

Abstract
Context
Smoking remains a major public health problem. Twin studies indicate that the ability to quit smoking is substantially heritable, with genetics that overlap modestly with the genetics of vulnerability to dependence on addictive substances.
Objectives
To identify replicated genes that facilitate smokers’ abilities to achieve and sustain abstinence from smoking (hereinafter referred to as quit-success genes) found in more than 2 genome-wide association (GWA) studies of successful vs unsuccessful abstainers, and, secondarily, to nominate genes for selective involvement in smoking cessation success with bupropion hydrochloride vs nicotine replacement therapy (NRT).
Design
The GWA results in subjects from 3 centers, with secondary analyses of NRT vs bupropion responders.
Setting
Outpatient smoking cessation trial participants from 3 centers.
Participants
European American smokers who successfully vs unsuccessfully abstain from smoking with biochemical confirmation in a smoking cessation trial using NRT, bupropion, or placebo (N=550).
Main Outcome Measures
Quit-success genes, reproducibly identified by clustered nominally positive single-nucleotide polymorphisms (SNPs) in more than 2 independent samples with significant P values based on Monte Carlo simulation trials. The NRT-selective genes were nominated by clustered SNPs that display much larger t values for NRT vs placebo comparisons. The bupropion-selective genes were nominated by bupropion-selective results.
Results
Variants in quit-success genes are likely to alter cell adhesion, enzymatic, transcriptional, structural, and DNA, RNA, and/or protein-handling functions. Quit-success genes are identified by clustered nominally positive SNPs from more than 2 samples and are unlikely to represent chance observations (Monte Carlo P < .0003). These genes display modest overlap with genes identified in GWA studies of dependence on addictive substances and memory.
Conclusions
These results support polygenic genetics for success in abstaining from smoking, overlap with genetics of substance dependence and memory, and nominate gene variants for selective influences on therapeutic responses to bupropion vs NRT. Molecular genetics should help match the types and/or intensity of anti-smoking treatments with the smokers most likely to benefit from them.
Cigarette smoking continues to contribute substantially to US morbidity and mortality, despite increasingly stringent public health measures that make it more difficult and expensive to smoke.1 Smokers may thus now include many who have considerable difficulty in achieving sustained abstinence from tobacco, despite the availability of therapeutic drugs that act at sites that include nicotinic receptors and monoamine transporters.2 Understanding the mechanisms that facilitate or impede smokers’ abilities to achieve and sustain abstinence from smoking could thus inform a major current US public health problem and augment understanding of mnemonic and other brain mechanisms that might influence abstinence.3 Understanding mechanisms that contribute to the abstinence-enhancing efficacies of specific therapeutic drugs could help match smokers with treatments that would be more likely to be effective for them.
Twin studies suggest that 40% to 60% of individual differences in the ability to successfully quit smoking are heritable.4 Nicotine dependence is also heritable in ways that overlap substantially with vulnerability to dependence on other addictive substances.5–8 Many of the heritable influences on successful smoking cessation, however, appear to differ from those that influence aspects of nicotine dependence.4
Several pharmacological approaches increase smoking cessation success. Nicotine replacement therapies (NRTs) and bupropion hydrochloride can each double effective rates of smoking cessation.9,10 These 2 approaches use different pharmacological mechanisms and might conceivably be expected to work differently in different individuals.11
Candidate gene and genome-wide association (GWA) studies can detect genes that harbor allelic variants with modest effects on disorders that display complex genetic architectures (http://www.niehs.nih.gov/oc/news/gei.htm). Some, although not all, candidate gene studies have associated variants in genes related to dopamine and opioid neurotransmission with success in smoking cessation.12–17 Genome-wide association studies can detect the effects of allelic variants in genes that were unanticipated, and have already identified previously unanticipated addiction-vulnerability genes that are likely to contain variants that contribute to interindividual differences in vulnerability to substance dependence.18–23 Convergence between addiction vulnerability and genes that facilitate smokers’ abilities to achieve and sustain abstinence from smoking (hereinafter referred to as quit-success genes) would provide enhanced confidence in the genes identified by both comparisons. Confidence in correct identification of quit-success genes for smoking that are not also addiction vulnerability genes, however, requires replicated results from multiple samples.
We herein report the results of such convergent, replicate GWA studies of smoking cessation. We included the study participants in independent clinical trials from 3 centers whose success in smoking cessation has been rigorously assessed by self-report and biochemical test results. We describe (1) substantial convergence of the results from these independent samples that is unlikely to be due to chance alone; (2) more modest, but significant, overlap with GWA data for dependence on nicotine and other substances of abuse; and (3) initial evidence of specific genetic influences on therapeutic responses to NRT vs bupropion.
METHODS
SAMPLES 1 THROUGH 3
European American individuals who smoked responded to newspaper, flyer, and television advertisements and/or to physician referrals for help in quitting smoking.16,24 Subjects aged 18 to 65 years who provided informed consent, were not pregnant or lactating, had no DSM-IV Axis I psychiatric disorder, reported no current use of study medications (eg, bupropion or nicotine-containing products other than cigarettes), and described no contraindications for use of these medications were included. Individuals who were dependent on other addictive substances, current users of psychotropic medications, or diagnosed as having cancer in the last 6 months were excluded.
