

Abstract
Background
The systemic response to injury is characterized by massive release of norepinephrine (NE) into the circulation as a result of global sympathetic activation. We have recently demonstrated that NE modulates the recruitment of macrophages to the cutaneous wound. We hypothesized that NE suppresses wound macrophage phagocytic function through canonical adrenergic signaling pathways.
Methods
Murine wound macrophages were harvested at five days after injury and treated with physiologic and pharmacologic dose norepinephrine. Phagocytosis of green fluorescent protein-labeled E.coli was assayed by flow cytometry. The signaling pathways mediating NE modulation of wound macrophage phagocytosis were interrogated by pharmacologic manipulation of alpha- and beta-ARs, intracellular cAMP and PKA. Tissue specificity was determined by comparison of wound macrophages to splenic macrophages.
Results
Both physiologic and pharmacologic dose NE suppressed wound macrophage phagocytic efficiency. This effect was mediated by alpha- and beta-ARs in a dose-dependent fashion. Direct stimulation of cAMP suppressed phagocytic efficiency and blockade of PKA signaling prevented NE-mediated suppression of phagocytic efficiency. Splenic macrophage phagocytic efficiency was less than that of wound macrophages and was not altered by NE.
Conclusions
NE has a profound immunosuppressive effect on wound macrophage function that is tissue specific and appears to be mediated through adrenergic receptors and their canonical downstream signaling pathway. Attenuation of post-injury immunosuppression represents another potential mechanism by which beta-AR blockade may reduce morbidity and mortality following severe injury.
Background
Trauma is the leading killer of Americans 1-44 years old and results in more years of potential life lost than any other disease process(1). The incidence of infectious complications, both systemic and local, is markedly increased following trauma and sepsis accounts for the majority of deaths after the initial 48 hours post-injury(2, 3). A growing body of literature supports the concept of neuroimmunomodulation, or nervous system modulation of immune function(4). Following severe injury there is destruction of noradrenergic neurons innervating the injured tissue and massive release of norepinephrine into the peripheral circulation(5, 6). Alterations in the post-trauma catecholamine milieu are often compounded by the clinical use of vasopressor agents for hemodynamic support.
Macrophages play a critical role in successful tissue repair as key mediators of the inflammatory phase of wound healing(7-9). Following their recruitment to the site of injury, macrophages function in phagocytosis and the production of growth factors and cytokines to coordinate wound repair(10). Depletion of monocytes and macrophages resulted in impaired healing, with deficits ranging from delay in clearance of debris to lasting alterations in later phases of wound healing as evidenced by decreased fibroblast proliferation and fibrogenesis(11). Through the process of phagocytosis, wound macrophages perform an antimicrobial innate immune function and link to the adaptive immune response through antigen presentation(12).
Numerous studies have demonstrated functional α- and β-adrenoreceptors on macrophages(13-17). The production of pro-inflammatory cytokines appears to be inversely related to intracellular cyclic adenosine monophosphate (cAMP) levels, which are regulated by α2-adrenoreceptor mediated inhibition and β2-adrenoreceptor mediated activation of adenylate cyclase. For example, low levels of norepinephrine result in enhanced production of TNF-α(15, 17), whereas high levels of norepinephrine inhibit TNF-α production(17, 18). Additionally, high levels of norepinephrine appear to inhibit macrophage proliferation and promote apoptosis through non-receptor mediated mechanisms, likely through the production of cytotoxic reactive oxygen species(13).
While a clear role for norepinephrine in modulation of cytokine production by macrophages exists(15-18), there has been comparatively little investigation of catecholamine-mediated alterations in phagocytosis, a critical step in the wound healing process(19-21). The present study examines the role of norepinephrine in modulation of wound macrophage phagocytic function using E.coli bacteria, a biologically relevant target. We demonstrate that norepinephrine negatively modulates wound macrophage phagocytic efficiency through α- and β-adrenoreceptors in a cAMP and PKA-dependent fashion. Additionally, we demonstrate that this response has tissue-bed specificity within a given host.
Methods
Isolation of wound macrophages
All animal procedures were reviewed and approved by the Loyola University Institutional Animal Use and Care Committee. To isolate wound macrophages for in vitro analysis, a standard subcutaneous sponge model was employed(22). Mice were anesthetized by intraperitoneal injection of ketamine (100 mg/kg body weight) and xylazine (10 mg/kg). The dorsal side of the animals was shaved and scrubbed with betadine. A 2 cm longitudinal skin incision was made through the dermis and panniculus carnosus in a paraspinal location and two polyvinyl alcohol (PVA) sponges (20 × 5 × 2 mm, Rippey, El Dorado Hill, CA) were placed in dorsal subcutaneous pockets. The skin edges were approximated and closed with surgical clips. At 120 hours post-injury, the animals were sacrificed and the sponges retrieved and placed into 1 mL of Dulbecco's PBS (D-PBS, Gibco BRL, Grand Island, NY) in a 17 × 100 mm polypropylene tube. Sponges were manually compressed to free inflammatory cells from the sponge matrix. The barrel of a 3 mL syringe was inserted into the top of each tube and the sponges placed into the syringe barrel. The syringe and tube were centrifuged at 200x g for 1-2 minutes to remove any residual liquid and cells from the sponges. Cells were counted using a hemocytometer and adjusted to a concentration of 1 × 106 cells/mL in Roswell Park Memorial Institute (RPMI)-1640 (Gibco BRL) media supplemented with 10% fetal bovine serum and placed in Teflon-coated tissue culture vials to maintain them in a non-adherent state. In order to maximally simulate the wound environment and minimize post-harvest phenotypic alterations in cells, no further separation was undertaken and wound macrophages were kept in suspension with other wound inflammatory cells (F4/80+ Macrophages 41 ± 2.0%, Gr-1+/F4/80− Neutrophils 6.9 ± 0.68%).
