

Abstract
The use of mass spectrometry to study protein-ligand interactions is expanding into more complex systems including protein-DNA interactions. The excess amount of a model DNA, or more typically an oligodeoxynucleotide (ODN), needed to study such interactions in an amide H/D exchange experiment, for example, causes serious signal suppression in the protein analysis. We describe here a modification of the traditional H/D exchange protocol whereby we utilize a strong anion exchange column to remove rapidly the ODN from solution before MS analysis. We showed the successful incorporation of such a column in a study of two protein-ODN systems: (1) the DNA-binding domain of human telomeric repeat binding factor 2 with a telomeric oligodeoxynucleotide and (2) thrombin with the thrombin-binding aptamer. The approach gave no appreciable difference in back-exchange compared to a method in which no SAX is used.
Introduction
Hydrogen-deuterium (H/D) exchange mass spectrometry (MS) [1] is an emerging tool for protein-ligand interactions. Although the final measurement is in the gas phase of a mass spectrometer, H/D exchange enables the interactions to be studied under 'biologically relevant' solution conditions (i.e., neutral pH, strong buffers, and high ionic strengths) and at relatively low concentrations of protein. The amide exchange is monitored by measuring the mass change of a protein, in either a bound or unbound state, after incubation in deuterated buffer. If amide sites are affected by ligand binding, their exchange rates may decrease in the bound state. Differences are caused by global or local conformational changes associated with ligand binding, formation of protein-ligand interfaces, and/or formation of more stabilized secondary structure elements throughout the protein. Both kinetics of exchange and affinity of the protein can be determined, the latter by either PLIMSTEX (Protein-Ligand Interactions by Mass Spectrometry, Titration, and H/D Exchange) [2] or SUPREX (Stability of Unpurified Proteins from Rates of H/D Exchange) [3].
In many H/D exchange protocols, an aqueous protein solution is diluted with deuterated buffer to make an exchange solution high in D2O content. After an appropriate exchange time, the reaction is quenched by lowering the pH to 2–3 and the temperature to near 0 °C. The quenched solution is loaded onto a reverse-phase column and washed with chilled water (pH 2–3). The wash step removes salts, buffers, and back-exchanges the fast-exchanging deuterated sites. The protein is eluted into the mass spectrometer, where its mass is measured to reveal the extent of deuterium uptake. Ideally the steps from quench to detection are sufficiently rapid (within 3 min) to ensure that few deuteria are lost to back-exchange.
Although H/D exchange has been successfully applied to many protein-ligand interactions, there are few reports of those involving an oligodeoxynucleotide [4–5]. While investigating two protein-ODN interactions using electrospray ionization (ESI), we discovered that the protein signal can be entirely suppressed by the presence of an ODN, especially when its concentration exceeds that of the protein by a factor of three or more. Given that ODN:protein ratios greater than 10:1 are required to ensure nearly complete binding, a means of reducing the concentration of the ODN prior to MS analysis is needed. One possibility is to add ion exchange chromatography; another is to change to matrix-assisted laser desorption ionization (MALDI), which is less susceptible to signal suppression [4].
Ion exchange chromatography [6–8] separates species based upon their charge in solution. An anion exchange column should trap ODN anions but minimally affect the protein cations, taking advantage that, under quench conditions, the ODN is anionic and the protein cationic. Here we describe the use of a dual column approach for H/D exchange of protein-ODN complexes and test the approach by using two model systems.
Experimental
Reagents
The sodium chloride (NaCl) salt, potassium chloride (KCl) salt, HEPES buffer (N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid and sodium salt), isotopically labeled water (D2O, 99.999% deuterium replacement), formic acid, hydrochloric acid, and acetonitrile reagents were purchased from Sigma Aldrich (St. Louis, MO) at the highest purity available and were used without further purification. The α-thrombin protein was purchased from Haematologic Technologies Inc (Essex Junction, VT) as a 167 µM solution in 50% glycerol (aq) and was stored at −4 °C upon receipt without further purification. The thrombin-binding aptamer (5′ GGT TGG TGT GGT TGG 3′) was purchased from Integrated DNA Technologies (Coralville, IA) as an ammonium salt. This sample was purified by the manufacturer using reverse phase HPLC to exclude salts as much as possible. The DNA-binding domain of human TRF2 was prepared as described previously [5]. The model of telomeric DNA (5′ GTT AGG GTT AGG G 3′) and its complement were also purchased from Integrated DNA Technologies (Coralville, IA), analyzed by mass spectrometry for purity, and used without further purification.
