

Abstract
The low density lipoprotein (LDL) oxidation hypothesis has generated considerable interest in oxidative stress and how it might affect atherosclerosis. However, the failure of antioxidants, particularly vitamin E, to affect the progression of the disease in humans has convinced even staunch supporters of the hypothesis to take a step backwards and reconsider alternatives.
Preponderant evidence for the hypothesis came from animal antioxidant intervention studies. In this review we point out basic differences between animal and human atherosclerosis development and suggest that human disease starts where animal studies end. While initial oxidative steps in the generation of early fatty streak lesions might be common, the differences might be in the steps involved in the decomposition of peroxidized lipids into aldehydes and their further oxidation into carboxylic acids. We suggest that these steps may not be amenable to attenuation by antioxidants and antioxidants might actually counter the stabilization of plaque by preventing the formation of carboxylic acids which are anti-inflammatory in nature. The formation of such dicarboxylic acids may also be conducive to plaque stabilization by trapping calcium.
We suggest that agents that would prevent the decomposition of lipid peroxides and promote the formation and removal of lipid hydroxides, such as paraoxonase (PON 1) or apo A1/high density lipoprotein (HDL) might be more conducive to plaque regression.
Keywords: Atherosclerosis, aldehydes, dicarboxylation, inflammation, calcification
Oxidation hypothesis of atherosclerosis
Atherosclerosis is the major manifestation of cardiovascular diseases. The role of lipoproteins in atherosclerosis development has been a topic of interest for over five decades. It is now established beyond doubt that high levels of low density lipoprotein (LDL) and low levels of high density lipoprotein (HDL) contribute significantly to the development and progression of cardiovascular diseases [1–5].
The biochemical processes that contributed to the formation of early atherosclerotic lesions, the fatty streak lesions, are still under debate. The LDL oxidation hypothesis was put forward in the eighties to explain the formation of fatty streak lesions and over 4000 publications and countless reviews have appeared on the topic to date [6–11] providing evidence for the presence of oxidative processes in human disease. The potential steps involved in the development of early atherosclerotic lesion as outlined by the hypothesis are listed in table 1.
Table 1.
Steps involved in fatty streak lesion formation:
| 1. | Increased plasma and intimal LDL. |
| 2. | Oxidation of lipids associated with LDL |
| 3. | Chemotactic recruitment of monocytes and their differentiation into macrophages. |
| 4. | Modification of apolipoprotein B100 to a negatively charged form (Oxidatively modified LDL) by products derived from lipid peroxidation. |
| 5. | Uptake of oxidatively modified LDL by macrophages. |
| 6. | Retention of lipid laden macrophages in the intima. |
The oxidized LDL
The hypothesis originated from a simple observation that in vitro incubation of macrophages with oxidized LDL and not with native LDL led to intracellular cholesterol ester accumulation [12–15]. The oxidation of LDL is a complex process during which both the protein and lipids undergo oxidative changes and form complex products. The peroxidized lipids decompose generating both free and core aldehydes and ketones that covalently modify ε- amino groups of lysine residues of the protein moiety [16–17]. The latter not only generates Schiff’s bases, thus modifying charges on the amino acids, but also results in both intra- and inter- molecular cross-links between proteolyzed apo B. The oxidative modification is not unique to LDL as other lipoproteins such as very low density lipoprotein (VLDL), beta very low density lipoprotein (β-VLDL), and even high density lipoprotein (HDL) have been suggested to undergo similar oxidative changes [18–22] accompanied by changes in their pro- or anti-atherosclerotic behavior. The take home message from these studies was that the modification of LDL might be the key determinant of lipid uptake by macrophages.
Potential mechanisms of oxidation
The mechanism(s) by which oxidation of LDL might occur in vivo or the extent of oxidation are subject to speculation and are predominantly derived from in vitro studies (Table 2). Practically every type of oxidative system has been suggested to contribute to the oxidation of LDL. In vivo evidence in support or against any of these mechanisms is ambiguous. For example, overexpression/knockout strategies involving specific enzymes often yielded conflicting results. Overexpression of arachidonate 12/15 lipoxygenase in rabbits protected against atherosclerosis [23], while overexpression and knock out of this enzyme in mice yielded results in favor of the involvement of this enzyme in oxidative processes [24, 25]. Similarly, myeloperoxidase (MPO) gene knockout in mice was expected to protect against atherosclerosis, while the results showed an enhanced atherosclerosis [26]. However, overexpression of human MPO in mice appeared to marginally increase atherosclerosis susceptibility [27]. These studies were often interpreted as evidence in favor of distinct human MPO protein; however, it is hard to understand how the MPO activity could be different from different sources although variations might exist in protein amino acid sequence and subunits unless the inference is that only in humans MPO plays a role and not in mice. Similarly, conflicting results have emerged from studies that have manipulated antioxidant enzymes such as superoxide dismutase [28].
Table 2.
Potential mechanisms for the oxidation of LDL
| Mechanism | Ref. |
|---|---|
| 1. Lipoxygenase reaction | 23,24,25 |
| 2. Copper and ceruloplasmin mediated oxidation | 28, 30 |
| 3. Iron mediated oxidation | 235, 236 |
| 4. Peroxidase mediated oxidation including Myeloperoxidase and heme | 26,27,35 |
| 5. Peroxynitrite mediated oxidation | 29,30 |
| 6. Thiol-dependent oxidation | 38,39 |
| 7. Xanthine Oxidase, NADPH Oxidase and other superoxide generators | 28 |
| 8. AAPH or other means of radical generation including cytochromes | 29, 30 |
The oxidation systems vastly differ in their properties and many of them require co-oxidants. MPO mediated oxidations require co-oxidants such as H2O2 or lipid peroxides and thus over expression or knockout of MPO alone would have little effect on the oxidative status. Some oxidants damage the proteins more readily than others. For example treatment of LDL with 2,2′-azobis(2-amidinopropane) dihydrochloride (AAPH), a radical generator or peroxynitrite results in more protein oxidation than lipid peroxidation [29, 30]. Similarly, peroxidase catalyzed oxidation generate very little aldehyde products as compared to metal catalyzed oxidations [31]. Lipoxygenase reactions which are exclusively intracellular, might require additional reactions or transfers before or after lipoxygenase reactions occur [32]. To our knowledge, no one has characterized oxidized LDL isolated from animals or humans to the extent that mechanistic insights could be derived, although there are numerous claims of the presence of an electronegative LDL (presumably minimally oxidized LDL) in human and animal plasma [33, 34].
Antioxidants and atherosclerosis-prooxidant nature is ignored
While all these reactions are oxidative in nature, they are not uniformly amenable to inhibition by traditional antioxidants. Vitamin E or simple phenols such as tyrosine or estradiol actually enhance peroxidase-mediated oxidation of LDL [35–37]. This has been taken as evidence for an intermediate pro-oxidant role for antioxidants. Similarly, many thiols may enhance or inhibit the oxidation of LDL depending on the peroxide/metal content of LDL [38–39]. The bottom line is we neither know of the oxidation system(s) that might be involved nor the effects of specific antioxidant chemicals on such reactions.
Antioxidants and animal models of atherosclerosis
One of the strongest evidence for the oxidation hypothesis was derived from animal studies that tested the effects of antioxidants on atherosclerosis. A variety of antioxidants have been tested using a number of different animal models. These include WHHL rabbits, cholesterol-fed rabbits, hypercholesterolemic hamsters and monkeys, and various mouse models of atherosclerosis. Animal models were developed to study the formation of early atherosclerotic lesion in isolated environment so that contribution of individual risk factors and associated biochemical events could be segregated and studied. The human atherosclerosis is a chronic disease associated with the ageing process. During the lifelong process spanning decades, numerous risk factors contribute to atherosclerosis. The necessity to study the disease in a short and meaningful period of time (combined with the human impatience and the pressure of the time constraints of the scientific grant funding process) has created numerous models that have shrunk the atherogenic process to a mere few months. All these models depend on the increases in plasma cholesterol levels beyond what one would see in a human being. These models were developed with the sole purpose of studying the contribution of cholesterol carrying lipoproteins or other associated processes to the formation of macrophage-rich fatty streak lesions and have performed remarkably well to this end. In fact, the end points of these studies are often lesion size/areas or lesion lipid parameters.
These animal models and the use of numerous antioxidants (Table 3) have suggested that antioxidants do attenuate the atherogenic process. However, problems related to the very high concentrations of antioxidants (sometimes even more than that of cholesterol in the diet itself!) and toxicity of many different types of antioxidants (BHT, DPPD) were often overlooked. There was a race to demonstrate that every chemical with known antioxidant property was anti-atherosclerotic.
Table 3.
Animal models and antioxidants in the prevention of atherosclerosis:
| Model | Antioxidant(s) used |
|---|---|
| LDL Receptor Knockout mice | Vitamin E, beta-carotene, vitamin C, probucol, S-nitroso-N-acetylcysteine, nitric oxide-releasing aspirin, 2,3-dihydro-5-hydroxy-2, 2-dipentyl-4,6-di-tert-butylbenzofuran. |
| Apolipoprotein E knockout mice | N-acetylcysteine, vitamin E, probucol, N,N'-diphenyl 1,4-phenylenediamine (DPPD), calcium channel blockers- lacidipine, disodium ascorbyl phytostanyl phosphate, taurine, tocotrienol, catechins. |
| WHHL Rabbits | Probucol, vitamin E, and fluvastatin, an HMG-CoA (hydroxy-3-methylglutaryl coenzyme A), ubiquinone-10, N-acetylcysteine, astaxanthin, water-soluble vitamin E derivative, N,N'-diacetyl-L-cystine. |
| Cholesterol-fed NZW rabbits | Probucol, vitamin E, polyphenol, U74006F, BHT, N,N'-diphenyl 1,4-phenylenediamine (DPPD) |
| Cholesterol-fed hamsters | Isoflavones, vegetable extract (Oxxynea), alpha-lipoic acid (ALP), Vitamin E, Green tea, probucol, grape extracts. |
| Cholesterol-fed guinea pigs | Vitamin C, Vitamin E, Huajiao volatile oil |
| Monkeys | Vitamin E, probucol, various soy components. |
Why did human trials using antioxidant fail?
Combined with epidemiological evidence and success in several animal trials in a number of species using a variety of antioxidants, the hypothesis appeared to be on solid ground. This euphoria of initial success led to clinical trials that were planned mostly to be the "first" to validate the hypothesis. These trials have been reviewed in many articles [40–42] and will not be discussed here. These ill-conceived clinical trials were performed without understanding the a) enzymes and factors involved in the oxidative process including those contributed by associated risk factors, b) nature of the oxidative process and associated changes in the lipid and protein moieties, c) whether other lipoproteins, particularly HDL was affected by these processes, d) whether antioxidants ameliorated any specific steps in the oxidative process, e) the compatibility of the oxidative process/antioxidant action with current therapy, f) the steps involved in the atherogenic process and the role(s) of oxidative processes, if any, on these steps, g) the distinction between the early fatty streak lesions and the advanced vulnerable lesions that lead to plaque rupture, thrombosis, and acute coronary events, and h) above all the distinction between the animal models and human atherosclerotic development. The latter is of great significance as the antioxidant trials completely ignored pathological findings and clinical outcomes.
Tocopherol-vitamin or antioxidant?
Obviously vitamin E was the chosen antioxidant as the expectation of the general public and scientists was that a natural antioxidant (such as vitamin E) would have less undesirable effects. Accordingly, a number of clinical trials were performed using vitamin E alone or combined with vitamin C or other antioxidants at pharmacological doses often disregarding the definition of the term “vitamin” that suggested a requirement at miniscule doses. Some of these trials were piggyback studies on cancer and other diseases. These trials with vitamin E have not been overwhelmingly supportive of the hypothesis. Understanding the sequence of events that led to the hypothesis, the molecular basis of antioxidant action, the potential interaction with other drugs, and the stage of the disease process to which oxidation was suggested to contribute, dosage, the transport mechanisms involved in the release and utilization of vitamin E in the body, its interaction(s) with cytochrome systems, and its role as a prooxidant in peroxidase-mediated oxidations, played little role in the design of the study. More importantly, little if any attention was paid to factors that would affect and control oxidation in vivo.
Oxidized LDL in humans
While the potential use of antioxidants in controlling the disease process has lost its supporters, more and more evidence supports a role for an oxidative process in atherosclerosis and associated risk factors. The prevalence of antibodies that recognize epitopes of Ox-LDL seem to correlate well with the disease process [43–48]. These antibodies also detect such LDL in the plasma of subjects who have higher risk associated with atherosclerosis [49–52] and seem to recognize oxidative tailored lipid epitope. Interestingly, such antibodies are not available commercially to study animal atherosclerosis and the human oxidized LDL kit does not appear to work with animal blood samples. This does not appear to be related to the specific antibodies used in the kit sounding yet another ominous bell of caution that the fatty streak lesions seen in animals or in children might not involve the generation of such epitopes.
Myeloperoxidase products in humans
Oxidative enzymes such as MPO and products derived from their actions seem to predict the vulnerable population and seem to correlate with the severity of the disease [53–57]. A variety of products, such as chloro and nitro tyrosines have been detected at increased levels in the plasma of subjects with established coronary artery disease as compared to control subjects. Considering that Vitamin E might actually enhance oxidation [35, 58–60], the clinical trials might have actually endorsed the hypothesis rather than shooting it down. Perhaps, when inhibitors of specific oxidases are available, additional trials might be warranted to induce the development of anti-atherosclerotic pharmacological agents.