Isolation of splenic macrophages
Intact spleens were harvested from wounded animals at the time of subcutaneous sponge retrieval (120 hours post-injury). Total splenocytes were obtained by mechanical homogenization of the spleen in 1 mL of sterile PBS on ice. The cell suspension was passed through a cell strainer (100μm, Falcon) to remove residual connective tissue and resuspended in 30 mL of PBS. Total splenocytes were pelleted (200x g × 5 min) and resuspended in 5 mL of PBS. Splenic macrophages were then separated by density gradient centrifugation (Ficoll-Paque PLUS, Amersham Biosciences, Piscataway, NJ). Cells were counted by hemocytometer and adjusted to a concentration of 1 × 106 cells/mL in RPMI 1640 supplemented with 10% fetal bovine serum and placed in Teflon-coated tissue culture vials.
Cell treatment protocols
(A) To determine if norepinephrine modulates phagocytosis by wound macrophages, cells were initially divided into three groups (cells from n=6 animals per group) and received either no treatment (Control), physiologic norepinephrine (10−9 M, Sigma, St. Louis, MO), or pharmacologic norepinephrine (10−6 M, Sigma). Cells were then maintained in non-adherent culture overnight (18 hours) prior to assaying phagocytosis. (B) To determine if norepinephrine-mediated changes in phagocytosis were adrenoreceptor dependent, cells were divided into three groups (n=5-6 animals per group) and received either non-selective α-adrenergic blockade (phentolamine, 10−6 M, Sigma), non-selective β-adrenergic blockade (propranolol, 10−6 M, Sigma), or combined α- and β-adrenergic blockade (phentolamine and propranolol). Cells were pre-treated with adrenergic blockade for two hours prior to norepinephrine treatment as above (total of nine treatment groups). (C) The role of cAMP in modulation of wound macrophage phagocytosis was determined by treatment of cells (n=6 animals) with forskolin (5×10−5 M, Sigma) in the presence of 3-isobutyl-1-methylxanthine (IBMX, 5×10−5 M, Sigma), a non-specific phosphodiesterase inhibitor, overnight (18 hours) prior to assaying phagocytosis. (D) To determine if norepinephrine-mediated changes in phagocytosis involve the protein kinase A (PKA) signaling pathway, cells were incubated with H-89 (10−5 M, Sigma), a specific inhibitor of PKA, for two hours prior to norepinephrine treatment as above. The dosages and treatment durations employed were chosen to allow ease of comparison with previous studies(20, 21, 23).
E.coli Phagocytosis Assay
E.coli K-12 bacteria constitutively expressing green fluorescent protein (GFP-E.coli, a kind gift from ConjuGon, Inc., Madison, WI) were grown in LB broth containing chloramphenicol (20 μg/mL) in a shaking incubator at 37°C overnight. The following morning, bacteria were pelleted (1850x g for 15 minutes) and washed in 1X PBS (30 mL) three times. The bacteria were then re-suspended and diluted in 1X PBS to a final O.D. of 1.3-1.5 at λ=665 nm (∼108 bacteria/mL). The bacteria were kept protected from light at room temperature until used to assay phagocytosis.
On the day prior to assay, inflammatory cells were isolated from retrieved subcutaneous sponges, as described above. Cells were incubated overnight at 37°C in Teflon-coated tissue culture vials. On the day of the assay, cells were washed twice with D-PBS and re-suspended in 1 mL of D-PBS with 0.1% glucose. An aliquot (100 μL) of the bacterial suspension was added to the cells, the cell-bacteria mixture pelleted (200x g × 5 min) to obtain maximal contact, and allowed to incubate at 37°C for 1 hour. From the time of addition of bacteria, all steps were carried out protected from light. The phagocytic process was arrested by the addition of 1 mL ice-cold PBS. The cells were washed twice with ice-cold PBS, resuspended in lysozyme (1 mg/mL in PBS, Amresco, Solon, OH) and incubated at 4°C for 20 minutes on an orbital shaker to remove adherent extracellular bacteria. Cells were then washed twice with ice-cold PBS.
For quantitative analysis, cells were stained with rat anti-mouse F4/80-APC (1:50 dilution, eBioscience, San Diego, CA) and maintained on ice until analyzed by flow cytometry (FACSCalibur or FACSCanto, Becton Dickinson, San Jose, CA). Flow cytometry data analysis (FlowJo v6.4.2, TreeStar, Ashland, OR) included comparison of the percentage of cells that engaged in phagocytosis as well as comparison of the mean fluorescence intensity (MFI) of the GFP channel in the phagocytosis-positive cells (Phagocytic Index; proportional to number of bacteria ingested per cell; normalized to 100% in the control group to allow comparisons of data from multiple experiments).
