

Summary
CCA addition to the 3′ end is an essential step in tRNA maturation. High-resolution crystal structures of the CCA enzymes reveal primary enzyme contact with the tRNA minihelix domain, consisting of the acceptor stem and T stem-loop. RNA and DNA minihelices are efficient substrates for CCA addition in steady-state kinetics. However, in contrast to structural models and steady-state experiments, we show here by single turnover kinetics that minihelices are insufficient substrates for the E. coli CCA enzyme, but that only the full-length tRNA is kinetically competent. Even a nick in the full-length tRNA backbone in the T loop, or as far away from the minihelix domain as in the anticodon loop, prevents efficient CCA addition. These results suggest a kinetic quality control provided by the CCA enzyme to inspect the integrity of the tRNA molecule and to discriminate against nicked or damaged species from further maturation.
Keywords: kinetic quality control, tRNA surveillance, single turnover kinetics
Many tRNA molecules must acquire the CCA sequence at their 3′ end as an essential step in the maturation process.1 Also, tRNAs that are synthesized with the CCA sequence must be repaired in the event of CCA degradation by nucleases. The enzyme that catalyzes the synthesis and repair of the CCA sequence is ATP(CTP):tRNA nucleotidyl transferase (the CCA enzyme), which is present in all three domains of life and is essential for growth in organisms that lack the CCA sequence in their tRNA genes.2 According to sequence motifs in the catalytic domains, the CCA enzymes are divided into two classes, where the archaeal enzymes are assigned to class I of the nucleotidyl transferase superfamily, while the bacterial and eukaryotic enzymes are assigned to class II.3 Both classes of CCA enzymes use a single catalytic site to perform sequential nucleotide addition without copying a nucleic acid template, 4; 5; 6; 7; 8; 9; 10; 11 in contrast to template-dependent polynucleotide polymerases. The generally accepted model of the template-independent CCA addition assumes that both classes of CCA enzymes only bind to the acceptor-T stem loop of tRNA (the minihelix domain) for recognition.6 This model is based on co-crystal structures of the class I Archaeoglobus fulgidus CCA enzyme (AfCCA) bound to a full-length tRNA (Fig.1A) and to minihelices.9; 11 In these structures, the anticodon stem-loop (ASL) domain is projected outward while the minihelix domains are in superimposable positions among various substrates and are held in fixed location by the enzyme. Consistent with footprint and biochemical analyses,12; 13; 14 these structures reveal that the 3′ end of the minihelix domain refolds at each step of nucleotide addition to place the terminus at the active site. The minihelix domain is accommodated in an extended class I cleft that has a counterpart in class II enzymes with similar shape, charge, and dimensions.6; 9 Indeed, tRNA minihelices are efficient substrates for both classes of CCA enzymes in steady-state kinetics.15; 16; 17 However, we show here by fast kinetics of the class II E. coli CCA enzyme (EcCCA) that the minihelix domain is insufficient to account for the kinetics of CCA addition, but that the full-length tRNA is necessary. Even a backbone break in ASL, distal from the acceptor end, causes a delay in CCA addition. These results suggest the possibility that the CCA addition serves to kinetically discriminate against nicked or damaged tRNA in the quality control of tRNA maturation.
Figure 1.
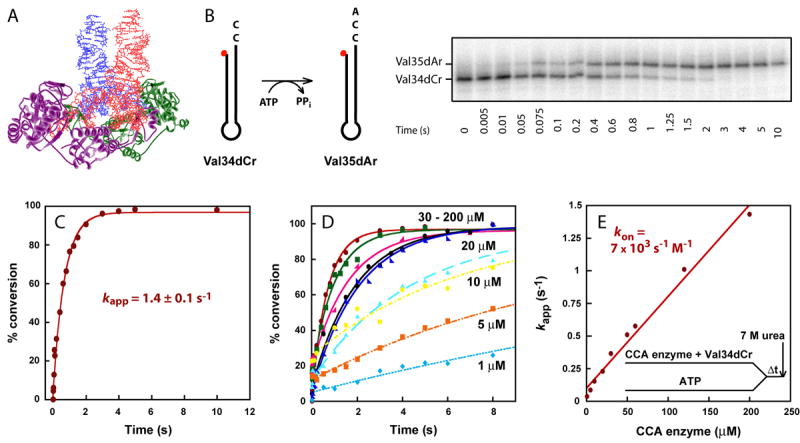
(A) Crystal structure of the dimer of AfCCA-tRNA complex (PDB 1SZ1). The tRNA backbone is highlighted in red and blue, whereas AfCCA subunits are represented by purple and green ribbons. (B) A schematic drawing of the Val34dCr minihelix substrate for A76 addition, using ATP as the nucleotide donor. The minihelix is 5′-labeled with 32P (50 nM), indicated by a red dot, and the time course of conversion to Val35dCr catalyzed by EcCCA (200 μM) is analyzed by 12% PAGE/7M urea. The nomenclature of minihelices is established by the identity of the tRNA (Val), the length of the minihelices (from a 34- to 35-mer), the DNA backbone (d), and the terminal ribose residue (from Cr to Ar). (C) Fitting the data of the time course in (B) to a single exponential equation to derive the kapp = 1.4 ± 0.1 s-1. (D) Representative time courses of A76 addition to Val34dCr by increasing concentration of EcCCA (1-200 μM, as shown). Reactions were performed by pre-incubating EcCCA with the minihelix substrate, followed by rapid mixing with ATP (1 mM) over various time periods, then terminated by mixing with 7 M urea as shown in (E). (E) The linear increase of kapp as a function of EcCCA concentration based on data of (D), giving an estimated kon of 7 × 103 s-1M-1.
Fast kinetics provides insights for understanding enzyme mechanisms that are unattainable from steady-state kinetics. The steady-state kcat and Km parameters are complex terms of all of the reactions occurring on the enzyme, such that individual steps are buried within these terms and are not resolvable. We addressed the limitations of steady-state kinetics by developing an assay for measuring transient kinetics of CCA addition, treating the enzyme as a stoichiometric reactant relative to the substrate to allow for the isolation of specific rate and equilibrium constants. Because both the enzyme and substrate were examined at μM concentrations, similar to those in cell physiology, the information obtained by transient kinetics is biologically relevant.
Results
Kinetics of Minihelices
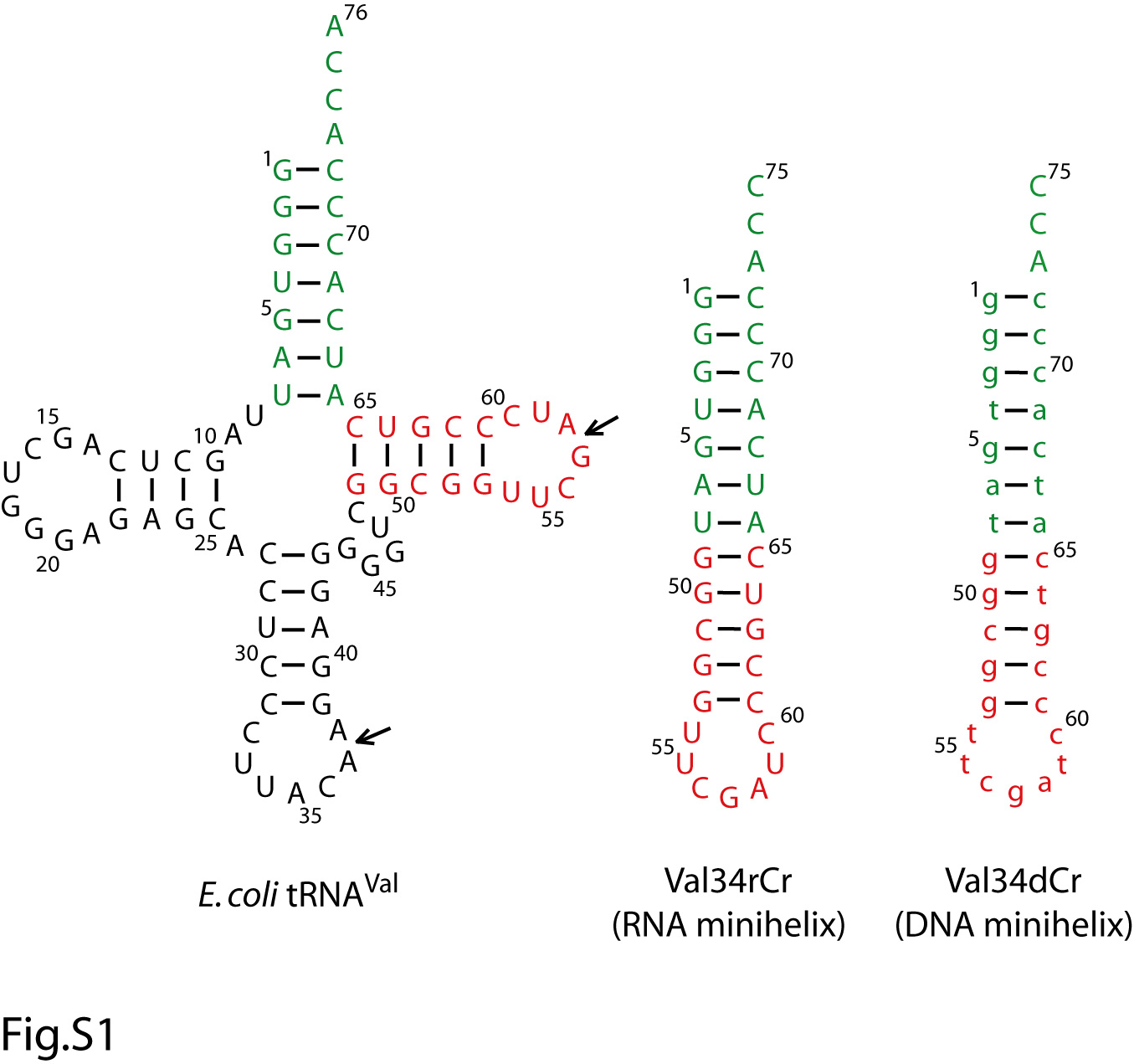
Extensive steady-state kinetics of minihelices has been performed with the class II EcCCA. These studies support the minihelix recognition model, showing that the dihydrouridine and ASL regions are dispensable, and that the only ribose requirement for nucleotide addition is the last one in the minihelix domain.15 For example, a 34-mer DNA minihelix based on the sequence of E. coli tRNAVal that terminates with C75 is an efficient substrate for A76 addition, exhibiting kcat of 1.3 min-1 similar to the kcat of 1.1 min-1 of a reconstituted full-length tRNA.16 This DNA minihelix, designated as Val34dCr (Fig.S1), was chemically synthesized with a deoxyribose backbone up to the 1-72 base pair and a ribose backbone at positions 73-75. Similarly, DNA minihelices Val33dCr and Val32dAr that terminate with C74 and A73, respectively, are functional substrates for C75 and C74 addition 15, although in these cases oligoC synthesis is observed when CTP is present (without ATP) as the only nucleotide triphosphate.16; 17; 18
To extend steady-state studies of EcCCA, single turnover kinetics was developed with minihelix substrates. The reaction of A76 addition to the DNA minihelix Val34dCr was performed on a rapid chemical quench instrument and was monitored by following conversion of the 5′-32P-labeled minihelix to the Val35dAr product. EcCCA was maintained in molar excess of the substrate to ensure only one turnover, while ATP was present at a saturating concentration (1 mM) relative to its Km (3 μM).17 Further increase in the ATP concentration did not increase the reaction rate. The labeled substrate and product minihelices were separated by denaturing polyacrylamide gel electrophoresis and quantified by image analysis (Fig.1B). The product conversion was nearly 100% and the time course of the conversion was well fit to a single exponential equation that describes the nature of the transient kinetics, permitting determination of the apparent rate constant kapp (Fig.1C). A set of controls was performed to vary systematically the reaction pH, and concentrations of Mg+2 and NaCl, to establish the condition that yielded the highest rate. It was also determined that quenching the reaction by 7 M urea (resulting in 5.6 M final concentration), as used in our experiments, is as effective in quenching as 500 mM EDTA.