Human atherosclerosis and animal models
The human form of atherosclerotic disease differs considerably from the disease models in animals. As mentioned before, the animal models are predominantly setup to develop and study the very early fatty streak lesions with macrophage foam cells. Such lesions start very early in life in humans (even in infants and children) below the age of 15 [61–68]. Maternal hypercholesterolemia seems to strongly influence the development of atherosclerotic lesions in the young. Again, interestingly, while plasma LDL levels positively correlate to the severity of coronary artery disease, infants and children have much lower LDL levels as compared to adults and the elderly population suggesting that the correlation between age and plasma LDL levels could be a statistical coincidence rather than one of clinical significance except when specific sub-fractions with increased atherogenic potency such as oxidized LDL or small dense LDL account for the increase. However, the presence of fatty streak atherosclerotic lesions alone in the absence of significant other risk factors might be clinically inconsequential especially considering lower plasma lipid levels and compounding risk factors in children as compared to adults. Also, quantitatively, adults may have greater lesion surface area as compared to children, in addition to differences in the type of lesions. Acquisition of additional risk factors might significantly affect the progression or regression of the early childhood lesions.
Progression of human disease-Calcification
The progression of human atherosclerotic lesions has been well studied by several pioneers [69–73]. They unanimously conclude that the macrophage-lipid-rich early fatty streak lesions progress to extra-cellular lipid rich raised complex lesions later in adult life with or without calcification [69–71]. Atherosclerotic calcification begins as early as the second decade of life, just after fatty streak formation. The lesions of younger adults have revealed small aggregates of crystalline calcium among the lipid particles of lipid cores. Calcium deposits are found more frequently and in greater amounts in elderly individuals and in more advanced lesions. In most advanced lesions, when calcification dominates, components such as lipid deposits and increased fibrous tissue may also be present. The extent of calcification also has been correlated with plaque burden. It has been demonstrated that coronary calcification is detected in the vast majority of patients with a first MI. Age-related calcium score is highly predictive for myocardial infarction event risk in prospective studies and the association between coronary calcium and CHD events usually remains significant after adjusting for other CHD risk factors. These study results support the concept that coronary calcium is associated with a relative profound independent increased risk of CHD events in women and men. However, there has been a great deal of debate about the role of calcification in plaque rupture [74–79].
Calcification has been noted as both intra- and extra- cellular deposits [78], the latter predominantly is associated with extracellular lipids. Many researchers believe that coronary arterial calcification may represent an attempt to protect the weakened atherosclerotic plaque prone to rupture [77, 79]. Calcified lesions and fibrotic lesions are much stiffer than cellular lesions and are unlikely to be associated with sites of plaque rupture [77, 80]. In fact, a recent study showed that less than 15% of the ruptured human lesions showed evidence of associated calcification [80]. It is speculated that the plaque rupture often occurs at the interface between a calcified and non-calcified atherosclerotic areas of the lesion. Thus, calcification could be seen as a potential stabilizing force that may increase the biochemical stability of the plaque by imparting rigidity while at the same time decreasing the plaque’s mechanical stability.
Calcified human lesions
Neither the mechanism(s) involved in atherosclerotic calcification nor the nature of the calcium deposits has been positively identified. Calcification is identified mostly by Von Kossa staining or by physical imaging techniques such as computerized tomography. While they give valuable information, they fail to indicate the chemical form in which calcium is deposited. It is generally believed that calcium phosphate (hydroxyapatite, Ca3[−PO4]2− ×Ca[OH]2), precipitates in diseased coronary arteries by a mechanism similar to that found in active bone formation and remodeling [77, 81–87]. Others believe that specific sulfated proteoglycans with high affinity for calcium are involved in the deposition of calcium in lesion areas [88–91]. Support for this comes from the variety of proteoglycans, such as decorin, that are synthesized and secreted by smooth muscle cells (cells that are proximally associated with calcification). The synthesis of these proteins is also regulated by factors that influence the atherosclerotic process thus, providing credibility to their involvement.
Inflammation and atherosclerosis
Yet another major difference between human and animal atherosclerosis is the contribution of inflammatory cytokines and matrix digesting enzymes (MMPs) to the advancement of the vulnerable plaque [92–98]. Most of these cytokines and MMPs are induced by oxidative stress [99–125] suggesting that ongoing oxidative processes might be involved in the generation of the vulnerable plaque. Inflammatory cytokines, matrix disruption, release of lytic lipids such as lyso phosphatidylcholine, aldehydes, release of reactive oxygen species, and proteolytic enzymes have been suggested to play major roles in plaque vulnerability and rupture.
Risk factors and oxidative stress
Well-known risk factors might contribute to such vulnerability by promoting the formation of inflammatory cytokines and the generation of oxygen radicals (Fig. 1). Diabetes, ageing, hypertension, smoking, physical inactivity and obesity, hyperlipidemia, particularly hyper cholesterolemia including the prevalence of small dense LDL, hyperhomocysteinemia, are recognized risk factors for atherosclerosis. It is interesting to note that most of the risk factors for atherosclerosis also are risk factors for heart failure suggesting a chronic perturbation of the immune system and endothelial activation. Innumerable studies have documented that every risk factor for atherosclerosis is associated with an increased oxidative stress. One would assume these genetic, environmental, and life style associated risk factors somehow would increase oxidative and inflammatory stress and promote the progression of early lesions to the vulnerable plaque leading to plaque rupture.
Figure 1.
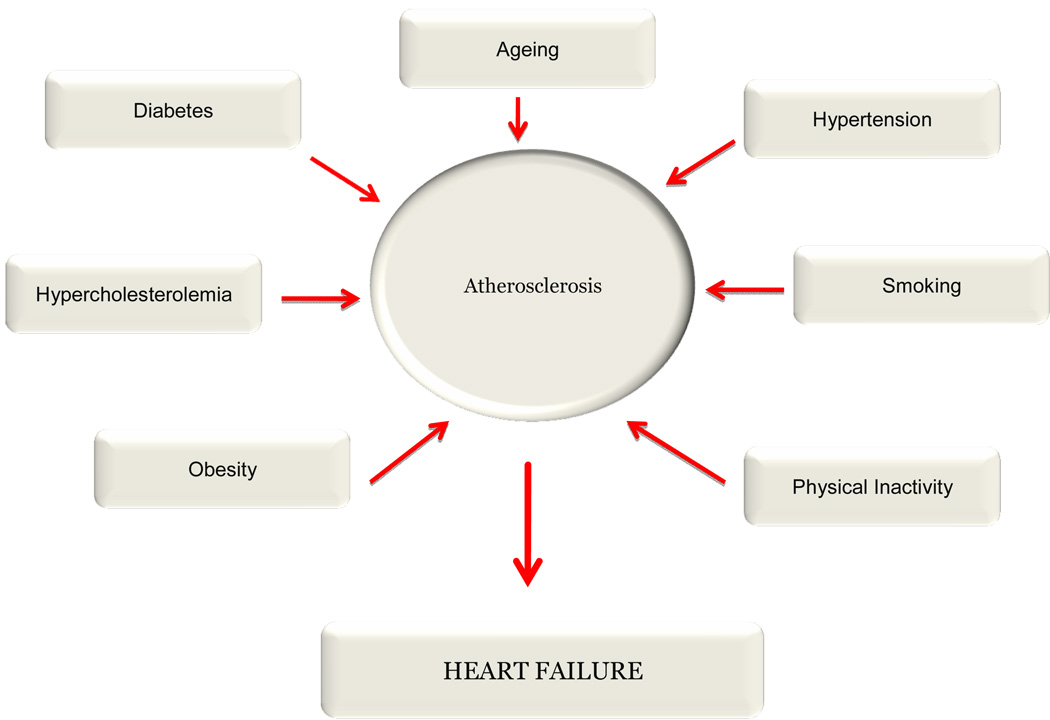
a) Diabetes and oxidative stress
Oxidative stress has long been associated with diabetes and hypertension. Both clinical and experimental studies show the excessive reactive oxygen species generation in hyperglycemic conditions. The toxicity of diabetic serum has long been attributed to the presence of oxidized lipids. Hyperglycemic conditions are associated with high levels of oxidant production [126–128]. The ability of glucose to generate oxygen radicals and H2O2 non-enzymatically and enzymatically has been known for a long time [128–130]. In addition, hyperglycemia appears to induce lipid oxidative enzymes such as 12/15 lipoxygenases and NADPH oxidases [131, 132]. In addition, glucose also reacts with amino groups of proteins and lipids to generate advanced glycation end products (AGEs) [133, 134]. These products among other properties also induce oxygen radical production thus amplifying the production of H2O2 and LOOH [135, 136].
b) Hypertension and oxidative stress
Similarly, molecules associated with hypertension, for example nitric oxide and angiotensin II are intimately associated with oxidative stress [137–140]. While the former’s beneficial vaso dilatating action is cancelled by oxygen radicals generating a more virulent form of oxidant, peroxynitrite [141], angiotensin II potentiates such inactivation by generating superoxide radicals by way of enzymes that utilize NADPH oxidation [137].
c) Cigarette smoke and oxidation
The fundamental mechanism by which cigarette smoke is thought to contribute to atherosclerosis has been suggested to be mediated by causing oxidative stress [142–144]. Innumerable articles have alluded to the presence of free radicals in the gas/tar phase of cigarette smoke [142–144]. In addition, the ability of cigarette smoke to deplete plasma antioxidants and induce the oxidation of LDL in vitro and ex vivo has also been well documented [145–148].
d) Physical activity and oxidative stress
Recently there have been several exciting findings in relation to obesity/physical inactivity and oxidative stress. Leptin has been shown to induce oxidative stress as well as being induced by oxidative stress [149, 150]. While vigorous physical activity in early stages might deplete body’s antioxidants and promote oxidative stress, sustained and long term physical activity has been noted to raise antioxidant defense and reduce oxidative stress [151–158].
The propensity of small dense LDL to undergo increased oxidation has also been observed [159–161]. While small dense LDL may well represent one of the most atherogenic forms of LDL, it remains to be seen whether its oxidation specifically has any relevance in vivo.
In many of the above considerations, the underlying assumption is that the oxidative stress contributes to the formation of oxidized LDL and that the components of oxidized LDL could act to stimulate the production of inflammatory mediators that are hallmarks of subjects with advanced atherosclerotic lesions. There is also an underlying expectation that new lesions are continuously generated and oxidative stress is not only involved in their generation but somehow promote their conversion to advanced vulnerable plaque.
Oxidative stress and body’s antioxidant defense
The failure of antioxidants (at least vitamin E containing supplements) to benefit human subjects with advanced disease might suggest that either the hypothesis is flawed or some form of oxidative stress might actually be beneficial. Could oxidative stress be beneficial? Some of the arguments in favor of a “beneficial” oxidative stress are illustrated in Table 4. First, oxidative stress has been known to induce body’s defense mechanisms by triggering the synthesis of antioxidant enzymes. The induction of catalase [162], GSH [163] and GSH peroxidase [164], nitric oxide synthase [165], MnSOD [166,167] and heme oxygenase [168,169] by oxidants has been well described in literature. We described the ability of oxidized fatty acids to induce apo A1 (a major component of HDL) synthesis by liver and intestinal cells [170] which also could signify a beneficial effect. We also recently described the induction of apo AV gene expression and a decrease in plasma triglycerides by oxidized fatty acids [171]. Some of these effects could be attributed to PPAR ligand like actions of oxidized lipids [170, 172–174], a point that is conveniently ignored when considering the deleterious actions of oxidized LDL. Oxidation also has been suggested to play a key role in the generation and removal of senescent apoptotic cells [175–177] and perhaps involved in the detoxification of many cardiovascular drugs including statins [178].
Table 4.
Potential benefits of oxidative stress:
| 1. | Oxidative stress and oxidized lipids have been shown to induce antioxidant enzymes such as, nitric oxide synthase, catalase, MnSOD, GSH synthesis, and heme oxygenase. |
| 2. | Oxidized lipids induce apo A1 gene expression and synthesis. |
| 3. | Induce apo AV gene expression and decrease plasma triglyceride levels. Also act as PPAR ligands and induce fatty acid oxidation |
| 4. | Might be involved in senescent cell clearance and cellular signaling mechanisms. |
| 5. | Might be involved and essential for the detoxification of many cardiovascular drugs. |
| 6. | Might actually be involved in the beneficial actions of exercise, long chain polyunsaturated fat, and estradiol. |
Exercise, PUFA, and Estradiol
More importantly, the three well-known modalities that are deterrents of cardiovascular diseases, exercise, the consumption of polyunsaturated fatty acids (PUFA), and the estrogenic hormones are intimately associated with oxidative stress. Vigorous physical activity has been shown to deplete antioxidants and promote oxidant production [151–158]. Similarly, PUFA is more readily oxidized as compared to saturated or monounsaturated fatty acids [179–181], and estradiol has been shown to potentiate the actions of neutrophils and MPO action [36, 37]. Consistently and repeatedly, physical activity and PUFA have been shown not only to prevent the development of atherosclerotic lesions but also to promote regression [182–187]. While the precise mechanisms of action of these are yet unknown, oxidative stress-induced antioxidant defense has been suggested to be an important factor [157].
Antioxidant activities of cardiovascular drugs
However, such actions must be weighed together with the fact that many of the currently available cardiovascular drugs are antioxidants in nature (Table 5). These drugs include statins [187, 188], calcium channel blockers [189–193], angiotensin II receptor antagonists [194], ACE inhibitors [195–199], compounds such as dobutamine [200], nitric oxide donors [201–202], salicylate [203, 204] and many others. Regardless of their action as antioxidants in vivo, their chemical nature as antioxidants cannot be ignored. It is likely that the metabolic profiles of these compounds are more favorable than that of vitamin E.
Table 5.
Cardiovascular drugs that have antioxidant properties
| 1. | Nitric oxide donors. |
| 2. | Calcium channel blockers |
| 3. | Angiotensin type II receptor blockers. |
| 4. | Dobutamine. |
| 5. | Statins. |
| 6. | Certain ACE inhibitors. |
| 7. | Salicylate and related compounds. |
Too much aldehydes from peroxidized lipids?
