

Abstract
Intrinsic or acquired chemoresistance is a major cause of treatment failure in patients with malignant brain tumors. Alkylating agents, the mainstay of treatment for brain tumors, damage the DNA and induce apoptosis, but the cytotoxic activity of these agents is dependent on DNA repair pathways. For example, O6-methylguanine DNA adducts can cause double-strand breaks, but this is dependent on a functional mismatch repair (MMR) pathway. Thus, tumor cell lines deficient in MMR are resistant to alkylating agents. Perhaps the most important mechanism of resistance to alkylating agents is the DNA repair enzyme O6-methylguanine methyltransferase (MGMT), which can eliminate the cytotoxic O6-methylguanine DNA adduct before it causes harm. Another mechanism of resistance to alkylating agents is the base excision repair (BER) pathway. Consequently, efforts are ongoing to develop effective inhibitors of BER. Poly(ADP-ribose)polymerase (PARP-1) plays a pivotal role in BER and is an important therapeutic target. Developing effective strategies to overcome chemoresistance requires the identification of reliable preclinical models that recapitulate human disease and can be used to facilitate drug development. This manuscript describes the diverse mechanisms of chemoresistance operating in malignant glioma and efforts to develop reliable preclinical models and novel pharmacologic approaches to overcome resistance to alkylating agents.
Keywords: Glioma, chemoresistance, MGMT, temozolomide, PARP inhibitor
INTRODUCTION
Intrinsic or acquired resistance to cytotoxic agents remains the greatest obstacle to successful treatment of human cancer, and a variety of mechanisms of drug resistance have been identified in human tumors. High-grade malignant gliomas, including glioblastoma (GBM) and anaplastic astrocytoma, are among the most rapidly growing and devastating cancers. Standard treatment consists of surgery followed by radiotherapy and chemotherapy with alkylating agents. Alkylating agents, including carmustine (BCNU), lomustine (CCNU), and temozolomide, readily cross the blood-brain barrier and have demonstrated the most activity against malignant glioma. However, despite the ability of these agents to achieve therapeutic concentrations in the brain, malignant gliomas are often resistant to alkylating agents. Identifying and understanding the diverse mechanisms of chemoresistance operating in human tumors and developing effective strategies to overcome resistance is the focus of ongoing preclinical and clinical research.
One important mechanism of resistance to alkylating agents is mediated by the DNA repair enzyme O6-methylguanine methyltransferase (MGMT) (1,2), which repairs O6-alkylguanine adducts. BCNU and CCNU are bifunctional alkylating agents that form lethal double-strand crosslinks (3). In contrast, the activity of methylating agents such as temozolomide and dacarbazine results in persistent O6-methylguanine adducts that initiate futile cycling of the DNA mismatch repair (MMR) pathway and ultimately causes DNA double-strand breaks and induction of apoptosis (4,5). The methylation damage induced by these agents can be reversed by MGMT. Although several studies have shown that a deficiency of MGMT can increase the sensitivity of high-grade glioma to alkylating agents (6-8), many tumors with low levels of MGMT are nevertheless chemoresistant (1,2). This suggests that other mechanisms are also involved in the resistance of some tumors to chemotherapy. For example, deficiency in MMR can make tumor cells tolerant to mismatched base pairing and mutations in p53, or overexpression of the anti-apoptotic protein Bcl-2 or Bcl-XL can cause resistance to pro-apoptotic signals. The base excision repair (BER) pathway can also render alkylating agents ineffective by repairing other DNA adducts that would otherwise be cytotoxic.
This review will summarize the mechanisms of chemoresistance in malignant glioma and will focus on the development of agents designed to overcome resistance to alkylating agents, with particular emphasis on novel molecules designed to inhibit the BER pathway. An important component of this research involves the development of clinically relevant xenograft models for testing novel treatment strategies to overcome chemoresistance. Herein, we will describe a nude mouse serially transplantable xenograft model of GBM that represents an important tool for testing new agents and treatment strategies.
MECHANISMS OF CHEMORESISTANCE
O6-Methylguanine Methyltransferase
The DNA repair enzyme MGMT appears to play a major role in resistance to temozolomide and other alkylating agents by removing alkyl groups from the O6 position of guanine (9). The protective effect of MGMT has been demonstrated in human cell lines (10) and in human xenograft models (11). For example, studies with MGMT knockout mice revealed that fibroblasts and bone marrow cells were significantly more sensitive to the toxic effects of temozolomide than cells from MGMT wild-type mice (12). Moreover, retrovirus-mediated transfer of the human MGMT gene to cells that are deficient in endogenous MGMT activity results in high levels of resistance to temozolomide and other alkylating agents (13). In vivo studies that used mixtures of MGMT-positive and MGMT-negative colon cancer cell lines injected into nude mice and exposed to BCNU demonstrated that the extent of MGMT expression in mixed xenografts correlated directly with their sensitivity to BCNU (14). Xenografts with 0% to 10% MGMT-positive cells were highly sensitive to BCNU and exhibited a significant growth delay, whereas xenografts with increasing percentages of MGMT-positive cells grew rapidly in the presence of BCNU (Fig 1) (14). Pretreatment with a selective MGMT inhibitor, O6-benzylguanine (O6-BG), has been shown to suppress MGMT activity for approximately 6 hours and sensitize xenografts containing MGMT-positive cells to BCNU (14). Similarly, other studies showed that inhibition of MGMT with O6-BG promotes increased antitumor activity of temozolomide both in vitro (15-17) and in vivo (11,18). Thus, direct removal of O6-methylguanine by MGMT appears to be a major mechanism of resistance to the cytotoxic effects of alkylating agents.
Fig 1.

Growth of tumor xenografts containing various percentages of O6-methylguanine methyltransferase-positive cells (indicated by numbers above each curve) in animals treated with 23 mg/kg bischloroethyl nitrosourea (BCNU). Adapted with permission from Phillips et al, Cancer Res 57:4817-4823, 1997 (14).
In human cancer, the MGMT gene is not commonly inactivated by mutation; loss of MGMT function is most frequently caused by promoter-region hypermethylation (19,20). Preclinical studies have shown that methylation of discrete regions of the MGMT promoter is associated with epigenetic silencing of the gene, loss of MGMT expression (21,22), and diminished DNA repair activity. Tumors with methylated MGMT promoters are more sensitive to alkylating agents, whereas tumors with unmethylated MGMT promoters express high levels of the enzyme and are more resistant to alkylating agents. From a clinical standpoint, MGMT promoter methylation is associated with improved response to alkylating agents, as evidenced by improved survival in patients with high-grade gliomas who are treated with BCNU or temozolomide (23-25). Therefore, MGMT promoter methylation may predict the therapeutic response to alkylating agents. Several ongoing studies are investigating the correlation between MGMT methylation status and temozolomide sensitivity. These studies are also investigating whether increasing the dose intensity of temozolomide treatment can overcome resistance mediated by MGMT.
