

Abstract
Cancer therapeutics include an ever-increasing array of tools at the disposal of clinicians in their treatment of this disease. However, cancer is a tough opponent in this battle and current treatments which typically include radiotherapy, chemotherapy and surgery are not often enough to rid the patient of his or her cancer. Cancer cells can become resistant to the treatments directed at them and overcoming this drug resistance is an important research focus. Additionally, increasing discussion and research is centering on targeted and individualized therapy. While a number of approaches have undergone intensive and close scrutiny as potential approaches to treat and kill cancer (signaling pathways, multidrug resistance, cell cycle checkpoints, anti-angiogenesis, etc.), much less work has focused on blocking the ability of a cancer cell to recognize and repair the damaged DNA which primarily results from the front line cancer treatments; chemotherapy and radiation. More recent studies on a number of DNA repair targets have produced proof-of-concept results showing that selective targeting of these DNA repair enzymes has the potential to enhance and augment the currently used chemotherapeutic agents and radiation as well as overcoming drug resistance. Some of the targets identified result in the development of effective single-agent anti-tumor molecules. While it is inherently convoluted to think that inhibiting DNA repair processes would be a likely approach to kill cancer cells, careful identification of specific DNA repair proteins is increasingly appearing to be a viable approach in the cancer therapeutic cache.
Keywords: DNA repair, translation, DNA repair inhibition, cancer therapeutics
INTRODUCTION
Cancer therapeutics include an ever-increasing array of tools at the disposal of clinicians in their treatment of this disease. However, cancer is a tough opponent in this battle, and current treatments are too frequently not enough to rid the patient of his or her cancer. In some cancers, such as pediatric leukemia, there have been dramatic improvements in survival, however in others, such as pancreatic cancer, the survival rate remains a dismal four percent [1]. Clearly new and innovative approaches need to be investigated. One such approach that is gaining interest and enthusiasm in the treatment of cancer is rationally designed therapy toward specific molecular targets such as growth factor receptors, cell cycle proteins, etc. While much more is known about intervention into other pathways (signaling pathways, multidrug resistance, cell cycle checkpoints, anti-angiogenesis, etc.) as potential approaches to treat and kill cancer, much less work has focused on blocking the ability of a cancer cell to recognize and repair the damaged DNA which results from front line cancer treatments; chemotherapy and radiation. Both types of treatments induce DNA damage which is acted upon by a variety of DNA repair pathways. Critical steps within these pathways are emerging as potential targets and could be utilized to either enhance the current standard of care i.e. chemotherapy and radiation or to partner with other small molecules directed at specific targets and pathways mentioned above. While agents that inhibit DNA repair have not received a significant amount of attention as a cancer therapeutic, more recently it has become a major area of research. Studies in DNA repair have long been conducted by outstanding biochemists and molecular biologists, but translating these findings to the clinic for cancer therapeutics has lagged significantly. Moreover, the field of DNA repair has extended dramatically from its original description as enzymes that interact directly with the damaged base or DNA to include numerous signaling pathways such as BRCA1 and 2 (breast cancer gene 1 and 2), FANC (fanconi anemia complementation group C), ATM (Ataxia telangiectasia) and others. The main focus of this review will be on the enzymes in DNA repair pathways that act upon the damaged DNA substrate. While it is inherently convoluted to think that inhibiting DNA repair processes would be a feasible approach to kill cancer cells, careful identification of DNA repair proteins with crucial roles in preserving DNA integrity is a seemingly viable approach in the cancer therapeutic cache.
The major DNA repair pathways include direct (O6-alkylguanine DNA methyltransferase, AGT), base excision (BER), nucleotide excision (NER), mismatch (MMR), homologous (HR) and non-homologous end-joining (NHEJ) repair. While all pathways will be discussed, the main focus of this review will be on BER and direct repair as pathways with rationale therapeutic targets. The reason for this is straight-forward; existing evidence suggests that both pathways contain proteins that are reasonable, clinically relevant targets. Other pathways, such as MMR and NER, for example, do not have as much compelling data to suggest clinical applicability. Furthermore, given previous findings that knocking out, down or mutating members of the MMR or NER pathways can lead to serious diseases such as hereditary non-polyposis colon cancer (HNPCC) [2] or Xeroderma pigmentosum (XP) [3], respectively, the rationale for targeting members of these pathways is more strained. That being said, new tumor cell targeting approaches could overcome these difficulties. For a comprehensive review of the other pathways and ancillary pathways related to DNA repair, readers are encouraged to investigate the review by Belzile, et al. [4]. Additionally, some other careful reviews have appeared relating to DNA repair and therapeutics and readers of this review are encouraged to read them [5-8].
DIRECT REPAIR
The direct repair pathway is typified by O6-alkylguanine DNA methyltransferase (AGT) which plays an important biological role in directly repairing O6-alkylguanine in DNA [5]. Elevated levels of AGT in tumors correlate to resistance [9] making AGT one of the first DNA repair targets identified for cancer therapeutics [10]. Additionally, AGT has potential usefulness in gene therapy protocols to protect against side effects of cancer chemotherapy such as myelosuppression [9-11]. Ideally, inhibition of this protein in tumors would sensitize the tumors to chemotherapeutic agents that generate lesions at the O6-position of guanine. AGT is a protein and not an enzyme since it preferentially removes one alkyl group from the O6-position of guanine to its active site, rendering it inactive, and is subsequently degraded (Fig. 1A).
Fig. 1.
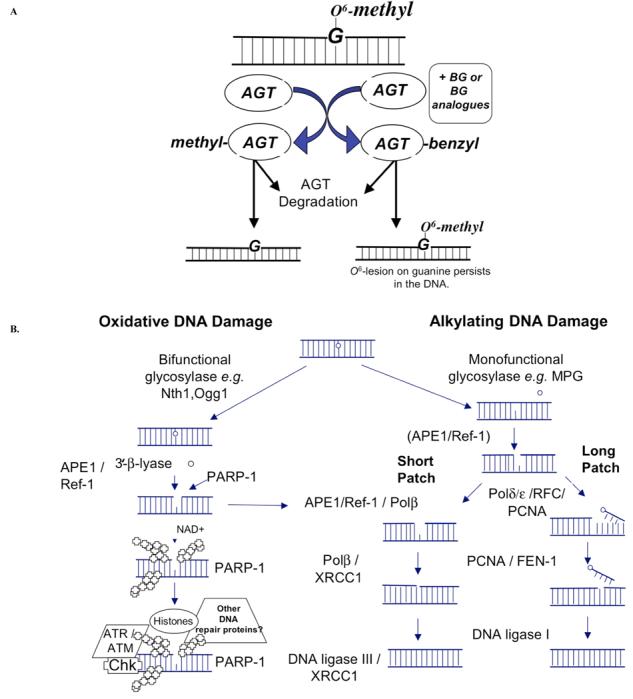
Cartoon of two of the most promising DNA repair pathways for cancer therapeutics. A) The direct reversal pathway is made up of the AGT protein and recognizes O6-alkylguanine lesions in DNA. Clinically used inhibitor, O6-Benzylguanine (BG) acts as a pseudo-substrate and inactivates AGT resulting in a persistence of DNA lesions and an increase in sensitivity to agents that generate O6-lesions. B) The DNA base excision repair (BER) pathway is involved in repairing DNA damage caused by alkylating and oxidizing DNA damaging agents as well as ionizing radiation (IR). The DNA BER pathway is made of up both the short- and long-patch sub-pathways with APE1/Ref-1 playing a crucial role in both. Proteins that are currently good or potential targets for cancer therapeutics are shown in red letters.