Model system: thrombin interaction
The master solution of the thrombin-binding aptamer was prepared by dissolving weighed amounts of solid sample in deionized water. The master protein solution was prepared by diluting the solution received from the manufacturer with HEPES buffer solution. The concentrations of these master solutions were determined by UV absorption spectrophotometry; for each sample, a calibration plot was prepared to give the molar absorptivity. For the aptamer, the experimentally determined value agreed to within 1% of the value calculated by the nearest neighbor method for the given sequence and agreed with the molar absorptivity reported by the manufacturer. For the protein, the experimentally determined value agreed to within 5% of the value reported by the manufacturer. The "working master" apo thrombin solution was 2.5 µM in 10 mM HEPES, pH 6.8 and contained 25 mM NaCl and 0.8% glycerol (v/v) while the "working master" holo solution was 2.5 µM thrombin and 25 µM thrombin-binding aptamer in 10 mM HEPES, pH 6.8 containing 25 mM NaCl, and 0.8% glycerol (v/v).
Model system: human TRF2 interaction
Double-strand telomeric DNA was prepared by mixing equimolar amounts of the complementary, single-stranded ODNs in a 10 mM HEPES buffer containing 150 mM KCl. Duplex formation was monitored by UV absorption spectrophotometry (260 nm) by decreasing the solution temperature from 95 °C to 25 °C by 5 °C increments every 30 minutes. The apo protein stock solution was prepared so that it contained 41 µM hTRF2 in 10 mM HEPES (pH 7.4) and 150 mM KCl. The holo protein stock solution was prepared so that it contained 41 µM protein and 410 µM of the telomeric DNA in 10 mM HEPES (pH 7.4) and 150 mM KCl. The preparation of these samples was discussed previously [5].
Mass spectrometry
Mass spectra were acquired using a Waters quadrupole-time of flight (Q-ToF) Ultima (Milford, MA) mass spectrometer equipped with an ESI source and operated in the positive-ion mode. The instrument conditions were optimized for each protein analyzed; briefly the capillary voltage was 3.0–3.2 kV, the cone-voltage readback was 100–105 V, the cone-gas flow rate was 40 L/hr, the desolvation gas flow rate was 400 L/hr, the source temperature was 80 °C, and the desolvation temperature was 180 °C. The MS profile used for quadrupole transmission was: from m/z 500, dwell for 5% of the scan time, ramp to m/z 1000 for 45% of the scan time, then dwell at m/z 1000 for the remaining 50% of the scan time. The scan time for the instrument was 2 sec. The protein-ODN solution was passed through the strong anion exchange column (1×15 mm OPTI-GUARD, Optimize Technologies Inc., Oregon City, OR), to trap the ODN, and then onto a C8 reverse-phase guard column (1×15 mm OPTI-GUARD, Optimize Technologies Inc., Oregon City, OR), pre-equilibrated with 0.2% formic acid in water, to trap the protein. The reverse-phase column was washed with 300 µL of 0.2% formic acid in water (0 °C) and eluted using an isocratic flow of 30% solvent B (hTRF2) or 50% solvent B (thrombin) at 40 µL/min by a Waters CapLC (Milford, MA). The HPLC solvents were (A) 94.7% deionized water, 5% acetonitrile, and 0.3% formic acid and (B) 94.7% acetonitrile, 5% deionized water, 0.3% formic acid.
Results and Discussion
To minimize ionization suppression effects of an oligodeoxynucleotide (ODN) in protein studies, we considered capturing the protein on a reverse-phase column and washing away the ODN. Although such gradient elution may be successful, its use increases the experimental time and, consequently, the extent of back-exchange. Further, a gradient elution approach would not be amenable for studies in which the deuterated protein is proteolyzed to gain higher resolution data because retention of the peptides formed in proteoloysis and ODN will be similar; creating the potential for peptide-signal suppression. These disadvantages prompted us to consider including a strong anion exchange (SAX) column to remove the ODN.