For qualitative analysis, cells were stained with rat anti-mouse F4/80-PE (1:50 dilution, eBioscience, San Diego, CA). The cell suspension was centrifuged in a cytocentrifuge (Shandon Cytospin 2; Shandon, Oakland, CA), mounted with an anti-fade mounting medium (Vectashield, Vector Laboratories, Burlingame, CA) and maintained at 4°C until imaged by confocal microscopy (Zeiss LSM 510 microscope, Carl Zeiss, Thornwood, NJ).
Statistical Analysis
The mean and standard error of mean were calculated for each experimental group. Statistical analysis was performed using GraphPad Prism (Version 4.0, GraphPad Software, San Diego, CA). Data that were described over time were analyzed by analysis of variance (ANOVA) followed by Bonferonni post-comparison testing. Data described at single time points were analyzed by Student's unpaired t test. Results with a p value less than 0.05 were considered statistically significant.
Results
Norepinephrine suppresses wound macrophage phagocytic efficiency
To determine if norepinephrine modulates wound macrophage phagocytic ability, macrophages were isolated from 120-hour wounds and received no treatment (Control), physiologic-dose norepinephrine (10−9 M), or pharmacologic-dose norepinephrine (10−6 M) for 18 hours prior to assaying phagocytosis of GFP-E.coli by flow cytometry. Norepinephrine treatment did not affect the viability of macrophages in vitro (data not shown). Both physiologic and pharmacologic doses of norepinephrine decreased wound macrophage phagocytic index, indicating fewer bacteria ingested per cell, a measure of phagocytic efficiency (Control 100 ± 4.6, Physiologic NE 70 ± 7.0, Pharmacologic NE 68 ± 3.7, p<0.001, Figure 1A and B). However, norepinephrine treatment did not affect the percentage of cells that engaged in phagocytosis (Control 71 ± 9.8, Physiologic NE 63 ± 7.7, Pharmacologic NE 59 ± 6.8, p=NS, Figure 1C). Visualization of macrophages and bacteria by confocal microscopy following phagocytosis confirmed that the ingested bacteria were intracellular (Figure 1D).
Figure 1. Norepinephrine suppresses wound macrophage phagocytic efficiency.




Macrophages isolated from 120-hour wounds were placed in culture for 18 hours with media alone (Control, red), physiologic norepinephrine (Phys NE, 10−9 M, blue), or pharmacologic norepinephrine (Pharm NE, 10−6 M, green) and phagocytosis of E.coli was assessed. (A) Norepinephrine-treatment did not alter the percentage of macrophages that engaged in phagocytosis (p=NS). (B) Representative phagocytosis histograms for control (red), physiologic NE-treated (blue) and pharmacologic NE-treated (green) wound macrophages. Both physiologic- (blue) and pharmacologic-dose norepinephrine treatment (green) resulted in a shift of the phagocytosis curve to the left, indicating fewer bacteria ingested per cell. The geometric mean of each curve was calculated to provide the phagocytic index (C) and was analyzed by two-way ANOVA followed by Bonferonni post-comparison testing. Both physiologic- and pharmacologic-dose norepinephrine suppressed the phagocytic efficiency of 120-hour wound macrophages (*p<0.001). (D) Representative confocal microscopy image demonstrating phagocytosis of GFP-E.coli (green, upper right) by a macrophage (phycoerythrin-F4/80, red, upper left). False-DIC (white, lower left) delineates the boundaries of the macrophage, demonstrating that the visualized bacteria are intracellular (merged image, lower right).
Norepinephrine-mediated suppression of wound macrophage phagocytosis is tissue-bed specific
To determine if tissue-bed variability exists in the norepinephrine response of macrophages, both splenic and wound macrophages were isolated from the same animals at 120 hours post-wounding. Splenic and wound macrophages received physiologic norepinephrine or pharmacologic norepinephrine prior to phagocytosis and were compared to untreated controls. After 18 hours of pre-treatment, phagocytosis of GFP-E.coli was assayed by flow cytometry. Expression of cell-surface F4/80 was used as a marker of maturity to ensure comparison of similar macrophages between the spleen (67 ± 0.6%) and wounds (100%). At baseline, splenic macrophages demonstrated less phagocytic capacity than wound macrophages from the same animal (Wound/Untreated 100 ± 3.6 vs. Spleen/Untreated 67.8 ± 2.5, p<0.001, Figure 2). In contrast to wound macrophages, norepinephrine did not suppress phagocytosis by splenic macrophages (Spleen/Physiologic NE 64.2 ± 2.5, Spleen/Pharmacologic NE 70.3 ± 3.0, p=NS, Figure 2).
Figure 2. Norepinephrine-mediated suppression of wound macrophage phagocytosis is tissue-bed specific.

Splenic macrophages isolated from animals 120-hours post-wounding were treated with physiologic norepinephrine (Phys NE, 10−9 M), or pharmacologic norepinephrine (Pharm NE, 10−6 M) for 18 hours and phagocytosis of E.coli was assessed in comparison to wound macrophages from the same animals. Splenic macrophage phagocytic function was less than that of wound macrophages at baseline(*p<0.001). Additionally, unlike wound macrophages (*p<0.001), norepinephrine treatment did not suppress splenic macrophage phagocytosis (p=NS).