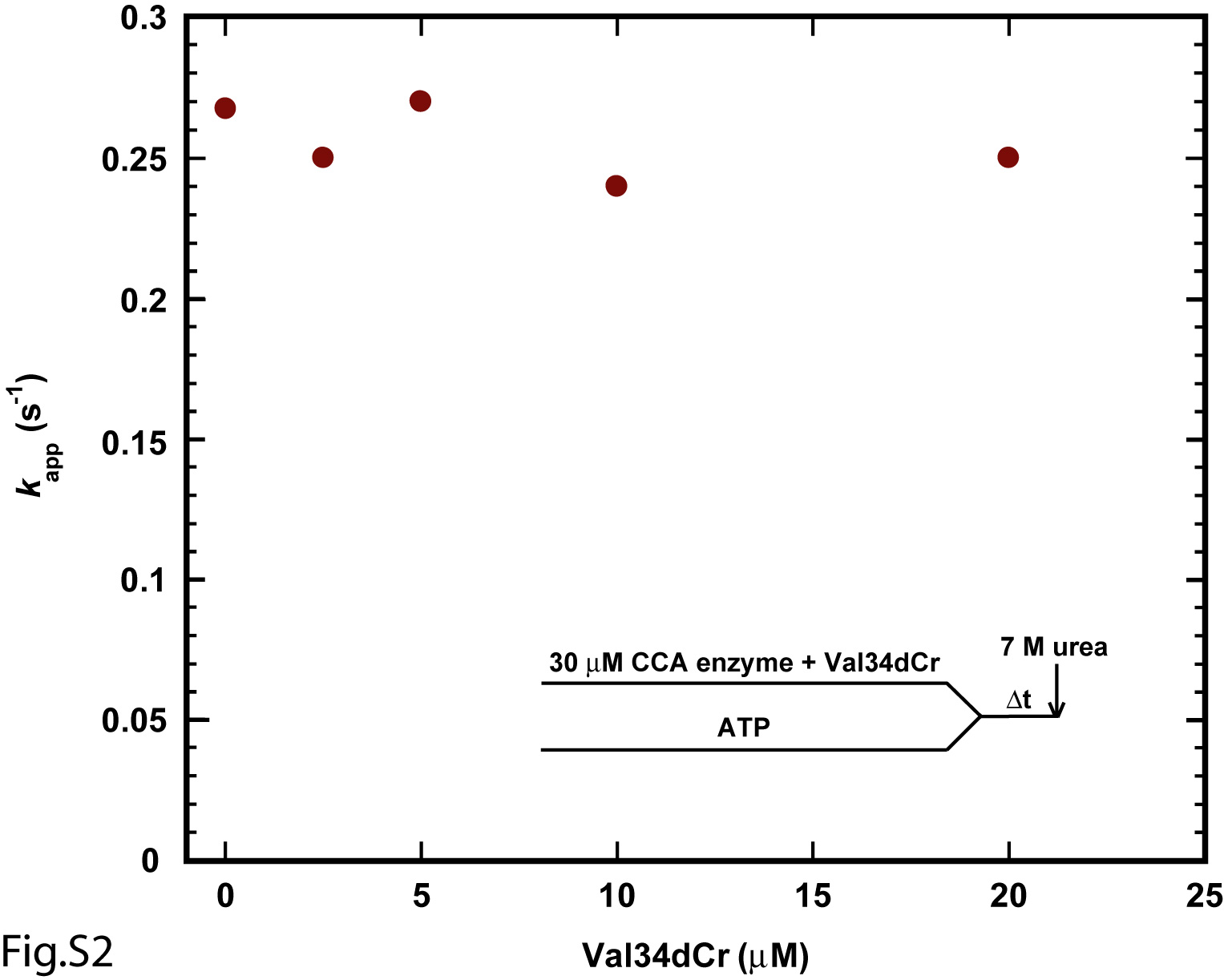
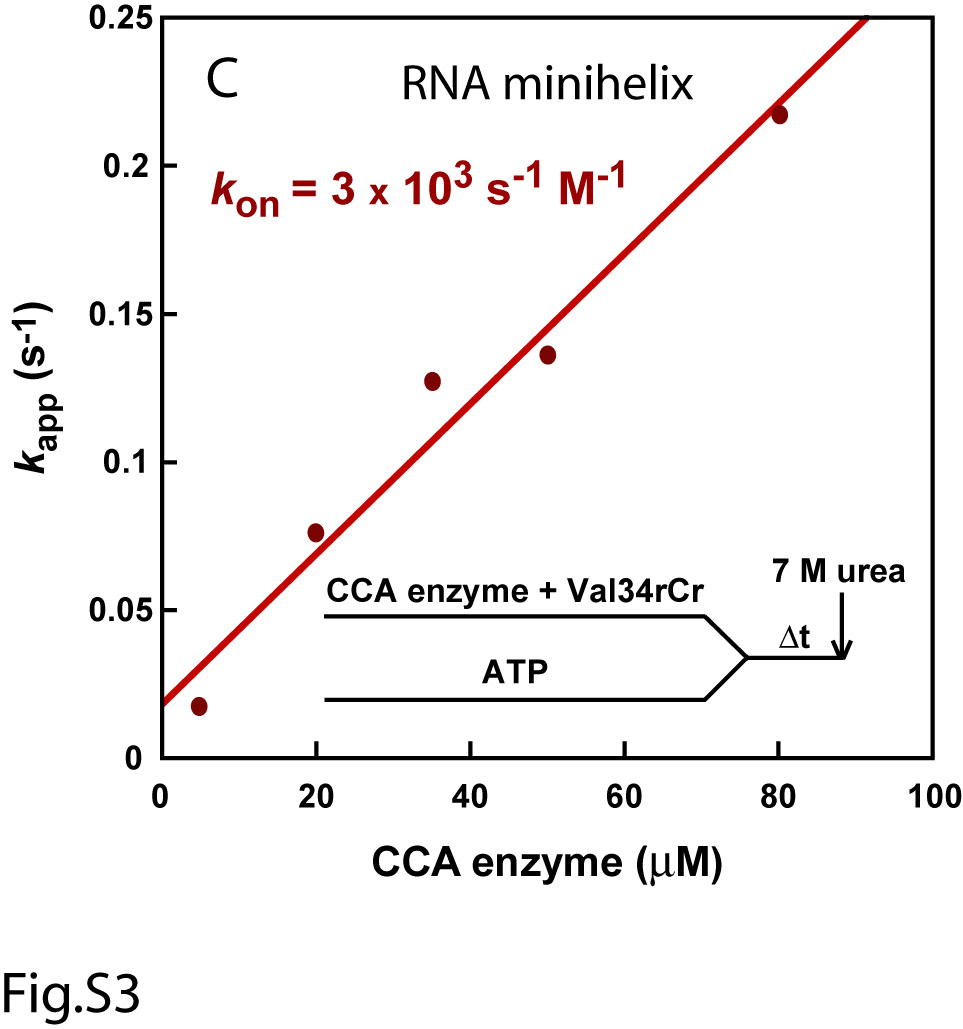
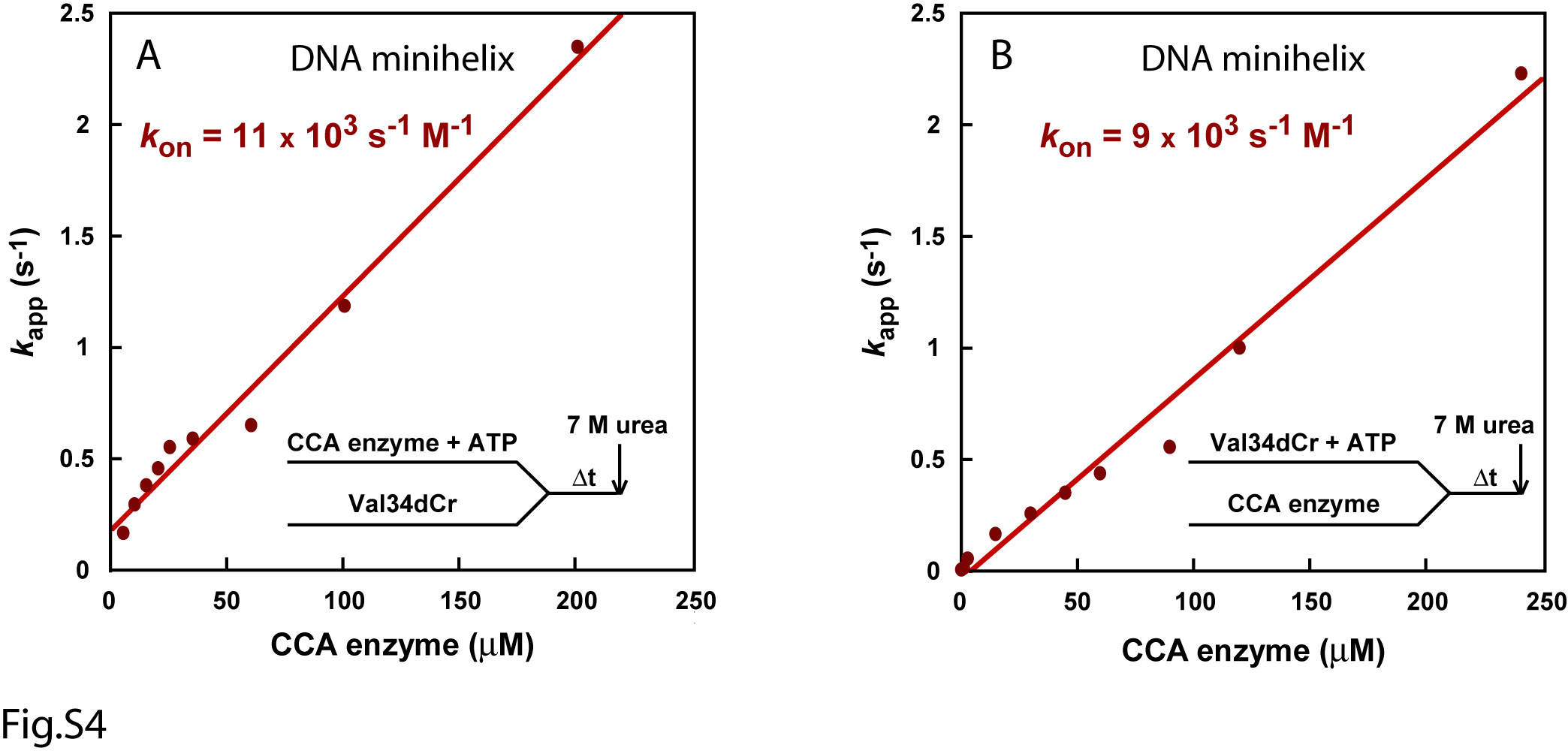
In single turnover kinetics with enzyme in excess of the RNA substrate, the rate of nucleotide addition was found to depend on concentration of the enzyme, but not the RNA substrate. For example, pre-incubation of EcCCA with Val34dCr, followed by rapid mixing with ATP, yielded time courses that increased in kapp with increasing concentrations of the enzyme (Fig.1D). In contrast, fitting the time course of the reaction to a single exponential equation (%product = A×(1 − e−kapp×t)) over a range of different Val34dCr substrate concentrations (0.01-20 μM) in the presence of excess of enzyme did not change the first-order rate constant kapp (Fig.S2), while the initial rate (V = kapp [ES]) of the reaction increased with increasing substrate concentrations. However, despite the dependence of kapp on EcCCA concentration, kapp increased linearly as a function of enzyme concentration up to as high as 200 μM (Fig.1E). The linear kappvs. concentration plot suggests that the rate of formation of the productive enzyme-substrate complex was unfavorable, and that the conditions were far away from saturation even at the highest attainable enzyme concentration. Thus, the likely interpretation of the linear plot of kappvs. concentration is that the reaction rate is dependent on the rate of enzyme-substrate association. Under these circumstances, the slope of the plot gives an approximation of the enzyme-substrate association rate (kon). This kon was determined to be 7 × 103 s-1M-1 (Fig.1E), which was indeed significantly slower than the diffusion-controlled limit of 107-108 s-1M-1. The y intercept gives the koff of the enzyme-substrate complex, which was determined to be 0.1 s-1. The overall Kd of the enzyme-substrate complex was calculated from koff/kon, which yielded 14 μM, indicating a poor affinity. The slow kon of the enzyme-minihelix association did not improve upon conversion of the DNA backbone of Val34dCr to the RNA backbone of Val34rCr (Fig.S3). Also, alteration of the order of premixing reactants did not improve the kinetics. For example, pre-incubation of EcCCA with ATP, followed by rapid mixing with Val34dCr, yielded a linear kappvs. concentration plot with an estimated kon of 11 × 103 s-1M-1, koff of 0.2 s-1, and Kd of 18 μM (Fig.S4A). Similarly, pre-incubation of Val34dCr with ATP, followed by rapid mixing with the enzyme, also yielded a linear kappvs. concentration plot with an estimated kon of 9 × 103 s-1M-1 (Fig.S4B). Here, the koff cannot be determined with precision to calculate Kd.