Subjects were enrolled in 1 of 4 randomized clinical trials for smoking cessation that were approved by the appropriate institutional review boards. Each participant received standardized behavioral counseling.
Sample 1
Sample 1 consisted of subjects enrolled in (1) a double-blind placebo-controlled trial of bupropion hydrochloride, 300 mg/d, or matching placebo for 10 weeks (bupropion trial) or (2) an open-label trial of a nicotine nasal spray vs a nicotine patch for 8 weeks (NRT trial).16 Smoking status was assessed by telephone interview at 0, 8, and 24 weeks after the targeted quit date using validated timeline follow-back methods.25 Abstinence was also assessed by measuring levels of cotinine (<15 μg/L [to convert to nanomoles per liter, multiply by 5.675]; bupropion trial) or carbon monoxide (NRT trial). The 126 individuals with biochemically confirmed abstinence for at least the 7 days before the end of treatment (8 weeks) and at the 24-week assessment were contrasted with the 140 individuals who were not abstinent at either measurement. Sample 1 was 55% female, averaged 45 years of age, reported smoking an average of 21 cigarettes a day, and displayed average Fagerström Test for Nicotine Dependence (FTND) scores of 5.4 before treatment. Eighty-nine percent described at least 1 previous effort to quit smoking.
Sample 2
Sample 2 consisted of subjects enrolled in a trial of the clinical effectiveness of NRT. Participants received active nicotine skin patches, 21 mg/d, or placebo patches for 2 weeks before the targeted quit date. Participants also received oral mecamylamine hydrochloride, 10 mg/d, before the targeted quit date to attenuate the reinforcing effects of smoking.26 After the quit date, participants were randomly assigned to groups that received mecamylamine hydrochloride, 10 mg/d, vs matching placebo and nicotine skin patches at dosages of 21 or 42 mg every 24 hours. Fifty-five individuals reported continuous abstinence from smoking at assessment 6 weeks after the quit date with confirmation by means of carbon monoxide levels; 79 were not abstinent. Sample 2 was 48% female, averaged 44 years of age, reported smoking an average of 30 cigarettes a day, and displayed average pretreatment FTND scores of 6.4. Most of these individuals reported at least 1 previous attempt to quit smoking (average, 4.4 attempts). Initial analysis of the data from these individuals has been reported previously.19
Sample 3
Sample 3 consisted of individuals enrolled in a 10-week double-blind, placebo-controlled trial of placebo or bupropion hydrochloride (150 mg/d for the first 3 days, then 300 mg/d) with a targeted quit date 1 week after initiation of drug or placebo therapy.24 Smoking cessation was assessed using point abstinence, defined by self-report and a saliva cotinine levels of less than 15 μg/L. The 60 individuals with biochemically confirmed abstinence for at least the 7 days before the end of treatment and at the 24-week assessment were contrasted with the 90 individuals who were not abstinent at either measurement. Sample 3 was 51% female, averaged 45 years of age, reported smoking an average of 25 cigarettes a day, and displayed an average pretreatment FTND score of 7.5. Most of these individuals reported at least 1 previous attempt to quit smoking (average, 5 attempts).
DNA PREPARATION, POOLING, AND ANALYSIS
Genomic DNA was prepared from blood samples,20 carefully quantitated, combined into pools representing 13 to 20 subjects with successful or unsuccessful attempts to quit smoking (eTable 1; available at: http://www.archgenpsychiatry.com), and analyzed as described by scientists blinded to the quit success phenotype20 (described in the supplementary text; available at: http://www.archgenpsychiatry.com). Allele frequencies for each single-nucleotide polymorphism (SNP) in each DNA pool were assessed on the basis of hybridization to the perfect-match cells from replicate experiments as described elsewhere.18,22 Nominally positive SNPs with 2-tailed t values for successful vs unsuccessful abstainer differences of P < .01 were identified as described.18,21,22
Analyses focused on nominally positive SNPs that cluster in small chromosomal regions in at least 2 samples because this pattern of results would not be anticipated by chance but would be anticipated if haplotype blocks that contained multiple SNPs were present at differing frequencies in successful vs unsuccessful abstainers. The Table thus identifies SNPs that (1) display t values of P < .01 in comparing successful vs unsuccessful abstainers; (2) cluster so that at least 3 such SNPs on at least 2 array types (sample 1) or at least 2 such SNPs (samples 2 and 3) lie within 0.1 megabase (Mb) of each other; (3) identify annotated genes; and (4) identify genes that contain clustered SNPs with P < .01 compared with at least 1 other sample. Genes that provide replicated results based on clustered SNPs with nominal P < .01 findings and that lie within the annotated exons of the genes listed, ±10 kilobase, are listed in eTable 1 and eTable 2. The location of clustered SNPs within the genes’ exons and introns, within the genes’ flanks, and outside the annotated genes are listed in eTable 2 and eTable 3.