Norepinephrine-mediated suppression of wound macrophage phagocytosis is adrenoreceptor dependent
To determine if norepinephrine-mediated suppression of wound macrophage phagocytic efficiency was mediated through adrenoreceptor binding, inflammatory cells were isolated from subcutaneous sponges 120 hours post-wounding and treated with either α- or β-adrenergic blockade for two hours prior to treatment with physiologic norepinephrine or pharmacologic norepinephrine. After 18 hours of pre-treatment, phagocytosis of GFP-E.coli was assayed by flow cytometry. Neither α- nor β-adrenergic blockade alone or in combination altered wound macrophage phagocytosis (data not shown). Suppression of wound macrophage phagocytosis by physiologic dose norepinephrine was partially blocked by pre-treatment with α-adrenergic blockade (Untreated Control 100 ± 5.1, α-Blockade/Physiologic NE 68 ± 13, p=NS, Figure 3) and almost completely blocked by β-adrenergic blockade (Untreated Control 100 ± 5.1, β-Blockade/Physiologic NE 85 ± 16, p=NS; Figure 3) as well as combined α- and β-adrenergic blockade (Untreated Control 100 ± 5.1, α+β-Blockade/Physiologic NE 93 ± 7.6, p=NS, Figure 3). Suppression of wound macrophage phagocytosis by pharmacologic dose norepinephrine was almost completely blocked by pre-treatment with α-adrenergic blockade (Untreated Control 100 ± 5.1, α-Blockade/Pharmacologic NE 80 ± 15, p=NS, Figure 3) but was not effected by β-adrenergic blockade (Untreated Control 100 ± 5.1, β-Blockade/Pharmacologic NE 56 ± 9, p<0.01; Figure 3). Combined α- and β-adrenergic blockade blocked pharmacologic norepinephrine mediated suppression of phagocytosis (Untreated Control 100 ± 5.1, α+β-Blockade /Pharmacologic NE 90 ± 9.3, p=NS, Figure 3).
Figure 3. Norepinephrine-mediated suppression of wound macrophage phagocytosis is adrenoreceptor dependent.


Macrophages isolated from 120-hour wounds were pre-treated with either α- or β-adrenergic blockade for two hours prior to treatment with physiologic norepinephrine (NE +, 10−9 M) or pharmacologic norepinephrine (NE ++, 10−6 M) for 18 hours and assessment of phagocytosis of E.coli. Neither α- nor β-adrenergic blockade alone altered wound macrophage phagocytosis (p=NS, data not shown). α-adrenergic blockade partially prevented the suppression of wound macrophage phagocytosis by physiologic norepinephrine and completely blocked pharmacologic norepinephrine-mediated suppression (p=NS vs. Untreated Control). β-adrenergic blockade completely blocked physiologic norepinephrine-mediated suppression (p=NS) but did not prevent the suppression of wound macrophage phagocytosis by pharmacologic norepinephrine (*p<0.05). Combined α- and β-adrenergic blockade prevented both physiologic and pharmacologic norepinephrine-mediated suppression of wound macrophage phagocytosis (p=NS).
Norepinephrine-mediated suppression of wound macrophage phagocytosis is cAMP and protein kinase A (PKA) dependent
Classically, adrenoreceptor activation by catecholamines results in increased intracellular cAMP, which leads to downstream activation of PKA. To determine if this pathway was involved in the norepinephrine/adrenoreceptor-mediated changes in phagocytosis by wound macrophages, isolated cells from 120-hour wounds received either physiologic norepinephrine (10−9 M), pharmacologic norepinephrine (10−6 M), or forskolin (5×10−5 M) in the presence of IBMX (5×10−5 M) for 18 hours prior to assaying phagocytosis of GFP-E.coli by flow cytometry. Treatment with forskolin, which directly elevates cAMP, decreased wound macrophage phagocytic efficiency, although to a lesser extent than treatment with physiologic or pharmacologic doses of norepinephrine (Control 100 ± 6.2, Physiologic NE 61 ± 3.5, Pharmacologic 59 ± 2.8, Forskolin 81 ± 4.0, p<0.05, Figure 4).
Figure 4. The adenylate cyclase activator forskolin suppresses wound macrophage phagocytosis.

Macrophages isolated from 120-hour wounds were treated with physiologic norepinephrine, pharmacologic norepinephrine, or forskolin in the presence of phosphodiesterase inhibitor (IBMX) for 18 hours and phagocytosis of E.coli was assessed. Treatment with forskolin suppressed wound macrophage phagocytic efficiency(*p<0.05), although to a lesser degree than treatment with physiologic or pharmacologic doses of norepinephrine (#p<0.05).
Next, macrophages isolated from 120-hour wounds were treated with PKA inhibitor for two hours prior to treatment with physiologic norepinephrine or pharmacologic norepinephrine. Following overnight incubation, phagocytosis of GFP-E.coli was assayed by flow cytometry. PKA-inhibition alone had no effect on wound macrophage phagocytosis (data not shown). Pre-treatment with PKA inhibitor blocked the suppression of wound macrophage phagocytosis by physiologic norepinephrine and partially prevented pharmacologic norepinephrine-mediated suppression (Control 100 ± 4.6, PKA Blockade/Physiologic NE 95 ± 9, PKA Blockade/Pharmacologic NE 83 ± 7, p=NS, Figure 5).