Kinetics of full-length tRNAs
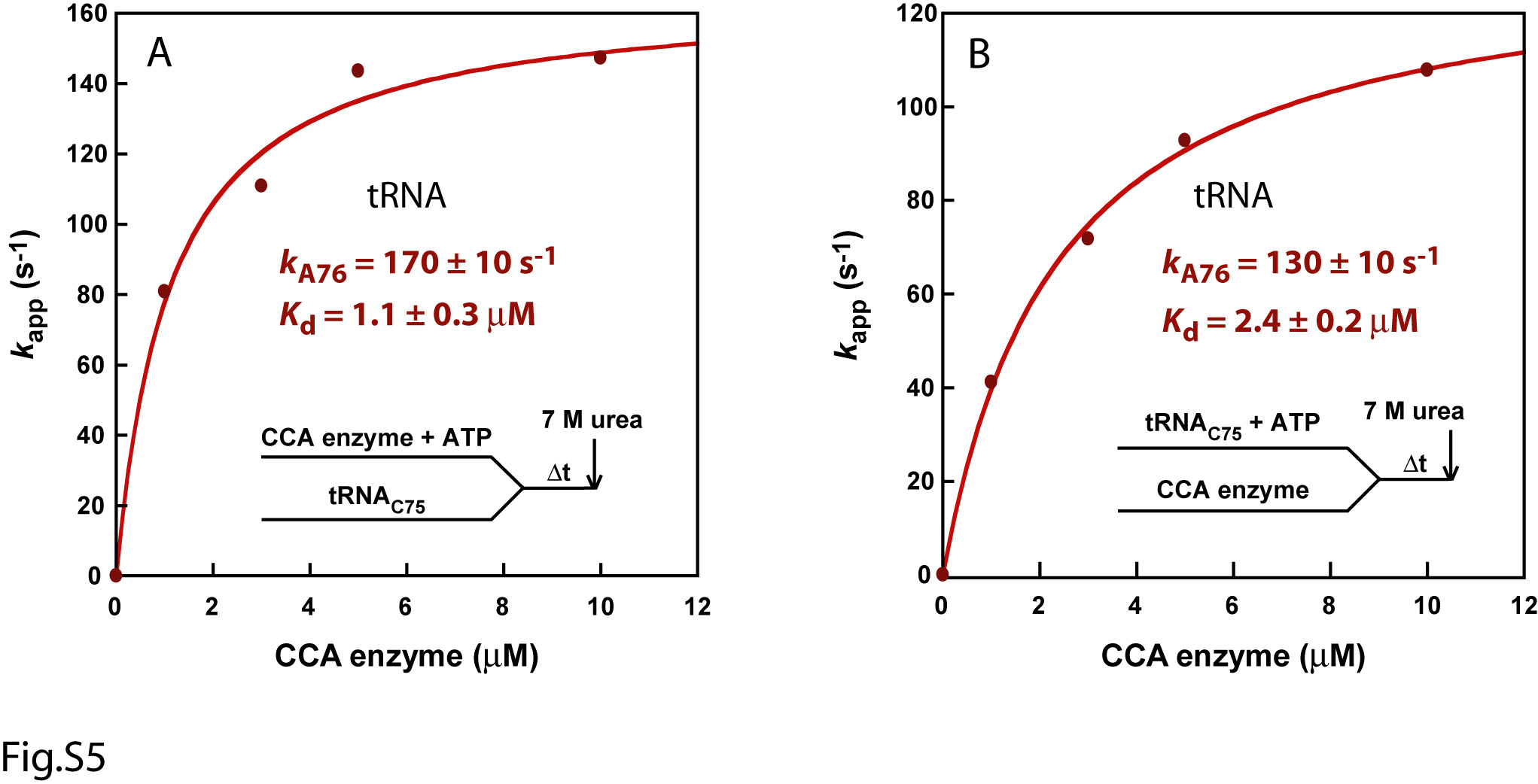
The above results show that, although the minihelix domain was competent for steady-state kinetics, it is inadequate for fast kinetics. To eliminate the possibility that the observed deficiency with minihelix substrates was due to defects caused by the applied reaction conditions, the full-length tRNAC75 substrate was tested using the same reaction conditions. The full-length tRNAC75 substrate was synthesized and labeled by run-off transcription (Fig.2A). Formation of the enzyme-tRNAC75 complex, followed by rapid mixing with ATP, yielded time courses that were well fit to a single-exponential equation (Fig.2B). Because the time courses showed no lags or delays, and because the plot of kappvs. EcCCA concentration showed saturation (Fig.2C), this suggested rapid equilibrium binding between EcCCA, ATP, and the tRNA substrate, which is in contrast with the kinetics of minihelices. Fitting the plot of kappvs EcCCA concentration to a hyperbolic equation revealed the saturating kapp at 160 ± 10 s-1, which is defined as the rate constant kA76 for A76 addition. This kA76 is a composite term that includes all of the events up to and including nucleotidyl transfer. The curve fitting also revealed the kinetic Kd of 0.8 ± 0.2 μM for the affinity of EcCCA with tRNAC75 (Fig.2C). Here the kinetic Kd is the Km of the single turnover reaction (Km(sto)). In rapid equilibrium condition, the thermodynamics of binding controls the proportion of the tRNA substrate actually bound to the enzyme and thus the reaction rate kapp. The kinetic Kd (or Km(sto)) includes the possibility that a rearrangement of the initial enzyme-NTP-tRNA complex may occur to form the productive enzyme-substrate interaction. Similar results were obtained by altering the premixing conditions. For example, premixing EcCCA with ATP, followed by rapid mixing with tRNAC75, showed saturation kinetics with kA76 = 170 ± 10 s-1 and kinetic Kd = 1.1 ± 0.3 μM (Fig.S5A). Premixing tRNAC75 with ATP, followed by rapid mixing with EcCCA, showed saturation kinetics with kA76 = 130 ± 10 s-1 and kinetic Kd = 2.4 ± 0.2 μM (Fig.S5B).