Using Monte Carlo simulation trials, we calculated P values for (1) the clustering of nominally positive SNPs from a single sample that lie within annotated genes and (2) the convergence between these clusters from a single sample and similar data from at least 1 other sample based on 10 000 or 100 000 simulation trials, as noted in the presented P values. Each trial sampled a random set of SNPs from the database that contains the results from these studies and applied the same procedure that had been followed for the actual data analysis. The number of trials for which the results from the randomly selected set of SNPs matched or exceeded the results actually observed from the SNPs identified in the present study was tabulated. We calculated empirical P values by dividing the number of trials for which the observed results were matched or exceeded by the total number of Monte Carlo simulation trials performed. This method examines the properties of the SNPs in the present data set and thus should be relatively robust, despite the uneven distribution of SNP markers across the genome in the Affymetrix microarrays (Affymetrix Inc, Santa Clara, California), the slightly different complement of SNPs represented on the early access and commercial versions of the arrays, and the differing criteria for clustering applied to the larger sample 1 and smaller samples 2 and 3.
We estimated statistical power using the observed standard deviations and mean abuser/control differences from each sample using the program PS, version 2.1.3132 (α=.05).
To control for the possibility that differences between successful and unsuccessful abstainers were due to occult ethnic differences, we compared the clustered SNPs that displayed nominally positive results in the present study with SNPs that displayed the largest differences among African American vs European American control individuals previously studied in this laboratory, or the largest differences among subsets of self-reported white controls recruited from different sites within the United Kingdom.33 To control for the possibility that differences were due to noisy assays, we compared the largest variation between pools in data from this and other studies that used the same arrays, as described previously.18
Secondary analyses pooled data from placebo-, bupropion-, and NRT-treated individuals from all 3 samples. For each SNP, t values for allele frequency differences between successful vs unsuccessful abstainers in bupropion vs placebo and NRT vs placebo comparisons were plotted (Figure 1). Although there is no single statistical approach to such data, we sought SNPs that displayed an effect on responses to at least 1 of these treatments that was of nominally high significance, and divided these SNPs into those that displayed bupropion-selective, NRT-selective, or nonselective effects. We identified SNPs that displayed t values corresponding to P < .005 for NRT (t > 3.69), bupropion (t > 3.58), or both. We calculated the differences between the t values for NRT vs placebo and the t values for the differences between bupropion vs placebo for each SNP. We defined as NRT specific the one-third of SNPs for which t values for NRT provided the largest positive differences from those for bupropion; as nonspecific the one-third of SNPs for which t values for nicotine replacement were similar to those for bupropion; and as bupropion specific the one-third of SNPs for which t values for bupropion provided the largest positive differences from those for NRT. We then tallied the genes that were identified by at least 2 such SNPs. Genes for which most SNPs were NRT specific, those for which most SNPs were bupropion specific, and those for which there was no clear treatment specificity are listed in eTable 4.
Figure 1.
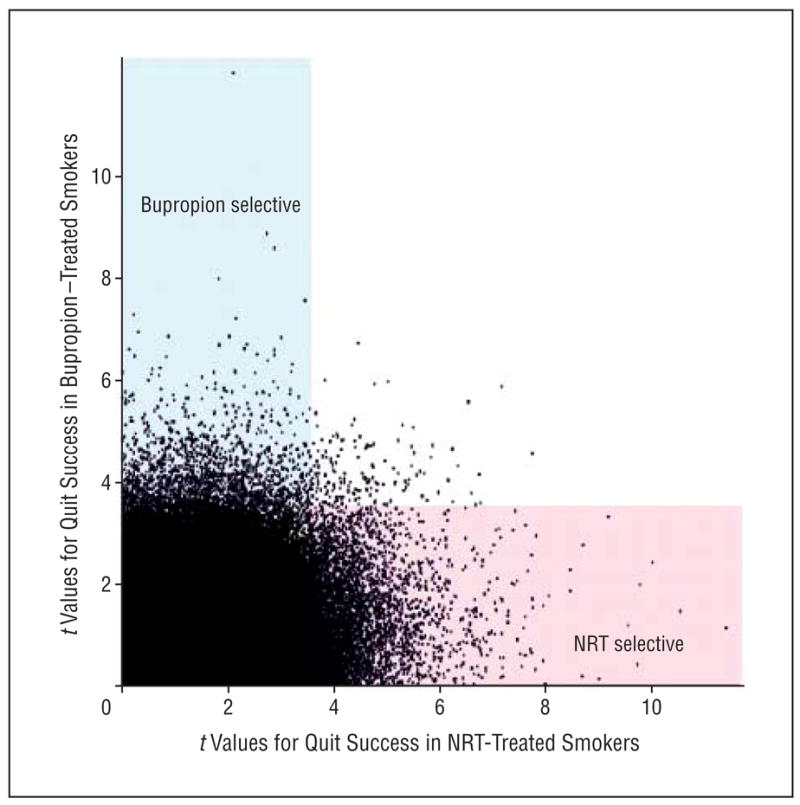
Scatterplot of the distributions of t values for comparisons between individuals with successful vs unsuccessful attempts to quit smoking in pooled samples from those receiving nicotine replacement therapy (NRT) (x-axis) vs bupropion hydrochloride (y-axis) for secondary analyses seeking candidate genes with treatment-specific effects. If no genes provided treatment-specific effects, values would cluster on a 45° line from the origin. We highlight single-nucleotide polymorphisms (SNPs) that provide NRT-selective (pink shading) or bupropion-selective (blue shading) effects (see also eTable 4). The t values of 3.6 and 3.7 for NRT and bupropion, respectively, correspond to P < .005. These data combine individuals from samples 1 and 2 who received NRT and individuals from samples 1 and 3 who received bupropion.