We considered the possibility that the oxidation hypothesis did not extend “far” enough to include the decomposition of peroxidized lipids. The oxidation hypothesis took into consideration that the aldehyde products generated by the decomposition of peroxidized lipids covalently modify the €-amino groups of lysine residues to generate the oxidatively modified LDL. Considering that even the simplest PUFA, linoleic acid, could generate several aldehyde products (e.g. 2-hydroxy hexanal, 4-hydroxy nonenal, oxo-nonanoic acid), one would end up with a glut of such products. Earlier studies by Haberland and coworkers have suggested that derivatization of as little as 15% of lysine residues of apoprotein B100 are enough to impart macrophage scavenger receptor recognition [16, 17]. A mole of LDL particle is expected to have about 350 mols of free amino groups. About 15% modification of these (excluding other oxidative structural changes) would be equal to the modification of approximately 50 amino groups. Considering that about 1 mol equivalent of LDL would have about or more than 0.5 mol equivalent of PUFA and each PUFA is capable of generating at least two more aldehyde products, one would have 20 or more equivalent of aldehydes per modifiable lysine residues. In addition, the lysosomal proteolysis would release aldehydes bound lysine peptides as well as free aldehydes due to the instability of Schiff bases in such environments. Of course such arguments consider that all oxidizable PUFA will undergo oxidation once oxidation is initiated and there would be a glut of free and core aldehydes.
Pro-atherogenic effects of aldehydes
The pro-atherogenic effects of aldehyde products have received relatively little attention as compared to the oxidized lipids themselves. The initial burst of activities centered on the modification process per se and the results seemed to indicate that small molecular water-soluble aldehydes such as malondialdehyde (MDA) readily diffused from the lipoprotein particle [205]. Pioneering studies by Esterbauer and coworkers brought attention to more reactive and lipophilic aldehydes such as 4-hydroxy nonenal (4-HNE) in to focus [206–209]. Besides being capable of protein modification and crosslinking 4-HNE is now recognized to affect the atherogenic process by a number of different ways. Aldehydes such as 4-HNE might cause enzyme inactivation, apoptosis, modify proteins associated with cell signaling, and induce pro-inflammatory cytokines [206–217].
Aldehydes to carboxylic acids
While 4-HNE attracted considerable attention, the other half of the decomposition product of oxidized linoleic acid (oxo-valeric acid, oxo-nonanoic acid and other oxo-acids and core aldehydes) has attracted little attention [218]. The core aldehydes (which are esterified to lipids such as cholesterol or lysophospholipids) are seen only as markers of oxidation or as antigenic lipids or as substrates for enzymes such as platelet activating factor acetyl hydrolase (PAF-acetyl hydrolase) or phospholipases. One such product (oxo-nonanoic acid) that is derived from the oxidation of linoleic acid is of great biological significance as it gets readily oxidized to nonane dioic acid (azelaic acid). Azelaic acid is a lipophilic dicarboxylic acid as opposed to short chain dicarboxylic acids such as malonic acid, despite the reactive methylene group reactivity of the latter. Therefore, it would be expected to be formed and to accumulate in greater amounts in lipid-rich domains.
Azelaic acid, a lipid peroxidation-derived lipophilic dicarboxylic acid
Azelaic acid would have two important anti-atherogenic properties: one, as other dicarboxylic acids, it would have great affinity for calcium and precipitate calcium in lipid-rich domains [219–222]. This might account for the unusually high calcification associated with lipid core domains. Second, azelaic acid has been noted to have anti-inflammatory properties and has been noted to decrease cytokine production by macrophages and inflammatory cells [223–225]. It also has been noted to reduce oxidant production by leukocytes [223]. Thus, it has been added as an active ingredient in topical skin medications, particularly as anti-acne agent [224–226].
The oxidation of oxo-nonanoic acid to azelaic acid is a simple oxidation step and is inhibited by antioxidants [227] analogous to the oxidation of other aldehydes [228–230]. The oxidation of aldehydes into acids is catalyzed by both enzymatic (aldehydes oxidase, xanthine oxidase) as well as non-enzymatic oxidation.
Could antioxidants inhibit the conversion of aldehydes to carboxylic acids?
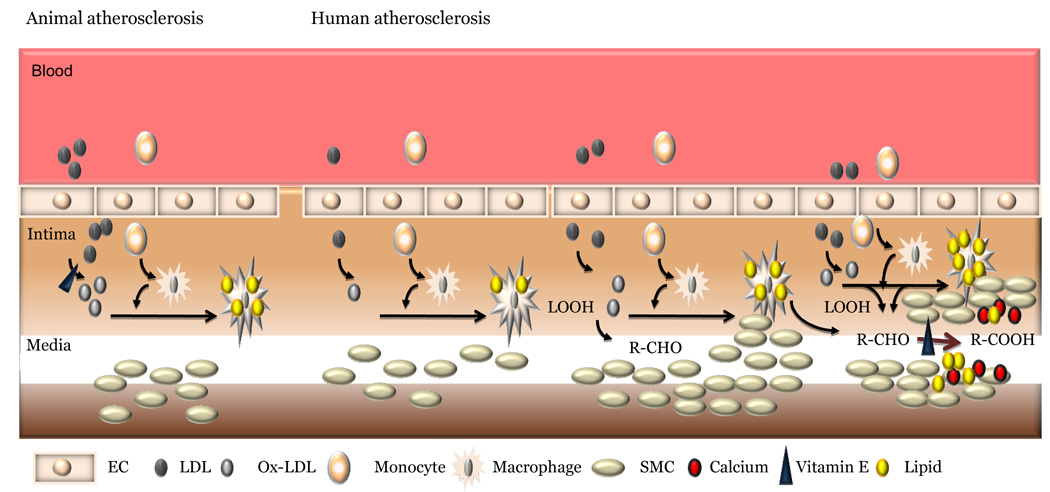
Thus, in human secondary prevention trials with antioxidants, there is a distinct possibility that the high levels of antioxidants could have prevented the generation of anti-atherogenic and anti-inflammatory dicarboxylic acids. Such dicarboxylic acids also might be involved in calcification process and the inhibition of their formation by antioxidants would be conducive to the formation of non-calcified vulnerable plaque (Fig. 2). Until the specific nature of calcium deposits are identified in human lesions, one could only speculate the role of such dicarboxylic acids in calcification. Either they can complex calcium and thus could represent calcified lesions, or they could destabilize the calcium phosphate/calcium extracellular proteoglycan complex thus acting against calcification. It remains to be seen whether agents that would prevent the decomposition of lipid peroxides into long chain aldehydes or enhance the conversion of aldehydes into dicarboxylic acids would have a better cardiovascular outcome. The carboxylic acids, including dicarboxylic acids are readily oxidized in the mitochondria and peroxisomes and thus could be readily eliminated. Molecules such as apo A1/HDL or enzymes such as paraoxonases or glutathione peroxidase might be inherently anti-atherogenic by not only promoting the conversion of hydroperoxides into hydroxides as well as by complexing with the aldehyde products but also by removing the hydroxides from the artery for elimination [231–234] as suggested by Fogelman and associates.
Figure 2.
Summary
In this review we point out the differences between animal and human atherosclerosis and suggest that human atherosclerosis might have progressed beyond the point of oxidation. We suggest that the end products of lipid peroxidation, carboxylic acids (and not aldehydes) might influence calcification and inflammation. Antioxidants might prevent the formation of innocuous carboxylic acids from toxic aldehydes.
Acknowledgments
This work was supported by NIH grants HL69038 and DK056353.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.National Cholesterol Education Program. Third Report of the Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (ATP III Final Report) Bethesda, MD: National Heart, Lung, and Blood Institute; 2002. [Google Scholar]
- 2.National Cholesterol Education Program. Adult Treatment Panel III, Update 2004: Implications of Recent Clinical Trials for the ATP III Guidelines. Bethesda, MD: National Heart, Lung, and Blood Institute; 2004. [DOI] [PubMed] [Google Scholar]
- 3.Rosin BL. The progression of cardiovascular risk to cardiovascular disease. Cardiovasc. Med. 2007;8:s3–s8. [PubMed] [Google Scholar]
- 4.Le NA, Walter MF. The role of hypertriglyceridemia in atherosclerosis. Curr. Atheroscler. Rep. 2007;9:110–115. doi: 10.1007/s11883-007-0006-7. [DOI] [PubMed] [Google Scholar]
- 5.Cromwell WC, Otvos JD. Low-density lipoprotein particle number and risk for cardiovascular disease. Curr. Atheroscler. Rep. 2004;6:381–387. doi: 10.1007/s11883-004-0050-5. [DOI] [PubMed] [Google Scholar]
- 6.Steinberg D, Parthasarathy S, Crew TE, Khoo JC, Witztum JL. Beyond cholesterol: modification of low-density lipoprotein that increase its atherogenecity. N. Engl. J. Med. 1989;320:915–924. doi: 10.1056/NEJM198904063201407. [DOI] [PubMed] [Google Scholar]
- 7.Parthasarathy S. Modified Lipoproteins in the Pathogenesis of Atherosclerosis. Austin, TX: R. G. Landes Co.; 1994. [Google Scholar]
- 8.Steinberg D, Lewis A. Conner Memorial Lecture. Oxidative modification of LDL and atherogenesis. Circulation. 1997;95:1062–1071. doi: 10.1161/01.cir.95.4.1062. [DOI] [PubMed] [Google Scholar]
- 9.Chisolm GM, Steinberg D. The oxidative modification hypothesis of atherogenesis: an overview. Free Radic. Biol. Med. 2000;28:1815–1826. doi: 10.1016/s0891-5849(00)00344-0. [DOI] [PubMed] [Google Scholar]
- 10.Berliner J. Introduction. Lipid oxidation products and atherosclerosis. Vascul. Pharmacol. 2002;38:187–191. doi: 10.1016/s1537-1891(02)00168-4. [DOI] [PubMed] [Google Scholar]
- 11.Matsuura E, Kobayashi K, Tabuchi M, Lopez LR. Oxidative modification of low-density lipoprotein and immune regulation of atherosclerosis. Prog. Lipid Res. 2006;45:466–486. doi: 10.1016/j.plipres.2006.05.001. [DOI] [PubMed] [Google Scholar]
- 12.Henriksen T, Mahoney EM, Steinberg D. Enhanced macrophage degradation of biologically modified low density lipoprotein. Arteriosclerosis. 1983;3:149–159. doi: 10.1161/01.atv.3.2.149. [DOI] [PubMed] [Google Scholar]
- 13.Steinbrecher UP, Parthasarathy S, Leake DS, Witztum JL, Steinberg D. Modification of low density lipoprotein by endothelial cells involves lipid peroxidation and degradation of low density lipoprotein phospholipids. Proc. Natl. Acad. Sci. USA. 1984;81:3883–3887. doi: 10.1073/pnas.81.12.3883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hessler JR, Morel DW, Lewis LJ, Chisolm GM. Lipoprotein oxidation and lipoprotein-induced cytotoxicity. Arteriosclerosis. 1983;3:215–222. doi: 10.1161/01.atv.3.3.215. [DOI] [PubMed] [Google Scholar]
- 15.Morel DW, DiCorleto PE, Chisolm GM. Endothelial and smooth muscle cells alter low density lipoprotein in vitro by free radical oxidation. Arteriosclerosis. 1984;4:357–364. doi: 10.1161/01.atv.4.4.357. [DOI] [PubMed] [Google Scholar]
- 16.Haberland ME, Olch CL, Fogelman AM. Role of lysines in mediating interaction of modified low density lipoproteins with the scavenger receptor of human monocyte macrophages. J. Biol. Chem. 1984;259:11305–11311. [PubMed] [Google Scholar]
- 17.Haberland ME, Fogelman AM, Edwards PA. Specificity of receptor-mediated recognition of malondialdehyde-modified low density lipoproteins. Proc. Natl. Acad. Sci. USA. 1982;79:1712–1716. doi: 10.1073/pnas.79.6.1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Young IS, McFarlane C, McEneny J. Oxidative modification of triacylglycerol-rich lipoproteins. Biochem. Soc. Trans. 2003;31:1062–1065. doi: 10.1042/bst0311062. [DOI] [PubMed] [Google Scholar]
- 19.Francis GA. High density lipoprotein oxidation: in vitro susceptibility and potential in vivo consequences. Biochim. Biophys. Acta. 2000;1483:217–235. doi: 10.1016/s1388-1981(99)00181-x. [DOI] [PubMed] [Google Scholar]
- 20.Shao B, Oda M, Vaisar T, Oram J, Heinecke J. Pathways for oxidation of high-density lipoprotein in human cardiovascular disease. Curr. Opin. Mol. Ther. 2006;8:198–205. [PubMed] [Google Scholar]