Figure 2.
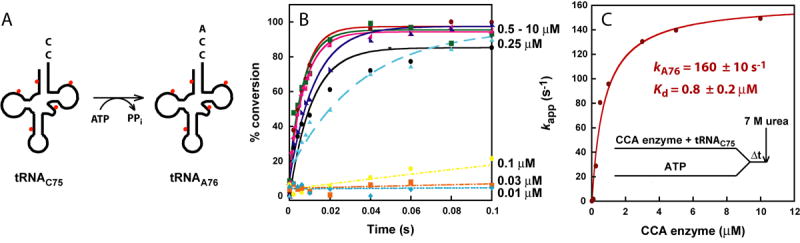
(A) A schematic drawing of the tRNA75C substrate for A76 addition, using ATP as the nucleotide donor. The tRNA is labeled during transcription by incorporation of [α-32P]AMP, indicated by red dots. (B) Representative time courses of A76 addition to tRNA75C by increasing concentration of EcCCA (0.01-10 μM, as shown). Reactions were performed by pre-incubating EcCCA with tRNA75C (maintained at 1/10 below the concentration of EcCCA), followed by rapid mixing with ATP (1 mM) over various time periods, then terminated by mixing with 7 M urea as shown in (C). (C) Fitting the data of (B) to a hyperbolic equation to derive kA76 and Kd for tRNAC75.
The contrast between the full-length tRNA75C and minihelices in fast kinetics emphasized that the tRNA whole body is necessary for high affinity and rapid productive binding to the enzyme. Moreover, the kA76 value in the range of 130-170 s-1 for tRNAC75 is comparable to the kpol value of 220 s-1 of the template-dependent T7 RNA polymerase,19 indicating that the rate-determining steps of these enzymes take place in similar time scales. The kinetic Kd of 0.8-2.4 μM for the enzyme affinity to tRNAC75 is in the range of the physiological concentration of tRNA precursors, estimated at ∼2 μM.3
Characterization of nicked tRNAs
An earlier study showed that EcCCA has a phosphohydrolase activity that repairs aberrant tRNA 3′ ends before nucleotide addition.20 The ability of EcCCA to discriminate against minihelices suggested the further possibility that the enzyme might inspect the tRNA whole body to reject nicks in the tRNA phosphodiester backbone. To test this possibility, two nicked substrates were prepared (Fig.S1). One was formed by hybridization of the G1-G57 fragment of E. coli tRNAVal with the chemically synthesized A58-C75 fragment to produce a nick in the T loop of the minihelix domain. Such a T-nicked substrate has been shown to be functional for recognition by EcCCA in steady-state kinetics.16 Also, a similar approach was used earlier to create a T-nicked substrate in the sequence framework of E. coli tRNAPro, which was shown to be functional for aminoacylation by E. coli prolyl-tRNA synthetase,21 an enzyme that recognizes the tRNA global structure.22 The other nicked substrate of E. coli tRNAVal was formed by hybridization of the G1-A37 fragment of the tRNA with the G38-C75 fragment to produce a nick in the ASL. In this case, the G38-C75 fragment replaced the natural A38 with G38 to allow efficient transcription in vitro. A similar ASL-nicked tRNA in the sequence framework of E. coli tRNAGln has been previously constructed, where the backbone nick was readily sealed by T4 RNA ligase to generate an intact tRNA that is recognized by E. coli glutaminyl-tRNA synthetase,23 which also recognizes the tRNA global structure for aminoacylation.
In preparation of the two nicked substrates of E. coli tRNAVal, the 3′ fragment was labeled with 32P to allow analysis of hybridization by gel electrophoresis in native conditions containing 10 mM MgCl2. Hybridization analysis revealed two products in the generation of the T-nicked substrate (Fig.3A, left), and one major and one minor product in the generation of the ASL-nicked substrate (Fig.3A, right). In both cases, the major product (indicated by red arrows) exhibited the same gel mobility as that of the intact and full-length tRNA and was isolated from the native gel and used as the nicked substrate, whereas the minor product (indicated by black arrows) exhibited aberrant gel mobility and was not used. Both the gel purified T- and ASL-nicked substrates were determined for their concentrations by UV absorption.
Figure 3.
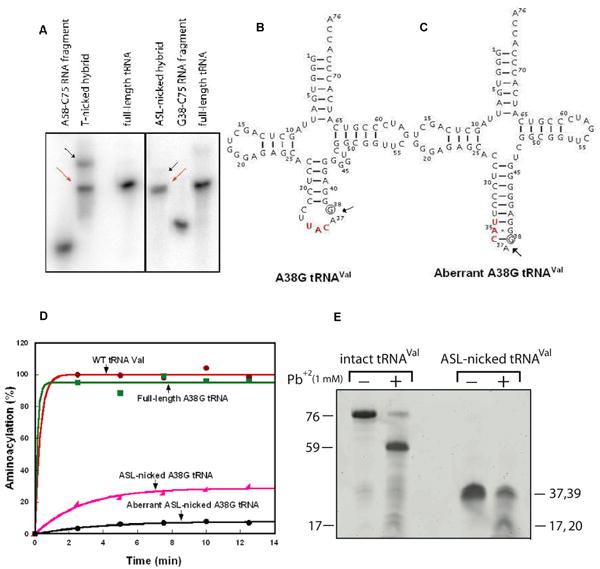
Characterization of nicked tRNAs. (A) Formation of the T-nicked (left) and ASL-nicked (right) tRNAC75 substrates by hybridization of an unlabeled 5′-fragment with a 32P-labeled 3′ fragment. Full-length and intact 32P-tRNAC75 was run as a marker. The nicked substrates (indicated by the red arrows) that co-migrated with the intact tRNA marker were isolated from gels and used for analysis of CCA addition, while the aberrant hybridization products that migrated slower than the intact tRNA were marked by black arrows and were not further analyzed for CCA addition. (B) Sequence and cloverleaf structure of the A38G mutant of E. coli tRNAVal, where the A38G substitution is indicated by a circle. (C) The aberrant structure of the ASL-nicked A38G tRNAVal. (D) Aminoacylation of tRNAVal transcripts (2 μM) by purified E. coli ValRS (4 μM). (E) Lead cleavage of intact A38G tRNA and the ASL-nicked A38G tRNA analyzed by a 12% denaturing PAGE. The numbers on the sides of the gel indicate the lengths of tRNA or fragments.