Chemotherapeutic agent, BCNU (1,3-bis (2-chloroethyl)-1-nitrosourea) generates chloroethyl modifications at O6, which can rearrange and lead to an interstrand cross-link [12]. Interstrand DNA crosslinks are particularly cytotoxic because they block DNA replication. AGT repairs DNA damage following chloroethylating and methylating agents such as BCNU which generates several DNA adducts including O6-methylguanine (O6-meG). O6-meG preferentially pairs with thymine and leads to GC to AT transitions [10,13]. Inhibition of AGT leads to sensitization to BCNU as the cells are unable to repair the lesion on the O6-postion of guanine which then leads to subsequent interstrand crosslink formation. However, more recently, interest has centered on the role of AGT in the response of patients to temozolomide (TMZ) treatment which is a recently developed chemotherapeutic alternative to BCNU for patients with astrocytoma, glioblastoma, metastatic brain tumors and malignant melanoma [14]. TMZ methylates DNA at the N7 and O6-positions of guanine and N3 of adenine [15] generating lesions that are repaired by both the AGT and BER pathways [16]. Inhibition of AGT in conjunction with TMZ treatment also leads to tumor cell sensitization [10,17-19]. Furthermore, several research groups have focused their efforts on the manipulation of AGT to sensitize tumor cells to alkylating agents while protecting the bone marrow compartment from the characteristic myelosuppression associated with chemotherapy. Hematopoietic bone marrow progenitor cells are protected by gene transfer of mutant AGT protein(s) that are resistant to inhibition due to an alteration of their active site [20-22]. This chemo-sensitive compartment is protected while inhibiting the tumor cell AGT and thereby increasing alkylating agent sensitivity and efficacy.
As mentioned, agents such as streptozotocin, procarbazine, dacarbazine, BCNU, CCNU (1-2-chloroethyl-3-cyclohexyl-1-nitrosourea) and TMZ generate lesions at the O6-position of guanine making AGT a target to inhibit in combination with these drugs [17]. The therapeutic approach has been to treat tumor cells or patients with an agent such as O6-benzyguanine (BG) which acts as a pseudo-substrate and renders AGT inactive to repair the DNA damage in the tumor cell (Fig. 1A). An orally bioavailable BG analogue, O6-(4-bromothenyl)-guanine (O6-BTG) appears to be more than ten times more potent than BG [23]. Recently, a clinical trial using [6-(4-bromo-2-thienyl) methoxy]purin-2-amine] or Lomeguatrib along with TMZ demonstrated significant efficacy in the reduction of AGT levels in the 23 patients with advanced solid tumors, with one complete response and 12 patients with stable disease for at least three months [24]. As expected, the dose limiting toxicity of the combination of lomeguatrib and TMZ was myelosuppression. Other molecules with similar mechanisms of action as BG include O6-benzyl-2'-deoxyguanosine which has improved potency and solubility and O6-4-bromothenylguanine-C8-B-d-glucoside which is a glucose conjugate with tumor-targeting action due to the selective uptake of glucose by tumors. The latter has been shown to have activity against TMZ and fotemustine in vitro [25]. A number of clinical trials have opened using BG and analogues on a wide variety of tumors including glioma, lymphoma, myeloma, melanoma, colon cancer, sarcoma and others [5, 17, 9].
Finally, unexpected or off-target effects of BG have recently been identified [26-28]. In an elegant series of experiments, head and neck tumor cell lines pretreated with BG resulted in a 2-fold decrease in the ED50 of cisplatin and a concomitant increase in DNA damage and in the percentage of cells undergoing apoptosis. The enhancement was independent of AGT activity. Similar enhancement was observed with carboplatin, but no enhancement was seen in AGT-deficient cell lines with radiation or TMZ, demonstrating the dependence of the effect on bifunctional, cross-linking agents [26]. Additional studies have ruled out the role of glutathione (GSH) or NER pathways for this increase in cytoxicity observed using BG with cisplatin [27, 28]. However, it was recently shown that cyclin-dependent kinase (CDK)2 pathway is affected by a number of guanine derivatives which inhibit CDK2 and enhances cisplatin cytotoxic and apoptotic effects. This group includes BG although it was not the most potent enhancer of cisplatin-induced cytotoxicity [27].
BG is currently being used as a modulator of BCNU, Gliadel, and TMZ in Phase II and III clinical trials. To date, there have been no significant toxicities associated with BG when it is administered alone to humans [29-33]. The development of BG as a DNA repair inhibitor is a great example of bench research resulting in clinical application.
BASE EXCISION REPAIR (BER)
Within mammalian BER there are two branches: long patch (LP) and short patch (SP), and they differ in the proteins involved as well as in the length of DNA that is synthesized during the repair process. The mechanism of which pathway is chosen in the cell is still under investigation but the type of damage and the glycosylase that removes the damage are involved [34,35]. The predominant BER pathway, SP-BER repairs an oxidized or alkylated base beginning with a DNA glycosylase (e.g. methylpurine DNA glycosylase or MPG also called AAG) which removes the damaged base, leaving the DNA backbone intact. The second enzyme in this pathway, Apurinic endonuclease 1 / Redox factor-1 (APE1/Ref-1), hydrolyzes the phosphodiester backbone immediately 5' to an abasic site (reviewed in [36]). Oxidation and alkylation of DNA bases as well as abasic sites are generated following chemotherapeutic treatment. The APE1/Ref-1 incision generates a normal 3'-hydroxyl group and an abasic deoxyribose-5-phosphate, which is processed by subsequent enzymes of the BER pathway, β-polymerase (β-pol) and DNA Ligase III/XRCC1. LP-BER functions to resolve oxidized or reduced abasic sites and involves APE1/Ref-1 incision activity as well. Polymerases δ and ε, PCNA, and RFC can provide gap filling DNA synthesis as well as βpol. Flap endonuclease (FEN1) cleaves the displaced oligonucleotide, and Ligase I seals the nicks (Fig. 1B) (reviewed in [35]. Both pathways are important for the survival and integrity of DNA sequence to cells. Likewise, manipulation of both pathways by a tumor cell could impart resistance to chemotherapeutic agents that generate alkylation and oxidative damage. Some examples of chemotherapeutic agents that generate lesions that BER would repair include: temozolomide [37], melphalan [38], thiotepa [39], methyl-lexitropsin (Me-lex) [40], dacarbazine / procarbazine [41], and streptozotocin [42] (Table 1). Some chemotherapeutic agents generate reactive oxygen species (ROS) as a “by-product” such as platinum-based drugs i.e. oxaliplatin and cisplatin [43,44], anthracyclines i.e. epirubicin, daunorubicin, doxorubicin [45] and paclitaxel [46,47] indicating that BER may be important in the response to these drugs as well (Table 1). Ionizing radiation (IR) creates lesions on DNA that are acted upon by the BER pathway and is used extensively in cancer therapy regimens (reviewed in [48]), yet another indication that inhibition of the BER pathway will have clinical implications and efficacy.
Table 1.