Mismatch Repair
The MMR pathway is critical for mediating the cytotoxic effect of O6-methylguanine. The MMR pathway is comprised of several proteins (hMLH1, hPMS2, hMSH2, hMSH3, and hMSH6) and is programmed to correct errors in DNA base pairing arising during DNA replication. Defects in this system cause resistance to temozolomide, presumably because the cell becomes tolerant to the mispairing of O6-methylguanine with thymine. During DNA replication, DNA polymerase mispairs O6-methylguanine with thymine, which triggers MMR-dependent removal of the mispaired thymine. However, the O6-methylguanine remains, and subsequent mispairing of O6-methylguanine with yet another thymine leads to repetitive rounds of MMR (15,26,27). It has been proposed that this futile cycling of the MMR system induces double-strand breaks, which in turn triggers p53-dependent cell cycle arrest and apoptosis (28). Consequently, tumors that are deficient in MMR are relatively resistant to the cytotoxic effects of alkylating agents such as temozolomide. In fact, many tumors are known to be deficient in MMR. For example, hereditary nonpolyposis colon cancer is caused by mutations in the hMLH1 or hMSH2 genes (29). Therefore, the cytotoxicity of temozolomide is low in colon cancer cell lines harboring these mutations (30). Deficiency in MMR also occurs in a number of sporadic human cancers due to methylation of the hMLH1 gene promoter. Of the 60 cell lines in the National Cancer Institute tumor panel, five are deficient in hMLH1 activity, and these cell lines are all resistant to temozolomide (31).
Poly(ADP-ribose)polymerase
Another important mechanism of resistance to alkylating agents is the BER pathway, which is thought to be involved in repair of N7-methylguanine and N3-methyladenine DNA adducts. These are the most common DNA adducts created by temozolomide, accounting for approximately 90% of all methylation events. Poly(ADP-ribose)polymerase (PARP-1) plays a pivotal role in the BER pathway. This enzyme is activated by DNA strand breaks and is expressed at high levels in most human cell lines. This led to the investigation of PARP inhibitors as a means of increasing the cytotoxic activity of temozolomide. Inhibition of BER would be expected to increase the cytotoxicities of N7-methylguanine and N3-methyladenine lesions, which are otherwise readily repaired in most normal cells and tumor cells with an intact BER pathway. In particular, PARP inhibitors would be expected to dramatically enhance the cytotoxicity of temozolomide in MMR-deficient cells because these cells tolerate O6-methylguanine DNA adducts (32).
Apoptosis-Regulating Genes
In addition to the resistance mechanisms described above that involve DNA repair, chemoresistance in gliomas may be influenced by dysregulation of apoptosis-regulating genes and proteins. For example, loss of normal p53 function, up-regulation of Bcl-2 or Bcl-XL, or overexpression of epidermal growth factor receptor (EGFR) can disrupt the normal apoptotic response to DNA damage. It is well known that p53 plays a central role in detecting DNA damage and regulating the signaling pathways required to mediate apoptosis. Murine tumors deficient in Trp53 (the gene that encodes p53 in mice) and cell lines that express mutant Trp53 are less sensitive to gamma-irradiation or chemotherapy (33). Reintroduction of wild-type Trp53 by adenoviral transfer restores sensitivity to cytotoxic agents resulting in apoptosis and suppression of tumor growth in nude mice (34,35). Wild-type p53 interacts with the promoters of a variety of genes, including EGFR, MDM2, and Bcl-2. Wild-type p53 increases transcriptional activity of the EGFR and MDM2 genes and represses transcription of Bcl-2 (36). Based on the analysis of patient tumor samples, inactivation of p53 occurs in more than 50% of primary GBMs, and this is associated with up-regulation of Bcl-2 (15,37,38). Consistent with these findings, up-regulation of either Bcl-2 or EGFR expression in glioma cell lines has been correlated with drug resistance and decreased apoptotic response in vitro (39,40). EGFR is the most frequently amplified and mutated oncogene in de novo malignant glioma, and several clinical as well as histopathologic studies have suggested that the presence of EGFR amplification might affect the response of malignant gliomas to chemotherapy (41). In one study, overexpression of a tumor-derived mutant form of EGFR known as variant III (EGFRvIII) in wild-type p53-expressing human glioma cells resulted in resistance to cisplatin (39). The mechanism involved was suggested to be suppression of drug-induced apoptosis, which was largely mediated by increased expression of Bcl-XL and subsequent inhibition of caspase-3–like protease activation. Although dysregulated expression of EGFR, MDM2, and Bcl-2 frequently occurs in GBM, none of these changes in gene expression has been clearly identified as an independent prognostic factor for either the biologic behavior of gliomas or patient survival (42). Additional studies into the mechanisms regulating apoptosis and sensitivity to DNA damage may reveal new strategies for overcoming the resistant phenotype of GBM.
Given the important role of temozolomide in the treatment of gliomas and the probable dependence of its activity on MGMT activity, a recent study has sought to better characterize the molecular pathways mediating temozolomide cytotoxicity in human malignant glioma cell lines in terms of MGMT, Bcl-2, and p53 expression (40). This study showed that forced expression of Bcl-XL protein attenuates temozolomide cytotoxicity in MGMT-negative LNT-229 cells but not in MGMT-positive LN-18 cells. Neither irradiation nor dexamethasone modulated MGMT activity or temozolomide sensitivity in this paradigm. Abrogation of p53 wild-type function with RNA interference or expression of a dominant-negative p53 mutant also conferred resistance to temozolomide. Conversely, agents such as CP-31398 that stabilize the wild-type conformation of p53 sensitize glioma cells to temozolomide (43). These data suggest that wild-type p53 activity is necessary for sensitivity to alkylating agents. Further, since low-grade gliomas have a high incidence of p53 mutations, they may be less sensitive to temozolomide. However, these conclusions are limited by the preclinical paradigm applied and may not accurately reflect the clinical situation.
Transporter Proteins
Molecular transporter proteins that actively expel chemotherapeutic drugs from tumor cells have been implicated in resistance to chemotherapy, and this may be another mechanism that could result in resistance to temozolomide (44). The two transporters commonly associated with drug resistance in cancer cells are the MDR1 gene products P-glycoprotein and the multidrug resistance-associated protein (45). Multidrug resistance-associated protein overexpression has been shown to have a direct effect on the response of neuroblastomas to chemotherapy and to be associated with poor clinical outcome (46). The role of these drug transporters in resistance to alkylating agents in gliomas is an area of active interest.