Because the ASL-nicked substrate has the A38G substitution (Fig.3B), which can induce an aberrant folding (Fig.3C), this nicked substrate was characterized further. The following three experiments support the notion that the ASL-nicked substrate, as isolated from Fig.3A, existed in the proposed canonical structure (Fig.3B). First, the ASL-nicked substrate is readily ligated by T4 RNA ligase 2 (ref24) to 55% (data not shown). This high efficiency of ligation is not expected for the putative aberrant structure, which is unfavorable for ligation. Second, the ASL-nicked substrate is aminoacylated by E. coli ValRS with better efficiency than the minor species, which might represent the aberrant ASL-nicked tRNA (designated as such in Fig.3D). The rationale of this experiment was based on extensive structural and biochemical studies of the tRNAVal-ValRS interaction, which must depend on direct enzyme recognition of the tRNA anticodon nucleotides.25 In the canonical structure, the anticodon nucleotides are accessible for recognition, although the presence of the nick adjacent to the anticodon triplet is expected to reduce the aminoacylation activity relative to that of the intact tRNA. However, in the putative aberrant structure, the anticodon nucleotides are inaccessible, preventing recognition by ValRS. Importantly, aminoacylation analysis also showed that the intact A38G mutant retained the activity of the wild-type tRNA, indicating that the A38G substitution did not significantly alter the tRNA structure. Third, the ASL-nicked substrate was competent for lead cleavage (Fig.3E). The metal ion lead induces cleavage between nucleotide 17 and the conserved G18 in the canonical tRNA tertiary core.26 This cleavage is coordinated by precise positioning of lead in a pocket formed by conserved residues in the D and T loops.27 Substitutions in the tRNA tertiary core can eliminate lead cleavage.27 Notably, the intact A38G mutant of tRNAVal was a functional substrate for lead cleavage, generating a large fragment of 59-mer and a smaller fragment of 17-mer. These fragments were well resolved by a 12% denaturing PAGE (Fig.3E). The ASL-nicked A38G mutant dissociated in the denaturing PAGE into a mixture of two co-migrating fragments, a 39-mer and a 37-mer. Upon incubation of the ASL-nicked substrate with lead ions under native conditions, one of the co-migrating fragments (the 37-mer) was cleaved into 17- and 20-mer fragments, while the 39-mer fragment remained intact. This cleavage pattern is consistent with the presence of a prominent Pb+2 cleavage site in the D/T loop region, suggesting that the ASL-nicked tRNA basically adopts the canonical L-shaped tRNA fold. Together, the above results support the notion that the ASL-nicked tRNA adopts the normal tRNA structure that is recognized for ligation, for aminoacylation, and for lead cleavage.
Kinetics of nicked tRNAs
Single turnover kinetics of A76 addition to both the T-nicked and ASL-nicked substrates was measured by mixing a preformed EcCCA-substrate complex with ATP. Both nicked substrates were competent for CCA addition, with more than 90% of substrate converted to product at the end of the reaction. However, the T-nicked substrate exhibited slow kinetics, with kapps ∼1000-fold lower than those of the intact tRNAC75 at equivalent enzyme concentrations (Fig.4A). A plot of kappvs. enzyme concentration was linear up to 100 μM EcCCA with a slow kon of 7 × 103 s-1M-1 (Fig.4A, inset) in a pattern similar to that of minihelices. The ASL-nicked substrate also exhibited a slower rate, but the kinetics of kappvs. enzyme concentration showed saturation, permitting the determination of kA76 (170 ± 10 s-1), similar to that of the intact tRNA, but a 10-fold higher kinetic Kd (8.6 ± 1.3 μM) for enzyme-substrate association (Fig.4A). Thus, EcCCA discriminates against the ASL nick by reducing the affinity to the nicked substrate. Once stably bound to the nicked substrate, the enzyme catalyzes A76 addition at a rate identical to that of the intact tRNA.
Figure 4.
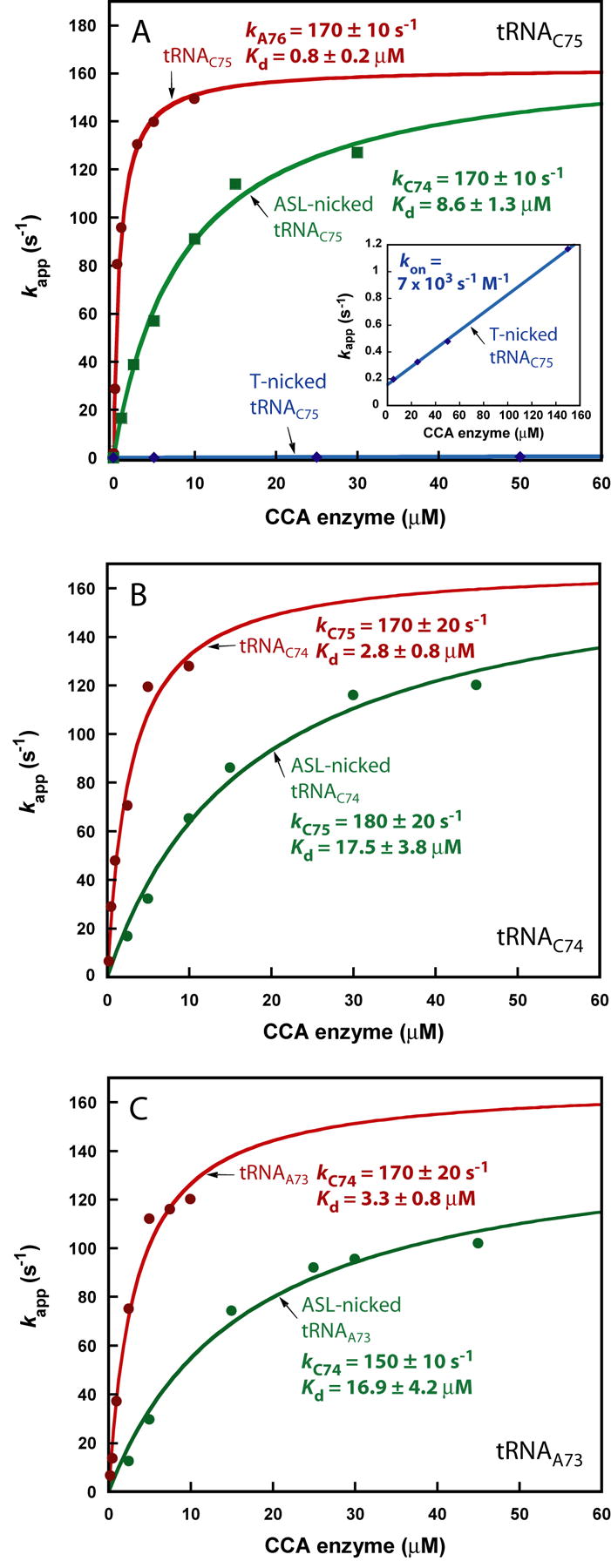
Kinetics of nicked tRNA. (A) Kinetics of A76 addition to the intact (red), ASL-nicked (green), and T-nicked (blue) tRNAC75 as a function of EcCCA concentration. Inset: an expanded view of the kappvs. EcCCA plot for the T-nicked tRNAC75. (B) Kinetics of C75 addition to the intact (red) and ASL-nicked (green) tRNAC74 as a function of EcCCA concentration. (C) Kinetics of C74 addition to the intact (red) and ASL-nicked (green) tRNAA73 as a function of EcCCA concentration. Single turnover rates were monitored by incubating a tRNA substrate with EcCCA, followed by rapid mixing with the correct nucleotide donor.
The kinetic discrimination of EcCCA against minihelices and nicked tRNA substrates was further demonstrated for C75 and C74 addition. C75 addition to the minihelix Val33dCr, performed in the presence of a saturating concentration of CTP (5 mM), produced oligoC (Fig.S6A,B), consistent with previous observations.17; 18 In this case, the rate of C75 addition based on summing all the products of oligoC synthesis was the same as the rate based on substrate consumption. However, measurement of kapp as a function of EcCCA concentration showed a linear increase of the rate with a rather slow kon of 250 s-1M-1 (Fig.S6C,D). In contrast, C75 addition to the full-length tRNAC74 manifested two major improvements. First, the addition was precisely one nucleotide without synthesis of oligoC (Fig.S7A,B), indicating that the presence of the tRNA whole body strengthened the specificity of the enzyme. Second, the kinetics of C75 addition was saturable (Fig.S7C,D) with kC75 = 170 ± 20 s-1 and kinetic Kd = 2.8 ± 0.8 μM of the tRNAC74 substrate. Furthermore, introduction of a backbone nick between positions 37-38 in the ASL increased the kinetic Kd to 17.5 ± 3.8 μM but had little effect on kC75 (180 ± 20 s-1) (Fig.4B), indicating discrimination at the binding step. These observations were recapitulated for C74 addition. In the presence of CTP only, the Val32dAr minihelix was a substrate for both C74 addition and oligoC synthesis (Fig.S8A,B), and the kinetics of nucleotide addition revealed a linear increase of kapp as a function of enzyme concentration, exhibiting an even slower kon of 25 s-1M-1 (Fig.S8C,D) than that for C75 addition. In contrast, the full-length tRNAA73 was an efficient substrate for C74 addition, which was rapidly followed by C75 addition (Fig.S9A,B). Kinetic analysis revealed a saturating kC74 of 170 ± 20 s-1 and a kinetic Kd of 3.3 ± 0.8 μM for the tRNAA73 substrate (Fig.S9C,D). Also, introduction of a backbone nick at positions 37-38 raised the kinetic Kd to 16.9 ± 4.2 μM, but had only a small effect on kC74 (150 ± 10 s-1) (Fig.4C).
Discussion
The kinetic parameters of A76, C75, and C74 addition to the full-length and ASL-nicked tRNAVal substrates by EcCCA are summarized in Table 1. With the full-length tRNA, the enzyme catalyzes rapid nucleotide addition with a similar rate at all three steps and has a physiologically relevant high affinity. However, introduction of a backbone break to the ASL, the domain that is separate from the CCA end by the longest distance (∼75 Å) in tRNA, reduces the enzyme affinity to the nicked-substrate, suggesting that the proper activity of the minihelix domain for CCA addition is dependent on the integrity of the ASL. The overall discrimination against the nick for each nucleotide addition is manifested by the ratio of the specificity factor kN/Kd for the intact tRNA over the factor for the nicked substrate. This discrimination increases from 5.7-fold for C74 addition, to 6.1-fold for C75 addition, to 10.6-fold for A76 addition, indicating a progressively closer scrutiny of the tRNA substrate as the enzyme approaches completion of CCA synthesis. The discrimination against the T-nicked substrate is even stronger, because reactions did not reach saturation even at high enzyme concentrations (∼100 μM). This is rationalized by the fact that the nick is localized in the miniehelix domain, which is in direct contact with the enzyme.