To test whether these nicotine-specific, nonspecific, and bupropion-specific SNPs were clustered on chromosomal regions in ways that we would anticipate if they identified haplotypes that contained such gene variants but not if they represented random noise, we sought clustering of the SNPs that appeared to identify these bupropion- or NRT-selective genes. Tests for the significance of the clustering of the bupropion- and NRT-selective SNPs were performed as noted for tests of the clustering of the quit-success SNPs described in this section.
RESULTS
VARIABILITY AND CORRESPONDING POWER CALCULATIONS
There was modest variability in the allele frequency assessments that were based on hybridization to the perfect-match cells on each of 4 arrays for DNA from each pool.18,22 Standard errors of the mean (SEMs) for the variation among the 4 replicate studies of each DNA pool were 0.037, 0.035, and 0.048 for samples 1 through 3, respectively. The SEMs for the variation among the pools studied for each phenotype group were 0.029, 0.028, and 0.034. These results support modest variation. They are consistent with results of validation studies that show correlations of 0.95 between pooled and individually determined genotype frequencies using these arrays18,20,21,27–29 (see also the supplementary text). Based on these variances, the power to detect allele frequency differences in samples 1, 2, and 3 was 0.71, 0.45, and 0.46, respectively, for 5% allele frequency differences, and 0.99, 0.95, and 0.96, respectively, for 10% differences.
SUCCESSFUL VS UNSUCCESSFUL ABSTAINERS IN EACH OF THE SAMPLES
In comparing data from successful vs unsuccessful abstainers from samples 1, 2, and 3, we identified 5411, 4539, and 4894 SNPs, respectively, whose allele frequencies differ between the 2 groups with nominal P < .01 (Figure 2). The nominally positive SNPs from comparisons between successful vs unsuccessful abstainers in each of these samples cluster together to extents much greater than expected by chance if their allelic frequencies were independent of each other (Monte Carlo P < .00001). For sample 1, 1434 of these 5411 nominally positive SNPs lay in 308 clusters in which each positive SNP lay within 0.1 Mb of at least 2 other positive SNPs, with representation from SNPs on both array types (Figure 2). For samples 2 and 3, 2258 of the 4539 nominally positive SNPs and 2184 of the 4894 nominally positive SNPs, respectively, lie in 820 and 861 clusters in which each positive SNP lies within 0.1 Mb of at least 1 other positive SNP (Figure 2).
Figure 2.
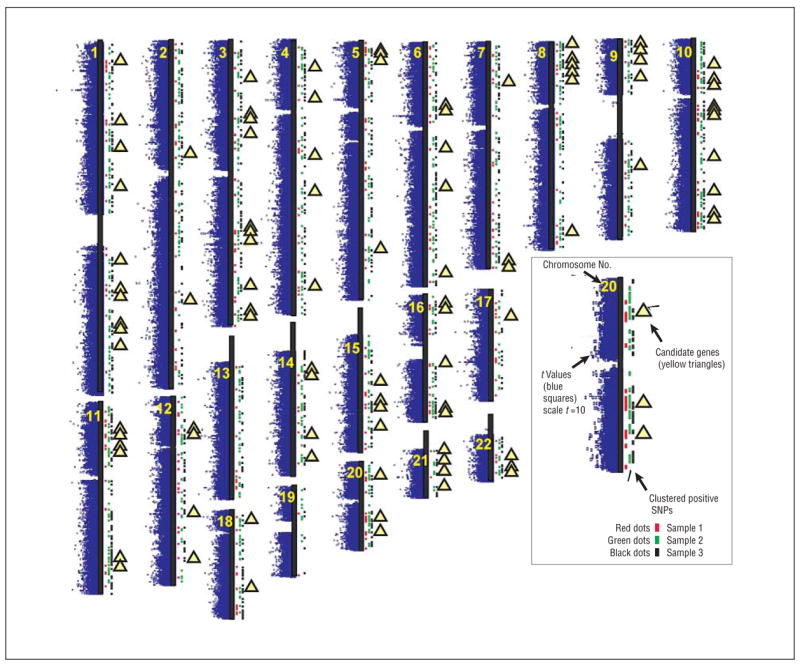
Distributions of data from the present study mapped onto cartoons of human chromosomes 1 through 10 (top row), 11 through 17 (second row), and 18 through 22 (third row). Blue dots to the left of the (black) main axis for each chromosome represent the t values for all single-nucleotide polymorphisms (SNPs) in the comparison of allele frequencies in individuals with successful vs unsuccessful attempts to quit smoking from sample 1 with the chromosomal position of each corresponding SNP. Red dots to the right of the (black) main axis for each chromosome mark sites for clustered positive SNPs from sample 1 for which successful vs unsuccessful abstainer differences display P < .01, lie within 0.1 megabase (Mb) of at least 1 other SNP that displays P < .01, and cluster so that