription factor coupling hepatic carbohydrate utilization and lipid synthesis. Cell Metabolism. 2006;4:107–110. doi: 10.1016/j.cmet.2006.06.008. [DOI] [PubMed] [Google Scholar]
- Wang H, Storlien LH, Huang XF. Effects of dietary fat types on body fatness, leptin, and ARC leptin receptor, NPY, and AgRP mRNA expression. Am J Physiol Endocrinol Metab. 2002;282(6):E1352–E1359. doi: 10.1152/ajpendo.00230.2001. [DOI] [PubMed] [Google Scholar]
- Wang PY, Liu P, Wang J, Sontag E, Anderson RG. A cholesterol-regulated PP2A/HePtP complex with dual specificity Erk1/2 phosphatase activity. EMBO J. 2003;22:2258–2267. doi: 10.1093/emboj/cdg255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Botolin D, Christian B, Busik C, Xu J, Jump DB. Tissue-specific, nutritional and developmental regulation of rat fatty acid elongases. J Lipid Res. 2005;46:706–715. doi: 10.1194/jlr.M400335-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Botolin D, Xu Jinghua, Christian B, Mitchell E, Jayaprakasam B, Nair M, Peters JM, Busik J, Olson LK, Jump DB. Regulation of hepatic fatty acid elongase and desaturase expression in diabetes and obesity. J. Lipid. Res. 2006;47:2028–2041. doi: 10.1194/jlr.M600177-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsh GI, Wilson C, Proud CG. GSK3: a Shaggy frog story. Trends in Cell Biology. 1996;6:274–279. doi: 10.1016/0962-8924(96)10023-4. [DOI] [PubMed] [Google Scholar]
- Winzell MS, Pacini G, Ahren B. Insulin secretion after dietary supplementation with conjugated linoleic acids and n-3 polyunsaturated fatty acids in normal and insulin-resistant mice. Am J Physiol Endocrinol Metab. 2006;290(2):E347–E354. doi: 10.1152/ajpendo.00163.2005. [DOI] [PubMed] [Google Scholar]
- Wisely GB, et al. Hepatocyte nuclear factor 4 is a transcription factor that constitutively binds fatty acids. Structure (Camb) 2002;10(9):1225–1234. doi: 10.1016/s0969-2126(02)00829-8. [DOI] [PubMed] [Google Scholar]
- Wolfrum C, Borrmann CM, Borchers T, Spener F. Fatty acids and hypolipidemic drugs regulate peroxisome proliferator-activated receptors alpha- and gamma-mediated gene transcription via liver fatty acid binding protein: a signaling path to the nucleus. Proc. Natl. Acad. Sci. U.S.A. 2001;98:2323–2328. doi: 10.1073/pnas.051619898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worgall TS, Sturley SL, Seo T, Osborne TF, Deckelbaum RJ. Polyunsaturated fatty acids decrease expression of promoters with sterol regulatory elements by decreasing levels of mature sterol regulatory element-binding protein. J Biol Chem. 1998;273(40):25537–25540. doi: 10.1074/jbc.273.40.25537. [DOI] [PubMed] [Google Scholar]
- Wu M, Harvey KA, Ruzmetov N, Welch ZR, Sech L, Jackson K, Stillwell W, Zaloga GP, Siddiqui RA. Omega 3-PUFA attenuate breast cancer cell growth through activation of neutral sphingomyelinase-mediated pathway. Int. J. Cancer. 2005;117:340–348. doi: 10.1002/ijc.21238. [DOI] [PubMed] [Google Scholar]
- Xu HE, et al. Molecular recognition of fatty acids by peroxisome proliferator-activated receptors. Mol Cell. 1999;3(3):397–403. doi: 10.1016/s1097-2765(00)80467-0. [DOI] [PubMed] [Google Scholar]
- Xu J, Christian B, Jump DB. Regulation of rat hepatic L-pyruvate kinase promoter composition and activity by glucose, N-3 PUFA and peroxisome proliferator activated receptor-alpha agonist. J Biol Chem. 2006;281:18351–18362. doi: 10.1074/jbc.M601277200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Ji S, Venable DY, Franklin JL, Messina JL. Prolonged insulin treatment inhibits GH signaling via STAT3 and STAT1. J Endocrinol. 2005;184(3):481–492. doi: 10.1677/joe.1.05977. [DOI] [PubMed] [Google Scholar]
- Xu J, Nakamura MT, Cho HP, Clarke SD. Sterol regulatory element binding protein-1 expression is suppressed by dietary polyunsaturated fatty acids. A mechanism for the coordinate suppression of lipogenic genes by polyunsaturated fats. J Biol Chem. 1999;274(33):23577–23583. doi: 10.1074/jbc.274.33.23577. [DOI] [PubMed] [Google Scholar]
- Xu J, Teran-Garcia M, Park JH, Nakamura MT, Clarke SD. Polyunsaturated fatty acids suppress hepatic sterol regulatory element-binding protein-1 expression by accelerating transcript decay. J Biol Chem. 2001;276(13):9800–9801. doi: 10.1074/jbc.M008973200. [DOI] [PubMed] [Google Scholar]
- Yabe D, Komuro R, Liang G, Goldstein JL, Brown MS. Liver-specific mRNA for Insig-2 down-regulated by insulin: implications for fatty acid synthesis. Proc Natl Acad Sci U S A. 2003;100(6):3155–3160. doi: 10.1073/pnas.0130116100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yahagi N, et al. A crucial role of sterol regulatory element-binding protein-1 in the regulation of lipogenic gene expression by polyunsaturated fatty acids. J Biol Chem. 1999;274(50):35840–35844. doi: 10.1074/jbc.274.50.35840. [DOI] [PubMed] [Google Scholar]
- Yan W, Tai HH. Glycogen synthase kinase-3 phosphorylation, T-cell factor signaling activation, and cell morphology change following stimulation of thromboxane receptor alpha. J Pharmacol Exp Ther. 2006;317(1):267–274. doi: 10.1124/jpet.105.096826. [DOI] [PubMed] [Google Scholar]
- Youdim KA, Martin A, Joseph MA. Essential fatty acids and the brain: possible health implications. Int. J. Dev. Neurosci. 2000;18:383–399. doi: 10.1016/s0736-5748(00)00013-7. [DOI] [PubMed] [Google Scholar]