Table 1.
| Intact tRNA | ASL-nicked tRNA | Discrimination | |||||
|---|---|---|---|---|---|---|---|
| kN (s-1) | Kd (tRNA, μM) | kN/Kd (s-1μM-1) | kN (s-1) | Kd (tRNA, μM) | kN/Kd (s-1μM-1) | kN/Kd (intact)/kN/Kd (nicked) | |
| A76 | 170 ± 10 | 0.8 ± 0.2 | 212 | 170 ± 10 | 8.6 ± 1.3 | 20 | 10.6 |
| C75 | 170 ± 20 | 2.8 ± 0.8 | 61 | 180 ± 20 | 17.5 ± 3.8 | 10 | 6.1 |
| C74 | 170 ± 20 | 3.3 ± 0.8 | 51 | 150 ± 10 | 16.9 ± 4.2 | 8.9 | 5.7 |
The reported kinetic Kd (or Km(sto)) values represent the monomer concentration of EcCCA. In crystal structures of the class I AfCCA, the enzyme exists as a dimer and binds two molecules of tRNA or the minihelix domain.6; 7; 9; 11 In the crystal structure of the class II Bacillus stearothermophilus CCA enzyme, the enzyme also exists as a dimer and is modeled to bind two molecules of tRNA.5; 6
Values reported are based on experiments performed with pre-incubating EcCCA with a tRNA substrate in one syringe of the rapid quench instrument, followed by rapid mixing with NTP from the other syringe.
The discrimination against backbone nicks by EcCCA is likely shared in common by all class II CCA enzymes (including those of eukaryotes), given the similar properties of substrate recognition among members of this class.29 In the lifetime of tRNA biosynthesis, backbone nicks can be generated in a number of ways. For example, misincorporation of nucleotides, inappropriate processing and folding, or incomplete post-transcriptional modifications in tRNA30 can lead to aberrant tertiary structures that are prone to nuclease cleavage. Some bacterial and eukaryotic tRNAs are also subject to cleavage at the ASL by specific toxins.31; 32 In eukaryotes, the ASL is the site that carries introns for certain tRNA precursors (usually between positions 37-38 as designated in Fig.S1). Splicing intermediates without properly religated backbones that accumulate in cells have been identified.33 Discrimination of these nicked tRNAs from CCA addition offers a quality control mechanism that is manifested at the kinetic level. Based on the estimated cellular concentration of CCA enzyme at ∼0.8 μM [calculated from ref3], the kinetic quality control ensures that only full-length and intact tRNAs are subject to rapid CCA addition at a rate of ∼30, 40, and 80 s-1 for addition of C74, C75, and A76, respectively (e.g., ). As such, tRNAs with the CCA sequence can quickly continue on in the maturation process or proceed to reactions for protein synthesis, which in turn provide protein-binding partners (such as aminoacyl-tRNA synthetases, elongation factors, and the ribosome) that protect the CCA sequence. In contrast, the backbone-nicked tRNAs are rejected by the CCA enzyme and, without protein protection, are rapidly degraded by the various tRNA surveillance machineries that have been discovered in both E. coli and yeast.30; 34; 35
The synthesis and maintenance of the CCA sequence confers “eligibility” to a tRNA for participation in protein synthesis, even if the tRNA is still at an early stage in the maturation process (such as in the nucleus). Protein synthesis is a multi-step process that requires energy consumption for aminoacylation of a matured tRNA, for accommodation of the aminoacylated tRNA at the ribosome, and for translocation of the tRNA through the ribosome after peptide bond formation. Thus, in the overall maturation process of a tRNA, CCA addition represents an important checkpoint prior to becoming eligible for protein synthesis. The quality control of CCA addition at the checkpoint can ensure that only backbone-intact tRNAs remain in the active processing pathway as substrates for both the repair and de novo biosynthesis functions of the CCA enzyme. For example, in E. coli, where the CCA sequence is encoded in all tRNA genes, EcCCA functions to regenerate missing nucleotides from the CCA sequence that are removed by nucleases. In bacteria (such as Bacillus subtilis) that do not encode the CCA sequence in some of their tRNA genes, precursor tRNAs must be processed by endonucleases to remove 3′ trailer sequences,36 and the CCA sequence added de novo as a prerequisite for efficient processing of the 5′ end.37; 38 A similar situation exists in the eukaryotic nucleus, where the CCA sequence is a determinant for nuclear tRNA aminoacylation and for export of tRNA with correctly processed 5′ and 3′ ends.39; 40; 41
This study demonstrates that, although tRNA minihelices are competent in steady-state kinetics and appear to embody all of the binding interactions with both classes of CCA enzymes, they are not sufficient for fast CCA addition. The minihelix domain is thought to have evolved earlier than the anticodon domain to serve as a tag for the replication of primitive RNA genomes.42; 43 Indeed, minihelices that carry major determinants for various tRNA functions have been reported. For example, the minihelix domain is recognized by RNase P for processing 5′ leader sequences44, and it provides the primary contact site for recognition of aminoacyl-tRNA by bacterial elongation factor EF-Tu.45; 46 The minihelix of tRNAAla serves as the aminoacylation domain of the bacterial tmRNA molecule that tags proteins synthesized from damaged mRNAs to degradation.47 Importantly, the minihelix domain of tRNAAla contains the G3:U70 major determinant for aminoacylation with alanine.48; 49 Although the domain by itself retains the specificity of aminoacylation, its catalytic efficiency (kcat/Km) is reduced from that of the full-length tRNA.50 Upon analysis by single turnover kinetics, the minihelix domain is likely to be further reduced relative to that of the full-length tRNA. The inefficient interaction of the isolated minihelix domain with EcCCA, and perhaps with other enzymes as well, may arise from the fact that, although the isolated minihelix domain recapitulates the sequence of the coaxially stacked acceptor stem and T stem-loop, it lacks the support from interactions with the D loop. This could affect the conformation of the T loop and/or reduce the conformational rigidity of the acceptor-T stem module. Also, as seen in the crystal structure of yeast tRNAPhe (ref 9), the coaxial stacking of the acceptor stem and T stem-loop has a small angle of 14° (ref 28), which may not be properly maintained in the absence of the D and T loop interactions.
Our results here demonstrate that tRNA domains distal from the minihelix domain (such as ASL) are determinants for rapid activity of CCA addition. Similarly, a recent study demonstrates that, although the ASL of a tRNA provides all the information necessary for the anticodon-codon base pairing interaction on the ribosome, the region of the tRNA distal from the ASL, which involves the D stem of the tRNA tertiary core, influences the fidelity of the anticodon-codon base pairing.51 These studies, together with those mentioned above of minihelices, emphasize the notion that individual tRNA domains exhibit their biologically relevant functions only in the context of the entire tRNA molecule, even though these domains in isolation appear to possess the sequence and structural information required for function. This notion is likely to hold in the interaction of tRNA with other protein-binding partners, such as those required for tRNA modification and processing. In the context of the tRNA whole body, we further show that EcCCA effectively discriminates against backbone nicks or damage at each step of CCA addition, offering a previously unrecognized surveillance step before committing tRNA molecules to protein synthesis.
Materials and Methods
Preparation of EcCCA
A previous over-expression clone of wild-type E. coli cca gene was constructed by inserting the gene between the NdeI and BamH1 sites of pET-22b(+) (Novagen) and by changing the TGA stop codon to the CTA leucine codon.18 Site-directed mutagenesis by QuikChange (Stratagene) was performed on this clone to remove most of the C-terminal extension sequence, leaving only a glycine codon between the leucine codon of the gene and the His-tag. The modified plasmid was transformed into E. coli BL21(DE3) and enzyme expression was induced for 4 h with 0.3 mM IPTG at 37°C. The collected cells were sonicated and lysed in 20 mM Tris-HCl pH 8.0, 500 mM NaCl, 6 mM MgCl2, 5 mM imidazole, 3 mM β-mercaptoethanol, 5% glycerol). EcCCA was bound to the Co2+-chelated TALON resin (ClonTech) and eluted with 25-50 mM of imidazole. The fractions with the highest contents of EcCCA were pooled, concentrated, and exchanged into storage buffer of 100 mM glycine pH 9.0, 50 mM NaCl, 2 mM MgCl2, 1 mM β-mercaptoethanol, and 40% glycerol. Enzyme concentration was determined by Bradford assay.
Substrate preparation
DNA minihelices Val34dCr, Val33dCr, and Val32dAr, containing ribose moieties starting at position 73, and RNA minihelix Val34rCr were chemically synthesized by IDT (Coralville) and used directly. The full-length substrates of E. coli tRNAVal, tRNAC75, tRNAC74, and tRNAA73 were prepared by run-off transcription by T7 RNA polymerase based on template sequences synthesized from overlapping oligonucleotides. The full-length transcripts were separated from their templates and unincorporated NTPs by 12% denaturing PAGE/7M urea, localized by UV shadowing, and extracted from gels. The T-nicked substrate was prepared by annealing the G1-G57 fragment (transcribed by T7 RNA polymerase) with the A58-C75 RNA fragment (synthesized by IDT). The ASL-nicked substrate was prepared by annealing the G1-A37 fragment (transcribed by T7 RNA polymerase) with the G38-C75 RNA fragment (which contained the A38G substitution and was transcribed by T7 RNA polymerase). Both nicked substrates was formed by heating a mixture of the 5′ fragment in 2-fold molar excess of the 3′ fragment at 90°C for 3 minutes, followed by slow cooling to 37°C for 10 minutes. The annealed mixtures were separated by a non-denaturing 12% PAGE/90 mM Tris pH 8.0, 90 mM borate and 10 mM MgCl2 (TBM) gel, and the annealed hybrids that co-migrated with a full-length tRNA standard were extracted from gels and eluted in 10 mM Tris, pH 8.0. The nicked tRNA substrates were precipitated by ethanol and stored in 10 mM Tris-HCl, pH 8.0 and 1 mM MgCl2. Such nicked substrates migrated to the same electrophoretic position as the intact full-length tRNA on native gel analysis, indicating that the fragments in the nicked substrate did not dissociate during gel elution. Concentrations of full-length and nicked substrates were determined by UV absorption.