- 21.Bergt C, Pennathur S, Fu X, Byun J, O'Brien K, McDonald T, Singh P, Anantharamaiah GM, Chait A, Brunzell J, Geary RL, Oram JF, Heinecke JW. The myeloperoxidase product hypochlorous acid oxidizes HDL in the human artery wall and impairs ABCA1-dependent cholesterol transport. Proc. Natl. Acad. Sci. U S A. 2004;101:13032–13037. doi: 10.1073/pnas.0405292101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Parthasarathy S, Printz D, Boyd D, Joy L, Steinberg D. Macrophage oxidation of low density lipoprotein generates a modified form recognized by the scavenger receptor. Arteriosclerosis. 1986;6:505–510. doi: 10.1161/01.atv.6.5.505. [DOI] [PubMed] [Google Scholar]
- 23.Shen J, Herderick E, Cornhill JF, Zsigmond E, Kim HS, Kuhn H, Guevara NV, Chan L. Macrophage-mediated 15-lipoxygenase expression protects against atherosclerosis development. J. Clin. Invest. 1996;98:2201–2208. doi: 10.1172/JCI119029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Harats D, Shaish A, George J, Mulkins M, Kurihara H, Levkovitz H, Sigal E. Overexpression of 15-lipoxygenase in vascular endothelium accelerates early atherosclerosis in LDL receptor-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2000;20:2100–2105. doi: 10.1161/01.atv.20.9.2100. [DOI] [PubMed] [Google Scholar]
- 25.Huo Y, Zhao L, Hyman MC, Shashkin P, Harry BL, Burcin T, Forlow SB, Stark MA, Smith DF, Clarke S, Srinivasan S, Hedrick CC, Pratico D, Witztum JL, Nadler JL, Funk CD, Ley K. Critical role of macrophage 12/15-lipoxygenase for atherosclerosis in apolipoprotein E-deficient mice. Circulation. 2004;110:2024–2031. doi: 10.1161/01.CIR.0000143628.37680.F6. [DOI] [PubMed] [Google Scholar]
- 26.Brennan ML, Anderson MM, Shih DM, Qu XD, Wang X, Mehta AC, Lim LL, Shi W, Hazen SL, Jacob JS, Crowley JR, Heinecke JW, Lusis AJ. Increased atherosclerosis in myeloperoxidase-deficient mice. J. Clin. Invest. 2001;107:419–430. doi: 10.1172/JCI8797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McMillen TS, Heinecke JW, LeBoeuf RC. Expression of human myeloperoxidase by macrophages promotes atherosclerosis in mice. Circulation. 2005;111:2798–2804. doi: 10.1161/CIRCULATIONAHA.104.516278. [DOI] [PubMed] [Google Scholar]
- 28.Tribble DL, Gong EL, Leeuwenburgh C, Heinecke JW, Carlson EL, Verstuyft JG, Epstein CJ. Fatty streak formation in fat-fed mice expressing human copper-zinc superoxide dismutase. Arterioscler. Thromb. Vasc. Biol. 1997;17:1734–1740. doi: 10.1161/01.atv.17.9.1734. [DOI] [PubMed] [Google Scholar]
- 29.Gieseg S, Duggan S, Gebicki JM. Peroxidation of proteins before lipids in U937 cells exposed to peroxyl radicals. Biochem. J. 2000;350:215–218. [PMC free article] [PubMed] [Google Scholar]
- 30.Patel R, Diczfalusy U, Dzeletovic S, Wilson M, Darley-Usmar V. Formation of oxysterols during oxidation of low density lipoprotein by peroxynitrite, myoglobin, and copper. J. Lipid Res. 1996;37:2361–2371. [PubMed] [Google Scholar]
- 31.Wieland E, Parthasarathy S, Steinberg D. Peroxidase-dependent metal-independent oxidation of low density lipoprotein in vitro: a model for in vivo oxidation? Proc. Natl. Acad. Sci. USA. 1993;90:5929–5933. doi: 10.1073/pnas.90.13.5929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rankin SM, Parthasarathy S, Steinberg D. Evidence for a dominant role of lipoxygenase(s) in the oxidation of LDL by mouse peritoneal macrophages. J. Lipid Res. 1991;32:449–456. [PubMed] [Google Scholar]
- 33.Damasceno NR, Sevanian A, Apolinario E, Oliveira JM, Fernandes I, Abdalla DS. Detection of electronegative low density lipoprotein (LDL-) in plasma and atherosclerotic lesions by monoclonal antibody-based immunoassays. Clin. Biochem. 2006;39:28–38. doi: 10.1016/j.clinbiochem.2005.09.014. [DOI] [PubMed] [Google Scholar]
- 34.Barros MR, Bertolami MC, Abdalla DS, Ferreira WP. Identification of mildly oxidized low-density lipoprotein (electronegative LDL) and its auto-antibodies IgG in children and adolescents hypercholesterolemic offsprings. Atherosclerosis. 2006;184:103–107. doi: 10.1016/j.atherosclerosis.2004.11.027. [DOI] [PubMed] [Google Scholar]
- 35.Santanam N, Parthasarathy S. Paradoxical actions of antioxidants in the oxidation of low density lipoprotein by peroxidases. J. Clin. Invest. 1995;95:2594–2600. doi: 10.1172/JCI117961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Santanam N, Shem-Brewer R, McClatchey R, Castellano PZ, Murphy AA, Voelkel S, Parthasarathy S. Estradiol as an antioxidant: incompatible with its physiological concentrations and function. J. Lipid Res. 1998;39:2111–2118. [PubMed] [Google Scholar]
- 37.Chiang K, Parthasarathy S, Santanam N. Estrogen, neutrophils and oxidation. Life Sciences. 2004;75:2425–2438. doi: 10.1016/j.lfs.2004.04.035. [DOI] [PubMed] [Google Scholar]
- 38.Parthasarathy S. Oxidation of low-density lipoprotein by thiol compounds leads to its recognition by the acetyl LDL receptor. Biochim. Biophys. Acta. 1987;917:337–340. doi: 10.1016/0005-2760(87)90139-1. [DOI] [PubMed] [Google Scholar]
- 39.Sparrow CP, Olszewski J. Cellular oxidation of low density lipoprotein is caused by thiol production in media containing transition metal ions. J. Lipid Res. 1993;34:219–228. [PubMed] [Google Scholar]
- 40.Harris A, Devaraj S, Jialal I. Oxidative stress, alpha-tocopherol therapy, and atherosclerosis. Curr. Atheroscler. Rep. 2002;4:373–380. doi: 10.1007/s11883-002-0075-6. [DOI] [PubMed] [Google Scholar]
- 41.Witztum JL, Steinberg D. The oxidative modification hypothesis of atherosclerosis: does it hold for humans? Trends. Cardiovasc Med. 2001;11:93–102. doi: 10.1016/s1050-1738(01)00111-6. [DOI] [PubMed] [Google Scholar]
- 42.Williams KJ, Fisher EA. Oxidation, lipoproteins, and atherosclerosis: which is wrong, the antioxidants or the theory? Curr. Opin. Clin. Nutr. Metab. Care. 2005;8:139–146. doi: 10.1097/00075197-200503000-00006. [DOI] [PubMed] [Google Scholar]
- 43.Palinski W, Hörkko S, Miller E, Steinbrecher UP, Powell HC, Curtiss LK, Witztum JL. Cloning of monoclonal autoantibodies to epitopes of oxidized lipoproteins from apolipoprotein E-deficient mice: demonstration of epitopes of oxidized low density lipoprotein in human plasma. J. Clin. Invest. 1996;98:800–814. doi: 10.1172/JCI118853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Itabe H, Yamamoto H, Imanaka T, Shimamura TK, Uchiyama H, Kimura J, Sanaka T, Hata Y, Takano T. Sensitive detection of oxidatively modified low density lipoprotein using a monoclonal antibody. J. Lipid Res. 1996;37:45–53. [PubMed] [Google Scholar]
- 45.Itabe H, Yamamoto H, Suzuki M, Kawai Y, Nakagawa Y, Suzuki A, Imanaka T, Takano T. Oxidized phosphatidylcholines that modify proteins: analysis by monoclonal antibody against oxidized low density lipoprotein. J. Biol.Chem. 1996;271:33208–33217. doi: 10.1074/jbc.271.52.33208. [DOI] [PubMed] [Google Scholar]
- 46.Fraley AE, Tsimikas S. Clinical applications of circulating oxidized low-density lipoprotein biomarkers in cardiovascular disease. Curr. Opin. Lipidol. 2006;17:502–509. doi: 10.1097/01.mol.0000245255.40634.b5. [DOI] [PubMed] [Google Scholar]
- 47.Horkko S, Bird DA, Miller E, Itabe H, Leitinger N, Subbanagounder G, Berliner JA, Friedman P, Dennis EA, Curtiss LK, Palinski W, Witztum JL. Monoclonal autoantibodies specific for oxidized phospholipids or oxidized phospholipid-protein adducts inhibit macrophage uptake of oxidized low-density lipoproteins. J. Clin Invest. 1999;103:117–128. doi: 10.1172/JCI4533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Holvoet P, Vanhaecke J, Janssens S, Van de Werf F, Collen D. Oxidized LDL and malondialdehyde-modified LDL in patients with acute coronary syndromes and stable coronary artery disease. Circulation. 1998;98:1487–1494. doi: 10.1161/01.cir.98.15.1487. [DOI] [PubMed] [Google Scholar]
- 49.Holvoet P, Stassen JM, Van Cleemput J, Collen D, Vanhaecke J. Oxidized low density lipoproteins in patients with transplant-associated coronary artery disease. Arterioscler. Thromb. Vasc. Biol. 1998;18:100–107. doi: 10.1161/01.atv.18.1.100. [DOI] [PubMed] [Google Scholar]
- 50.Holvoet P, Collen D, Van de Werf F. Malondialdehyde-modified LDL as a marker of acute coronary syndromes. JAMA. 1999;281:1718–1721. doi: 10.1001/jama.281.18.1718. [DOI] [PubMed] [Google Scholar]
- 51.Shoji T, Nishizawa Y, Fukumoto M, Shimamura K, Kimura J, Kanda H, Emoto M, Kawagishi T, Morii H. Inverse relationship between circulating oxidized low density lipoprotein (oxLDL) and anti-oxLDL antibody levels in healthy subjects. Atherosclerosis. 2000;148:171–177. doi: 10.1016/s0021-9150(99)00218-x. [DOI] [PubMed] [Google Scholar]
- 52.Salonen JT, Ylä-Herttuala S, Yamamoto R, Butler S, Korpela H, Salonen R, Nyyssönen K, Palinski W, Witztum JL. Autoantibody against oxidised LDL and progression of carotid atherosclerosis. Lancet. 1992;339:883–887. doi: 10.1016/0140-6736(92)90926-t. [DOI] [PubMed] [Google Scholar]
- 53.Hazen SL, Heinecke JW. 3-Chlorotyrosine, a specific marker of myeloperoxidase-catalyzed oxidation, is markedly elevated in low density lipoprotein isolated from human atherosclerotic intima. J. Clin. Invest. 1997;99:2075–2081. doi: 10.1172/JCI119379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sugiyama S, Okada Y, Sukhova GK, Virmani R, Heinecke JW, Libby P. Macrophage myeloperoxidase regulation by granulocyte macrophage colony-stimulating factor in human atherosclerosis and implications in acute coronary syndromes. Am. J. Pathol. 2001;158:879–891. doi: 10.1016/S0002-9440(10)64036-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Thukkani AK, McHowat J, Hsu FF, Brennan ML, Hazen SL, Ford DA. Identification of alpha-chloro fatty aldehydes and unsaturated lysophosphatidylcholine molecular species in human atherosclerotic lesions. Circulation. 2003;108:3128–3133. doi: 10.1161/01.CIR.0000104564.01539.6A. [DOI] [PubMed] [Google Scholar]
- 56.Meuwese MC, Stroes ES, Hazen SL, Van Miert JN, Kuivenhoven JA, Schaub RG, Wareham NJ, Luben R, Kastelein JJ, Khaw KT, Boekholdt SM. Serum myeloperoxidase levels are associated with the future risk of coronary artery disease in apparently healthy individuals: the EPIC-Norfolk Prospective Population Study. J. Am. Coll. Cardiol. 2007;50:159–165. doi: 10.1016/j.jacc.2007.03.033. [DOI] [PubMed] [Google Scholar]
- 57.Parastatidis I, Thomson L, Fries DM, Moore RE, Tohyama J, Fu X, Hazen SL, Heijnen HF, Dennehy MK, Liebler DC, Rader DJ, Ischiropoulos H. Increased protein nitration burden in the atherosclerotic lesions and plasma of apolipoprotein A-I deficient mice. Circ. Res. 2007;107:368–376. doi: 10.1161/CIRCRESAHA.107.157537. [DOI] [PubMed] [Google Scholar]
- 58.Bowry VW, Stocker R. Tocopherol-mediated peroxidation: the pro-oxidant effect of vitamin E on the radical-initiated oxidation of human low-density lipoprotein. J. Am. Chem. Soc. 1993;115:6029–6044. [Google Scholar]
- 59.Bowry VW, Mohr D, Cleary J, Stocker R. Prevention of tocopherol-mediated peroxidation of ubiquinol-10-free human low density lipoprotein. J Biol Chem. 1995;270:5756–5763. doi: 10.1074/jbc.270.11.5756. [DOI] [PubMed] [Google Scholar]
- 60.Upston JM, Terentis AC, Stocker R. Tocopherol-mediated peroxidation (TMP) of lipoproteins: implications for vitamin E as a potential anti-atherogenic supplement. FASEB J. 1999;13:977–994. doi: 10.1096/fasebj.13.9.977. [DOI] [PubMed] [Google Scholar]
- 61.Stary HC. Evolution and progression of atherosclerotic lesions in coronary arteries of children and young adults. Arteriosclerosis. 1989;9:I19–I32. [PubMed] [Google Scholar]
- 62.Zieske AW, Malcom GT, Strong JP. Natural history and risk factors of atherosclerosis in children and youth: the PDAY study. Pediatr. Pathol. Mol. Med. 2002;21:213–237. doi: 10.1080/15227950252852104. [DOI] [PubMed] [Google Scholar]