PRECLINICAL MODELS TO TEST STRATEGIES TO OVERCOME CHEMORESISTANCE
Efforts to develop new strategies to overcome chemoresistance underscore the need for reliable preclinical models to test these strategies. Ideally, robust preclinical models would facilitate the development of optimal therapeutic regimens so that only the most effective treatment regimens are moved forward into clinical testing. These model systems could be used to develop predictive markers for treatment response that would guide the development of new agents or individualized therapies, thereby minimizing patients' exposure to ineffective treatment approaches.
Cell Culture Models
Several cell culture models have been used to demonstrate the role of MGMT in resistance to temozolomide. For example, studies with Chinese hamster ovary cells lacking endogenous MGMT expression have demonstrated that overexpression of MGMT confers resistance to temozolomide (47). Similarly, established glioma cell lines have provided a model for studying the correlation between MGMT levels and sensitivity to various alkylating agents (10). Although cell culture models are valuable for the design of cancer therapies, these models do not consistently predict tumor response to cytotoxic or biologic agents in the clinical setting. Cultured cell lines do not necessarily reflect the biology or heterogeneity of primary tumors nor their therapeutic response to treatment and may have less relevance in the clinical setting than in vivo models.
Transgenic Mouse Models
Transgenic mouse models are genetically defined animal models that allow for testing of therapies that are targeted specifically at the affected pathways (48). For example, transgenic mice have been engineered to recapitulate key genetic features of GBM such as mutant EGFR; such mice are excellent for the evaluation of specific molecular mechanistic questions. However, because of limited genetic diversity, these models may be less useful for identifying the multiple mechanisms of sensitivity and resistance found in the spectrum of genetically diverse tumors found in patients.
Xenograft Models
In xenograft models, established cell lines are often grown as subcutaneous flank (heterotopic) or intracranial (orthotopic) tumors. However, orthotopic xenografts created from established GBM cell lines generally fail to recapitulate the infiltrative growth pattern of primary human GBM; instead, they tend to form solid masses at the injection site, compressing rather than invading the parenchyma (49-52). In addition, growth of tumor cell lines in culture can lead to a high degree of selection, causing the loss of important gene alterations that are common among primary GBMs such as EGFR gene amplification (53,54). Glioblastoma cell lines established from primary tumors with amplified EGFR and/or EGFR mutations invariably lose this amplification and mutation through prolonged cell culture. This observation suggests that the growth advantage associated with increased EGFR expression and the constitutively active mutant form of the receptor (eg, EGFRvIII) is specific to the in vivo environment and that high-level EGFR signaling may in fact be a disadvantage for in vitro tumor cell growth (53,54). Although the use of heterotopically grown cell lines offers some insight into tumor biology, prolonged selection of these cell lines in cell culture limits their clinical relevance for understanding patient tumor responsiveness to chemotherapeutic or biologic agents.
Recently, the amenability of a serially transplantable nude mouse xenograft model was indicated for testing novel therapeutics and for answering key biologic and clinical questions regarding mechanisms of resistance, drug dosing, and combination strategies (55,56). In this model (Fig 2), GBM tissue samples are obtained from patients and serially propagated in the flank of athymic nude mice. This method of propagation allows preservation of key morphologic and histopathologic features, such as the presence of necrosis, similar to those of the primary GBMs from which they were derived (Fig 3). By establishing and maintaining multiple GBM xenografts in this manner, a genetically diverse panel of GBM tumors is available for testing novel therapeutic strategies.
Fig 2.

Methodology for generating a murine serially transplantable xenograft model. Human glioblastomas are propagated in the flank of mice and then briefly cultured in vitro before stereotactic injection into the brains of nude mice.
Fig 3.
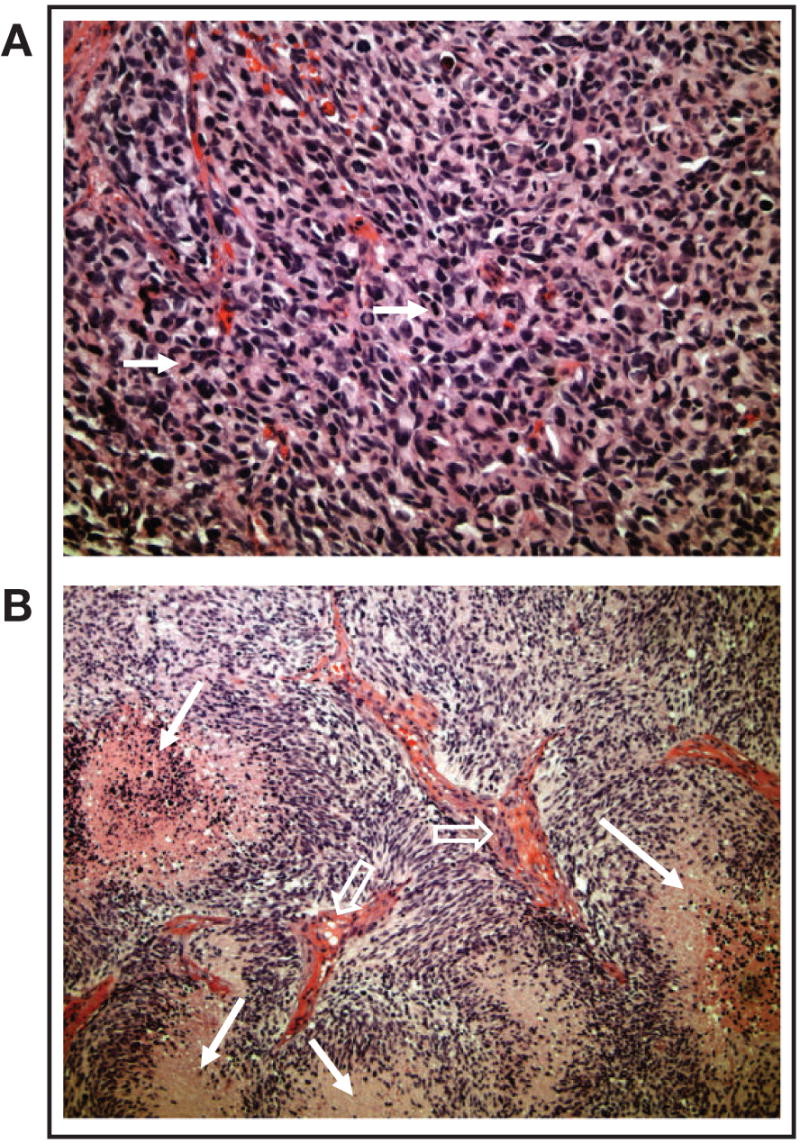
Morphology and histopathology of glioblastomas grown in the flank. Flank tumors recapitulated human tumor morphology as evidenced by the presence of (A) high cellularity and mitotic activity (arrows) and (B) necrosis (closed arrows) and vascular endothelial proliferation (open arrows).