Clinical Agents that Create DNA Lesions that are Repaired by Direct and Base Excision Repair
| Type of Damage | Pathway | Ref. | |
|---|---|---|---|
| Direct-Acting Agents | |||
| Temozolomide |
O6-meG N3-meA N7-meG |
Direct BER BER |
[37] |
| Melphalan | N3-meA DNA cross-links |
BER NER |
[38] |
| Thiotepa | Formamidopyrimidine 7-Methyl-formamidopyrimidine |
BER BER |
[39] |
| Radiation | SSBs DSBs ROS |
BER HR / NHEJ BER |
[113] |
| BCNU / CCNU | Chloroethyl adduct at O6-G DNA cross-links |
Direct NER/HR |
[42] |
| Dacarbazine / Procarbazine |
O6-meG N7-meG |
Direct BER |
[41] |
| Streptozotocin |
O6-meG N3-meA N7-meG |
Direct BER BER |
[42] |
| Indirect-Acting Agents | |||
| Platinum Agents | |||
| Oxaliplatin | Inter-and intrastrand DNA crosslinks ROS |
NER / MMR / HR BER |
[114] [43] |
| Cisplatin | Inter- and intrastrand DNA crosslinks ROS |
NER / MMR / HR BER |
[114] [43] |
| Anthracyclines | [45] | ||
| Epirubicin | ROS | BER | |
| Daunorubicin | ROS | BER | |
| Doxorubicin | ROS | BER | [115] |
| Nucleoside analogs | |||
| Gemcitabine / Troxacitabine | L-configuration nucleoside analogs | BER | [116] |
| Other | |||
| Paclitaxel | Microtubule stabilization ROS |
BER | [47] |
Glycosylases
The first step in repairing a single base lesion in DNA is the removal of the damaged base and is achieved by numerous glycosylases which are specific for the type of DNA lesion that is in need of repair. For example, methylpurine DNA glycosylase (MPG, AAG), 8-oxoguanine DNA glycosylase (OGG1), uracil DNA glycosylase (UNG), Thymine DNA glycosylase (TDG), etc remove specific lesions as their names suggest (reviewed in [49]). Based on the diversity of lesions and glycosylases that recognize them, this may not be the ideal step to target in attempts to sensitize cancer cells to DNA damaging agents. There is a report that divalent ions, Cd2+, Ni2+, and Zn2+ can inhibit MPG [50]. In addition, reduced levels of MPG protein using siRNA increased the sensitivity of cervical carcinoma cell lines to TMZ, MMS, N-methyl-N-nitrosourea, and BCNU [51]. However there are several reports that overexpressing this glycosylase is deleterious to cancer cells demonstrating that there is a delicate balance within the BER pathway that can be exploited to kill tumor cells [52-55]. Abasic sites are generated following glycosylase removal of a damaged base and there is only one enzyme that processes abasic sites – APE1/Ref-1, indicating that it may be a better therapeutic target than the glycosylases.
APE1/Ref-1
The DNA repair activity of APE1/Ref-1 is fundamental to the maintenance of the genome and survival of cells. Even in the absence of exogenous DNA damage, siRNA directed against APE1/Ref-1 results in a decrease in proliferation, an increase in AP sites and increased levels of apoptosis [56].The expression of APE1/Ref-1 is altered in numerous cancers including prostate, ovarian, cervical, germ cell tumor, rhabdomyosarcoma, and colon [36]. APE1/Ref-1 is an essential enzyme in the BER pathway which is responsible for the repair of DNA caused by oxidative and alkylation damage and thus protects cells against the toxic effects of endogenous and exogenous agents including chemotherapeutic agents [57]. As importantly, APE1/Ref-1 also functions as a reduction-oxidation (redox) factor maintaining transcription factors in an active reduced state (reviewed in [36] and [58]). APE1/Ref-1 stimulates the DNA binding activity of numerous transcription factors that are involved in cancer promotion and progression such as AP-1 (fos/jun), NFκB, HIF-1α, CREB, p53 and others [36,58]. We believe both of these activities have potential in sensitizing cancer cells to chemotherapeutic agents. Without a capacity to repair their DNA, the tumor cells are forced to undergo apoptosis, and without APE1/Ref-1's reducing activity on transcription factors, transcription factors are unable to bind DNA thus preventing the propagation of growth signals.
APE1/Ref-1 is active in both LP- and SP-BER and interacts with several proteins involved in both sides of the BER pathway including 8-oxoguanine DNA glycosylase (OGG1), X-ray cross complementing-1 (XRCC1), proliferating cell nuclear antigen (PCNA), Flap endonuclease 1 (FEN1), and polymerase β (reviewed in [49]). Inhibitors of APE1/Ref-1's DNA repair function is hypothesized to sensitize tumor cells to alkylating and oxidizing agents based on many reports in the literature that demonstrate a reduction in the protein does just that, sensitize the cells to agents that generate damage that APE1/Ref-1 acts upon. Targeted reduction of APE1/Ref-1 protein by specific anti-sense oligonucleotides or siRNA renders mammalian cells hypersensitive to methylmethane sulfonate (MMS), H2O2, bleomycin, TMZ, melphalan, and gemcitabine [59-63]. The importance of APE1/Ref-1 to the cell is demonstrated by the lethality of APE1/Ref-1 mouse knockouts at E3.5 to E9.5 and the lack of viable cell lines completely deficient for APE1/Ref-1[64] and reviewed in[65]. Knocking out APE1/Ref-1 in the mouse or in cancer cell lines using RNA interference or siRNA methodology emphasizes the importance of its function to the survival and propagation of cells.
There are three compounds that have been reported as APE1/Ref-1 DNA repair inhibitors: methoxyamine (MX), 7-Nitroindole-2-Carboxylic Acid (NCA), and lucanthone (Table 2). All of these compounds enhance the cytotoxic potential of temozolomide in numerous cancer cell lines (ovarian, breast, colon, fibrosarcoma) [53,54,66-69]. The mechanism behind the enhancement of TMZ-induced cytotoxicity by MX is well-characterized and is not through direct inhibition of APE1/Ref-1, but by a blockade of the BER pathway. Following the glycosylase-driven removal of a damaged nucleotide, an abasic site is generated. MX, an alkoxyamine derivative can bind to the acyclic sugar left in the DNA abasic site thereby blocking the downstream members of the BER pathway, APE1/Ref-1 and β pol [70,71]. Perhaps even more dramatic results could be achieved with a direct inhibitor of the DNA repair activity of APE1/Ref-1 with a reduction in the amount of off-target effects. Using a high through-put screen, the NCA compound was discovered. In this published study, NCA potentiates the activity of MMS, TMZ, H2O2, and Zeocin in HT1080 human fibrosarcoma cells presumably through inhibition of the AP endonuclease, 3′-phosphodiesterase, 3′-phosphatase, and 3′ to 5′-exonuclease activities of APE1/Ref-1 [68]. Finally a compound, lucanthone known as a topoisomerase II inhibitor [72] also may have effects on the DNA repair activity of APE1/Ref-1. A dose-dependent increase in AP sites following lucanthone treatment in breast cancer cell lines [73] and potentiation of the cell killing effect of MMS and TMZ [69] support an inhibition of APE1/Ref-1 by lucanthone. However, cytotoxicity following lucanthone treatment is presumably due more to inhibition of Topoisomerase II based on the doses that are necessary for in vitro APE1/Ref-1 inhibition.
Table 2.
DNA Repair Inhibitors
| Direct (AGT) | BER | Others Related to DNA Repair | ||
|---|---|---|---|---|
| 06BG | Methoxyamine | Ape1 repair | IC 87361 | DNA-PK |
| 06-benzyl-2'-deoxyguanosine | E3330 | Ape1 redox | IC87102 | DNA-PK |
| 06-4 Bromethenyl-guanine (06-BTG) | Lucanthone | Ape1 repair / Topo II | NU7441 | DNA-PK |
| 06-4 Bromethenyl-guanine-(C8-β-d-glucoside) | KJA | β-polymerase | NU7026 | DNA-PK |
| Lomeguatrib [6-(4-bromo-2-thienyl) methoxy]-purin-2-amine] |
Pamoic Acid | β-polymerase | OK-1035 | DNA-PK |
| CRT0044876 | Ape1 repair | SU11752 | DNA-PK | |
| Nu1025 | Vanillin | DNA-PK | ||
| Nu1064 | Salvicine | DNA-PKcs | ||
| Nu1085 | Ku55933 | ATM Kinase | ||
| AG14361 | LY294002 | ATM Kinase | ||
| AG14699 | PARP | Wortmannin | ATM Kinase | |
| GPI15427 | ||||
| CEP6800 | PARP | |||
| INO-1001 | PARP | |||
Not nearly as many efforts have been put toward discovering a redox inhibitor of APE1/Ref-1. Partly because the importance and relevance of its redox function are just becoming apparent. The anti-cancer potential of a redox inhibitor of APE1/Ref-1 is great. Transcription factors including HIF-1α, p53, NFκB, CREB, and AP-1 have all been implicated in important aspects of cancer including angiogenesis and tumor promotion and progression. Through inhibition of APE1/Ref-1's redox activity these transcription factors would be unable to bind to DNA and thereby tumor cell signaling of angiogenesis and uncontrolled growth would be slowed and/or stopped. The following compounds have an effect on the redox activity of APE1/Ref-1: soy isoflavones, resveratrol, and E3330 [43,74-77]. The dual functions of APE1/Ref-1 point to it as an important target to sensitize tumor cells to chemotherapy [78]. Perhaps a drug that caused a reduction in APE1/Ref-1 mRNA and/or protein levels would be potent and effective against a growing tumor.