Minihelices and the 3′ fragment of the T-nicked substrate were labeled at the 5′ end with γ-32P-ATP (3000 Ci/mmol, Perkin Elmer) by T4 polynucleotide kinase (NEB) for 45 minutes at 37°C. After heat inactivation of the kinase, the labeled substrates were separated from free ATP by a Centrispin-20 column (Princeton Separation). The full-length tRNAs and 3′ fragments of the ASL-nicked substrates were labeled internally by using α-32P-ATP (3000 Ci/mmol, Perkin Elmer) during the T7 transcription and were purified by 12% PAGE/7M urea.
Single turnover kinetics
Single turnover assays were performed using an RQF-3 (KinTek). The reaction buffer for minihelices contained 1 mM DTT, 50 or 100 mM glycine (pH 9.0), 5 or 10 mM MgCl2 and 10% glycerol (including glycerol contributed by the enzyme storage buffer), and the saturating concentration of a specific nucleoside triphosphate (1 mM ATP or 5 mM CTP). Increasing concentrations of EcCCA, as annotated in the figures, were mixed with 50 nM of a minihelix substrate. For the full-length tRNA substrate, both the enzyme and substrate concentrations increased but maintained at a fixed 10:1 molar ratio. For each reaction, 10 μl of each syringe was loaded into the instrument. After different reaction times (0.1 to 300 s for the minihelices and the T-nicked tRNA, 2 to 70 ms for the tRNAs), the reactions were quenched with 80 μl of 7 M urea quench solution. The reaction samples were separated on a 12% PAGE/7M urea gel, exposed to a phosphor screen, scanned with Storm 820 and analyzed with ImageQuant 5.2 software (GE Healthcare).
The fraction of product formation was calculated based on the % of substrate that was converted to product. The fraction of substrate conversion over time was fit to a single exponential equation: %product = A×(1-e−kapp×t), where A is the amplitude, t is the time and kapp is the observed rate constant of the A76, C75, or C74 incorporation, respectively. The data of kappvs. enzyme concentration were fit to a hyperbolic equation: , where k76 is the catalytic rate constant and Kd is the dissociation constant. Alternatively, a linear fit was obtained: kapp = koff + kon×[Enz], where koff is the intercept and kon is the association rate constant.
Aminoacylation and lead cleavage analysis
Aminoacylation of tRNAVal transcripts (2 μM) by purified E. coli ValRS (4 μM) at 37 °C in 50 mM Tris-HCl, pH 7.5, 20 mM KCl, 4.0 mM DTT, 0.2 mg/mL BSA, 10 mM MgCl2, 2.0 mM ATP, and 20 μM [3H]-valine (15,600 dpm/pmole). The activity of aminoacylation was measured by monitoring the enzyme-catalyzed synthesis of [3H]-val-tRNAVal collected on filter pads in 5% TCA (trichloroacetic acid). Lead cleavage was performed by incubating lead acetate (1 mM) with intact or ASL-nicked A38G mutant of E. coli tRNAVal (1.0 and 2.5 μg, respectively) in 25 mM MOPS, pH 7.0, 1.5 mM spermine, 15 mM MgCl2 for 20 min at 42 °C. The reaction was terminated by the denaturing loading dye and the cleavage products were resolved by 12% denaturing PAGE.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
We thank Catherine Joyce for advice, Alan Weiner for encouragement, and Eric Phizicky, Murray Deutscher, Ken Johnson, and Jon Lorsch for comments on the manuscript. We also thank the anonymous reviewers for very helpful suggestions. This work was supported by NIH grant GM068561 to YMH.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Deutscher MP. tRNA nucleotidyltransferase. Enzymes. 1982;15:183–215. [Google Scholar]
- 2.Aebi M, Kirchner G, Chen JY, Vijayraghavan U, Jacobson A, Martin NC, Abelson J. Isolation of a temperature-sensitive mutant with an altered tRNA nucleotidyltransferase and cloning of the gene encoding tRNA nucleotidyltransferase in the yeast Saccharomyces cerevisiae. J Biol Chem. 1990;265:16216–20. [PubMed] [Google Scholar]
- 3.Yue D, Maizels N, Weiner AM. CCA-adding enzymes and poly(A) polymerases are all members of the same nucleotidyltransferase superfamily: characterization of the CCA-adding enzyme from the archaeal hyperthermophile Sulfolobus shibatae. Rna. 1996;2:895–908. [PMC free article] [PubMed] [Google Scholar]
- 4.Yue D, Weiner AM, Maizels N. The CCA-adding enzyme has a single active site. J Biol Chem. 1998;273:29693–700. doi: 10.1074/jbc.273.45.29693. [DOI] [PubMed] [Google Scholar]
- 5.Li F, Xiong Y, Wang J, Cho HD, Tomita K, Weiner AM, Steitz TA. Crystal structures of the Bacillus stearothermophilus CCA-adding enzyme and its complexes with ATP or CTP. Cell. 2002;111:815–24. doi: 10.1016/s0092-8674(02)01115-7. [DOI] [PubMed] [Google Scholar]
- 6.Xiong Y, Li F, Wang J, Weiner AM, Steitz TA. Crystal structures of an archaeal class I CCA-adding enzyme and its nucleotide complexes. Mol Cell. 2003;12:1165–72. doi: 10.1016/s1097-2765(03)00440-4. [DOI] [PubMed] [Google Scholar]
- 7.Okabe M, Tomita K, Ishitani R, Ishii R, Takeuchi N, Arisaka F, Nureki O, Yokoyama S. Divergent evolutions of trinucleotide polymerization revealed by an archaeal CCA-adding enzyme structure. Embo J. 2003;22:5918–27. doi: 10.1093/emboj/cdg563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Augustin MA, Reichert AS, Betat H, Huber R, Morl M, Steegborn C. Crystal Structure of the Human CCA-adding Enzyme: Insights into Template-independent Polymerization. J Mol Biol. 2003;328:985–94. doi: 10.1016/s0022-2836(03)00381-4. [DOI] [PubMed] [Google Scholar]
- 9.Xiong Y, Steitz TA. Mechanism of transfer RNA maturation by CCA-adding enzyme without using an oligonucleotide template. Nature. 2004;430:640–5. doi: 10.1038/nature02711. [DOI] [PubMed] [Google Scholar]
- 10.Tomita K, Fukai S, Ishitani R, Ueda T, Takeuchi N, Vassylyev DG, Nureki O. Structural basis for template-independent RNA polymerization. Nature. 2004;430:700–4. doi: 10.1038/nature02712. [DOI] [PubMed] [Google Scholar]
- 11.Tomita K, Ishitani R, Fukai S, Nureki O. Complete crystallographic analysis of the dynamics of CCA sequence addition. Nature. 2006;443:956–60. doi: 10.1038/nature05204. [DOI] [PubMed] [Google Scholar]
- 12.Shi PY, Maizels N, Weiner AM. CCA addition by tRNA nucleotidyltransferase: polymerization without translocation? Embo J. 1998;17:3197–206. doi: 10.1093/emboj/17.11.3197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cho HD, Chen Y, Varani G, Weiner AM. A model for C74 addition by CCA-adding enzymes: C74 addition, like C75 and A76 addition, does not involve tRNA translocation. J Biol Chem. 2006;281:9801–11. doi: 10.1074/jbc.M512603200. [DOI] [PubMed] [Google Scholar]
- 14.Cho HD, Verlinde CL, Weiner AM. Archaeal CCA-adding enzymes: central role of a highly conserved beta-turn motif in RNA polymerization without translocation. J Biol Chem. 2005;280:9555–66. doi: 10.1074/jbc.M412594200. [DOI] [PubMed] [Google Scholar]
- 15.Shi PY, Weiner AM, Maizels N. A top-half tDNA minihelix is a good substrate for the eubacterial CCA- adding enzyme. Rna. 1998;4:276–84. [PMC free article] [PubMed] [Google Scholar]
- 16.Seth M, Thurlow DL, Hou YM. Poly(C) synthesis by class I and class II CCA-adding enzymes. Biochemistry. 2002;41:4521–32. doi: 10.1021/bi0120953. [DOI] [PubMed] [Google Scholar]
- 17.Hou YM, Gu SQ, Zhou H, Ingerman L. Metal-Ion-Dependent Catalysis and Specificity of CCA-Adding Enzymes: A Comparison of Two Classes. Biochemistry. 2005;44:12849–59. doi: 10.1021/bi0509402. [DOI] [PubMed] [Google Scholar]
- 18.Hou YM. Unusual synthesis by the Escherichia coli CCA-adding enzyme [In Process Citation] Rna. 2000;6:1031–43. doi: 10.1017/s1355838200000686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Anand VS, Patel SS. Transient state kinetics of transcription elongation by T7 RNA polymerase. J Biol Chem. 2006;281:35677–85. doi: 10.1074/jbc.M608180200. [DOI] [PubMed] [Google Scholar]
- 20.Yakunin AF, Proudfoot M, Kuznetsova E, Savchenko A, Brown G, Arrowsmith CH, Edwards AM. The HD domain of the Escherichia coli tRNA nucleotidyltransferase has 2′,3′-cyclic phosphodiesterase, 2′-nucleotidase, and phosphatase activities. J Biol Chem. 2004;279:36819–27. doi: 10.1074/jbc.M405120200. [DOI] [PubMed] [Google Scholar]