Figure 5. Norepinephrine-mediated suppression of wound macrophage phagocytosis is mediated by protein kinase A (PKA).

Macrophages isolated from 120-hour wounds were treated with the PKA inhibitor H-89 for two hours prior to treatment with physiologic norepinephrine (NE +, 10−9 M) or pharmacologic norepinephrine (NE ++, 10−6 M) for 18 hours and assessment of phagocytosis of E.coli. PKA blockade alone had no effect on wound macrophage phagocytosis (p=NS, data not shown). PKA inhibition prevented physiologic norepinephrine-mediated suppression of wound macrophage phagocytosis and partially prevented pharmacologic norepinephrine-mediated suppression (p=NS vs. Untreated Control).
Conclusions
Phagocytosis is a phylogenetically conserved process by which cells ingest microbial pathogens and debris(24). While a number of different cell types can undertake phagocytosis, macrophages are professional phagocytes, a specialized subset of immune cells that respond to activation and differentiation signals by modulating their phagocytic efficiency(25). Norepinephrine, the primary neurotransmitter of the peripheral sympathetic nervous system, is released in large quantities from peripheral nerves into the circulation following injury and has previously been shown to be involved in wound healing(26-29). While a number of studies report catecholamine-mediated alterations in the phenotype of splenic and peritoneal macrophages(20, 21, 30), the present study is the first to delineate a role for norepinephrine in modulation of wound macrophage phagocytic function.
We have demonstrated that exogenous norepinephrine, in both physiologic and pharmacologic doses, decreased wound macrophage phagocytic efficiency. Interestingly, there was no alteration in the number of macrophages that engaged in phagocytosis, a quantity that is often reported in the literature to be altered by norepinephrine treatment(31). Our results also indicate that this norepinephrine-mediated effect acts through both the α- and β-adrenergic receptor pathways in a dose-defined fashion. We observed that lower doses of norepinephrine acted primarily through the β-adrenergic receptors while higher doses had a predominantly α-adrenergic mechanism of transmission (Figure 6). This result is in contrast to previous reports of norepinephrine modulation of cytokine function(15, 17) but is consistent with the previous observation that norepinephrine competes for β-adrenoreceptor binding with relatively low potency(14) and evidence that high doses of catecholamines may lead to internalization of the β-adrenoreceptor(32).
Figure 6. Putative model of norepinephrine-mediated alterations in wound macrophage phagocytosis.
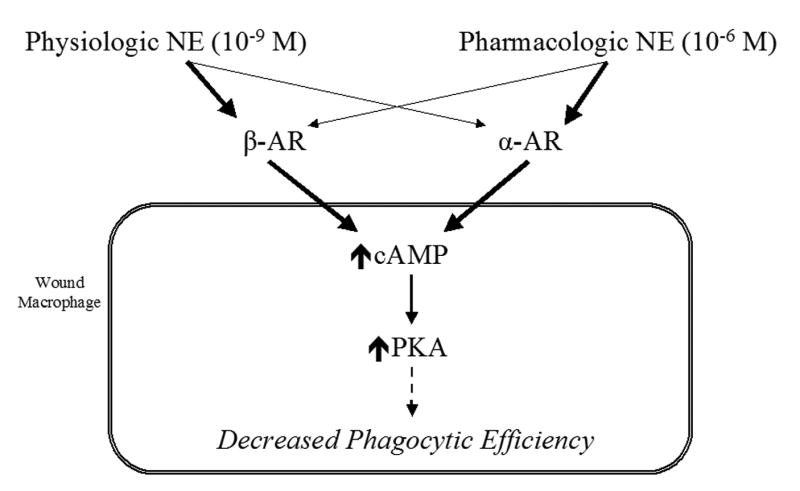
A putative model of norepinephrine-mediated modulation of wound macrophage phagocytic efficiency is proposed. At physiologic doses of norepinephrine, extracellular effects appear to be mediated primarily by β-adrenergic receptor binding (heavy line), with a smaller contribution from α-adrenergic receptor stimulation (light line). At the pharmacologic dose of norepinephrine, the reverse is true; the extracellular effects appear to be mediated primarily by α-adrenergic receptor binding (heavy line), with a smaller contribution from β-adrenergic receptor stimulation (light line). Regardless of dose, the intracellular alterations leading to decreased phagocytic efficiency involve increased levels of cAMP and its downstream effector, PKA.
A distinction between resident and recruited macrophages is recognized in the literature(33, 34). We examined the tissue-bed specificity of norepinephrine-mediated modulations in macrophage phagocytosis by comparing wound and splenic macrophages isolated from the same animals at the same time point. We observed that, at baseline, splenic macrophages demonstrate lower phagocytic efficiency than wound macrophages. Additionally, norepinephrine did not alter phagocytosis by splenic macrophages from wounded animals. The underlying reasons for tissue-bed variation in macrophage responsiveness are unclear and multiple possibilities exist. Alterations in surface receptor expression during the process of monocyte to macrophage differentiation may result in varied phagocytic ability of macrophage populations in different anatomic sites(35). Evidence for this theory includes data that appearance of αVβ3 integrin on the surface of cells is temporally connected to their transition from monocytes to macrophages in vitro, and this parallels the appearance of phagocytic capacity(36). Another possibility is that wound macrophages represent a subset of monocytic cells that are able to respond to norepinephrine, and that splenic macrophages with this ability have been mobilized from the spleen to the site of injury(37). The activation state of macrophages is influenced by changes in the wound microenvironment, and may result in alterations of phagocytic capacity as well(38). Additionally, alterations in relative density of α- and β-adrenoreceptors on the macrophage surface could contribute to the observed differences(39), but this concept has not been adequately explored in the literature.