β-Pol
The polymerase responsible for DNA synthesis in SP-BER is β-pol. β-pol has associated lyase activity that is important and often rate-limiting in BER, and β-pol also plays a role in single nucleotide gap filling in LP-BER (reviewed in [79,80]), therefore it is a potential target for potentiation of tumor cells to DNA damaging agents. Natural product, koetjapic acid interacts with residues in the 8-kDa domain of β-pol which is responsible for its lyase activity and in combination with NMR chemical shift mapping, was used to discover pamoic acid as a β-pol inhibitor, both its lyase and polymerase activities [81]. Inhibition of β-pol with pamoic acid sensitizes mouse fibroblasts to laboratory agent, MMS. β-pol is most likely not the most attractive protein to target in cancer therapy based on the lack of sensitization of β-pol-null cells to several chemotherapeutic agents, including melphalan, mitozolomide, BCNU, and IR [82]. However these null cells are sensitive to TMZ [82-84].
PARP-1
Although not a direct part of the damage removal and base repair, poly (ADP) ribose polymerase-1 (PARP-1) interacts with several BER proteins including β-pol, XRCC1, and DNA Ligase III (reviewed in [49]) and acts as a sensor of DNA strand breaks. PARP-1 functions by cleavage of NAD+ to add ADP-ribose units to DNA, histones, and various DNA repair enzymes. This addition acts as a signal to many different cellular processes including replication, transcription, protein degradation, spindle maintenance and others (Fig. 1B) (reviewed in [85] and [86]). PARP-1 is just one member of a superfamily of poly (ADP-ribosyl) polymerase enzymes with 17 members to date [87]. Knockout of PARP-1 is not embryonic lethal, however double knockout of PARP-1 and -2 results in embryonic lethality around day 8 demonstrating the redundancy and overlapping roles of PARP-1 and -2 and their importance to the developing embryo [88]. Overactivation of PARP can lead to cell death via necrosis and apoptosis. Cellular depletion of NAD+ results in a necrotic cell death while PARP activation also plays a role in apoptosis-inducing factor (AIF)'s release from the mitochondria [89]. When PARP-1 is deficient or absent in cells, BER efficiency is greatly reduced [90]. Additionally, pharmacologic inhibition of PARP-1 in cancer cell lines and knocking out PARP-1 and PARP-2 in the mouse demonstrate that sensitization to alkylating agents, N-methyl-N-nitrosourea (MNU) and chemotherapeutics including TMZ and IR is possible [8,88,91,92]. Several PARP-1 inhibitors are now under investigation as single agents and in combination with TMZ (Table 2) (reviewed in [91]). In cells deficient in BRCA1 and BRCA2 (breast cancer susceptibility proteins), PARP inhibitors are reported to be extremely efficacious [93-95] presumably due to the combination of the lack of homologous recombination as a result of BRCA deficiency and the lack of PARP signaling to repair the DNA strand breaks. These findings suggest that selective tumor cell killing is possible in cancer cells that are defective in BRCA1 and BRCA2. One report contradicts these data indicating that specific cell killing in BRCA2 deficient cells is not achieved with PARP inhibitors, [96] however different cell lines and PARP inhibitors with differing potency were used in these studies, which could potentially lead to different effects on cell sensitivity [97]. Regardless of the specificity of PARP inhibitors to kill only cancer cells with a predisposed DNA repair deficiency, PARP inhibitors are an emerging class of chemotherapeutic agents that hopefully will have efficacy against a range of cancers.
NUCLEOTIDE EXCISION REPAIR (NER) AND MISMATCH REPAIR (MMR)
There is little information on targeting the MMR or NER pathways for cancer therapeutics (Fig. 2). One explanation for this centers on the relationship of genetic diseases that have been shown to be linked to defects in NER (Xeroderma pigmentosum, Cockayne's Syndrome) and MMR (Hereditary Non-polyposis Colon Cancer; HNPCC). Additionally, defective MMR has demonstrated an increase in tumor cell resistance to cisplatin [98]. These findings make a case that inhibiting NER or MMR enzymes may not be an appropriate approach. Alternatively, the reactivation of MMR enzymes may be one approach to re-sensitize tumors to chemotherapy. An example of this is the use of 2′-deoxy-5-azacytadine (decitabine) to reverse the methylation of MMR genes such as MLH1. In one study, restoring MLH1 expression results in the acquisition of cisplatin sensitivity [8,99]. This was also observed with 5-fluorouracil (5-FU) treatments of colon cancer cells [100]. Clearly, MLH1 reactivation by dacitabine treatment is not the only gene being reactivated so the actual mechanism of this newly acquired cisplatin sensitivity may not be totally attributable to MLH1 re-activation.
Fig. 2.
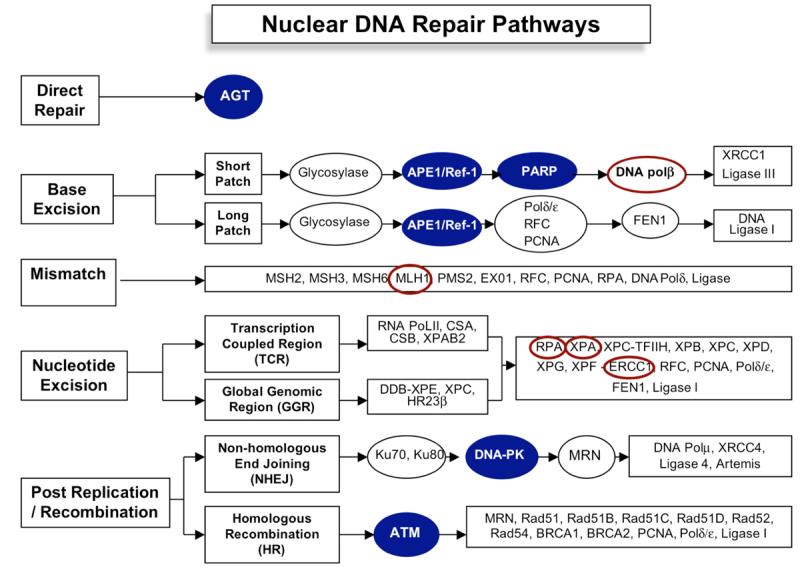
Schematic overview of mammalian DNA repair pathways. Proteins that are emerging as strong lead targets for cancer therapeutics are shown with a blue background. Those that have little data or data only generated in cell lines are shown circled in red. Not all members of each pathway are shown.