With the test panel approach, therapies are routinely evaluated against intracranial tumors that are established from short-term cultures of freshly resected flank tumors. Single-cell suspensions are stereotactically injected into the brains of nude mice (Fig 2), and this results in a nearly 100% rate of successful engraftment. Analysis of the resulting intracranial tumors shows that these tumors maintain key histopathologic features of the original tumor, such as a highly invasive growth pattern and nuclear pleomorphism (Fig 4). Therefore, it is expected that the chemosensitivity of the serially propagated human tumor likely would be comparable to that of the patient tumor from which it was derived and that this model would predict the activity of chemotherapies used alone or in combinations. Indeed, MGMT promoter methylation is faithfully maintained from the original patient tumor specimen. Among a panel of xenografts developed at the Mayo Clinic, the proportion of xenografts that exhibit MGMT promoter methylation (approximately 45%) is similar to that reported in series of primary tumors (24). In contrast, 80% of GBM cell lines have a methylated MGMT promoter. This xenograft model demonstrates that primary human GBMs, which are propagated in the flank and transplanted orthotopically, closely resemble the human microenvironment, particularly in relation to growth characteristics and invasive properties.
Fig 4.
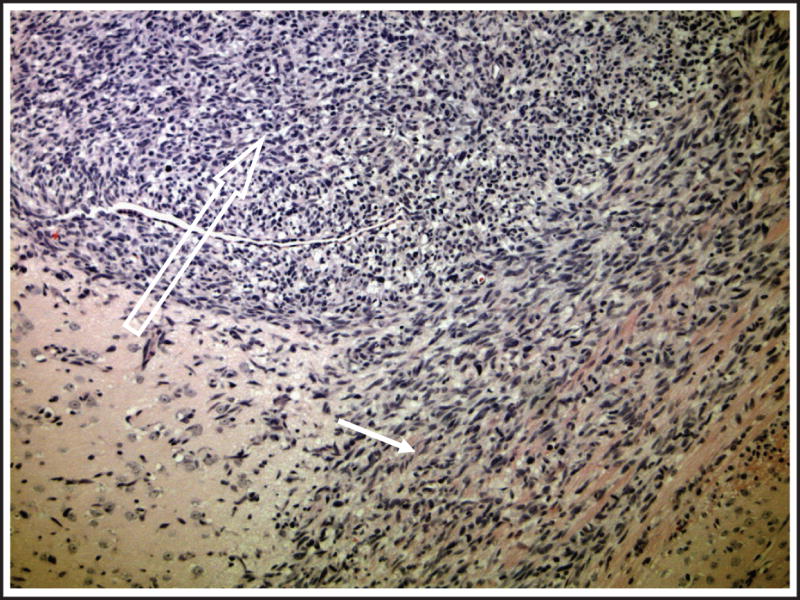
Growth of intracranial tumors established from flank xenografts. A representative section of an orthotopic tumor displays a close resemblance to primary tumors regarding advanced stage of growth. The tumor revealed high cellularity (open arrow) and invasion into corpus callosum (closed arrow).
PARP INHIBITORS
There is considerable interest in modulating the activity of PARP-1 in the clinical setting both for the treatment of cancer and nonmalignant diseases. Potential applications in the nonmalignant setting have recently been reviewed (57,58). In the realm of cancer therapy, preclinical models have shown that inhibition of PARP-1 can potentiate the activity of both radiotherapy and a variety of chemotherapeutic agents (59-65), and PARP-1 inhibitors have also demonstrated single-agent activity against certain tumor cell lines (66,67). These observations have stimulated considerable interest in these agents as anticancer therapy. AG-14361 is a potent inhibitor of PARP-1 with an inhibition constant in the nanomolar range, and AG-014699, a potent tricyclic indole with an inhibition constant of < 5 nM, is a close analogue and the first PARP inhibitor to enter clinical trials in cancer patients (68). AG-014699 was developed through a collaborative program between the University of Newcastle upon Tyne, UK, and Pfizer.
The effects of AG-14361 on the cytotoxicity of temozolomide were studied using two genetically matched cell lines; the parent cell line was MMR deficient, and the matched cell line was MMR proficient by virtue of the transfer of chromosome 3 (69). As expected, AG-14361 enhanced temozolomide cytotoxicity in both MMR-proficient and MMR-deficient cell lines, but the degree of enhancement was greater in the MMR-deficient cell lines (3.7- to 5.2-fold increase compared with only a 1.5- to 3.3-fold increase in the MMR-proficient cell lines). The enhanced potentiation of temozolomide activity in MMR-deficient cells compared with MMR-proficient cells suggests that it might be possible to selectively enhance the antitumor activity of temozolomide in the clinical setting using a PARP inhibitor because normal tissue (particularly the hematopoietic system) would be MMR proficient, whereas tumors may be MMR deficient. Although, as with any agent that enhances the cytotoxic potential of a chemotherapy drug, addition of a PARP inhibitor may cause significant added toxicity. The phase I study with AG-014699 demonstrated that a normal dose of temozolomide could be safely administered in combination with a PARP inhibitor (70). However, in the phase II study in patients with metastatic melanoma, a significant proportion of patients required a 25% reduction of the temozolomide dose or treatment delays because of myelosuppression (71).
The phase I trial of AG-014699 in combination with temozolomide for the treatment of solid tumors (primarily metastatic melanoma) was divided into two parts (70). The first part of the study was designed with a pharmacodynamic endpoint to demonstrate significant (at least 50%) PARP inhibition in peripheral blood lymphocytes 24 hours after the first dose of AG-014699. Temozolomide 100 mg/m2 (50% of the standard dose) was given in combination with AG-014699 (escalating doses from 1 to 12 mg/m2) daily for 5 days every 28 days. The reduced dose of temozolomide was initially used in the event that inhibition of PARP potentiated the known myelosuppressive effects of temozolomide. Significant inhibition of PARP-1 was demonstrated at all dose levels investigated. Inhibition was profound and sustained for 24 hours at doses > 8 mg/m2 (Fig 5). Moreover, the combination of 12 mg/m2 AG-014699 with 100 mg/m2 temozolomide was not toxic. In the second part of the study, patients with metastatic melanoma (exclusively) were treated with 12 mg/m2 AG-014699 and escalating doses of temozolomide. All patients consented to tumor biopsies to allow assessment of target tissue PARP inhibition. During this part of the study, it was possible to administer a full dose of temozolomide (200 mg/m2) with 12 mg/m2 AG-014699 without additional toxicity being observed while achieving significant tumor PARP inhibition (∼60% below baseline activity) in tumor tissue 5 hours after dosing. The 5-hour time point was selected because at that time temozolomide is known to cause maximum DNA strand breaks (72); this would be the optimal time to have maximal PARP inhibition in order to achieve effective chemopotentiation. Maximum PARP inhibition in tumor biopsies has been observed with the highest dose of AG-014699 investigated (18 mg/m2); however, this dose also enhanced the myelosuppressive effects of temozolomide, leading to dose reductions and delays.