at least 3 clustered outlier SNPs from 2 array types (StyI, NspI) are found in the cluster. Green dots to the right of the black main axis for each chromosome mark sites for clustered positive SNPs from sample 2 for which successful vs unsuccessful abstainer differences display P < .01, lie within 0.1 Mb of at least 1 other SNP that displays P < .01, and cluster so that at least 2 clustered outlier SNPs are found in the cluster. Black dots to the right of the black main axis for each chromosome mark sites for clustered positive SNPs from sample 3 for which successful vs unsuccessful abstainer differences display P < .01, lie within 0.1 Mb of at least 1 other SNP that displays P < .01, and cluster so that at least 2 clustered outlier SNPs are found in the cluster. Yellow triangles to the right of the black main axis for each chromosome mark sites for genes identified by clustered positive SNPs from at least 2 samples (see the Table). Chromosomal positions are based on National Center for Biotechnology Information Map Viewer Build 36.1 coordinates (http://www.ncbi.nlm.nih.gov/mapview/) and supplemental data from NetAffx Analysis Center (http://www.science.fau.edu/genechip/NetAffx_analysis_center.html).
In each of these independent samples, we thus observed clustering that we would anticipate if many of these nominally positive SNPs identified haplotypes that were present in different frequencies in our samples of successful vs unsuccessful abstainers but not if they represented chance independent observations.
NOMINALLY POSITIVE SNPs THAT MIGHT RESULT FROM OCCULT RACIAL/ETHNIC STRATIFICATION OF THE SAMPLES OR FROM NOISY SNP ASSAYS
Controls for occult racial/ethnic stratification and poor technical quality do not explain the nominally positive SNPs from these comparisons of successful vs unsuccessful abstainers. The SNPs that display the largest allele frequency differences between European and African American controls in previous assessments18 (G.R.U., T.D., and C.J., unpublished data, 2006), and between self-reported white samples from the United Kingdom33 and the SNPs that display the largest between-pool variances in the present data set do not overlap with those that distinguish comparisons of successful vs unsuccessful abstainers more than anticipated by chance (40 vs 135, 0 vs 0, and 103 vs 105, respectively [sample 1]; 39 vs 114, 0 vs 0, and 115 vs 120, respectively [sample 2]; and 35 vs 122, 0 vs 0, and 112 vs 95, respectively [sample 3]).
GENES IDENTIFIED BY REPLICATED GWA RESULTS FROM MORE THAN 2 INDEPENDENT SAMPLES
Genes in which 3 or more (sample 1) or 2 or more (samples 2 and 3) nominally positive SNPs cluster and where clustered nominally positive SNPs from at least 1 other sample are also present are listed in eTable 2.
Nominally positive clustered SNPs from comparisons of successful vs unsuccessful abstainers in samples 1 through 3 thus cluster together on small chromosomal regions to extents much greater than those expected by chance. The Monte Carlo values for the replication/convergence for samples 1 vs 2, 1 vs 3, and 2 vs 3 are P=.00054, P=.0016, and P=.00063, respectively.
Eight genes are identified by clustered positive SNPs that come from samples 1, 2, and 3 (boldfaced in the Table), 23 from samples 1 and 2, 29 from samples 1 and 3, and 61 from samples 2 and 3 (Monte Carlo P=.0003 for overall convergence).
Table.