NER and MMR have been implicated in cross-link DNA damage repair and are the presumed major pathways for this type of DNA damage (Fig. 2). Most studies have focused on increasing the sensitization of tumor cells to cisplatin or other platinum agents because they are so widely used to treat many different kinds of cancer. One study used anti-sense RNA to ERCC1, a NER enzyme, in combination with cisplatin and demonstrated an increase in ovarian cancer cell killing [101]. Cisplatin-induced cytotoxicity is also enhanced with the addition of gemcitabine presumably due to the blockage of cross-link repair. However, the link between this finding and NER is unclear. A more direct link was observed using a global cyclin-dependent kinase inhibitor (UCN-01; 7-hydroxystaurosporine) which has enhanced cisplatin tumor sensitivity [102] and is in Phase I clinical trials [103]. This drug blocks the interaction between XPA (Xeroderma pigmentosum group A) and ERCC1 (excision repair cross-complementing group 1) which has been implicated in the damage-recognition and/or incision steps of NER [104]. Similarly, the RPA protein (replication protein A) which is a single-stranded DNA binding protein that is involved in chromosomal DNA replication, repair and recombination pathways is being investigated as a potential target to enhance cisplatin sensitivity [105]. A number of other small molecule inhibitors that target proteins involved in pathways other than DNA repair result in the reduction in DNA repair response. For example, ZD1839 (Gefitinib or IRESSA) which is an epidermal growth factor receptor tyrosine kinase (EGFR-TK) inhibitor actually alters the DNA repair response and increases cisplatin cytotoxicity [106,107]. A similar result is observed for VEGF (vascular epidermal growth factor) inhibitor, SU5416. This compound causes a decrease in ERCC1 expression and increases cisplatin sensitivity [108,109]. How a “specific” VEGF or EGFR-TK inhibitor causes altered DNA repair response or decreased ERCC1 protein expression leading to enhanced cisplatin cytotoxicity is completely unknown at this time [108]. As the signaling pathways that respond to DNA damage are elucidated, the underlying mechanisms of a decrease in NER and/or MMR proteins following targeted therapies such as SU5416 or ZD1839 will become more apparent. Perhaps it will then be possible to design better inhibitors for proteins in the NER and MMR pathways.
HOMOLOGOUS RECOMBINATION (HR), NON-HOMOLOGOUS END-JOINING (NHEJ) AND DOUBLE-STRAND BREAK (DSB) REPAIR
HR corrects chromosome breaks that occur during the S/G2 portion of the cell cycle. Double strand breaks are recognized by the MRN complex (Mre11, Rad50 and Nbs1) and recruits the Ataxia telangiectasia (ATM) protein which regulates HR and cell-cycle checkpoints [4[. Cells that are defective in ATM are easily killed by direct DSB forming agents such as IR, radiomimetics, reactive oxygen species (ROS) or indirectly by alkylating agents. A number of inhibitors such as Wortmannin, LY2940002 and caffeine (Table 2) inhibit ATM function, but are not very specific, have off-target effects and inhibit a number of members of the PI3K (phosphatidylinositol 3-kinase) family with numerous side effects [8]. A newer compound, KU55933 (2-morpholin-4yl-6-thiaanthren-1-ylpyran-4-one) which appears to be a more specific inhibitor works as an ATP competitive inhibitor of ATM. KU55933 blocks the phosphorylation of numerous ATM targets including SMC1 (structural maintenance of chromosome 1), NBS1 (Nijmegen Breakage Syndrome), p53 and histone-H2AX [110]. H2AX has also become a target for therapeutics as initial data demonstrated that peptide inhibitors of the γ̃H2AX demonstrated increased sensitivity to IR [111]. Initial data supports the use of KU55933 in conjunction with chemotherapeutic agents camptothecin, doxorubicin and etoposide [6,110] as well as IR [110].
The NHEJ pathway rejoins DNA double-strand breaks regardless of sequence homology or not at the ends of the broken DNA in contrast to HR which requires sequence homology [4]. Furthermore, NHEJ appears to be the main pathway for the repair of DNA double-strand breaks and is independent of cell cycle. Proteins involved in this pathway include Ligase 4, Artemis, XRCC4, the Ku proteins, Ku70 and Ku80 and most importantly for this review, DNA-protein kinase (DNA-PK). Most cells lacking DNA-PK, including mice, are hypersensitive to IR and numerous DNA cross-linking agents. Various inhibitors are being developed targeting DNA-PK such as NU7026 (Table 2) which sensitizes cells containing DNA-PK to IR [112]. DNA PK inhibitors, IC87361 and IC87102 are effective in sensitizing tumor microvasculature and tumor cells to IR [6]. Given the importance of repairing DNA double-strand breaks on tumor cell survival and their relationship to IR, the search for inhibitors of members of this pathway, including DNA-PK is important to pursue [5-8].
CONCLUSIONS
The identification of targets in pathways such as signaling pathways, multidrug resistance, cell cycle checkpoints, anti-angiogenesis, and others have been prime areas of focus for molecular biologists for a number of years. Attention to DNA repair pathways as potential targets has lagged significantly. However, recent findings are starting to cause a sea of change in this area of study. As demonstrated by the list of known DNA repair protein/enzyme inhibitors (Table 2), this field is clearly not saturated and ripe for further study and innovation. Lead targets such as those that actually interact with the DNA and/or the DNA adduct including AGT, PARP and APE1/Ref-1 show encouraging promise in translational studies. Other less direct targets, such as the signaling molecules in DSB repair pathways, ATM and DNA-PK are also clearly viable targets. Furthermore, numerous studies are identifying small nucleotide polymorphisms (SNPs) in DNA repair enzymes and signaling molecules such that potential DNA repair biomarkers can be coupled with therapeutic targets. This growing area is also showing great promise. This accumulating body of evidence clearly demonstrates that selective targeting of these DNA repair enzymes has the potential to enhance and augment the currently used chemotherapeutic agents and radiation as well as overcoming drug resistance.