In the modified procedure, we placed the SAX column in the flow path after the syringe injection port and in front of the injection valve (see Figure 1). At the start of an experiment, the injection valve was in LOAD for injection of the protein/ODN mixture with a standard syringe. The solution passed sequentially through the SAX column, the injection loop, the reverse-phase column (typically C8), and finally through the waste port of the valve. Because the quenched solution’s pH is 2–3, the protein remains positively charged whereas the ODN is negative. Therefore, the ODN adheres to the SAX column while the protein passes on and is trapped on the reverse-phase column. Having separated the ODN and protein molecules, the protein is washed with chilled water (pH 2–3) and eluted isocratically into the mass spectrometer by switching the injector valve to INJECT. While in this mode, the ODN was eluted from the SAX column with 300 µL of aqueous NaOH (pH 9–10).
Figure 1.
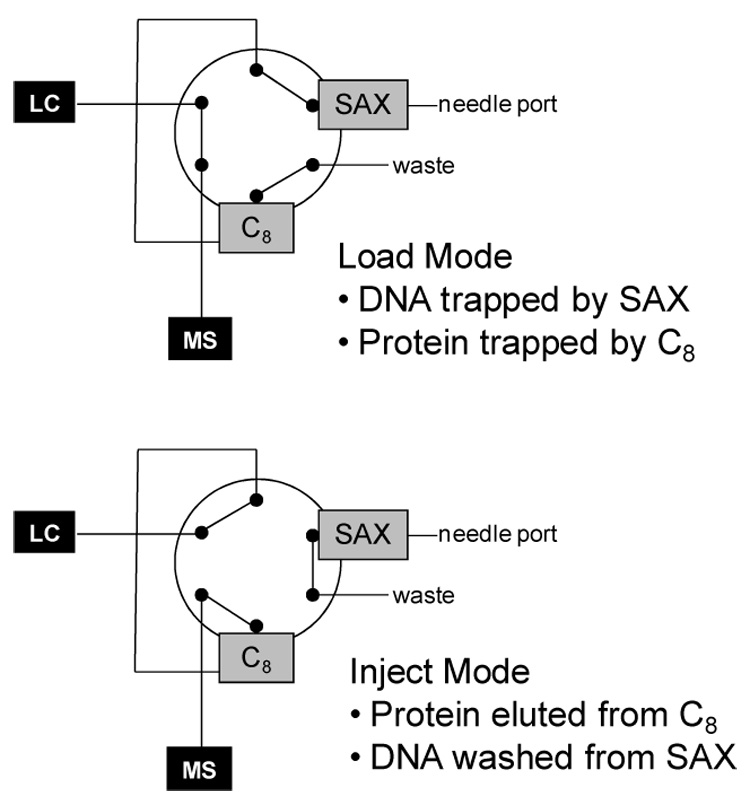
Diagram of the dual column setup in the load and inject mode.
One model system to test the effectiveness of strong anion exchange involves the 7.5 kDa DNA-binding domain of the human telomeric repeat factor 2 (hTRF2) that interacts with a double stranded telomeric ODN [5]. The second model is the thrombin-thrombin binding aptamer where a 36 kDa protein interacts with a single-stranded DNA aptamer that is folded into a non-canonical quadruplex structure [9].
The mass spectrum (Figure 2a) is of 10 pmol of hTRF2 injected onto a C8 column and eluted into the mass spectrometer without the use of an SAX column. That in Figure 2b, however, is of 10 pmol of hTRF2 with 100 pmol of telomeric ODN injected onto and eluted from a C8 column without the use of an SAX column. This 10:1 ratio ensured nearly complete binding of the protein in solution. We observed no protein ions owing to excessive ion suppression from the ODN. Observed instead are the positive ions of the C-rich strand of telomeric DNA (the ion at m/z 956.7 is the +4 charge state and the ion at m/z 1275.3 is the +3). When we incorporated the SAX column and injected the same 10:1 sample, we found a significant increase in the protein signal and no ions from the ODN (Figure 2c). The relative intensity of the protein signal in Figure 2c is 10–15% that of Figure 2a.