Fig 5.

Pattern of ex vivo PARP inhibition observed in PBLs from patient treated with a PARP inhibitory dose of AG-014699 27.8 mg (12 mg/m2). Abbreviations: PAR, poly(ADP)-ribose; PBL, peripheral blood lymphocytes.
The encouraging PARP inhibition observed in this phase I study provided the rationale for a phase II study of the combination of AG-014699 and temozolomide in patients with metastatic melanoma. Forty-six patients were treated with temozolomide (200 mg/m2) and AG-014699 (12 mg/m2) daily for 5 days every 28-day cycle (71). This study confirmed that temozolomide can be administered in the presence of a PARP inhibitor with only minor dose reductions being required because of hematologic toxicity; 18 (39%) patients required a dose reduction of temozolomide to 150 mg/m2. Moreover, this study met its primary objective (ie, an increase in the response rate compared with historical data with temozolomide alone), and this combination also appears to improve time to progression.
Other PARP-1 inhibitors are now entering early clinical development in a range of malignant indications. For example, there are plans for INO-010 to enter clinical studies in combination with temozolomide both in patients with metastatic melanoma and in patients with malignant glioma (Slazman A, personal communication, PARP2005 conference). Although PARP inhibitors for clinical use were initially developed as chemo-potentiating agents, contemporary data suggest a role for single-agent treatment with a PARP inhibitor in tumors that are deficient in homologous recombination repair. It has recently been shown that cells homozygous for nonfunctional BRCA1 or BRCA2 are exquisitely sensitive to PARP inhibitors (67,73). BRCA1 and BRCA2 are involved in homologous recombination repair, leading to the hypothesis that single-strand breaks that accumulate due to inhibition of PARP are converted to double-strand breaks during DNA replication, which, in the absence of homologous recombination repair, cannot be corrected and thus lead to apoptosis. This observation was made independently by two different groups using different PARP inhibitors and model cell lines, suggesting that this sensitivity is indeed because of PARP inhibition rather than any other phenomenon. Inherited mutations in BRCA1 and BRCA2 are associated with a very high lifetime risk of breast and ovarian cancer and may also be etiologic in some cases of prostate and pancreatic cancer. An orally available PARP inhibitor KU-0059436 (KuDOS, AstraZeneca) is in phase I development and has shown a suggestion of antitumor activity in BRCA defective tumors (74). A formal phase II study of AG-014699 in familial breast and ovarian cancer is also planned. Furthermore, suppression of BRCA protein expression because of promoter methylation has been reported, suggesting that some spontaneous malignancies may also be deficient in homologous recombination repair as a result of epigenetic gene silencing (75). Other acquired defects affecting homologous recombination repair have also been reported in human tumors and would be expected to confer sensitivity to PARP inhibitors (76,77).
CONCLUSIONS AND FUTURE DIRECTIONS
In recent years, our understanding of the pathways responsible for repairing DNA damage and rendering cancer cells resistant to chemotherapy has greatly improved; consequently, effective strategies to modulate clinical resistance to cytotoxic agents are evolving. DNA damage caused by temozolomide is repaired by BER, removed by MGMT, or tolerated in MMR-deficient cells. Modulation of the first two pathways of resistance is now becoming a clinical possibility.
Early clinical trials with MGMT inhibitors such as O6-BG have been completed (reviewed by Hegi et al in this supplement), and this article reviews the promising initial results of trials investigating modulation of temozolomide resistance with PARP-1 inhibitors. Inhibition of PARP activity would, in addition, allow PARP inhibitors to act as radiopotentiators because the DNA damage caused by radiotherapy consists of single- and double-strand breaks (63). Repair of both types of lesions is known to involve recruitment of PARP-1 (78). Therefore, the use of a potent PARP inhibitor as part of multimodality therapy with temozolomide and radiotherapy in patients with GBM may have particular clinical relevance and might significantly improve therapeutic benefit in this group of patients.
Developing effective treatment regimens is a major challenge in malignant glioma, and identification of a reliable preclinical model that could recapitulate human disease would facilitate this process. The murine serially transplantable xenograft model is simple and reproducible and maintains most of the characteristics of primary human GBM. This model preserves the molecular phenotype and invasiveness of GBM xenografts, which is critical for investigating molecularly targeted agents. The combination of flank tumor genetic characterizations with a procedure for initiating intracranial xenografts makes this model an invaluable tool for multiple neuro-oncology research applications. The versatility and applicability of this preclinical model will contribute to the elucidation of the molecular mechanisms of chemoresistance, which should ultimately result in the identification of more effective therapeutic strategies for malignant glioma.
Acknowledgments
Funding for medical editorial assistance was provided by Schering-Plough International. Thank you to Jeff Riegel, PhD, ProEd Communications, Inc., for his medical editorial assistance with this manuscript.