ACKNOWLEDGEMENTS
This work was supported by grants CA094025, CA106298, CA114574 and Riley Children's Foundation (M.R. Kelley) and a Marsha Rivkin Center for Ovarian Cancer Research grant (M.L. Fishel). Both authors were also supported on this work by the Ovar'coming Together foundation.
REFERENCES
- 1.Donghui LY, Li Li, Jiao DZ, Chang G, Beinart RA, Wolff DBEMMHJLA. International Journal of Cancer. 2007;120:1748. doi: 10.1002/ijc.22301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Akrami SM. Arch Iran Med. 2006;9:381. [PubMed] [Google Scholar]
- 3.Leibeling D, Laspe P, Emmert S. J Mol Histol. 2006;37:225. doi: 10.1007/s10735-006-9041-x. [DOI] [PubMed] [Google Scholar]
- 4.Belzile J-P, Choudhury SA, Cournoyer D, Chow TYK. Current Gene Therapy. 2006;6:111. doi: 10.2174/156652306775515538. [DOI] [PubMed] [Google Scholar]
- 5.Ding J, Miao Z-H, Meng L-H, Geng M-Y. Trends in Pharmacological Sciences. 2006;27:338. doi: 10.1016/j.tips.2006.04.007. [DOI] [PubMed] [Google Scholar]
- 6.Sanchez-Perez I. Clin Transl Oncol. 2006;8:642. doi: 10.1007/s12094-006-0034-8. [DOI] [PubMed] [Google Scholar]
- 7.Madhusudan S, Hickson ID. Trends Mol Med. 2005;11:503. doi: 10.1016/j.molmed.2005.09.004. [DOI] [PubMed] [Google Scholar]
- 8.Madhusudan S, Middleton MR. Cancer Treat Rev. 2005;31:603. doi: 10.1016/j.ctrv.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 9.Gerson SL. Nat Rev Cancer. 2004;4:296. doi: 10.1038/nrc1319. [DOI] [PubMed] [Google Scholar]
- 10.Gerson SL. J Clin Oncol. 2002;20:2388. doi: 10.1200/JCO.2002.06.110. [DOI] [PubMed] [Google Scholar]
- 11.Wadhwa PD, Zielske SP, Roth JC, Ballas CB, Bowman JE, Gerson SL. Annu Rev Med. 2002;53:437. doi: 10.1146/annurev.med.53.082901.104039. [DOI] [PubMed] [Google Scholar]
- 12.Gonzaga PE, Potter PM, Niu TQ, Yu D, Ludlum DB, Rafferty JA, Margison GP, Brent TP. Cancer Res. 1992;52:6052. [PubMed] [Google Scholar]
- 13.Friedberg EC, Walker GC, Siede W. DNA Repair and Mutagenesis. ASM Press; Washington, D. C.: 1995. [Google Scholar]
- 14.Nagasubramanian R, Dolan ME. Curr Opin Oncol. 2003;15:412. doi: 10.1097/00001622-200311000-00002. [DOI] [PubMed] [Google Scholar]
- 15.Denny BJ, Wheelhouse RT, Stevens MF, Tsang LL, Slack JA. Biochemistry. 1994;33:9045. doi: 10.1021/bi00197a003. [DOI] [PubMed] [Google Scholar]
- 16.Liu L, Yan L, Donze JR, Gerson SL. Mol Cancer Ther. 2003;2:1061. [PubMed] [Google Scholar]
- 17.Quinn JA, Desjardins A, Weingart J, Brem H, Dolan ME, Delaney SM, Vredenburgh J, Rich J, Friedman AH, Reardon DA, Sampson JH, Pegg AE, Moschel RC, Birch R, McLendon RE, Provenzale JM, Gururangan S, Dancey JE, Maxwell J, Tourt-Uhlig S, Herndon JE, 2nd, Bigner DD, Friedman HS. J Clin Oncol. 2005;23:7178. doi: 10.1200/JCO.2005.06.502. [DOI] [PubMed] [Google Scholar]
- 18.Kanzawa T, Bedwell J, Kondo Y, Kondo S, Germano IM. J Neurosurg. 2003;99:1047. doi: 10.3171/jns.2003.99.6.1047. [DOI] [PubMed] [Google Scholar]
- 19.Rabik CA, Njoku MC, Dolan ME. Cancer Treat Rev. 2006;32:261. doi: 10.1016/j.ctrv.2006.03.004. [DOI] [PubMed] [Google Scholar]
- 20.Zielske SP, Reese JS, Lingas KT, Donze JR, Gerson SL. J Clin Invest. 2003;112:1561. doi: 10.1172/JCI17922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zielske SP, Reese JS, Lingas KT, Donze JR, Gerson SL. J Clin Invest. 2003;112:1561. doi: 10.1172/JCI17922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bank A. J Clin Invest. 2003;112:1478. doi: 10.1172/JCI20336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Middleton MR, Margison GP. Lancet Oncol. 2003;4:37. doi: 10.1016/s1470-2045(03)00959-8. [DOI] [PubMed] [Google Scholar]
- 24.Ranson M, Middleton MR, Bridgewater J, Lee SM, Dawson M, Jowle D, Halbert G, Waller S, McGrath H, Gumbrell L, McElhinney RS, Donnelly D, McMurry TB, Margison GP. Clin Cancer Res. 2006;12:1577. doi: 10.1158/1078-0432.CCR-05-2198. [DOI] [PubMed] [Google Scholar]
- 25.Kaina B, Muhlhausen U, Piee-Staffa A, Christmann M, Garcia Boy R, Rosch F, Schirrmacher R. J Pharmacol Exp Ther. 2004;311:585. doi: 10.1124/jpet.104.071316. [DOI] [PubMed] [Google Scholar]
- 26.Fishel ML, Delaney SM, Friesen LD, Hansen RJ, Zuhowski EG, Moschel RC, Egorin MJ, Dolan ME. Mol Cancer Ther. 2003;2:633. [PubMed] [Google Scholar]
- 27.Fishel ML, Newell DR, Griffin RJ, Davison R, Wang LZ, Curtin NJ, Zuhowski EG, Kasza K, Egorin MJ, Moschel RC, Dolan ME. J Pharmacol Exp Ther. 2005;312:206. doi: 10.1124/jpet.104.073924. [DOI] [PubMed] [Google Scholar]
- 28.Fishel ML, Gamcsik MP, Delaney SM, Zuhowski EG, Maher VM, Karrison T, Moschel RC, Egorin MJ, Dolan ME. Cancer Chemother Pharmacol. 2005;55:333. doi: 10.1007/s00280-004-0901-3. [DOI] [PubMed] [Google Scholar]
- 29.Dolan ME, Roy SK, Fasanmade AA, Paras PR, Schilsky RL, Ratain MJ. J Clin Oncol. 1998;16:1803. doi: 10.1200/JCO.1998.16.5.1803. [DOI] [PubMed] [Google Scholar]
- 30.Friedman HS, Kokkinakis DM, Pluda J, Friedman AH, Cokgor I, Haglund MM, Ashley DM, Rich J, Dolan ME, Pegg AE, Moschel RC, McLendon RE, Kerby T, Herndon JE, Bigner DD, Schold SC., Jr. J Clin Oncol. 1998;16:3570. doi: 10.1200/JCO.1998.16.11.3570. [DOI] [PubMed] [Google Scholar]
- 31.Spiro TP, Gerson SL, Liu L, Majka S, Haaga J, Hoppel CL, Ingalls ST, Pluda JM, Willson JK. Cancer Res. 1999;59:2402. [PubMed] [Google Scholar]
- 32.Schilsky RL, Dolan ME, Bertucci D, Ewesuedo RB, Vogelzang NJ, Mani S, Wilson LR, Ratain MJ. Clin Cancer Res. 2000;6:3025. [PubMed] [Google Scholar]
- 33.Friedman HS, Pluda J, Quinn JA, Ewesuedo RB, Long L, Friedman AH, Cokgor I, Colvin OM, Haglund MM, Ashley DM, Rich JN, Sampson J, Pegg AE, Moschel RC, McLendon RE, Provenzale JM, Stewart ES, Tourt-Uhlig S, Garcia-Turner AM, Herndon JE, 2nd, Bigner DD, Dolan ME. J Clin Oncol. 2000;18:3522. doi: 10.1200/JCO.2000.18.20.3522. [DOI] [PubMed] [Google Scholar]