- 63.Palinski W, Napoli C. The fetal origins of atherosclerosis: Maternal hypercholesterolemia and cholesterol-lowering or antioxidant treatment during pregnancy influence in utero programming and postnatal susceptibility to atherogenesis. FASEB J. 2002;16:1348–1360. doi: 10.1096/fj.02-0226rev. [DOI] [PubMed] [Google Scholar]
- 64.Napoli C, Glass CK, Witztum JL, Deutsch R, D'Armiento FP, Palinski W. Influence of maternal hypercholesterolaemia during pregnancy on progression of early atherosclerotic lesions in childhood: Fate of Early Lesions in Children (FELIC) study. Lancet. 1999;354:1234–1241. doi: 10.1016/S0140-6736(99)02131-5. [DOI] [PubMed] [Google Scholar]
- 65.Napoli C, D'Armiento FP, Mancini FP, Postiglione A, Witztum JL, Palumbo G, Palinski W. Fatty streak formation occurs in human fetal aortas and is greatly enhanced by maternal hypercholesterolemia. Intimal accumulation of low density lipoprotein and its oxidation precede monocyte recruitment into early atherosclerotic lesions. J.Clin. Invest. 1997;100:2680–2690. doi: 10.1172/JCI119813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Homma S, Troxclair DA, Zieske AW, Malcom GT, Strong JP. The Pathobiological Determinants of Atherosclerosis in Youth (PDAY) Research Group. Histological topographical comparisons of atherosclerosis progression in juveniles and young adults. Atherosclerosis. 2007 doi: 10.1016/j.atherosclerosis.2007.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.McMahan CA, Gidding SS, Malcom GT, Schreiner PJ, Strong JP, Tracy RE, Williams OD, McGill HC. Pathobiological Determinants of Atherosclerosis in Youth (PDAY) Research Group. Comparison of coronary heart disease risk factors in autopsied young adults from the PDAY Study with living young adults from the CARDIA study. Cardiovasc. Pathol. 2007;16:151–158. doi: 10.1016/j.carpath.2006.12.003. [DOI] [PubMed] [Google Scholar]
- 68.McGill HC, Jr, Herderick EE, McMahan CA, Zieske AW, Malcolm GT, Tracy RE, Strong JP. Atherosclerosis in youth. Minerva Pediatr. 2002;54:437–447. [PubMed] [Google Scholar]
- 69.Ross R. Atherosclerosis--an inflammatory disease. N. Engl. J. Medicine. 1999;340:115–126. doi: 10.1056/NEJM199901143400207. [DOI] [PubMed] [Google Scholar]
- 70.Stary H, Chandler A, Glagov S, Guyton J, Insull W, Jr, Rosenfeld M, Schaffer SA, Schwartz CJ, Wagner WD, Wissler RW. A definition of initial, fatty streak, and intermediate lesions of atherosclerosis. A report from the Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart Association. Arterioscler. Thromb. 1994;14:840–856. doi: 10.1161/01.atv.14.5.840. [DOI] [PubMed] [Google Scholar]
- 71.Eggen DA, Strong JP, McGill HC. Coronary calcification: relationship to clinically significant coronary lesions and race, sex and topographic distribution. Circulation. 1965;32:948–955. doi: 10.1161/01.cir.32.6.948. [DOI] [PubMed] [Google Scholar]
- 72.Virmani R, Ladich ER, Burke AP, Kolodgie FD. Histopathology of carotid atherosclerotic disease. Neurosurgery. 2006;59:s219–s227. doi: 10.1227/01.NEU.0000239895.00373.E4. [DOI] [PubMed] [Google Scholar]
- 73.Virmani R, Burke AP, Farb A, Kolodgie FD. Pathology of the vulnerable plaque. J. Am. Coll. Cardiol. 2006;47:C13–C18. doi: 10.1016/j.jacc.2005.10.065. [DOI] [PubMed] [Google Scholar]
- 74.Johnson RC, Leopold JA, Loscalzo J. Vascular calcification: pathobiological mechanisms and clinical implications. Circ Res. 2006;99:1044–1059. doi: 10.1161/01.RES.0000249379.55535.21. [DOI] [PubMed] [Google Scholar]
- 75.Mackey RH, Venkitachalam L, Sutton-Tyrrell K. Calcifications, arterial stiffness and atherosclerosis. Adv. Cardiol. 2007;44:234–244. doi: 10.1159/000096744. [DOI] [PubMed] [Google Scholar]
- 76.Mazzini MJ, Schulze PC. Proatherogenic pathways leading to vascular calcification. Eur. J. Radiol. 2006;57:384–389. doi: 10.1016/j.ejrad.2005.12.025. [DOI] [PubMed] [Google Scholar]
- 77.Abedin M, Tintut Y, Demer LL. Vascular calcification: mechanisms and clinical ramifications. Arterioscler. Thromb. Vasc. Biol. 2004;24:1161–1170. doi: 10.1161/01.ATV.0000133194.94939.42. [DOI] [PubMed] [Google Scholar]
- 78.Stary HC. Natural history of calcium deposits in atherosclerosis progression and regression. Z Kardiol. 2000;89:28–35. doi: 10.1007/s003920070097. [DOI] [PubMed] [Google Scholar]
- 79.Vengrenyuk Y, Carlier S, Xanthos S, Cardoso L, Ganatos P, Virmani R, Einav S, Gilchrist L, Weinbaum S. A hypothesis for vulnerable plaque rupture due to stress-induced debonding around cellular microcalcifications in thin fibrous caps. Proc. Natl. Acad. Sci. U S A. 2006;103:14678–14683. doi: 10.1073/pnas.0606310103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Virmani R, Burke AP, Farb A, Kolodgie FD. Pathology of the vulnerable plaque. J. Am. Coll. Cardiol. 2006;47:c13–c18. doi: 10.1016/j.jacc.2005.10.065. [DOI] [PubMed] [Google Scholar]
- 81.Beadenkopf WG, Assaad DS, Love BM. Calcification in the coronary arteries and its relationship to arteriosclerosis and myocardial infarction. JAMA. 1964;92:865–871. [PubMed] [Google Scholar]
- 82.Speer MY, Giachelli CM. Regulation of cardiovascular calcification. Cardiovasc. Pathol. 2004;13:63–70. doi: 10.1016/S1054-8807(03)00130-3. [DOI] [PubMed] [Google Scholar]
- 83.Doherty TM, Detrano RC. Coronary arterial calcification as an active process: a new perspective on an old problem. Calcif. Tissue Int. 1994;54:224–230. doi: 10.1007/BF00301683. [DOI] [PubMed] [Google Scholar]
- 84.Watson KE. Pathophysiology of coronary calcification. J. Cardiovasc. Risk. 2000;7:93–97. doi: 10.1177/204748730000700202. [DOI] [PubMed] [Google Scholar]
- 85.Guo W, Morrisett J, DeBakey M, Lawrie G, Hamilton J. Quantification in situ of crystalline cholesterol and calcium phosphate hydroxyapatite in human atherosclerotic plaques by solid-state magic angle spinning NMR, Arterioscler. Thromb. Vasc. Biol. 2000;20:1630–1636. doi: 10.1161/01.atv.20.6.1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Nadra I, Mason J, Philippidis P, Florey O, Smythe C, McCarthy G, Landis RC, Haskard DO. Proinflammatory activation of macrophages by basic calcium phosphate crystals via protein kinase C and MAP kinase pathways: a vicious cycle of inflammation and arterial calcification? Circ. Res. 2005;96:1248–1256. doi: 10.1161/01.RES.0000171451.88616.c2. [DOI] [PubMed] [Google Scholar]
- 87.Cozzolino M, Dusso AS, Slatopolsky E. Role of calcium-phosphate product and bone-associated proteins on vascular calcification in renal failure. J. Am. Soc. Nephrol. 2001;12:2511–2516. doi: 10.1681/ASN.V12112511. [DOI] [PubMed] [Google Scholar]
- 88.Fischer JW, Steitz SA, Johnson PY, Burke A, Kolodgie F, Virmani R, Giachelli C, Wight TN. Decorin promotes aortic smooth muscle cell calcification and colocalizes to calcified regions in human atherosclerotic lesions. Arterioscler. Thromb. Vasc. Biol. 2004;24:2391–2396. doi: 10.1161/01.ATV.0000147029.63303.28. [DOI] [PubMed] [Google Scholar]
- 89.Bini A, Mann KG, Kudryk BJ, Schoen FJ. Noncollagenous bone matrix proteins, calcification, and thrombosis in carotid artery atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 1999;19:1852–1861. doi: 10.1161/01.atv.19.8.1852. [DOI] [PubMed] [Google Scholar]
- 90.Berenson G, Radhakrishnamurthy B, Srinivasan S, Vijayagopal P, Dalferes E. Proteoglycans and potential mechanisms related to atherosclerosis. Ann. N Y Acad. Sci. 1985;454:69–78. doi: 10.1111/j.1749-6632.1985.tb11845.x. [DOI] [PubMed] [Google Scholar]
- 91.Wight TN, Merrilees MJ. Proteoglycans in atherosclerosis and restenosis: key roles for versican. Circ. Res. 2004;94:1158–1167. doi: 10.1161/01.RES.0000126921.29919.51. [DOI] [PubMed] [Google Scholar]
- 92.Libby P. Changing concepts of atherogenesis. J. Intern. Med. 2000;247:349–358. doi: 10.1046/j.1365-2796.2000.00654.x. [DOI] [PubMed] [Google Scholar]
- 93.Croce K, Libby P. Intertwining of thrombosis and inflammation in atherosclerosis. Curr. Opin. Hematol. 2007;14:55–61. doi: 10.1097/00062752-200701000-00011. [DOI] [PubMed] [Google Scholar]
- 94.Viles-Gonzalez JF, Fuster V, Badimon JJ. Links between inflammation and thrombogenicity in atherosclerosis. Curr. Mol. Med. 2006;6:489–499. doi: 10.2174/156652406778018707. [DOI] [PubMed] [Google Scholar]
- 95.Yan ZQ, Hansson GK. Innate immunity, macrophage activation, and atherosclerosis. Immunol. Rev. 2007;219:187–203. doi: 10.1111/j.1600-065X.2007.00554.x. [DOI] [PubMed] [Google Scholar]
- 96.Hansson GK, Libby P. The immune response in atherosclerosis: a double-edged sword. Nat. Rev. Immunol. 2006;7:508–519. doi: 10.1038/nri1882. [DOI] [PubMed] [Google Scholar]
- 97.Ait-Oufella H, Salomon BL, Potteaux S, Robertson AK, Gourdy P, Zoll J, Merval R, Esposito B, Cohen JL, Fisson S, Flavell RA, Hansson GK, Klatzmann D, Tedgui A, Mallat Z. Natural regulatory T cells control the development of atherosclerosis in mice. Nat Med. 2006;12:178–180. doi: 10.1038/nm1343. [DOI] [PubMed] [Google Scholar]
- 98.Tobias PS, Curtiss LK. Toll-like receptors in atherosclerosis. Biochem. Soc. Trans. 2007;35:1453–1455. doi: 10.1042/BST0351453. [DOI] [PubMed] [Google Scholar]
- 99.Xing L, Remick DG. Mechanisms of oxidant regulation of monocyte chemotactic protein 1 production in human whole blood and isolated mononuclear cells. Shock. 2007;28:178–185. doi: 10.1097/shk.0b013e3180311cf4. [DOI] [PubMed] [Google Scholar]
- 100.Verhasselt V, Goldman M, Willems F. Oxidative stress up-regulates IL-8 and TNF-alpha synthesis by human dendritic cells. Eur J Immunol. 1998;28:3886–3890. doi: 10.1002/(SICI)1521-4141(199811)28:11<3886::AID-IMMU3886>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 101.Jaimes EA, Nath KA, Raij L. Hydrogen peroxide downregulates IL-1-driven mesangial iNOS activity: implications for glomerulonephritis. Am. J. Physiol. 1997;272:f721–f728. doi: 10.1152/ajprenal.1997.272.6.F721. [DOI] [PubMed] [Google Scholar]
- 102.Brenneisen P, Briviba K, Wlaschek M, Wenk J, Scharffetter-Kochanek K. Hydrogen peroxide (H2O2) increases the steady-state mRNA levels of collagenase/MMP-1 in human dermal fibroblasts. Free Radic. Biol. Med. 1997;22:515–524. doi: 10.1016/s0891-5849(96)00404-2. [DOI] [PubMed] [Google Scholar]
- 103.Schnurr K, Borchert A, Kuhn H. Inverse regulation of lipid-peroxidizing and hydroperoxyl lipid-reducing enzymes by interleukins 4 and 13. FASEB J. 1999;13:143–154. doi: 10.1096/fasebj.13.1.143. [DOI] [PubMed] [Google Scholar]
- 104.Hsu HY, Chiu SL, Wen MH, Chen KY, Hua KF. Ligands of macrophage scavenger receptor induce cytokine expression via differential modulation of protein kinase signaling pathways. J Biol Chem. 2001;276:28719–28730. doi: 10.1074/jbc.M011117200. [DOI] [PubMed] [Google Scholar]
- 105.Klouche M, Gottschling S, Gerl V, Hell W, Husmann M, Dorweiler B, Messner M, Bhakdi S. Atherogenic properties of enzymatically degraded LDL: selective induction of MCP-1 and cytotoxic effects on human macrophages. Arterioscler. Thromb. Vasc. Biol. 1998;18:1376–1385. doi: 10.1161/01.atv.18.9.1376. [DOI] [PubMed] [Google Scholar]
- 106.Frostegård J, Kjellman B, Gidlund M, Andersson B, Jindal S, Kiessling R. Induction of heat shock protein in monocytic cells by oxidized low density lipoprotein. Atherosclerosis. 1996;121:93–103. doi: 10.1016/0021-9150(95)05706-4. [DOI] [PubMed] [Google Scholar]
- 107.Thai SF, Lewis JG, Williams RB, Johnson SP, Adams DO. Effects of oxidized LDL on mononuclear phagocytes: inhibition of induction of four inflammatory cytokine gene RNAs, release of NO, and cytolysis of tumor cells. J. Leukoc. Biol. 1995;57:427–433. doi: 10.1002/jlb.57.3.427. [DOI] [PubMed] [Google Scholar]