Further evidence for the concept of tissue bed specificity of the macrophage response comes from studies of splenic, peritoneal and alveolar macrophages. Examination of catecholamine influence on phagocytosis by splenic macrophages reveals findings both complementary to and conflicting with ours(21). These investigators demonstrated a suppression of both percent phagocytosis and phagocytic efficiency in splenic macrophages isolated from animals that had experienced restraint stress. Additionally, in vitro norepinephrine treatment of splenic macrophages from unrestrained animals demonstrated a dose-response relationship, with enhancement of phagocytosis seen with low doses (10−11-10−15 M) and suppression seen with higher doses (10−5-10−7 M). These responses were abrogated by β-adrenergic blockade but were independent of α-adrenergic blockade. In contrast, previous studies of the role of catecholamines in modulating phagocytosis by peritoneal macrophages demonstrated a β-adrenoreceptor dependent enhancement of phagocytosis from doses of norepinephrine in the 10−5-10−4 M range(20, 30).
Studies of alveolar macrophages have demonstrated predominantly inhibitory effects of cAMP on phagocytosis, generation of reactive oxygen intermediates and the production of inflammatory cytokines(40, 41). More recently, investigation of the modulation of alveolar macrophage function has revealed differential roles for protein kinase A (PKA) and exchange protein directly activated by cAMP-1 (Epac-1), the downstream targets of cAMP(19). Specifically, suppression of phagocytosis appears to be mediated by Epac-1, but not PKA. The reverse is true of alveolar macrophage production of pro-inflammatory mediators, such as leukotriene B4 and TNF-α, which appears to be suppressed by PKA activation. In our study, suppression of phagocytic efficiency was observed with direct elevation of intracellular cAMP and a complete reversal of norepinephrine-mediated suppression was seen with PKA blockade. This would appear to point away from a role for Epac-1 in norepinephrine-mediated changes in phagocytosis by wound macrophages.
Taken as a whole, our results and those of other groups indicate a definite role for norepinephrine in the modulation of phagocytosis and phagocytic efficiency by macrophages. While heterogeneity in specific tissue-bed responses is present, the fundamental mechanisms involved include stimulation of either α- or β-adrenoreceptors followed by intracellular signaling via cAMP and its dependent protein kinases. Further evaluation of downstream changes in the gene transcription as a potential final common pathway for alterations in phagocytosis is warranted. These results reinforce the concept of norepinephrine and its intracellular signaling mediators as potential therapeutic targets in the manipulation of innate immunity.
Acknowledgments
Support: NIH T32-GM08750 (AG), R01-GM42577 (RLG), R01-GM55238 (LAD), Dr. Ralph and Marion C. Falk Medical Research Trust (AG, RLG, LAD)
Footnotes
Portions of this manuscript were or will be presented at:
- Surgical Forum of the 92nd Annual American College of Surgeons' Clinical Congress, Chicago, October 2006
- 2nd Annual Academic Surgical Congress, Society of University Surgeon's Program, Phoenix, Arizona, February 2007
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Ankush Gosain, Email: agosain@lumc.edu.
Kuzhali Muthu, Email: kmuthu@lumc.edu.
Richard L. Gamelli, Email: rgamell@lumc.edu.
Luisa A. DiPietro, Email: ldipiet@uic.edu.
References
- 1.Centers for Disease Control and Prevention: National Center for Injury Prevention and Control. Web-based Injury Statistics Query and Reporting System (WISQARS) www.cdc.gov/ncipc/wisqars 2005 [cited 2006 May 4]; Available from:
- 2.Baker CC, Oppenheimer L, Stephens B, Lewis FR, Trunkey DD. Epidemiology of trauma deaths. Am J Surg. 1980;140(1):144–50. doi: 10.1016/0002-9610(80)90431-6. [DOI] [PubMed] [Google Scholar]
- 3.Deitch EA. Infection in the compromised host. Surg Clin North Am. 1988;68(1):181–97. doi: 10.1016/s0039-6109(16)44439-7. [DOI] [PubMed] [Google Scholar]
- 4.Elenkov IJ, Wilder RL, Chrousos GP, Vizi ES. The sympathetic nerve--an integrative interface between two supersystems: the brain and the immune system. Pharmacol Rev. 2000;52(4):595–638. [PubMed] [Google Scholar]