- 34.Fortini P, Parlanti E, Sidorkina OM, Laval J, Dogliotti E. J Biol Chem. 1999;274:15230. doi: 10.1074/jbc.274.21.15230. [DOI] [PubMed] [Google Scholar]
- 35.Fortini P, Dogliotti E. DNA Repair (Amst) 2007;6:398. doi: 10.1016/j.dnarep.2006.10.008. [DOI] [PubMed] [Google Scholar]
- 36.Evans AR, Limp-Foster M, Kelley MR. Mutat Res. 2000;461:83. doi: 10.1016/s0921-8777(00)00046-x. [DOI] [PubMed] [Google Scholar]
- 37.Denny BJ, Wheelhouse RT, Stevens MF, Tsang LL, Slack JA. Biochemistry. 1994;33:9045. doi: 10.1021/bi00197a003. [DOI] [PubMed] [Google Scholar]
- 38.McHugh PJ, Gill RD, Waters R, Hartley JA. Nucleic Acids Res. 1999;27:3259. doi: 10.1093/nar/27.16.3259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xu Y, Hansen WK, Rosenquist TA, Williams DA, Limp-Foster M, Kelley MR. J Pharmacol Exp Ther. 2001;296:825. [PubMed] [Google Scholar]
- 40.Bobola MS, Varadarajan S, Smith NW, Goff RD, Kolstoe DD, Blank A, Gold B, Silber JR. Clin Cancer Res. 2007;13:612. doi: 10.1158/1078-0432.CCR-06-1127. [DOI] [PubMed] [Google Scholar]
- 41.Pletsa V, Valavanis C, van Delft JH, Steenwinkel MJ, Kyrtopoulos SA. Carcinogenesis. 1997;18:2191. doi: 10.1093/carcin/18.11.2191. [DOI] [PubMed] [Google Scholar]
- 42.Drablos F, Feyzi E, Aas PA, Vaagbo CB, Kavli B, Bratlie MS, Pena-Diaz J, Otterlei M, Slupphaug G, Krokan HE. DNA Repair (Amst) 2004;3:1389. doi: 10.1016/j.dnarep.2004.05.004. [DOI] [PubMed] [Google Scholar]
- 43.Yang S, Irani K, Heffron SE, Jurnak F, Meyskens FL., Jr. Mol Cancer Ther. 2005;4:1923. doi: 10.1158/1535-7163.MCT-05-0229. [DOI] [PubMed] [Google Scholar]
- 44.Meynard D, Le Morvan V, Bonnet J, Robert J. Oncol Rep. 2007;17:1213. [PubMed] [Google Scholar]
- 45.Minotti G, Menna P, Salvatorelli E, Cairo G, Gianni L. Pharmacol Rev. 2004;56:185. doi: 10.1124/pr.56.2.6. [DOI] [PubMed] [Google Scholar]
- 46.Fawcett H, Mader JS, Robichaud M, Giacomantonio C, Hoskin DW. Int J Oncol. 2005;27:1717. [PubMed] [Google Scholar]
- 47.Alexandre J, Hu Y, Lu W, Pelicano H, Huang P. Cancer Res. 2007;67:3512. doi: 10.1158/0008-5472.CAN-06-3914. [DOI] [PubMed] [Google Scholar]
- 48.Houtgraaf JH, Versmissen J, van der Giessen WJ. Cardiovasc Revasc Med. 2006;7:165. doi: 10.1016/j.carrev.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 49.Fan J, Wilson DM., 3rd Free Radic Biol Med. 2005;38:1121. doi: 10.1016/j.freeradbiomed.2005.01.012. [DOI] [PubMed] [Google Scholar]
- 50.Wang P, Guliaev AB, Hang B. Toxicol Lett. 2006;166:237. doi: 10.1016/j.toxlet.2006.06.647. [DOI] [PubMed] [Google Scholar]
- 51.Paik J, Duncan T, Lindahl T, Sedgwick B. Cancer Res. 2005;65:10472. doi: 10.1158/0008-5472.CAN-05-1495. [DOI] [PubMed] [Google Scholar]
- 52.Fishel ML, Seo YR, Smith ML, Kelley MR. Cancer Res. 2003;63:608. [PubMed] [Google Scholar]
- 53.Fishel ML, He Y, Smith ML, Kelley MR. Clin Cancer Res. 2007;13:260. doi: 10.1158/1078-0432.CCR-06-1920. [DOI] [PubMed] [Google Scholar]
- 54.Rinne M, Caldwell D, Kelley MR. Mol Cancer Ther. 2004;3:955. [PubMed] [Google Scholar]
- 55.Coquerelle T, Dosch J, Kaina B. Mutat Res. 1995;336:9. doi: 10.1016/0921-8777(94)00035-5. [DOI] [PubMed] [Google Scholar]
- 56.Fung H, Demple B. Mol Cell. 2005;17:463. doi: 10.1016/j.molcel.2004.12.029. [DOI] [PubMed] [Google Scholar]
- 57.Fleck O, Nielsen O. J Cell Sci. 2004;117:515. doi: 10.1242/jcs.00952. [DOI] [PubMed] [Google Scholar]
- 58.Tell G, Damante G, Caldwell D, Kelley MR. Antioxid Redox Signal. 2005;7:367. doi: 10.1089/ars.2005.7.367. [DOI] [PubMed] [Google Scholar]
- 59.Ono Y, Furuta T, Ohmoto T, Akiyama K, Seki S. Mutat Res. 1994;315:55. doi: 10.1016/0921-8777(94)90028-0. [DOI] [PubMed] [Google Scholar]
- 60.Walker LJ, Craig RB, Harris AL, Hickson ID. Nucl. Acids Res. 1994;22:4884. doi: 10.1093/nar/22.23.4884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bobola MS, Finn LS, Ellenbogen RG, Geyer JR, Berger MS, Braga JM, Meade EH, Gross ME, Silber JR. Clin Cancer Res. 2005;11:7405. doi: 10.1158/1078-0432.CCR-05-1068. [DOI] [PubMed] [Google Scholar]
- 62.Yang ZZ, Chen XH, Wang D. Clin Lymphoma Myeloma. 2007;7:296. doi: 10.3816/CLM.2007.n.006. [DOI] [PubMed] [Google Scholar]
- 63.Lau JP, Weatherdon KL, Skalski V, Hedley DW. Br J Cancer. 2004;91:1166. doi: 10.1038/sj.bjc.6602080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Xanthoudakis S, Curran T. Adv. Expt. Med. Biol. 1996;387:387. [PubMed] [Google Scholar]
- 65.Larsen E, Meza TJ, Kleppa L, Klungland A. Mutat Res. 2007;614:56. doi: 10.1016/j.mrfmmm.2006.01.023. [DOI] [PubMed] [Google Scholar]
- 66.Taverna P, Liu L, Hwang HS, Hanson AJ, Kinsella TJ, Gerson SL. Mutat Res. 2001;485:269. doi: 10.1016/s0921-8777(01)00076-3. [DOI] [PubMed] [Google Scholar]
- 67.Liu L, Nakatsuru Y, Gerson SL. Clin Cancer Res. 2002;8:2985. [PubMed] [Google Scholar]
- 68.Madhusudan S, Smart F, Shrimpton P, Parsons JL, Gardiner L, Houlbrook S, Talbot DC, Hammonds T, Freemont PA, Sternberg MJ, Dianov GL, Hickson ID. Nucleic Acids Res. 2005;33:4711. doi: 10.1093/nar/gki781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Luo M, Kelley MR. Anticancer Res. 2004;24:2127. [PubMed] [Google Scholar]
- 70.Talpaert-Borle M, Liuzzi M. Biochim Biophys Acta. 1983;740:410. doi: 10.1016/0167-4781(83)90089-1. [DOI] [PubMed] [Google Scholar]
- 71.Liuzzi M, Talpaert-Borle M. J Biol Chem. 1985;260:5252. [PubMed] [Google Scholar]
- 72.Bases RE, Mendez F. Int J Radiat Oncol Biol Phys. 1997;37:1133. doi: 10.1016/s0360-3016(97)00113-2. [DOI] [PubMed] [Google Scholar]
- 73.Mendez F, Goldman JD, Bases RE. Cancer Invest. 2002;20:983. doi: 10.1081/cnv-120005914. [DOI] [PubMed] [Google Scholar]
- 74.Raffoul JJ, Banerjee S, Singh-Gupta V, Knoll ZE, Fite A, Zhang H, Abrams J, Sarkar FH, Hillman GG. Cancer Res. 2007;67:2141. doi: 10.1158/0008-5472.CAN-06-2147. [DOI] [PubMed] [Google Scholar]
- 75.Hiramoto M, Shimizu N, Sugimoto K, Tang J, Kawakami Y, Ito M, Aizawa S, Tanaka H, Makino I, Handa H. J Immunol. 1998;160:810. [PubMed] [Google Scholar]
- 76.Mitomo K, Nakayama K, Fujimoto K, Sun X, Seki S, Yamamoto K. Gene. 1994;145:197. doi: 10.1016/0378-1119(94)90005-1. [DOI] [PubMed] [Google Scholar]