- 108.Brand K, Banka CL, Mackman N, Terkeltaub RA, Fan ST, Curtiss LK. Oxidized LDL enhances lipopolysaccharide-induced tissue factor expression in human adherent monocytes. Arterioscler. Thromb. 1994;14:790–797. doi: 10.1161/01.atv.14.5.790. [DOI] [PubMed] [Google Scholar]
- 109.Ku G, Thomas CE, Akeson AL, Jackson RL. Induction of interleukin 1 beta expression from human peripheral blood monocyte-derived macrophages by 9-hydroxyoctadecadienoic acid. J. Biol. Chem. 1992;267:14183–14188. [PubMed] [Google Scholar]
- 110.Frostegård J, Wu R, Giscombe R, Holm G, Lefvert AK, Nilsson J. Induction of T-cell activation by oxidized low density lipoprotein. Arterioscler Thromb. 1992;12:461–467. doi: 10.1161/01.atv.12.4.461. [DOI] [PubMed] [Google Scholar]
- 111.Lipton BA, Parthasarathy S, Ord VA, Clinton SK, Libby P, Rosenfeld ME. Components of the protein fraction of oxidized low density lipoprotein stimulate interleukin-1 alpha production by rabbit arterial macrophage-derived foam cells. J. Lipid Res. 1995;36:2232–2242. [PubMed] [Google Scholar]
- 112.Miller YI, Viriyakosol S, Worrall DS, Boullier A, Butler S, Witztum JL. Toll-like receptor 4-dependent and -independent cytokine secretion induced by minimally oxidized low-density lipoprotein in macrophages. Arterioscler. Thromb. Vasc. Biol. 2005;25:1213–1219. doi: 10.1161/01.ATV.0000159891.73193.31. [DOI] [PubMed] [Google Scholar]
- 113.Subbanagounder G, Wong JW, Lee H, Faull KF, Miller E, Witztum JL, Berliner JA. Epoxyisoprostane and epoxycyclopentenone phospholipids regulate monocyte chemotactic protein-1 and interleukin-8 synthesis. Formation of these oxidized phospholipids in response to interleukin-1beta. J. Biol. Chem. 2002;277:7271–7281. doi: 10.1074/jbc.M107602200. [DOI] [PubMed] [Google Scholar]
- 114.Xiao D, Wang Z, She M. Minimally modified low-density lipoprotein induces monocyte chemotactic protein-1 expression in vivo and a novel model for monocyte adhesion to arterial intima. Chin. Med. J. (Engl) 1999;112:438–442. [PubMed] [Google Scholar]
- 115.Cushing SD, Berliner JA, Valente AJ, Territo MC, Navab M, Parhami F, Gerrity R, Schwartz CJ, Fogelman AM. Minimally modified low density lipoprotein induces monocyte chemotactic protein 1 in human endothelial cells and smooth muscle cells. Proc. Natl. Acad. Sci. U S A. 1990;87:5134–5138. doi: 10.1073/pnas.87.13.5134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Zalba G, Fortuño A, Orbe J, San José G, Moreno MU, Belzunce M, Rodríguez, JA, Beloqui O, Páramo JA, Díez J. Phagocytic NADPH oxidase-dependent superoxide production stimulates matrix metalloproteinase-9: implications for human atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2007;27:587–593. doi: 10.1161/01.ATV.0000256467.25384.c6. [DOI] [PubMed] [Google Scholar]
- 117.Moshal KS, Sen U, Tyagi N, Henderson B, Steed M, Ovechkin AV, Tyagi SC. Regulation of homocysteine-induced MMP-9 by ERK1/2 pathway. Am. J. Physiol. Cell Physiol. 2006;290:C883–C891. doi: 10.1152/ajpcell.00359.2005. [DOI] [PubMed] [Google Scholar]
- 118.Scholz H, Aukrust P, Damås JK, Tonstad S, Sagen EL, Kolset SO, Hall C, Yndestad A, Halvorsen B. 8-isoprostane increases scavenger receptor A and matrix metalloproteinase activity in THP-1 macrophages, resulting in long-lived foam cells. Eur. J. Clin. Invest. 2004;34:451–458. doi: 10.1111/j.1365-2362.2004.01376.x. [DOI] [PubMed] [Google Scholar]
- 119.Kameda K, Matsunaga T, Abe N, Hanada H, Ishizaka H, Ono H, Saitoh M, Fukui K, Fukuda I, Osanai T, Okumura K. Correlation of oxidative stress with activity of matrix metalloproteinase in patients with coronary artery disease Possible role for left ventricular remodeling. Eur. Heart J. 2003;24:2180–2185. doi: 10.1016/j.ehj.2003.09.022. [DOI] [PubMed] [Google Scholar]
- 120.Kolev K, Skopál J, Simon L, Csonka E, Machovich R, Nagy Z. Matrix metalloproteinase-9 expression in post-hypoxic human brain capillary endothelial cells: H2O2 as a trigger and NF-kappa B as a signal transducer. Thromb. Haemost. 2003;90:528–537. doi: 10.1160/TH03-02-0070. [DOI] [PubMed] [Google Scholar]
- 121.Siwik DA, Pagano PJ, Colucci WS. Oxidative stress regulates collagen synthesis and matrix metalloproteinase activity in cardiac fibroblasts. Am. J. Physiol. Cell Physiol. 2001;280:c53–c60. doi: 10.1152/ajpcell.2001.280.1.C53. [DOI] [PubMed] [Google Scholar]
- 122.Kaplan M, Aviram M. Oxidized low density lipoprotein: atherogenic and proinflammatory characteristics during macrophage foam cell formation. An inhibitory role for nutritional antioxidants and serum paraoxonase. Clin. Chem. Lab. Med. 1999;37:777–787. doi: 10.1515/CCLM.1999.118. [DOI] [PubMed] [Google Scholar]
- 123.Rajavashisth TB, Yamada H, Mishra NK. Transcriptional activation of the macrophage-colony stimulating factor gene by minimally modified LDL. Involvement of nuclear factor-kappa B. Arterioscler. Thromb. Vasc. Biol. 1995;15:1591–1598. doi: 10.1161/01.atv.15.10.1591. [DOI] [PubMed] [Google Scholar]
- 124.Parhami F, Fang ZT, Fogelman AM, Andalibi A, Territo MC, Berliner JA. Minimally modified low density lipoprotein-induced inflammatory responses in endothelial cells are mediated by cyclic adenosine monophosphate. J. Clin. Invest. 1993;92:471–478. doi: 10.1172/JCI116590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Berliner JA, Schwartz DS, Territo MC, Andalibi A, Almada L, Lusis AJ, Quismorio D, Fang ZP, Fogelman AM. Induction of chemotactic cytokines by minimally oxidized LDL. Adv. Exp. Med. Biol. 1993;351:13–18. doi: 10.1007/978-1-4615-2952-1_2. [DOI] [PubMed] [Google Scholar]
- 126.Bemeur C, Ste-Marie L, Montgomery J. Increased oxidative stress during hyperglycemic cerebral ischemia. Neurochem Int. 2007;50:890–904. doi: 10.1016/j.neuint.2007.03.002. [DOI] [PubMed] [Google Scholar]
- 127.Ceriello A. Oxidative stress and diabetes-associated complications. Endocr. Pract. 2006;12:S60–S62. doi: 10.4158/EP.12.S1.60. [DOI] [PubMed] [Google Scholar]
- 128.Rolo AP, Palmeira CM. Diabetes and mitochondrial function: role of hyperglycemia and oxidative stress. Toxicol. Appl. Pharmacol. 2006;212:167–178. doi: 10.1016/j.taap.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 129.Bonnefont-Rousselot D. Glucose and reactive oxygen species. Curr. Opin. Clin. Nutr. Metab. Care. 2002;5:561–568. doi: 10.1097/00075197-200209000-00016. [DOI] [PubMed] [Google Scholar]
- 130.Hunt JV, Dean RT, Wolff SP. Hydroxyl radical production and autoxidative glycosylation. Glucose autoxidation as the cause of protein damage in the experimental glycation model of diabetes mellitus and ageing. Biochem J. 1988;256:205–212. doi: 10.1042/bj2560205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Natarajan R, Gerrity RG, Gu JL, Lanting L, Thomas L, Nadler JL. Role of 12-lipoxygenase and oxidant stress in hyperglycaemia-induced acceleration of atherosclerosis in a diabetic pig model. Diabetologia. 2002;45:125–133. doi: 10.1007/s125-002-8253-x. [DOI] [PubMed] [Google Scholar]
- 132.Natarajan R, Gu JL, Rossi J, Gonzales N, Lanting L, Xu L, Nadler J. Elevated glucose and angiotensin II increase 12-lipoxygenase activity and expression in porcine aortic smooth muscle cells. Proc Natl Acad Sci U S A. 1993;90:4947–4951. doi: 10.1073/pnas.90.11.4947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Masaki H, Okano Y, Sakurai H. Generation of active oxygen species from advanced glycation end-products (AGE) under ultraviolet light A (UVA) irradiation. Biochem. Biophys. Res. Commun. 1997;235:306–310. doi: 10.1006/bbrc.1997.6780. [DOI] [PubMed] [Google Scholar]
- 134.Schmidt AM, Hori O, Brett J, Yan SD, Wautier JL, Stern D. Cellular receptors for advanced glycation end products. Implications for induction of oxidant stress and cellular dysfunction in the pathogenesis of vascular lesions. Arterioscler. Thromb. 1994;14:1521–1528. doi: 10.1161/01.atv.14.10.1521. [DOI] [PubMed] [Google Scholar]
- 135.Al-Abed Y, Liebich H, Voelter W, Bucala R. Hydroxyalkenal formation induced by advanced glycosylation of low density lipoprotein. J. Biol. Chem. 1996;271:2892–2896. doi: 10.1074/jbc.271.6.2892. [DOI] [PubMed] [Google Scholar]
- 136.Bucala R, Makita Z, Koschinsky T, Cerami A, Vlassara H. Lipid advanced glycosylation: pathway for lipid oxidation in vivo. Proc Natl Acad Sci U S A. 1993;90:6434–6438. doi: 10.1073/pnas.90.14.6434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Mehta PK, Griendling KK. Angiotensin II cell signaling: physiological and pathological effects in the cardiovascular system. Am. J. Physiol. Cell Physiol. 2007;292:C82–C97. doi: 10.1152/ajpcell.00287.2006. [DOI] [PubMed] [Google Scholar]
- 138.Ushio-Fukai M, Alexander RW. Reactive oxygen species as mediators of angiogenesis signaling: role of NAD(P)H oxidase. Mol. Cell. Biochem. 2004;264:85–97. doi: 10.1023/b:mcbi.0000044378.09409.b5. [DOI] [PubMed] [Google Scholar]
- 139.Ushio-Fukai M. Redox signaling in angiogenesis: role of NADPH oxidase. Cardiovasc. Res. 2006;71:226–235. doi: 10.1016/j.cardiores.2006.04.015. [DOI] [PubMed] [Google Scholar]
- 140.Touyz RM. Reactive oxygen species as mediators of calcium signaling by angiotensin II: implications in vascular physiology and pathophysiology. Antioxid. Redox. Signal. 2005;7:1302–1314. doi: 10.1089/ars.2005.7.1302. [DOI] [PubMed] [Google Scholar]
- 141.Beckman JS, Beckman TW, Chen J, Marshall PA, Freeman BA. Apparent hydroxyl radical production by peroxynitrite: implications for endothelial injury from nitric oxide and superoxide. Proc Natl Acad Sci U S A. 1990;87:1620–1624. doi: 10.1073/pnas.87.4.1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Chalmers A. Smoking and oxidative stress. Am. J. Clin. Nutr. 1999;69:572. doi: 10.1093/ajcn/69.3.572. [DOI] [PubMed] [Google Scholar]
- 143.Cross CE, Van der Vliet A, Eiserich JP. Cigarette smokers and oxidant stress: a continuing mystery. Am. J. Clin. Nutr. 1998;67:184–185. doi: 10.1093/ajcn/67.2.184. [DOI] [PubMed] [Google Scholar]
- 144.Burke A, Fitzgerald GA. Oxidative stress and smoking-induced vascular injury. Prog. Cardiovasc. Dis. 2003;46:79–90. doi: 10.1016/s0033-0620(03)00076-8. [DOI] [PubMed] [Google Scholar]
- 145.Yokode M, Ueyama K, Arai NH, Ueda Y, Kita T. Modification of high- and low-density lipoproteins by cigarette smoke oxidants. Ann. N. Y. Acad. Sci. 1996;15:245–251. doi: 10.1111/j.1749-6632.1996.tb39067.x. [DOI] [PubMed] [Google Scholar]
- 146.Yamaguchi Y, Matsuno S, Kagota S, Haginaka J, Kunitomo M. Oxidants in cigarette smoke extract modify low-density lipoprotein in the plasma and facilitate atherogenesis in the aorta of Watanabe heritable hyperlipidemic rabbits. Atherosclerosis. 2001;156:109–117. doi: 10.1016/s0021-9150(00)00637-7. [DOI] [PubMed] [Google Scholar]
- 147.Bloomer RJ. Decreased blood antioxidant capacity and increased lipid peroxidation in youngcigarette smokers compared to nonsmokers: Impact of dietary intake. Nutr. J. 2007;6:39. doi: 10.1186/1475-2891-6-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Frei B, Forte TM, Ames BN, Cross CE. Gas phase oxidants of cigarette smoke induce lipid peroxidation and changes in lipoprotein properties in human blood plasma. Protective effects of ascorbic acid. Biochem. J. 1991;277:133–138. doi: 10.1042/bj2770133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Bouloumie A, Marumo T, Lafontan M, Busse R. Leptin induces oxidative stress in human endothelial cells. FASEB J. 1999;13:1231–1238. [PubMed] [Google Scholar]