Figure 2.
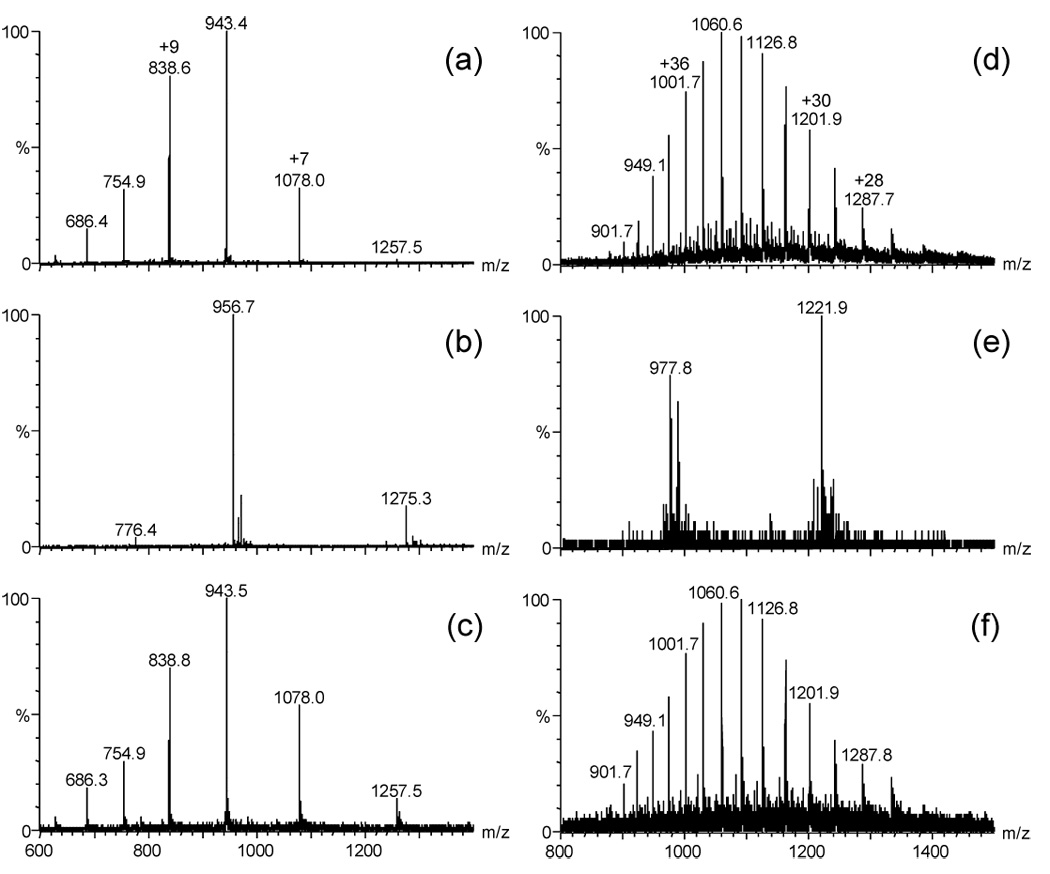
Mass spectra from two model systems. (a) 10 pmol hTRF2 without SAX column present, (b) 10 pmol hTRF2 and 100 pmol telomeric DNA without SAX column present, (c) 10 pmol hTRF2 and 100 pmol telomeric DNA with SAX column present, (d) 10 pmol thrombin without SAX column present, (e) 10 pmol thrombin and 100 pmol thrombin-binding aptamer without SAX column present, (f) 10 pmol thrombin and 100 pmol thrombin-binding aptamer with SAX column present.
Turning to the second model, the control is a mass spectrum (Figure 2d) of 10 pmol of thrombin loaded onto a C8 column and eluted into the mass spectrometer without an SAX column. That in Figure 2e, 10 pmol of thrombin and 100 pmol of its ODN ligand loaded onto a C8 column and eluted without an SAX column, shows the dramatic signal suppression of the ODN. The ions shown in Figure 2e are of the ODN ligand (the ion of m/z 977.9 is the +5 charge state and the ion of m/z 1221.9 is the +4 charge state). The salutary effect of incorporating an SAX column for the analysis of the 10:1 sample solution (Figure 2f) is revealed by an increase in the relative abundance of the protein signal, which is 10–15% of the relative abundance of the protein signal in Figure 2d.