Genes Harboring Allelic Variants That Distinguish Successful vs Unsuccessful Quitters in at Least 2 Samplesa
| Cluster SNPsb |
|||||
|---|---|---|---|---|---|
| Class/Gene | Description | 1 | 2 | 3 | P Valuec |
| Cell adhesion | |||||
| DAB1 | Disabled homologue 1 | 7 | 2 | .0117 | |
| ASTN | Astrotactin | 2 | 1 | .0334 | |
| USH2A | Usher syndrome 2A | 2 | 3 | .0421 | |
| CTNNA2 | Catenin 32 | 2 | 2 | .1254 | |
| CLSTN2 | Calsyntenin 2 | 1 | 4 | 3 | .0045 |
| SEMA5A | Semaphorin 5A | 5 | 1 | .0087 | |
| TRIO | Triple functional domain/PTPRF interacting protein | 2 | 2 | .0375 | |
| PTPRN2 | Receptor protein tyrosine phosphatase N2 | 5 | 5 | .0056 | |
| CSMD1 | Cub and Sushi multiple domains 1 | 10 | 5 | .0049 | |
| SGCZ | Sarcoglycan zeta | 3 | 5 | .0191 | |
| PTPRD | Receptor protein tyrosine phosphatase D | 2 | 8 | .0028 | |
| TEK | TEK receptor tyrosine kinase | 2 | 2 | 3 | .0011 |
| PCDH15 | Protocadherin 15 | 3 | 4 | .0175 | |
| NRG3 | Neuregulin 3 | 4 | 1 | .0561 | |
| DSCAML1 | Down syndrome cell adhesion molecule like 1 | 2 | 2 | .0365 | |
| NRXN3 | Neurexin 3 | 2 | 3 | .1002 | |
| ITGA11 | Integrin 311 | 2 | 1 | .0166 | |
| CDH13 | Cadherin 13 | 8 | 3 | 7 | .0020 |
| LAMA1 | Laminin 31 | 2 | 2 | .0186 | |
| PTPRT | Receptor protein tyrosine phosphatase T | 5 | 6 | 5 | .0024 |
| DSCAM | Down syndrome cell adhesion molecule | 10 | 2 | .0016 | |
| Enzymes | |||||
| ST6GALNAC3 | ST6 (3-N-acetyl-neuraminyl-2,3-3-galactosyl-1,3)-N-acetylgalactosaminide 3-2,6-sialyltransferase 3 | 2 | 4 | .0143 | |
| REN | Renin | 1 | 2 | .0067 | |
| CERKL | Ceramide kinase-like | 1 | 3 | .0079 | |
| NEK11 | NIMA (never in mitosis gene a)–related kinase 11 | 7 | 7 | .0016 | |
| TNIK | TRAF2 and NCK interacting kinase | 2 | 2 | .0377 | |
| LEPREL1 | Leprecan-like 1 | 4 | 2 | .0060 | |
| GALNT17 | Polypeptide N-acetylgalactosaminyltransferase 17 | 4 | 2 | .0580 | |
| ADCY2 | Adenylate cyclase 2 | 3 | 1 | .0175 | |
| PPP2R2B | Protein phosphatase 2 regulatory subunit B 3 isoform | 5 | 8 | .0007 | |
| DPP6 | Dipeptidyl peptidase 6 | 2 | 1 | .0310 | |
| PBK | PDZ binding kinase | 1 | 3 | .0038 | |
| ST3GAL1 | ST3 3-galactoside 3-2,3-sialyltransferase 1 | 2 | 1 | .0158 | |
| MOBKL2B | MOB1-like 2B | 1 | 2 | .0082 | |
| DAPK1 | Death-associated protein kinase 1 | 2 | 3 | .0105 | |
| LYZL1 | Lysozyme-like 1 | 2 | 2 | .0075 | |
| PRKG1 | cGMP-dependent protein kinase I | 7 | 3 | 4 | .0041 |
| ADAM12 | ADAM metallopeptidase domain 12 | 1 | 2 | .0457 | |
| FDX1 | Ferredoxin 1 | 2 | 1 | .0100 | |
| SERPINA2 | Serpin peptidase inhibitor A2 | 1 | 2 | .0078 | |
| SERPINA1 | Serpin peptidase inhibitor A1 | 1 | 2 | .0080 | |
| AKAP13 | A kinase anchor protein 13 | 5 | 2 | .0065 | |
| ALDH3A2 | Aldehyde dehydrogenase 3 family, member A2 | 1 | 1 | .0033 | |
| ATP9A | Adenosine triphosphatase class II, type 9A | 1 | 1 | 2 | .0026 |
| LARGE | Like-glycosyltransferase | 4 | 4 | .0067 | |
| Transcriptional regulation | |||||
| TDRD5 | Tudor domain containing 5 | 2 | 1 | .0064 | |
| RARB | Retinoic acid receptor 3 | 1 | 2 | .0218 | |
| SUPT3H | Suppressor of Ty 3 homologue | 3 | 2 | .0306 | |
| CREB5 | cAMP-responsive element binding protein 5 | 5 | 2 | .0082 | |
| GLIS3 | GLI-similar family zinc finger 3 | 3 | 2 | .0174 | |
| BNC2 | Basonuclin 2 | 3 | 6 | .0027 | |
| TACC2 | Transforming acidic coiled-coil containing protein 2 | 1 | 1 | .0141 | |
| SOX5 | SRY box 5 | 2 | 2 | .0514 | |
| NPAS3 | Neuronal PAS domain protein 3 | 5 | 1 | .0239 | |