- 150.Penumetcha M, Parthasarathy S. unpublished observations. [Google Scholar]
- 151.Balakrishnan SD, Anuradha CV. Exercise, depletion of antioxidants and antioxidant manipulation. Cell Biochem. Funct. 1998;16:269–275. doi: 10.1002/(SICI)1099-0844(1998120)16:4<269::AID-CBF797>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 152.Evans WJ. Muscle damage: nutritional considerations. Int J Sport Nutr. 1991;1:214–224. doi: 10.1123/ijsn.1.3.214. [DOI] [PubMed] [Google Scholar]
- 153.Clarkson PM. Micronutrients and exercise: anti-oxidants and minerals. J. Sports Sci. 1995;13:S11–S24. doi: 10.1080/02640419508732272. [DOI] [PubMed] [Google Scholar]
- 154.Ji LL. Oxidative stress during exercise: implication of antioxidant nutrients. Free Radic. Biol. Med. 1995;18:1079–1086. doi: 10.1016/0891-5849(94)00212-3. [DOI] [PubMed] [Google Scholar]
- 155.Johnston CS, Swan PD, Corte C. Substrate utilization and work efficiency during submaximal exercise in vitamin C depleted-repleted adults. Int. J. Vitam. Nutr. Res. 1999;69:41–44. doi: 10.1024/0300-9831.69.1.41. [DOI] [PubMed] [Google Scholar]
- 156.Wetzstein CJ, Shern-Brewer RA, Santanam N, Green NR, White-Welkley JE, Parthasarathy S. Does acute exercise affect the susceptibility of low density lipoprotein to oxidation? Free Radic. Biol. Med. 1998;24:679–682. doi: 10.1016/s0891-5849(97)00320-1. [DOI] [PubMed] [Google Scholar]
- 157.Shern-Brewer R, Santanam N, Wetzstein C, White-Welkley J, Parthasarathy S. Exercise and cardiovascular disease: a new perspective. Arterioscler. Thromb. Vasc. Biol. 1998;18:1181–1187. doi: 10.1161/01.atv.18.7.1181. [DOI] [PubMed] [Google Scholar]
- 158.Parthasarathy S, Khan-Merchant N, Penumetcha M, Santanam N. Oxidative stress in cardiovascular disease. J Nucl Cardiol. 2001;8:379–389. doi: 10.1067/mnc.2001.114150. [DOI] [PubMed] [Google Scholar]
- 159.Tribble DL, Holl LG, Wood PD, Krauss RM. Variations in oxidative susceptibility among six low density lipoprotein subfractions of differing density and particle size. Atherosclerosis. 1992;93:189–199. doi: 10.1016/0021-9150(92)90255-f. [DOI] [PubMed] [Google Scholar]
- 160.Chait A, Brazg RL, Tribble DL, Krauss RM. Susceptibility of small, dense, low-density lipoproteins to oxidative modification in subjects with the atherogenic lipoprotein phenotype, pattern B. Am. J. Med. 1993;94:350–356. doi: 10.1016/0002-9343(93)90144-e. [DOI] [PubMed] [Google Scholar]
- 161.Tribble DL, Rizzo M, Chait A, Lewis DM, Blanche PJ, Krauss RM. Enhanced oxidative susceptibility and reduced antioxidant content of metabolic precursors of small, dense low-density lipoproteins. Am. J. Med. 2001;110:103–110. doi: 10.1016/s0002-9343(00)00700-2. [DOI] [PubMed] [Google Scholar]
- 162.Meilhac O, Zhou M, Santanam N, Parthasarathy S. Lipid peroxides induce expression of catalase in cultured vascular cells. J. Lipid Res. 2000;41:1205–1213. [PubMed] [Google Scholar]
- 163.Moellering DR, Levonen AL, Go YM, Patel RP, Dickinson DA, Forman HJ, Darley-Usmar VM. Induction of glutathione synthesis by oxidized low-density lipoprotein and 1-palmitoyl-2-arachidonyl phosphatidylcholine: protection against quinone-mediated oxidative stress. Biochem J. 2002;362:51–59. doi: 10.1042/0264-6021:3620051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Lin F, Thomas JP, Girotti AW. Hyperexpression of catalase in selenium-deprived murine L1210 cells. Arch. Biochem. Biophys. 1993;305:176–185. doi: 10.1006/abbi.1993.1408. [DOI] [PubMed] [Google Scholar]
- 165.Ramasamy S, Parthasarathy S, Harrison DG. Regulation of endothelial nitric oxide synthase gene expression by oxidized linoleic acid. J Lipid Res. 1998;39:268–276. [PubMed] [Google Scholar]
- 166.Shatrov VA, Brüne B. Induced expression of manganese superoxide dismutase by non-toxic concentrations of oxidized low-density lipoprotein (oxLDL) protects against oxLDL-mediated cytotoxicity. Biochem J. 2003;374:505–511. doi: 10.1042/BJ20030420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Kinscherf R, Deigner HP, Usinger C, Pill J, Wagner M, Kamencic H, Hou D, Chen M, Schmiedt W, Schrader M, Kovacs G, Kato K, Metz J. Induction of mitochondrial manganese superoxide dismutase in macrophages by oxidized LDL: its relevance in atherosclerosis of humans and heritable hyperlipidemic rabbits. FASEB J. 1997;11:1317–1328. doi: 10.1096/fasebj.11.14.9409551. [DOI] [PubMed] [Google Scholar]
- 168.Agarwal A, Shiraishi F, Visner GA, Nick HS. Linoleoyl hydroperoxide transcriptionally upregulates heme oxygenase-1 gene expression in human renal epithelial and aortic endothelial cells. J. Am. Soc. Nephrol. 1998;9:1990–1997. doi: 10.1681/ASN.V9111990. [DOI] [PubMed] [Google Scholar]
- 169.Anwar AA, Li FY, Leake DS, Ishii T, Mann GE, Siow RC. Induction of heme oxygenase 1 by moderately oxidized low-density lipoproteins in human vascular smooth muscle cells: role of mitogen-activated protein kinases and Nrf2. Free Radic. Biol. Med. 2005;39:227–236. doi: 10.1016/j.freeradbiomed.2005.03.012. [DOI] [PubMed] [Google Scholar]
- 170.Rong R, Ramachandran S, Penumetcha M, Khan N, Parthasarathy S. Dietary oxidized fatty acids may enhance intestinal apolipoprotein A-I production. J. Lipid Res. 2002;43:557–564. [PubMed] [Google Scholar]
- 171.Garelnabi M, Selvarajan K, Litvinov D, Santanam N, Parthasarathy S. Dietary oxidized linoleic acid lowers triglycerides via APOA5/APOClll dependent mechanisms. Atherosclerosis. 2007 doi: 10.1016/j.atherosclerosis.2007.12.026. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Sülzle A, Hirche F, Eder K. Thermally oxidized dietary fat upregulates the expression of target genes of PPAR alpha in rat liver. J. Nutr. 2004;134:1375–1383. doi: 10.1093/jn/134.6.1375. [DOI] [PubMed] [Google Scholar]
- 173.Bull AW, Steffensen KR, Leers J, Rafter JJ. Activation of PPAR gamma in colon tumor cell lines by oxidized metabolites of linoleic acid, endogenous ligands for PPAR gamma. Carcinogenesis. 2003;24:1717–1722. doi: 10.1093/carcin/bgg131. [DOI] [PubMed] [Google Scholar]
- 174.Delerive P, Furman C, Teissier E, Fruchart J, Duriez P, Staels B. Oxidized phospholipids activate PPARalpha in a phospholipase A2-dependent manner. FEBS Lett. 2000;471:34–38. doi: 10.1016/s0014-5793(00)01364-8. [DOI] [PubMed] [Google Scholar]
- 175.Greenberg ME, Sun M, Zhang R, Febbraio M, Silverstein R, Hazen SL. Oxidized phosphatidylserine-CD36 interactions play an essential role in macrophage-dependent phagocytosis of apoptotic cells. J. Exp. Med. 2006;203:2613–2625. doi: 10.1084/jem.20060370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Matsura T, Togawa A, Kai M, Nishida T, Nakada J, Ishibe Y, Kojo S, Yamamoto Y, Yamada K. The presence of oxidized phosphatidylserine on Fasmediated apoptotic cell surface. Biochim. Biophys. Acta. 2005;1736:181–188. doi: 10.1016/j.bbalip.2005.08.011. [DOI] [PubMed] [Google Scholar]
- 177.Tyurina YY, Tyurin VA, Zhao Q, Djukic M, Quinn PJ, Pitt BR, Kagan VE. Oxidation of phosphatidylserine: a mechanism for plasma membrane phospholipid scrambling during apoptosis? Biochem. Biophys. Res. Commun. 2004;324:1059–1064. doi: 10.1016/j.bbrc.2004.09.102. [DOI] [PubMed] [Google Scholar]
- 178.Caron G, Ermondi G, Testa B. Predicting the oxidative metabolism of statins: an application of the MetaSite algorithm. Pharm. Res. 2007;24:480–501. doi: 10.1007/s11095-006-9199-7. [DOI] [PubMed] [Google Scholar]
- 179.Parthasarathy S, Khoo JC, Miller E, Barnett J, Witztum JL, Steinberg D. Low density lipoprotein rich in oleic acid is protected against oxidative modification: implications for dietary prevention of atherosclerosis. Proc Natl Acad Sci U S A. 1990;87:3894–3898. doi: 10.1073/pnas.87.10.3894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Reaven P, Parthasarathy S, Grasse BJ, Miller E, Steinberg D, Witztum JL. Effects of oleate-rich and linoleate-rich diets on the susceptibility of low density lipoprotein to oxidative modification in mildly hypercholesterolemic subjects. J. Clin. Invest. 1993;91:668–676. doi: 10.1172/JCI116247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Reaven PD, Grasse BJ, Tribble DL. Effects of linoleate-enriched and oleate-enriched diets in combination with alpha-tocopherol on the susceptibility of LDL and LDL subfractions to oxidative modification in humans. Arterioscler Thromb. 1994;14:557–566. doi: 10.1161/01.atv.14.4.557. [DOI] [PubMed] [Google Scholar]
- 182.Lada AT, Rudel LL. Dietary monounsaturated versus polyunsaturated fatty acids: which is really better for protection from coronary heart disease? Curr. Opin. Lipidol. 2003;14:41–46. doi: 10.1097/00041433-200302000-00008. [DOI] [PubMed] [Google Scholar]
- 183.Rudel LL, Kelley K, Sawyer JK, Shah R, Wilson MD. Dietary monounsaturated fatty acids promote aortic atherosclerosis in LDL receptor-null, human ApoB100-overexpressing transgenic mice. Arterioscler. Thromb. Vasc. Biol. 1998;18:1818–1827. doi: 10.1161/01.atv.18.11.1818. [DOI] [PubMed] [Google Scholar]
- 184.Rudel LL, Parks JS, Sawyer JK. Compared with dietary monounsaturated and saturated fat, polyunsaturated fat protects African green monkeys from coronary artery atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 1995;15:2101–2110. doi: 10.1161/01.atv.15.12.2101. [DOI] [PubMed] [Google Scholar]
- 185.Rudel LL, Johnson FL, Sawyer JK, Wilson MS, Parks JS. Dietary polyunsaturated fat modifies low-density lipoproteins and reduces atherosclerosis of nonhuman primates with high and low diet responsiveness. Am J Clin Nutr. 1995;62:S463–S470. doi: 10.1093/ajcn/62.2.463S. [DOI] [PubMed] [Google Scholar]
- 186.Wolfe MS, Sawyer JK, Morgan TM, Bullock BC, Rudel LL. Dietary polyunsaturated fat decreases coronary artery atherosclerosis in a pediatric-aged population of African green monkeys. Arterioscler. Thromb. 1994;14:587–597. doi: 10.1161/01.atv.14.4.587. [DOI] [PubMed] [Google Scholar]
- 187.Rikitake Y, Kawashima S, Takeshita S, Yamashita T, Azumi H, Yasuhara M, Nishi H, Inoue N, Yokoyama M. Anti-oxidative properties of fluvastatin, an HMG-CoA reductase inhibitor, contribute to prevention of atherosclerosis in cholesterol-fed rabbits. Atherosclerosis. 2001;154:87–96. doi: 10.1016/s0021-9150(00)00468-8. [DOI] [PubMed] [Google Scholar]
- 188.Stoll LL, McCormick ML, Denning GM, Weintraub NL. Antioxidant effects of statins. Drugs Today (Barc) 2004;40:975–990. doi: 10.1358/dot.2004.40.12.872573. [DOI] [PubMed] [Google Scholar]
- 189.Cominacini L, Fratta Pasini A, Garbin U, Pastorino AM, Davoli A, Nava C, Campagnola M, Rossato P, Lo Cascio V. Antioxidant activity of different dihydropyridines. Biochem. Biophys. Res. Commun. 2003;302:679–684. doi: 10.1016/s0006-291x(03)00158-x. [DOI] [PubMed] [Google Scholar]
- 190.Inouye M, Mio T, Sumino K. Nilvadipine protects low-density lipoprotein cholesterol from in vivo oxidation in hypertensive patients with risk factors for atherosclerosis. Eur. J. Clin. Pharmacol. 2000;56:35–41. doi: 10.1007/s002280050717. [DOI] [PubMed] [Google Scholar]
- 191.Kritz H, Oguogho A, Aghajanian AA, Sinzinger H, Semotiadil a new calcium antagonist, is a very potent inhibitor of LDL-oxidation. Prostaglandins Leukot. Essent. Fatty Acids. 1999;61:183–188. doi: 10.1054/plef.1999.0088. [DOI] [PubMed] [Google Scholar]