In the modified procedure, the time from quench to detection is not increased so it is unlikely that back-exchange is increased by inclusion of the SAX column. To test our hypothesis, we measured the extent of D uptake for a 1:1 mixture of hTRF2 and its ligand (double-stranded telomeric DNA) in the presence and absence of an SAX column. In this experiment, the protein-ODN mixture was incubated in deuterated buffer (97% D2O) for 10 minutes, quenched, loaded onto the C8 column (with and without SAX), washed, and eluted into the mass spectrometer. For triplicate experiments, the average uptake without SAX was 53.2 ± 0.9 Da whereas with SAX, the average was 53.7 ± 0.9 Da. Clearly, the presence of the SAX column does not affect the extent of back-exchange.
In summary, the application of H/D exchange to protein-ODN interactions requires decreasing the amount of signal suppression caused when the ODN coelutes with the protein into the ESI mass spectrometer. The simple incorporation of an SAX column into the experimental protocol enables trapping the ODN while passing most of the protein onto a reverse-phase column and into the ion source without increasing the extent of back-exchange. Although we do not report here the outcome and interpretation of the H/D exchange data, we can state that this modified approach worked for two protein-ODN systems: a TRF2 interaction, where the H/D exchange results were recently reported [5] and a thrombin interaction, where the H/D exchange study is the focus of current work.
Acknowledgments
These studies were supported by the National Institutes of Health and National Center for Research Resources (Grant P41RR000954).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Wales TE, Engen JR. Hydrogen exchange mass spectrometry for the analysis of protein dynamics. Mass Spectrometry Reviews. 2006;25:158–170. doi: 10.1002/mas.20064. [DOI] [PubMed] [Google Scholar]
- 2.Zhu MM, Rempel DL, Du Z, Gross ML. Quantification of protein-ligand interactions by mass spectrometry, titration, and H/D exchange: PLIMSTEX. Journal of the American Chemical Society. 2003;125:5252–5253. doi: 10.1021/ja029460d. [DOI] [PubMed] [Google Scholar]
- 3.Ghaemmaghami S, Fitzgerald MC. A quantitative, high-throughput screen for protein stability. Proceedings of the National Academy of Science. 2000;97(15):8296–8301. doi: 10.1073/pnas.140111397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ma L, Fitzgerald MC. A New H/D Exchange- and Mass Spectrometry-Based Method for Thermodynamic Analysis of Protein-DNA Interactions. Chemistry & Biology. 2003;10(12):1205–1213. doi: 10.1016/j.chembiol.2003.11.017. [DOI] [PubMed] [Google Scholar]
- 5.Sperry JB, Shi X, Rempel DL, Nishimura Y, Akashi S, Gross ML. A Mass Spectrometric Approach to the Study of DNA-Binding Proteins: Interaction of Human TRF2 with Telomeric DNA. Biochemistry. 2008;47(6):1797–1807. doi: 10.1021/bi702037p. [DOI] [PubMed] [Google Scholar]
- 6.Green GDJ, Prior CP. Method using anion exchange resin for removing DNA from protein preparations. 1990 WO 90/08159. [Google Scholar]
- 7.Willis TC, Goldberg B. A model system for DNA removal from protein solutions. American Biotechnology Laboratory. 1992;10(6):24. [PubMed] [Google Scholar]
- 8.Fritz JS, Gjerde DT. Ion Chromatography. 3rd edition. Weinheim: Wiley-VCH; 2000. [Google Scholar]
- 9.Padmanabhan K, Padmanabhan KP, Ferrara JD, Sadler JE, Tulinsky A. The structure of a-thrombin is inhibited by a 15-mer single-stranded DNA aptamer. Journal of Biological Chemistry. 1993;268:17651–17654. doi: 10.2210/pdb1hut/pdb. [DOI] [PubMed] [Google Scholar]