References
- 1.Belanich M, Randall T, Pastor MA, et al. Intracellular localization and intercellular heterogeneity of the human DNA repair protein O(6)-methylguanine-DNA methyltransferase. Cancer Chemother Pharmacol. 1996;37:547–55. doi: 10.1007/s002800050427. [DOI] [PubMed] [Google Scholar]
- 2.Friedman HS, McLendon RE, Kerby T, et al. DNA mismatch repair and O6-alkylguanine-DNA alkyltransferase analysis and response to Temodal in newly diagnosed malignant glioma. J Clin Oncol. 1998;16:3851–7. doi: 10.1200/JCO.1998.16.12.3851. [DOI] [PubMed] [Google Scholar]
- 3.Sariban E, Kohn KW, Zlotogorski C, et al. DNA cross-linking responses of human malignant glioma cell strains to chloroethylnitrosoureas, cisplatin, and diaziquone. Cancer Res. 1987;47:3988–94. [PubMed] [Google Scholar]
- 4.Karran P, Bignami M. Self-destruction and tolerance in resistance of mammalian cells to alkylation damage. Nucleic Acids Res. 1992;20:2933–40. doi: 10.1093/nar/20.12.2933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Karran P, Hampson R. Genomic instability and tolerance to alkylating agents. Cancer Surv. 1996;28:69–85. [PubMed] [Google Scholar]
- 6.Dolan ME, Moschel RC, Pegg AE. Depletion of mammalian O6-alkylguanine-DNA alkyltransferase activity by O6-benzylguanine provides a means to evaluate the role of this protein in protection against carcinogenic and therapeutic alkylating agents. Proc Natl Acad Sci U S A. 1990;87:5368–72. doi: 10.1073/pnas.87.14.5368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dolan ME, Pegg AE. O6-benzylguanine and its role in chemotherapy. Clin Cancer Res. 1997;3:837–47. [PubMed] [Google Scholar]
- 8.Friedman HS, Kokkinakis DM, Pluda J, et al. Phase I trial of O6-benzylguanine for patients undergoing surgery for malignant glioma. J Clin Oncol. 1998;16:3570–5. doi: 10.1200/JCO.1998.16.11.3570. [DOI] [PubMed] [Google Scholar]
- 9.Pegg AE, Byers TL. Repair of DNA containing O6-alkylguanine. FASEB J. 1992;6:2302–10. doi: 10.1096/fasebj.6.6.1544541. [DOI] [PubMed] [Google Scholar]
- 10.Baer JC, Freeman AA, Newlands ES, Watson AJ, Rafferty JA, Margison GP. Depletion of O6-alkylguanine-DNA alkyltransferase correlates with potentiation of temozolomide and CCNU toxicity in human tumour cells. Br J Cancer. 1993;67:1299–302. doi: 10.1038/bjc.1993.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Friedman HS, Dolan ME, Pegg AE, et al. Activity of temozolomide in the treatment of central nervous system tumor xenografts. Cancer Res. 1995;55:2853–7. [PubMed] [Google Scholar]
- 12.Glassner BJ, Weeda G, Allan JM, et al. DNA repair methyltransferase (MGMT) knockout mice are sensitive to the lethal effects of chemotherapeutic alkylating agents. Mutagenesis. 1999;14:339–47. doi: 10.1093/mutage/14.3.339. [DOI] [PubMed] [Google Scholar]
- 13.Wang G, Weiss C, Sheng P, Bresnick E. Retrovirus-mediated transfer of the human O6-methylguanine-DNA methyltransferase gene into a murine hematopoietic stem cell line and resistance to the toxic effects of certain alkylating agents. Biochem Pharmacol. 1996;51:1221–8. doi: 10.1016/0006-2952(96)00077-9. [DOI] [PubMed] [Google Scholar]
- 14.Phillips WP, Jr, Willson JK, Markowitz SD, et al. O6-methylguanine-DNA methyltransferase (MGMT) transfectants of a 1,3-bis(2-chloroethyl)-1-nitrosourea (BCNU)-sensitive colon cancer cell line selectively repopulate heterogenous MGMT+/MGMT- xenografts after BCNU and O6-benzylguanine plus BCNU. Cancer Res. 1997;57:4817–23. [PubMed] [Google Scholar]
- 15.Bobola MS, Tseng SH, Blank A, Berger MS, Silber JR. Role of O6-methylguanine-DNA methyltransferase in resistance of human brain tumor cell lines to the clinically relevant methylating agents temozolomide and streptozotocin. Clin Cancer Res. 1996;2:735–41. [PubMed] [Google Scholar]
- 16.Dolan ME, Mitchell RB, Mummert C, Moschel RC, Pegg AE. Effect of O6-benzylguanine analogues on sensitivity of human tumor cells to the cytotoxic effects of alkylating agents. Cancer Res. 1991;51:3367–72. [PubMed] [Google Scholar]
- 17.Wedge SR, Porteous JK, Newlands ES. 3-aminobenzamide and/or O6-benzylguanine evaluated as an adjuvant to temozolomide or BCNU treatment in cell lines of variable mismatch repair status and O6-alkylguanine-DNA alkyltransferase activity. Br J Cancer. 1996;74:1030–6. doi: 10.1038/bjc.1996.485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wedge SR, Newlands ES. O6-benzylguanine enhances the sensitivity of a glioma xenograft with low O6-alkylguanine-DNA alkyltransferase activity to temozolomide and BCNU. Br J Cancer. 1996;73:1049–52. doi: 10.1038/bjc.1996.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Esteller M, Herman JG. Generating mutations but providing chemosensitivity: the role of O6-methylguanine DNA methyltransferase in human cancer. Oncogene. 2004;23:1–8. doi: 10.1038/sj.onc.1207316. [DOI] [PubMed] [Google Scholar]
- 20.Middleton MR, Margison GP. Improvement of chemotherapy efficacy by inactivation of a DNA-repair pathway. Lancet Oncol. 2003;4:37–44. doi: 10.1016/s1470-2045(03)00959-8. [DOI] [PubMed] [Google Scholar]
- 21.Esteller M, Hamilton SR, Burger PC, Baylin SB, Herman JG. Inactivation of the DNA repair gene O6-methylguanine-DNA methyltransferase by promoter hypermethylation is a common event in primary human neoplasia. Cancer Res. 1999;59:793–7. [PubMed] [Google Scholar]
- 22.Qian XC, Brent TP. Methylation hot spots in the 5′ flanking region denote silencing of the O6-methylguanine-DNA methyltransferase gene. Cancer Res. 1997;57:3672–7. [PubMed] [Google Scholar]
- 23.Chen ZP, Yarosh D, Garcia Y, et al. Relationship between O6-methylguanine-DNA methyltransferase levels and clinical response induced by chloroethylnitrosourea therapy in glioma patients. Can J Neurol Sci. 1999;26:104–9. [PubMed] [Google Scholar]
- 24.Hegi ME, Diserens AC, Gorlia T, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352:997–1003. doi: 10.1056/NEJMoa043331. [DOI] [PubMed] [Google Scholar]
- 25.Jaeckle KA, Eyre HJ, Townsend JJ, et al. Correlation of tumor O6 methylguanine-DNA methyltransferase levels with survival of malignant astrocytoma patients treated with bischloroethylnitrosourea: a Southwest Oncology Group study. J Clin Oncol. 1998;16:3310–5. doi: 10.1200/JCO.1998.16.10.3310. [DOI] [PubMed] [Google Scholar]