- 5.Woolf PD, McDonald JV, Feliciano DV, Kelly MM, Nichols D, Cox C. The catecholamine response to multisystem trauma. Arch Surg. 1992;127(8):899–903. doi: 10.1001/archsurg.1992.01420080033005. [DOI] [PubMed] [Google Scholar]
- 6.Zetterstrom BE, Palmerio C, Fine J. Protection Of Functional And Vascular Integrity Of The Spleen In Traumatic Shock By Denervation. Proc Soc Exp Biol Med. 1964;117:373–6. doi: 10.3181/00379727-117-29584. [DOI] [PubMed] [Google Scholar]
- 7.Robson MC. Wound infection. A failure of wound healing caused by an imbalance of bacteria. Surg Clin North Am. 1997;77(3):637–50. doi: 10.1016/s0039-6109(05)70572-7. [DOI] [PubMed] [Google Scholar]
- 8.Martin P. Wound healing--aiming for perfect skin regeneration. Science. 1997;276(5309):75–81. doi: 10.1126/science.276.5309.75. [DOI] [PubMed] [Google Scholar]
- 9.Park JE, Barbul A. Understanding the role of immune regulation in wound healing. Am J Surg. 2004;187(5A):11S–16S. doi: 10.1016/S0002-9610(03)00296-4. [DOI] [PubMed] [Google Scholar]
- 10.Smith JW, Gamelli RL, Jones SB, Shankar R. Immunologic responses to critical injury and sepsis. J Intensive Care Med. 2006;21(3):160–72. doi: 10.1177/0885066605284330. [DOI] [PubMed] [Google Scholar]
- 11.Leibovich SJ, Ross R. The role of the macrophage in wound repair. A study with hydrocortisone and antimacrophage serum. Am J Pathol. 1975;78(1):71–100. [PMC free article] [PubMed] [Google Scholar]
- 12.Adams DO, Hamilton TA. The cell biology of macrophage activation. Annu Rev Immunol. 1984;2:283–318. doi: 10.1146/annurev.iy.02.040184.001435. [DOI] [PubMed] [Google Scholar]
- 13.Brown SW, Meyers RT, Brennan KM, Rumble JM, Narasimhachari N, Perozzi EF, et al. Catecholamines in a macrophage cell line. J Neuroimmunol. 2003;135(12):47–55. doi: 10.1016/s0165-5728(02)00435-6. [DOI] [PubMed] [Google Scholar]
- 14.Abrass CK, O'Connor SW, Scarpace PJ, Abrass IB. Characterization of the beta-adrenergic receptor of the rat peritoneal macrophage. J Immunol. 1985;135(2):1338–41. [PubMed] [Google Scholar]
- 15.Spengler RN, Allen RM, Remick DG, Strieter RM, Kunkel SL. Stimulation of alpha-adrenergic receptor augments the production of macrophage-derived tumor necrosis factor. J Immunol. 1990;145(5):1430–4. [PubMed] [Google Scholar]
- 16.Spengler RN, Chensue SW, Giacherio DA, Blenk N, Kunkel SL. Endogenous norepinephrine regulates tumor necrosis factor-alpha production from macrophages in vitro. J Immunol. 1994;152(6):3024–31. [PubMed] [Google Scholar]
- 17.Szelenyi J, Kiss JP, Vizi ES. Differential involvement of sympathetic nervous system and immune system in the modulation of TNF-alpha production by alpha2- and beta-adrenoceptors in mice. J Neuroimmunol. 2000;103(1):34–40. doi: 10.1016/s0165-5728(99)00234-9. [DOI] [PubMed] [Google Scholar]
- 18.Hu XX, Goldmuntz EA, Brosnan CF. The effect of norepinephrine on endotoxin-mediated macrophage activation. J Neuroimmunol. 1991;31(1):35–42. doi: 10.1016/0165-5728(91)90084-k. [DOI] [PubMed] [Google Scholar]
- 19.Aronoff DM, Canetti C, Serezani CH, Luo M, Peters-Golden M. Cutting edge: macrophage inhibition by cyclic AMP (cAMP): differential roles of protein kinase A and exchange protein directly activated by cAMP-1. J Immunol. 2005;174(2):595–9. doi: 10.4049/jimmunol.174.2.595. [DOI] [PubMed] [Google Scholar]
- 20.Garcia JJ, del Carmen Saez M, De la Fuente M, Ortega E. Regulation of phagocytic process of macrophages by noradrenaline and its end metabolite 4-hydroxy-3-metoxyphenyl-glycol. Role of alpha- and beta-adrenoreceptors. Mol Cell Biochem. 2003;254(12):299–304. doi: 10.1023/a:1027345820519. [DOI] [PubMed] [Google Scholar]
- 21.Roy B, Rai U. Dual mode of catecholamine action on splenic macrophage phagocytosis in wall lizard, Hemidactylus flaviviridis. Gen Comp Endocrinol. 2004;136(2):180–91. doi: 10.1016/j.ygcen.2003.12.023. [DOI] [PubMed] [Google Scholar]
- 22.Swift ME, Burns AL, Gray KL, DiPietro LA. Age-related alterations in the inflammatory response to dermal injury. J Invest Dermatol. 2001;117(5):1027–35. doi: 10.1046/j.0022-202x.2001.01539.x. [DOI] [PubMed] [Google Scholar]
- 23.Muthu K, Deng J, Gamelli R, Shankar R, Jones SB. Adrenergic modulation of cytokine release in bone marrow progenitor-derived macrophage following polymicrobial sepsis. J Neuroimmunol. 2005;158(12):50–7. doi: 10.1016/j.jneuroim.2004.08.003. [DOI] [PubMed] [Google Scholar]