| PAX9 | Paired box gene 9 | 1 | 2 | .0026 | |
| ZNF423 | Zinc finger protein 423 | 3 | 3 | .0044 | |
| SALL4 | Sal-like 4 | 1 | 1 | .0039 | |
| ERG | V-ets erythroblastosis virus E26 oncogene like | 2 | 2 | .0293 | |
| BCL2L13 | Bcl2-like 13 | 3 | 1 | .0037 | |
| Receptors | |||||
| GPR116 | G protein–coupled receptor 116 | 2 | 1 | .0114 | |
| CD109 | CD109 molecule | 1 | 1 | .0113 | |
| GRIK2 | Inotropic glutamate receptor kainate 2 | 2 | 2 | .0648 | |
| MTUS1 | Mitochondrial tumor suppressor 1 | 3 | 3 | .0049 | |
| GRIN2A | Inotropic glutamate receptor N-methyl D-aspartate 2A | 2 | 2 | .0427 | |
| GRIK1 | Inotropic glutamate receptor kainate 1 | 3 | 4 | .0028 | |
| Channels | |||||
| KCNN3 | N-type potassium channel 3 | 1 | 2 | .0224 | |
| KCNK2 | K-type potassium channel 2 | 4 | 2 | .0069 | |
| CACNA2D3 | Voltage-dependent calcium channel 32/3 subunit 3 | 3 | 8 | .0031 | |
| KCNIP4 | Kv channel interacting protein 4 | 6 | 2 | .0095 | |
| CACNB2 | Voltage-dependent calcium channel 32 | 2 | 2 | .0416 | |
| ITPR2 | Inositol 1,4,5-triphosphate receptor 2 | 6 | 3 | .0030 | |
| Transporters | |||||
| ATP8A1 | Aminophospholipid transporter class type 8A 1 | 1 | 1 | .0169 | |
| SLC1A2 | Solute carrier family 1 high-affinity glutamate transporter 2 | 3 | 3 | .0045 | |
| MATE2 | H3/organic cation antiporter | 1 | 1 | .0047 | |
| DNA/RNA processing | |||||
| RBM19 | RNA-binding motif protein 19 | 5 | 2 | .0033 | |
| A2BP1 | Ataxin 2–binding protein 1 | 14 | 12 | .0015 | |
| Ligands for receptors | |||||
| OLFM3 | Olfactomedin 3 | 1 | 7 | .0006 | |
| FGF12 | Fibroblast growth factor 12 | 3 | 2 | .0321 | |
| TMTC2 | Transmembrane and tetratricopeptide repeat containing 2 | 1 | 2 | .0234 | |
| THSD4 | Thrombospondin type I domain containing 4 | 3 | 2 | 4 | .0035 |
| Protein processing | |||||
| PARK2 | Parkin | 6 | 2 | .0236 | |
| SORCS1 | Sortilin-related VPS10 domain containing receptor 1 | 2 | 4 | .0168 | |
| Intracellular signaling pathways | |||||
| CHN2 | Chimerin 2 | 2 | 2 | .0314 | |
| PEBP4 | Phosphatidylethanolamine-binding protein 4 | 3 | 2 | .0065 | |
| NELL1 | NEL-like 1 | 2 | 2 | .0911 | |
| EFCBP2 | EF-hand calcium binding protein 2 | 1 | 1 | .0064 | |
| BIK | Bcl2-interacting killer | 1 | 2 | .0087 | |
| Structural proteins | |||||
| KIAA1026 | Kazrin | 3 | 2 | .0130 | |
| NPHS2 | Podocin | 1 | 2 | .0038 | |
| PKP1 | Plakophilin 1 | 1 | 1 | .0153 | |
| ERC2 | ELKS/RAB6-interacting/CAST family member 2 | 5 | 3 | .0129 | |
| MAGI1 | Membrane-associated guanylate kinase, WW and PDZ domain containing 1 | 7 | 2 | .0060 | |
| PARD3 | Partitioning defective 3 homologue | 4 | 2 | 2 | .0077 |
| UNC13C | Unc13 homologue C | 3 | 2 | .0354 | |
| SNAP25 | Synaptosomal-associated protein | 1 | 3 | .0068 | |
| MYO18B | Myosin XVIIIB | 5 | 3 | .0033 | |
| Unknown functions | |||||
| TMEM108 | Transmembrane protein 108 | 8 | 1 | .0009 | |
| C4orf22 | Chromosome 4 open reading frame 22 | 1 | 1 | .0836 | |
| FLJ20184 | Hypothetical protein FLJ20184 | 2 | 1 | .0139 | |
| C10orf64 | Chromosome 10 open reading frame 64 | 1 | 2 | .0078 | |
| BTBD16 | BTB (POZ) domain containing 16 | 1 | 1 | .0059 | |
| LUZP2 | Leucine zipper protein 2 | 3 | 1 | .0217 | |
| CCDC73 | Coiled-coil domain containing 73 | 3 | 3 | .0031 | |
| FLJ42220 | FLJ42220 protein | 2 | 2 | .0030 | |
| CCBE1 | Collagen- and calcium-binding EGF domains 1 | 1 | 2 | .0134 | |
Abbreviations: cAMP, cyclic adenosine monophosphate; cGMP, cyclic guanosine monophosphate; SNP, single-nucleotide polymorphism.