- 26.Middlemas DS, Stewart CF, Kirstein MN, et al. Biochemical correlates of temozolomide sensitivity in pediatric solid tumor xenograft models. Clin Cancer Res. 2000;6:998–1007. [PubMed] [Google Scholar]
- 27.Walker MC, Masters JR, Margison GP. O6-alkylguanine-DNA-alkyltransferase activity and nitrosourea sensitivity in human cancer cell lines. Br J Cancer. 1992;66:840–3. doi: 10.1038/bjc.1992.370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hickman MJ, Samson LD. Role of DNA mismatch repair and p53 in signaling induction of apoptosis by alkylating agents. Proc Natl Acad Sci U S A. 1999;96:10764–9. doi: 10.1073/pnas.96.19.10764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fink D, Aebi S, Howell SB. The role of DNA mismatch repair in drug resistance. Clin Cancer Res. 1998;4:1–6. [PubMed] [Google Scholar]
- 30.Liu L, Markowitz S, Gerson SL. Mismatch repair mutations override alkyltransferase in conferring resistance to temozolomide but not to 1,3-bis(2-chloroethyl)nitrosourea. Cancer Res. 1996;56:5375–9. [PubMed] [Google Scholar]
- 31.Taverna P, Liu L, Hanson AJ, Monks A, Gerson SL. Characterization of MLH1 and MSH2 DNA mismatch repair proteins in cell lines of the NCI anticancer drug screen. Cancer Chemother Pharmacol. 2000;46:507–16. doi: 10.1007/s002800000186. [DOI] [PubMed] [Google Scholar]
- 32.Bryant HE, Helleday T. Poly(ASP-ribose) polymerase inhibitors as potential chemotherapeutic agents. Biochem Soc Trans. 2005;32(Pt 6):959–61. doi: 10.1042/BST0320959. [DOI] [PubMed] [Google Scholar]
- 33.Lee JM, Bernstein A. p53 mutations increase resistance to ionizing radiation. Proc Natl Acad Sci U S A. 1993;90:5742–6. doi: 10.1073/pnas.90.12.5742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Asgari K, Sesterhenn IA, McLeod DG, et al. Inhibition of the growth of pre-established subcutaneous tumor nodules of human prostate cancer cells by single injection of the recombinant adenovirus p53 expression vector. Int J Cancer. 1997;71:377–82. doi: 10.1002/(sici)1097-0215(19970502)71:3<377::aid-ijc13>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- 35.Yang C, Cirielli C, Capogrossi MC, Passaniti A. Adenovirus-mediated wild-type p53 expression induces apoptosis and suppresses tumorigenesis of prostatic tumor cells. Cancer Res. 1995;55:4210–3. [PubMed] [Google Scholar]
- 36.Hall PA, Meek D, Lane DP. p53—integrating the complexity. J Pathol. 1996;180:1–5. doi: 10.1002/(SICI)1096-9896(199609)180:1<1::AID-PATH712>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 37.Hayashi Y, Ueki K, Waha A, Wiestler OD, Louis DN, von Deimling A. Association of EGFR gene amplification and CDKN2 (p16/MTS1) gene deletion in glioblastoma multiforme. Brain Pathol. 1997;7:871–5. doi: 10.1111/j.1750-3639.1997.tb00890.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Newcomb EW, Bhalla SK, Parrish CL, Hayes RL, Cohen H, Miller DC. bcl-2 protein expression in astrocytomas in relation to patient survival and p53 gene status. Acta Neuropathol (Berl) 1997;94:369–75. doi: 10.1007/s004010050721. [DOI] [PubMed] [Google Scholar]
- 39.Nagane M, Coufal F, Lin H, Bogler O, Cavenee WK, Huang HJ. A common mutant epidermal growth factor receptor confers enhanced tumorigenicity on human glioblastoma cells by increasing proliferation and reducing apoptosis. Cancer Res. 1996;56:5079–86. [PubMed] [Google Scholar]
- 40.Weller M, Malipiero U, Aguzzi A, Reed JC, Fontana A. Protooncogene bcl-2 gene transfer abrogates Fas/APO-1 antibody-mediated apoptosis of human malignant glioma cells and confers resistance to chemotherapeutic drugs and therapeutic irradiation. J Clin Invest. 1995;95:2633–43. doi: 10.1172/JCI117965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schlegel J, Merdes A, Stumm G, et al. Amplification of the epidermal-growth-factor-receptor gene correlates with different growth behaviour in human glioblastoma. Int J Cancer. 1994;56:72–7. doi: 10.1002/ijc.2910560114. [DOI] [PubMed] [Google Scholar]
- 42.Newcomb EW, Cohen H, Lee SR, et al. Survival of patients with glioblastoma multiforme is not influenced by altered expression of p16, p53, EGFR, MDM2 or Bcl-2 genes. Brain Pathol. 1998;8:655–67. doi: 10.1111/j.1750-3639.1998.tb00191.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hermisson M, Klumpp A, Wick W, et al. O6-methylguanine DNA methyltransferase and p53 status predict temozolomide sensitivity in human malignant glioma cells. J Neurochem. 2006;96:766–76. doi: 10.1111/j.1471-4159.2005.03583.x. [DOI] [PubMed] [Google Scholar]
- 44.Persidis A. Cancer multidrug resistance. Nat Biotechnol. 1999;17:94–5. doi: 10.1038/5289. [DOI] [PubMed] [Google Scholar]
- 45.Cole SP, Bhardwaj G, Gerlach JH, et al. Overexpression of a transporter gene in a multidrug-resistant human lung cancer cell line. Science. 1992;258:1650–4. doi: 10.1126/science.1360704. [DOI] [PubMed] [Google Scholar]
- 46.Norris MD, Bordow SB, Marshall GM, Haber PS, Cohn SL, Haber M. Expression of the gene for multidrug-resistance–associated protein and outcome in patients with neuroblastoma. N Engl J Med. 1996;334:231–8. doi: 10.1056/NEJM199601253340405. [DOI] [PubMed] [Google Scholar]
- 47.Cai Y, Wu MH, Xu-Welliver M, Pegg AE, Ludeman SM, Dolan ME. Effect of O6-benzylguanine on alkylating agent-induced toxicity and mutagenicity. In Chinese hamster ovary cells expressing wild-type and mutant O6-alkylguanine-DNA alkyltransferases. Cancer Res. 2000;60:5464–9. [PubMed] [Google Scholar]
- 48.Holland EC. A mouse model for glioma: biology, pathology, and therapeutic opportunities. Toxicol Pathol. 2000;28:171–7. doi: 10.1177/019262330002800122. [DOI] [PubMed] [Google Scholar]
- 49.Finkelstein SD, Black P, Nowak TP, Hand CM, Christensen S, Finch PW. Histological characteristics and expression of acidic and basic fibroblast growth factor genes in intracerebral xenogeneic transplants of human glioma cells. Neurosurgery. 1994;34:136–43. [PubMed] [Google Scholar]
- 50.Pilkington GJ, Bjerkvig R, De Ridder L, Kaaijk P. In vitro and in vivo models for the study of brain tumour invasion. Anticancer Res. 1997;17:4107–9. [PubMed] [Google Scholar]