- 192.Napoli C, Salomone S, Godfraind T, Palinski W, Capuzzi DM, Palumbo G, D'Armiento FP, Donzelli R, de Nigris F, Capizzi RL, Mancini M, Gonnella JS, Bianchi A. 1,4-Dihydropyridine calcium channel blockers inhibit plasma and LDL oxidation and formation of oxidation-specific epitopes in the arterial wall and prolong survival in stroke-prone spontaneously hypertensive rats. Stroke. 1999;30:1907–1915. doi: 10.1161/01.str.30.9.1907. [DOI] [PubMed] [Google Scholar]
- 193.Lupo E, Locher R, Weisser B, Vetter W. In vitro antioxidant activity of calcium antagonists against LDL oxidation compared with alpha-tocopherol. Biochem. Biophys. Res. Commun. 1994;203:1803–1808. doi: 10.1006/bbrc.1994.2396. [DOI] [PubMed] [Google Scholar]
- 194.Khan BV, Navalkar S, Khan QA, Rahman ST, Parthasarathy S. Irbesartan, an angiotensin type 1 receptor inhibitor, regulates the vascular oxidative state in patients with coronary artery disease. J. Am. Coll Cardiol. 2001;38:1662–1667. doi: 10.1016/s0735-1097(01)01615-1. [DOI] [PubMed] [Google Scholar]
- 195.Godfrey EG, Stewart J, Dargie HJ, Reid JL, Dominiczak M, Hamilton CA, McMurray J. Effects of ACE inhibitors on oxidation of human low density lipoprotein. Br. J. Clin. Pharmacol. 1994;37:63–66. doi: 10.1111/j.1365-2125.1994.tb04240.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Ziedén B, Wuttge DM, Karlberg BE, Olsson AG. Effects of in vitro addition of captopril on copper-induced low density lipoprotein oxidation. Br. J. Clin. Pharmacol. 1995;39:201–203. doi: 10.1111/j.1365-2125.1995.tb04433.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Hayek T, Attias J, Smith J, Breslow JL, Keidar S. Antiatherosclerotic and antioxidative effects of captopril in apolipoprotein E-deficient mice. J. Cardiovasc. Pharmacol. 1998;31:540–544. doi: 10.1097/00005344-199804000-00011. [DOI] [PubMed] [Google Scholar]
- 198.Van Antwerpen P, Boudjeltia KZ, Babar S, Legssyer I, Moreau P, Moguilevsky N, Vanhaeverbeek M, Ducobu J, Nève J. Thiol-containing molecules interact with the myeloperoxidase/H2O2/chloride system to inhibit LDL oxidation. Biochem. Biophys. Res. Commun. 2005;337:82–88. doi: 10.1016/j.bbrc.2005.09.013. [DOI] [PubMed] [Google Scholar]
- 199.Van Antwerpen P, Legssyer I, Zouaoui Boudjeltia K, Babar S, Moreau P, Moguilevsky N, Vanhaeverbeek M, Ducobu J, Nève J. Captopril inhibits the oxidative modification of apolipoprotein B-100 caused by myeloperoxidase in a comparative in vitro assay of angiotensin converting enzyme inhibitors. Eur J Pharmacol. 2006;537:31–36. doi: 10.1016/j.ejphar.2006.03.022. [DOI] [PubMed] [Google Scholar]
- 200.Miura T, Muraoka S, Ogiso T. Antioxidant activity of adrenergic agents derived from catechol. Biochem Pharmacol. 1998;55:2001–2006. doi: 10.1016/s0006-2952(98)00075-6. [DOI] [PubMed] [Google Scholar]
- 201.Hogg N, Struck A, Goss SP, Santanam N, Joseph J, Parthasarathy S, Kalyanaraman B. Inhibition of macrophage-dependent low density lipoprotein oxidation by nitric-oxide donors. J. Lipid Res. 1995;36:1756–1762. [PubMed] [Google Scholar]
- 202.Hogg N, Kalyanaraman B, Joseph J, Struck A, Parthasarathy S. Inhibition of low-density lipoprotein oxidation by nitric oxide. Potential role in atherogenesis. FEBS Lett. 1993;334:170–174. doi: 10.1016/0014-5793(93)81706-6. [DOI] [PubMed] [Google Scholar]
- 203.Hermann M, Kapiotis S, Hofbauer R, Exner M, Seelos C, Held I, Gmeiner B. Salicylate inhibits LDL oxidation initiated by superoxide/nitric oxide radicals. FEBS Lett. 1999;445:212–214. doi: 10.1016/s0014-5793(99)00043-5. [DOI] [PubMed] [Google Scholar]
- 204.Whiteman M, Kaur H, Halliwell B. Protection against peroxynitrite dependent tyrosine nitration and alpha 1-antiproteinase inactivation by some anti-inflammatory drugs and by the antibiotic tetracycline. Ann. Rheum. Dis. 1996;55:383–387. doi: 10.1136/ard.55.6.383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Steinbrecher UP, Witztum JL, Parthasarathy S, Steinberg D. Decrease in reactive amino groups during oxidation or endothelial cell modification of LDL. Correlation with changes in receptor-mediated catabolism. Arteriosclerosis. 1987;7:135–143. doi: 10.1161/01.atv.7.2.135. [DOI] [PubMed] [Google Scholar]
- 206.Jürgens G, Lang J, Esterbauer H. Modification of human low-density lipoprotein by the lipid peroxidation product 4-hydroxynonenal. Biochim. Biophys. Acta. 1986;875:103–114. doi: 10.1016/0005-2760(86)90016-0. [DOI] [PubMed] [Google Scholar]
- 207.Jessup W, Jurgens G, Lang J, Esterbauer H, Dean RT. Interaction of 4-hydroxynonenal-modified low-density lipoproteins with the fibroblast apolipoprotein B/E receptor. Biochem J. 1986;234:245–248. doi: 10.1042/bj2340245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Esterbauer H, Koller E, Slee RG, Koster JF. Possible involvement of the lipid-peroxidation product 4-hydroxynonenal in the formation of fluorescent chromolipids. Biochem J. 1986;239:405–409. doi: 10.1042/bj2390405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Quehenberger O, Koller E, Jürgens G, Esterbauer H. Investigation of lipid peroxidation in human low density lipoprotein. Free. Radic. Res. Commun. 1987;3:233–242. doi: 10.3109/10715768709069788. [DOI] [PubMed] [Google Scholar]
- 210.Hoff HF, Whitaker TE, O'Neil J. Oxidation of low density lipoprotein leads to particle aggregation and altered macrophage recognition. J. Biol. Chem. 1992;267:602–609. [PubMed] [Google Scholar]
- 211.Nadkarni DV, Sayre LM. Structural definition of early lysine and histidine adduction chemistry of 4-hydroxynonenal. Chem. Res. Toxicol. 1995;8:284–291. doi: 10.1021/tx00044a014. [DOI] [PubMed] [Google Scholar]
- 212.Requena JR, Fu MX, Ahmed MU, Jenkins AJ, Lyons TJ, Baynes JW, Thorpe SR. Quantification of malondialdehyde and 4- hydroxynonenal adducts to lysine residues in native and oxidized human low-density lipoprotein. Biochem. J. 1997;322:317–325. doi: 10.1042/bj3220317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Tsai L, Szweda PA, Vinogradova O, Szweda LI. Structural characterization and immunochemical detection of a fluorophore derived from 4-hydroxy-2-nonenal and lysine. Proc. Natl. Acad. Sci. U S A. 1998;95:7975–7980. doi: 10.1073/pnas.95.14.7975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Xu G, Liu Y, Sayre LM. Polyclonal antibodies to a fluorescent 4-hydroxy-2-nonenal (HNE)-derived lysine-lysine cross-link: characterization and application to HNE-treated protein and in vitro oxidized low-density lipoprotein. Chem. Res. Toxicol. 2000;13:406–413. doi: 10.1021/tx990200s. [DOI] [PubMed] [Google Scholar]
- 215.Marinari UM, Nitti M, Pronzato MA, Domenicotti C. Role of PKC-dependent pathways in HNE-induced cell protein transport and secretion. Mol. Aspects Med. 2003;24:205–211. doi: 10.1016/s0098-2997(03)00015-3. [DOI] [PubMed] [Google Scholar]
- 216.Awasthi Y, Sharma R, Cheng J, Yang Y, Sharma A, Singhal SS, Awasthi S. Role of 4-hydroxynonenal in stress-mediated apoptosis signaling. Mol. Aspects Med. 2003;24:219–230. doi: 10.1016/s0098-2997(03)00017-7. [DOI] [PubMed] [Google Scholar]
- 217.Yang Y, Sharma R, Sharma A, Awasthi S, Awasthi Y. Lipid peroxidation and cell cycle signaling: 4-hydroxynonenal, a key molecule in stress mediated signaling. Acta. Biochim. Pol. 2003;50:319–336. [PubMed] [Google Scholar]
- 218.Spiteller G. Linoleic acid peroxidation--the dominant lipid peroxidation process in low density lipoprotein--and its relationship to chronic diseases. Chem.Phys. Lipids. 1998;95:105–162. doi: 10.1016/s0009-3084(98)00091-7. [DOI] [PubMed] [Google Scholar]
- 219.Godfraind T, Sturbois X, Verbeke N. Calcium incorporation by smooth muscle microsomes. Biochim. Biophys. Acta. 1976;455:254–268. doi: 10.1016/0005-2736(76)90168-1. [DOI] [PubMed] [Google Scholar]
- 220.Nishio S, Kavanagh JP, Faragher EB, Garside J, Blacklock NJ. Calcium oxalate crystallisation kinetics and the effects of calcium and gamma-carboxyglutamic acid. Br. J. Urol. 1990;66:351–356. doi: 10.1111/j.1464-410x.1990.tb14953.x. [DOI] [PubMed] [Google Scholar]
- 221.Doyle IR, Ryall RL, Marshall VR. Inclusion of proteins into calcium oxalate crystals precipitated from human urine: a highly selective phenomenon. Clin. Chem. 1991;37:1589–1594. [PubMed] [Google Scholar]
- 222.Selvam R, Kalaiselvi P. Studies on calcium oxalate binding proteins: effect of lipid peroxidation. Nephron. 2001;88:163–167. doi: 10.1159/000045978. [DOI] [PubMed] [Google Scholar]
- 223.Akamatsu H, Komura J, Asada Y, Miyachi Y, Niwa Y. Inhibitory effect of azelaic acid on neutrophil functions: a possible cause for its efficacy in treating pathogenetically unrelated diseases. Arch. Dermatol. Res. 1991;283:162–166. doi: 10.1007/BF00372056. [DOI] [PubMed] [Google Scholar]
- 224.Iraji F, Sadeghinia A, Shahmoradi Z, Siadat AH, Jooya A. Efficacy of topical azelaic acid gel in the treatment of mild-moderate acne vulgaris. Indian J. Dermatol. Venereol. Leprol. 2007;73:94–96. doi: 10.4103/0378-6323.31892. [DOI] [PubMed] [Google Scholar]
- 225.Elewski B, Thiboutot D. A clinical overview of azelaic acid. Cutis. 2006;77:12–16. [PubMed] [Google Scholar]
- 226.Fleischer AB., Jr The evolution of azelaic acid. Cutis. 2006;77:4–6. [PubMed] [Google Scholar]
- 227.Lin CC, Wu SJ, Chang CH, Ng LT. Antioxidant activity of Cinnamomum cassia. Phyt. Other Res. 2003;7:726–730. doi: 10.1002/ptr.1190. [DOI] [PubMed] [Google Scholar]
- 228.Bolkenius FN. Leukocyte-mediated inactivation of alpha 1-proteinase inhibitor is inhibited by amino analogues of alpha-tocopherol. Biochim Biophys Acta. 1991;1095:23–29. doi: 10.1016/0167-4889(91)90040-5. [DOI] [PubMed] [Google Scholar]
- 229.Nègre-Salvayre A, Affany A, Hariton C, Salvayre R. Additional antilipoperoxidant activities of alpha-tocopherol and ascorbic acid on membrane-like systems are potentiated by rutin. Pharmacology. 1991;42:262–272. doi: 10.1159/000138807. [DOI] [PubMed] [Google Scholar]
- 230.Bindoli A, Valente M, Cavallini L. Inhibitory action of quercetin on xanthine oxidase and xanthine dehydrogenase activity. Pharmacol. Res. Commun. 1985;17:831–839. doi: 10.1016/0031-6989(85)90041-4. [DOI] [PubMed] [Google Scholar]
- 231.Navab M, Anantharamaiah GM, Reddy ST, Fogelman AM. Apolipoprotein A-I mimetic peptides and their role in atherosclerosis prevention. Nat. Clin. Pract. Cardiovasc. Medicine. 2006;3:540–547. doi: 10.1038/ncpcardio0661. [DOI] [PubMed] [Google Scholar]
- 232.Ansell B, Navab M, Watson K, Fonarow G, Fogelman A. Anti-inflammatory properties of HDL. Rev. Endocr. Metab Disord. 2004;5:351–358. doi: 10.1023/B:REMD.0000045107.71895.b2. [DOI] [PubMed] [Google Scholar]
- 233.Van Lenten B, Navab M, Shih D, Fogelman A, Lusis AJ. The role of high-density lipoproteins in oxidation and inflammation. Trends. Cardiovasc. Med. 2001;11:155–161. doi: 10.1016/s1050-1738(01)00095-0. [DOI] [PubMed] [Google Scholar]
- 234.Navab M, Berliner JA, Watson AD, Hama SY, Territo MC, Lusis AJ, Shih DM, Van Lenten BJ, Frank JS, Demer LL, Edwards PA, Fogelman AM. The Yin and Yang of oxidation in the development of the fatty streak. A review based on the 1994 George Lyman Duff Memorial Lecture. Arterioscler. Thromb. Vasc. Biol. 1996;16:831–842. doi: 10.1161/01.atv.16.7.831. [DOI] [PubMed] [Google Scholar]
- 235.Lamb DJ, Leake DS. Iron released from transferrin at acidic pH can catalyse the oxidation of low density lipoprotein. FEBS Lett. 1994 Sep 19;352(1):15–18. doi: 10.1016/0014-5793(94)00903-1. [DOI] [PubMed] [Google Scholar]
- 236.Lamb DJ, Hider RC, Leake DS. Hydroxypyridinones and desferrioxamine inhibit macrophage-mediated LDL oxidation by iron but not by copper. Biochem Soc Trans. 1993 Aug;21(Pt 3 3):234S. doi: 10.1042/bst021234s. [DOI] [PubMed] [Google Scholar]