- 77.Xanthoudakis S, Miao G, Wang F, Pan YC, Curran T. Embo J. 1992;11:3323. doi: 10.1002/j.1460-2075.1992.tb05411.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Fishel ML, Kelley MR. Molecular Aspects of Medicine. 2007 doi: 10.1016/j.mam.2007.04.005. In press. [DOI] [PubMed] [Google Scholar]
- 79.Beard WA, Wilson SH. Chem Rev. 2006;106:361. doi: 10.1021/cr0404904. [DOI] [PubMed] [Google Scholar]
- 80.Beard WA, Prasad R, Wilson SH. Methods Enzymol. 2006;408:91. doi: 10.1016/S0076-6879(06)08007-4. [DOI] [PubMed] [Google Scholar]
- 81.Hu HY, Horton JK, Gryk MR, Prasad R, Naron JM, Sun DA, Hecht SM, Wilson SH, Mullen GP. J Biol Chem. 2004;279:39736. doi: 10.1074/jbc.M402842200. [DOI] [PubMed] [Google Scholar]
- 82.Horton JK, Joyce-Gray DF, Pachkowski BF, Swenberg JA, Wilson SH. DNA Repair (Amst) 2003;2:27. doi: 10.1016/s1568-7864(02)00184-2. [DOI] [PubMed] [Google Scholar]
- 83.Sobol RW, Horton JK, Kuhn R, Gu H, Singhal RK, Prasad R, Rajewsky K, Wilson SH. Nature. 1996;379:183. doi: 10.1038/379183a0. [DOI] [PubMed] [Google Scholar]
- 84.Raaphorst GP, Ng CE, Yang DP. Int J Hyperthermia. 2002;18:33. doi: 10.1080/02656730110072352. [DOI] [PubMed] [Google Scholar]
- 85.Jagtap P, Szabo C. Nat Rev Drug Discov. 2005;4:421. doi: 10.1038/nrd1718. [DOI] [PubMed] [Google Scholar]
- 86.Hassa PO, Haenni SS, Elser M, Hottiger MO. Microbiol Mol Biol Rev. 2006;70:789. doi: 10.1128/MMBR.00040-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Otto H, Reche PA, Bazan F, Dittmar K, Haag F, Koch-Nolte F. BMC Genomics. 2005;6:139. doi: 10.1186/1471-2164-6-139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Menissier de Murcia J, Ricoul M, Tartier L, Niedergang C, Huber A, Dantzer F, Schreiber V, Ame JC, Dierich A, Le-Meur M, Sabatier L, Chambon P, de Murcia G. Embo J. 2003;22:2255. doi: 10.1093/emboj/cdg206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Yu SW, Wang H, Poitras MF, Coombs C, Bowers WJ, Federoff HJ, Poirier GG, Dawson TM, Dawson VL. Science. 2002;297:259. doi: 10.1126/science.1072221. [DOI] [PubMed] [Google Scholar]
- 90.Dantzer F, de La Rubia G, Menissier-De Murcia J, Hostomsky Z, de Murcia G, Schreiber V. Biochemistry. 2000;39:7559. doi: 10.1021/bi0003442. [DOI] [PubMed] [Google Scholar]
- 91.Ratnam K, Low JA. Clin Cancer Res. 2007;13:1383. doi: 10.1158/1078-0432.CCR-06-2260. [DOI] [PubMed] [Google Scholar]
- 92.de Murcia JM, Niedergang C, Trucco C, Ricoul M, Dutrillaux B, Mark M, Oliver FJ, Masson M, Dierich A, LeMeur M, Walztinger C, Chambon P, de Murcia G. Proc Natl Acad Sci U S A. 1997;94:7303. doi: 10.1073/pnas.94.14.7303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ, Helleday T. Nature. 2005;434:913. doi: 10.1038/nature03443. [DOI] [PubMed] [Google Scholar]
- 94.McCabe N, Lord CJ, Tutt AN, Martin NM, Smith GC, Ashworth A. Cancer Biol Ther. 2005;4:934. doi: 10.4161/cbt.4.9.2141. [DOI] [PubMed] [Google Scholar]
- 95.Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, Martin NM, Jackson SP, Smith GC, Ashworth A. Nature. 2005;434:917. doi: 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- 96.Gallmeier E, Kern SE. Cancer Biol Ther. 2005;4:703. doi: 10.4161/cbt.4.7.1909. [DOI] [PubMed] [Google Scholar]
- 97.Goetz M, Couch FJ. Cancer Biol Ther. 2005;4:707. doi: 10.4161/cbt.4.7.1998. [DOI] [PubMed] [Google Scholar]
- 98.Papouli E, Cejka P, Jiricny J. Cancer Res. 2004;64:3391. doi: 10.1158/0008-5472.CAN-04-0513. [DOI] [PubMed] [Google Scholar]
- 99.Plumb JA, Strathdee G, Sludden J, Kaye SB, Brown R. Cancer Res. 2000;60:6039. [PubMed] [Google Scholar]
- 100.Arnold CN, Goel A, Boland CR. Int J Cancer. 2003;106:66. doi: 10.1002/ijc.11176. [DOI] [PubMed] [Google Scholar]
- 101.Selvakumaran M, Pisarcik DA, Bao R, Yeung AT, Hamilton TC. Cancer Res. 2003;63:1311. [PubMed] [Google Scholar]
- 102.Mack PC, Gandara DR, Lau AH, Lara PN, Jr., Edelman MJ, Gumerlock PH. Cancer Chemother Pharmacol. 2003;51:337. doi: 10.1007/s00280-003-0571-6. [DOI] [PubMed] [Google Scholar]
- 103.Lara PN, Jr., Mack PC, Synold T, Frankel P, Longmate J, Gumerlock PH, Doroshow JH, Gandara DR. Clin Cancer Res. 2005;11:4444. doi: 10.1158/1078-0432.CCR-04-2602. [DOI] [PubMed] [Google Scholar]
- 104.Jiang H, Yang LY. Cancer Res. 1999;59:4529. [PubMed] [Google Scholar]
- 105.Shao RG, Cao CX, Zhang H, Kohn KW, Wold MS, Pommier Y. Embo J. 1999;18:1397. doi: 10.1093/emboj/18.5.1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Pujol JL, Viens P, Rebattu P, Laurie SA, Feld R, Deneulin A, Fandi A. J Thorac Oncol. 2006;1:417. [PubMed] [Google Scholar]
- 107.Judde JG, Rebucci M, Vogt N, de Cremoux P, Livartowski A, Chapelier A, Tran-Perennou C, Boye K, Defrance R, Poupon MF, Bras-Goncalves RA. Int J Cancer. 2007;120:1579. doi: 10.1002/ijc.22364. [DOI] [PubMed] [Google Scholar]
- 108.Zhong X, Li X, Wang G, Zhu Y, Hu G, Zhao J, Neace C, Ding H, Reed E, Li QQ. Int J Oncol. 2004;25:445. [PubMed] [Google Scholar]
- 109.Zhong X, Li QQ, Reed E. Cell Mol Life Sci. 2003;60:794. doi: 10.1007/s00018-003-3002-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Hickson I, Zhao Y, Richardson CJ, Green SJ, Martin NM, Orr AI, Reaper PM, Jackson SP, Curtin NJ, Smith GC. Cancer Res. 2004;64:9152. doi: 10.1158/0008-5472.CAN-04-2727. [DOI] [PubMed] [Google Scholar]
- 111.Kao J, Milano MT, Javaheri A, Garofalo MC, Chmura SJ, Weichselbaum RR, Kron SJ. Curr Cancer Drug Targets. 2006;6:197. doi: 10.2174/156800906776842957. [DOI] [PubMed] [Google Scholar]
- 112.Willmore E, de Caux S, Sunter NJ, Tilby MJ, Jackson GH, Austin CA, Durkacz BW. Blood. 2004;103:4659. doi: 10.1182/blood-2003-07-2527. [DOI] [PubMed] [Google Scholar]
- 113.Harrison L, Hatahet Z, Wallace SS. J Mol Biol. 1999;290:667. doi: 10.1006/jmbi.1999.2892. [DOI] [PubMed] [Google Scholar]
- 114.Rabik CA, Dolan ME. Cancer Treat Rev. 2007;33:9. doi: 10.1016/j.ctrv.2006.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Gajewski E, Gaur S, Akman SA, Matsumoto L, van Balgooy JN, Doroshow JH. Biochem Pharmacol. 2007;73:1947. doi: 10.1016/j.bcp.2007.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Chou KM, Kukhanova M, Cheng YC. J Biol Chem. 2000;275:31009. doi: 10.1074/jbc.M004082200. [DOI] [PubMed] [Google Scholar]