- 21.Yap LP, Stehlin C, Musier-Forsyth K. Use of semi-synthetic transfer RNAs to probe molecular recognition by Escherichia coli proline-tRNA synthetase. Chem Biol. 1995;2:661–6. doi: 10.1016/1074-5521(95)90029-2. [DOI] [PubMed] [Google Scholar]
- 22.Liu H, Peterson R, Kessler J, Musier-Forsyth K. Molecular recognition of tRNA(Pro) by Escherichia coli proline tRNA synthetase in vitro. Nucleic Acids Res. 1995;23:165–9. doi: 10.1093/nar/23.1.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sherlin LD, Bullock TL, Nissan TA, Perona JJ, Lariviere FJ, Uhlenbeck OC, Scaringe SA. Chemical and enzymatic synthesis of tRNAs for high-throughput crystallization. Rna. 2001;7:1671–8. [PMC free article] [PubMed] [Google Scholar]
- 24.Nandakumar J, Shuman S. Dual mechanisms whereby a broken RNA end assists the catalysis of its repair by T4 RNA ligase 2. J Biol Chem. 2005;280:23484–9. doi: 10.1074/jbc.M500831200. [DOI] [PubMed] [Google Scholar]
- 25.Fukai S, Nureki O, Sekine S, Shimada A, Tao J, Vassylyev DG, Yokoyama S. Structural basis for double-sieve discrimination of L-valine from L- isoleucine and L-threonine by the complex of tRNA(Val) and valyl-tRNA synthetase. Cell. 2000;103:793–803. doi: 10.1016/s0092-8674(00)00182-3. [DOI] [PubMed] [Google Scholar]
- 26.Krzyzosiak WJ, Marciniec T, Wiewiorowski M, Romby P, Ebel JP, Giege R. Characterization of the lead(II)-induced cleavages in tRNAs in solution and effect of the Y-base removal in yeast tRNAPhe. Biochemistry. 1988;27:5771–7. doi: 10.1021/bi00415a056. [DOI] [PubMed] [Google Scholar]
- 27.Behlen LS, Sampson JR, DiRenzo AB, Uhlenbeck OC. Lead-catalyzed cleavage of yeast tRNAPhe mutants. Biochemistry. 1990;29:2515–23. doi: 10.1021/bi00462a013. [DOI] [PubMed] [Google Scholar]
- 28.Holbrook SR, Sussman JL, Warrant RW, Kim SH. Crystal structure of yeast phenylalanine transfer RNA. II. Structural features and functional implications. J Mol Biol. 1978;123:631–60. doi: 10.1016/0022-2836(78)90210-3. [DOI] [PubMed] [Google Scholar]
- 29.Cho HD, Oyelere AK, Strobel SA, Weiner AM. Use of nucleotide analogs by class I and class II CCA-adding enzymes (tRNA nucleotidyltransferase): deciphering the basis for nucleotide selection. Rna. 2003;9:970–81. doi: 10.1261/rna.2110903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Alexandrov A, Chernyakov I, Gu W, Hiley SL, Hughes TR, Grayhack EJ, Phizicky EM. Rapid tRNA decay can result from lack of nonessential modifications. Mol Cell. 2006;21:87–96. doi: 10.1016/j.molcel.2005.10.036. [DOI] [PubMed] [Google Scholar]
- 31.Ogawa T, Tomita K, Ueda T, Watanabe K, Uozumi T, Masaki H. A cytotoxic ribonuclease targeting specific transfer RNA anticodons. Science. 1999;283:2097–100. doi: 10.1126/science.283.5410.2097. [DOI] [PubMed] [Google Scholar]
- 32.Lu J, Huang B, Esberg A, Johansson MJ, Bystrom AS. The Kluyveromyces lactis gamma-toxin targets tRNA anticodons. Rna. 2005;11:1648–54. doi: 10.1261/rna.2172105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ho CK, Rauhut R, Vijayraghavan U, Abelson J. Accumulation of pre-tRNA splicing ‘2/3’ intermediates in a Saccharomyces cerevisiae mutant. Embo J. 1990;9:1245–52. doi: 10.1002/j.1460-2075.1990.tb08232.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li Z, Reimers S, Pandit S, Deutscher MP. RNA quality control: degradation of defective transfer RNA. Embo J. 2002;21:1132–8. doi: 10.1093/emboj/21.5.1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kadaba S, Krueger A, Trice T, Krecic AM, Hinnebusch AG, Anderson J. Nuclear surveillance and degradation of hypomodified initiator tRNAMet in S. cerevisiae. Genes Dev. 2004;18:1227–40. doi: 10.1101/gad.1183804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Deutscher MP. Degradation of RNA in bacteria: comparison of mRNA and stable RNA. Nucleic Acids Res. 2006;34:659–66. doi: 10.1093/nar/gkj472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wegscheid B, Hartmann RK. The precursor tRNA 3′-CCA interaction with Escherichia coli RNase P RNA is essential for catalysis by RNase P in vivo. Rna. 2006;12:2135–48. doi: 10.1261/rna.188306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wegscheid B, Hartmann RK. In vivo and in vitro investigation of bacterial type B RNase P interaction with tRNA 3′-CCA. Nucleic Acids Res. 2007;35:2060–73. doi: 10.1093/nar/gkm005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lund E, Dahlberg JE. Proofreading and aminoacylation of tRNAs before export from the nucleus [see comments] Science. 1998;282:2082–5. doi: 10.1126/science.282.5396.2082. [DOI] [PubMed] [Google Scholar]
- 40.Sarkar S, Azad AK, Hopper AK. Nuclear tRNA aminoacylation and its role in nuclear export of endogenous tRNAs in Saccharomyces cerevisiae. Proc Natl Acad Sci U S A. 1999;96:14366–71. doi: 10.1073/pnas.96.25.14366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Grosshans H, Hurt E, Simos G. An aminoacylation-dependent nuclear tRNA export pathway in yeast. Genes Dev. 2000;14:830–40. [PMC free article] [PubMed] [Google Scholar]
- 42.de Duve C. Transfer RNAs: the second genetic code. Nature. 1988;333:117–8. doi: 10.1038/333117a0. [DOI] [PubMed] [Google Scholar]
- 43.Weiner AM, Maizels N. tRNA-like structures tag the 3′ ends of genomic RNA molecules for replication: implications for the origin of protein synthesis. Proc Natl Acad Sci U S A. 1987;84:7383–7. doi: 10.1073/pnas.84.21.7383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McClain WH, Guerrier-Takada C, Altman S. Model substrates for an RNA enzyme. Science. 1987;238:527–30. doi: 10.1126/science.2443980. [DOI] [PubMed] [Google Scholar]
- 45.Nissen P, Kjeldgaard M, Thirup S, Polekhina G, Reshetnikova L, Clark BF, Nyborg J. Crystal structure of the ternary complex of Phe-tRNAPhe, EF-Tu, and a GTP analog [see comments] Science. 1995;270:1464–72. doi: 10.1126/science.270.5241.1464. [DOI] [PubMed] [Google Scholar]
- 46.Nissen P, Thirup S, Kjeldgaard M, Nyborg J. The crystal structure of Cys-tRNACys-EF-Tu-GDPNP reveals general and specific features in the ternary complex and in tRNA. Structure. 1999;7:143–56. doi: 10.1016/s0969-2126(99)80021-5. [DOI] [PubMed] [Google Scholar]
- 47.Keiler KC, Waller PR, Sauer RT. Role of a peptide tagging system in degradation of proteins synthesized from damaged messenger RNA [see comments] Science. 1996;271:990–3. doi: 10.1126/science.271.5251.990. [DOI] [PubMed] [Google Scholar]
- 48.Hou YM, Schimmel P. A simple structural feature is a major determinant of the identity of a transfer RNA. Nature. 1988;333:140–5. doi: 10.1038/333140a0. [DOI] [PubMed] [Google Scholar]
- 49.McClain WH, Foss K. Changing the identity of a tRNA by introducing a G-U wobble pair near the 3′ acceptor end. Science. 1988;240:793–6. doi: 10.1126/science.2452483. [DOI] [PubMed] [Google Scholar]
- 50.Francklyn C, Schimmel P. Aminoacylation of RNA minihelices with alanine. Nature. 1989;337:478–81. doi: 10.1038/337478a0. [DOI] [PubMed] [Google Scholar]
- 51.Cochella L, Green R. An active role for tRNA in decoding beyond codon:anticodon pairing. Science. 2005;308:1178–80. doi: 10.1126/science.1111408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hauenstein S, Zhang CM, Hou YM, Perona JJ. Shape-selective RNA recognition by cysteinyl-tRNA synthetase. Nat Struct Mol Biol. 2004;11:1134–41. doi: 10.1038/nsmb849. [DOI] [PubMed] [Google Scholar]
- 53.Kim SH, Suddath FL, Quigley GJ, McPherson A, Sussman JL, Wang AH, Seeman NC, Rich A. Three-dimensional tertiary structure of yeast phenylalanine transfer RNA. Science. 1974;185:435–40. doi: 10.1126/science.185.4149.435. [DOI] [PubMed] [Google Scholar]
- 54.Robertus JD, Ladner JE, Finch JT, Rhodes D, Brown RS, Clark BF, Klug A. Structure of yeast phenylalanine tRNA at 3 A resolution. Nature. 1974;250:546–51. doi: 10.1038/250546a0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.