- 51.Saris SC, Bigner SH, Bigner DD. Intracerebral transplantation of a human glioma line in immunosuppressed rats. J Neurosurg. 1984;60:582–8. doi: 10.3171/jns.1984.60.3.0582. [DOI] [PubMed] [Google Scholar]
- 52.Tonn JC. Model systems in neurooncology. Acta Neurochir Suppl. 2002;83:79–83. doi: 10.1007/978-3-7091-6743-4_13. [DOI] [PubMed] [Google Scholar]
- 53.Bigner SH, Humphrey PA, Wong AJ, et al. Characterization of the epidermal growth factor receptor in human glioma cell lines and xenografts. Cancer Res. 1990;50:8017–22. [PubMed] [Google Scholar]
- 54.Pandita A, Aldape KD, Zadeh G, Guha A, James CD. Contrasting in vivo and in vitro fates of glioblastoma cell subpopulations with amplified EGFR. Genes Chromosomes Cancer. 2004;39:29–36. doi: 10.1002/gcc.10300. [DOI] [PubMed] [Google Scholar]
- 55.Giannini C, Sarkaria JN, Saito A, et al. Patient tumor EGFR and PDGFRA gene amplifications retained in an invasive intracranial xenograft model of glioblastoma multiforme. Neuro-Oncol. 2005;7:164–76. doi: 10.1215/S1152851704000821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sarkaria JN, Carlson BL, Schroeder MA, et al. Use of an orthotopic xenograft model for assessing the effect of epidermal growth factor receptor amplification on glioblastoma radiation response. Clin Cancer Res. 2006;12:2264–71. doi: 10.1158/1078-0432.CCR-05-2510. [DOI] [PubMed] [Google Scholar]
- 57.Beneke S, Diefenbach J, Burkle A. Poly(ADP-ribosyl)ation inhibitors: promising drug candidates for a wide variety of pathophysiologic conditions. Int J Cancer. 2004;111:813–8. doi: 10.1002/ijc.20342. [DOI] [PubMed] [Google Scholar]
- 58.Woon EC, Threadgill MD. Poly(ADP-ribose)polymerase inhibition—where now? Curr Med Chem. 2005;12:2373–92. doi: 10.2174/0929867054864778. [DOI] [PubMed] [Google Scholar]
- 59.Bowman KJ, White A, Golding BT, Griffin RJ, Curtin NJ. Potentiation of anti-cancer agent cytotoxicity by the potent poly(ADP-ribose) polymerase inhibitors NU1025 and NU1064. Br J Cancer. 1998;78:1269–77. doi: 10.1038/bjc.1998.670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Calabrese CR, Batey MA, Thomas HD, et al. Identification of potent nontoxic poly(ADP-ribose) polymerase-1 inhibitors: chemopotentiation and pharmacological studies. Clin Cancer Res. 2003;9:2711–8. [PubMed] [Google Scholar]
- 61.Miknyoczki SJ, Jones-Bolin S, Pritchard S, et al. Chemopotentiation of temozolomide, irinotecan, and cisplatin activity by CEP-6800, a poly(ADP-ribose) polymerase inhibitor. Mol Cancer Ther. 2003;2:371–82. [PubMed] [Google Scholar]
- 62.Tentori L, Leonetti C, Scarsella M, et al. Systemic administration of GPI 15427, a novel poly(ADP-ribose) polymerase-1 inhibitor, increases the antitumor activity of temozolomide against intracranial melanoma, glioma, lymphoma. Clin Cancer Res. 2003;9:5370–9. [PubMed] [Google Scholar]
- 63.Calabrese CR, Almassy R, Barton S, et al. Anticancer chemosensitization and radiosensitization by the novel poly(ADP-ribose) polymerase-1 inhibitor AG14361. J Natl Cancer Inst. 2004;96:56–67. doi: 10.1093/jnci/djh005. [DOI] [PubMed] [Google Scholar]
- 64.Curtin NJ. PARP inhibitors for cancer therapy. Expert Rev Mol Med. 2005;7:1–20. doi: 10.1017/S146239940500904X. [DOI] [PubMed] [Google Scholar]
- 65.Tentori L, Graziani G. Chemopotentiation by PARP inhibitors in cancer therapy. Pharmacol Res. 2005;52:25–33. doi: 10.1016/j.phrs.2005.02.010. [DOI] [PubMed] [Google Scholar]
- 66.Bryant HE, Schultz N, Thomas HD, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434:913–7. doi: 10.1038/nature03443. [DOI] [PubMed] [Google Scholar]
- 67.Farmer H, McCabe N, Lord CJ, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917–21. doi: 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- 68.Skalitzky DJ, Marakovits JT, Maegley KA, et al. Tricyclic benzimidazoles as potent poly(ADP-ribose) polymerase-1 inhibitors. J Med Chem. 2003;46:210–3. doi: 10.1021/jm0255769. [DOI] [PubMed] [Google Scholar]
- 69.Curtin NJ, Wang LZ, Yiakouvaki A, et al. Novel poly(ADP-ribose) polymerase-1 inhibitor, AG14361, restores sensitivity to temozolomide in mismatch repair-deficient cells. Clin Cancer Res. 2004;10:881–9. doi: 10.1158/1078-0432.ccr-1144-3. [DOI] [PubMed] [Google Scholar]
- 70.Plummer R, Middleton M, Wilson R, et al. First in human phase I trial of the PARP inhibitor AG-014699 with temozolomide (TMZ) in patients (pts) with advanced solid tumors. J Clin Oncol. 2005;23(suppl):208s. Abstract 3065. [Google Scholar]
- 71.Plummer R, Lorigan P, Evans J, et al. First and final report of a phase II study of the poly(ADP-ribose) polymerase (PARP) inhibitor, AG014699, in combination with temozolomide (TMZ) in patients with metastatic malignant melanoma (MM) J Clin Oncol. 2006;24(suppl):456s. Abstract 8013. [Google Scholar]
- 72.Danson SJ, Middleton MR. Temozolomide: a novel oral alkylating agent. Expert Rev Anticancer Ther. 2001;1:13–9. doi: 10.1586/14737140.1.1.13. [DOI] [PubMed] [Google Scholar]
- 73.Bryant HE, Schultz N, Thomas HD, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434:913–7. doi: 10.1038/nature03443. [DOI] [PubMed] [Google Scholar]
- 74.Fong PC, Spicer J, Reade S, et al. Phase I pharmacokinetic (PK) and pharmacodynamic (PD) evaluation of a small molecule inhibitor of Poly ADP-Ribose Polymerase (PARP), KU-0059436 (Ku) in patients (p) with advanced tumours. J Clin Oncol. 2006;24(suppl):126s. Abstract 3022. [Google Scholar]
- 75.Wei SH, Chen CM, Strathdee G, et al. Methylation microarray analysis of late-stage ovarian carcinomas distinguishes progression-free survival in patients and identifies candidate epigenetic markers. Clin Cancer Res. 2002;8:2246–52. [PubMed] [Google Scholar]
- 76.Kennedy RD, D'Andrea AD. DNA repair pathways in clinical practice: lessons from pediatric cancer susceptibility syndromes. J Clin Oncol. 2006;24:3799–808. doi: 10.1200/JCO.2005.05.4171. [DOI] [PubMed] [Google Scholar]
- 77.McCabe N, Turner NC, Lord CJ, et al. Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(ADP-ribose) polymerase inhibition. Cancer Res. 2006;66:8109–15. doi: 10.1158/0008-5472.CAN-06-0140. [DOI] [PubMed] [Google Scholar]
- 78.Christmann M, Tomicic MT, Roos WP, Kaina B. Mechanisms of human DNA repair: an update. Toxicology. 2003;193:3–34. doi: 10.1016/s0300-483x(03)00287-7. [DOI] [PubMed] [Google Scholar]