- 24.Greenberg S, Grinstein S. Phagocytosis and innate immunity. Curr Opin Immunol. 2002;14(1):136–45. doi: 10.1016/s0952-7915(01)00309-0. [DOI] [PubMed] [Google Scholar]
- 25.Rabinovitch M. Professional and non-professional phagocytes: an introduction. Trends Cell Biol. 1995;5(3):85–7. doi: 10.1016/s0962-8924(00)88955-2. [DOI] [PubMed] [Google Scholar]
- 26.Gosain A, Jones SB, Shankar R, Gamelli RL, DiPietro LA. Norepinephrine modulates the inflammatory and proliferative phases of wound healing. J Trauma. 2006;60(4):736–44. doi: 10.1097/01.ta.0000196802.91829.cc. [DOI] [PubMed] [Google Scholar]
- 27.Kim LR, Pomeranz B. The sympathomimetic agent, 6-hydroxydopamine, accelerates cutaneous wound healing. Eur J Pharmacol. 1999;376(3):257–64. doi: 10.1016/s0014-2999(99)00391-x. [DOI] [PubMed] [Google Scholar]
- 28.Kim LR, Whelpdale K, Zurowski M, Pomeranz B. Sympathetic denervation impairs epidermal healing in cutaneous wounds. Wound Repair Regen. 1998;6(3):194–201. doi: 10.1046/j.1524-475x.1998.60305.x. [DOI] [PubMed] [Google Scholar]
- 29.Souza BR, Cardoso JF, Amadeu TP, Desmouliere A, Costa AM. Sympathetic denervation accelerates wound contraction but delays reepithelialization in rats. Wound Repair Regen. 2005;13(5):498–505. doi: 10.1111/j.1067-1927.2005.00070.x. [DOI] [PubMed] [Google Scholar]
- 30.Ali RA, Qureshi MA, McCorkle FM. Profile of chicken macrophage functions after exposure to catecholamines in vitro. Immunopharmacol Immunotoxicol. 1994;16(4):611–25. doi: 10.3109/08923979409019742. [DOI] [PubMed] [Google Scholar]
- 31.Bassoe CF, Smith I, Sornes S, Halstensen A, Lehmann AK. Concurrent measurement of antigen- and antibody-dependent oxidative burst and phagocytosis in monocytes and neutrophils. Methods. 2000;21(3):203–20. doi: 10.1006/meth.2000.1001. [DOI] [PubMed] [Google Scholar]
- 32.Elenkov IJ, Hasko G, Kovacs KJ, Vizi ES. Modulation of lipopolysaccharide-induced tumor necrosis factor-alpha production by selective alpha- and beta-adrenergic drugs in mice. J Neuroimmunol. 1995;61(2):123–31. doi: 10.1016/0165-5728(95)00080-l. [DOI] [PubMed] [Google Scholar]
- 33.Geissmann F, Jung S, Littman DR. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity. 2003;19(1):71–82. doi: 10.1016/s1074-7613(03)00174-2. [DOI] [PubMed] [Google Scholar]
- 34.Stout RD, Suttles J. Functional plasticity of macrophages: reversible adaptation to changing microenvironments. J Leukoc Biol. 2004;76(3):509–13. doi: 10.1189/jlb.0504272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rossi AG, McCutcheon JC, Roy N, Chilvers ER, Haslett C, Dransfield I. Regulation of macrophage phagocytosis of apoptotic cells by cAMP. J Immunol. 1998;160(7):3562–8. [PubMed] [Google Scholar]
- 36.De Nichilo MO, Burns GF. Granulocyte-macrophage and macrophage colony-stimulating factors differentially regulate alpha v integrin expression on cultured human macrophages. Proc Natl Acad Sci U S A. 1993;90(6):2517–21. doi: 10.1073/pnas.90.6.2517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rice PA, Boehm GW, Moynihan JA, Bellinger DL, Stevens SY. Chemical sympathectomy alters numbers of splenic and peritoneal leukocytes. Brain Behav Immun. 2002;16(1):62–73. doi: 10.1006/brbi.2000.0611. [DOI] [PubMed] [Google Scholar]
- 38.Stein M, Keshav S, Harris N, Gordon S. Interleukin 4 potently enhances murine macrophage mannose receptor activity: a marker of alternative immunologic macrophage activation. J Exp Med. 1992;176(1):287–92. doi: 10.1084/jem.176.1.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wahle M, Neumann RP, Moritz F, Krause A, Buttgereit F, Baerwald CG. Beta2-adrenergic receptors mediate the differential effects of catecholamines on cytokine production of PBMC. J Interferon Cytokine Res. 2005;25(7):384–94. doi: 10.1089/jir.2005.25.384. [DOI] [PubMed] [Google Scholar]
- 40.Aronoff DM, Canetti C, Peters-Golden M. Prostaglandin E2 inhibits alveolar macrophage phagocytosis through an E-prostanoid 2 receptor-mediated increase in intracellular cyclic AMP. J Immunol. 2004;173(1):559–65. doi: 10.4049/jimmunol.173.1.559. [DOI] [PubMed] [Google Scholar]
- 41.Dent G, Giembycz MA, Rabe KF, Wolf B, Barnes PJ, Magnussen H. Theophylline suppresses human alveolar macrophage respiratory burst through phosphodiesterase inhibition. Am J Respir Cell Mol Biol. 1994;10(5):565–72. doi: 10.1165/ajrcmb.10.5.8179921. [DOI] [PubMed] [Google Scholar]