Chromosome number and initial chromosomal position for the genes come from National Center for Biotechnology Information Map Viewer Build 36.1 coordinates (http://www.ncbi.nlm.nih.gov/projects/mapview/F2). Monte Carlo P values for each gene come from 10 000 simulation trials that each begins with random sampling from a database that contains all gene segments ±10 kilobases (kb). These simulations assess the frequency of trials in which at least the observed numbers of nominally positive SNPs identified in each of the 3 samples studied herein was recorded to provide an empirical P value. Eight genes (noted in boldface) receive clustered, nominally highly significant association signals from each of the 3 samples studied herein.
Data are expressed as numbers of clustered SNPs from samples 1, 2, and 3 that display nominally highly significant (P < .01) allele frequency differences between individuals with successful and unsuccessful attempts to quit smoking within the gene’s exons or introns or in 10-kb 3′ or 5′ flanks of annotated exons. Empty cells reflect absence of clustered nominally significant SNPs in the sample.
Indicates the P values for each gene, based on Monte Carlo simulations.
PROBABILITIES AND CLASSES FOR INDIVIDUAL GENES
Simulation studies support the idea that many of the genes identified by the clustered positive results in more than 2 samples are unlikely to represent chance observations (Table). The probabilities that all of the genes identified in this study are random are substantially lower than the probability that any individual gene is listed in eTables 1 and 2 by chance. Nevertheless, most of these gene-by-gene P values indicate nominal statistical significance. Twenty-one of the genes relate to cell adhesion, 24 are enzymes, and 14 regulate transcription. Six encode receptors, 6 encode channels, 3 encode transporters, and 4 encode receptor ligands. Two genes are involved with RNA processing, 2 with protein processing, 5 with intracellular signaling, 9 with cell structure, and 9 with unknown functions. Clustered positive SNPs from several samples also nominate chromosomal areas in which no gene is currently annotated (eTable 3).
SECONDARY ANALYSES SEEKING CANDIDATE GENES WITH TREATMENT-SPECIFIC EFFECTS
We combined data from successful and unsuccessful abstainers from the groups treated with bupropion, NRT, or placebo from all 3 centers. As noted in Figure 1, the distribution of t values for the differences between bupropion vs placebo and the differences between NRT vs placebo support the idea that some SNPs identify gene variants that do not influence responses to each of these treatments in the same way. We identified SNPs that displayed t values corresponding to P < .005 for NRT, bupropion, or both and calculated the differences between the t values for NRT vs placebo and the t values for the differences between bupropion vs placebo for each SNP. The one-third of SNPs for which t values for NRT provided the largest positive differences from those for bupropion were considered NRT specific; the one-third for which t values for NRT were similar to those for bupropion were considered nonspecific; and the one-third for which t values for bupropion provided the largest positive differences from those for NRT were considered bupropion specific. Genes that are identified by atleast 2 SNPs in these groups are listed in eTable 4. The SNPs in the 41 NRT-specific, 66 nonspecific, and 26 bupropion-specific genes had mean t values of 4.28 vs 1.71,3.58 vs 2.67, and 1.71 vs 4.28 for NRT vs bupropion, respectively.
We sought chromosomal clustering of the SNPs that appeared to identify treatment-specific genes in ways that we would anticipate if they identified haplotypes that contained treatment-specific gene variants, but not if they represented random noise. Bupropion- and NRT-selective SNPs each clustered in small chromosomal regions with Monte Carlo P < .00001.
COMMENT
Convergent, replicated observations from these 3 GWA studies of smoking cessation are consistent with the substantial heritabilities for successful vs unsuccessful abstinence that are supported by classical genetic studies.4,7,34–37 The overlap between some of these quit-success genes with substance-dependence vulnerability genes provides an interesting list that we discuss in the following paragraphs (eTables 1 and 2). Nevertheless, this overlap between quit-success and substance-dependence genes is more modest than the overlaps that we have previously identified in GWA studies of dependence on different addictive substances.18,19,22,23
Cadherin 13 (CDH13) (Entrez Gene 1012) is a glycosyl-phosphatidylinositol–anchored cell adhesion molecule identified herein and also in multiple comparisons between substance-dependent vs control individuals. The CDH13 gene is expressed in neurons in interesting brain regions,38 can inhibit neurite extension,39 and can activate several signaling pathways,40–43 rendering it a strong candidate for roles in brain mechanisms important for both developing and quitting addictions.
Neurexin 3 (NRXN3) (Entrez Gene 9369) is a single-transmembrane domain cell adhesion molecule that has also been identified in association- and linkage-based comparisons between controls and individuals dependent on legal and illegal substances.21,44 Localization of this molecule to presynaptic aspects of classical synapses45 and to interesting brain regions provides sites at which NRXN3 gene variants could affect addictive processes.
Down syndrome cell adhesion molecule (DSCAM) (Entrez Gene 1826) is a single-transmembrane domain cell adhesion molecule that is expressed strongly in the brain46 in ways that are required for appropriate neuronal connections to form in memory-associated circuits in model organisms.47,48 Altered DSCAM expression in fruit flies changes memories for rewarded and punished behaviors.48
Cyclic guanosine monophosphate (cGMP)–dependent protein kinase 1 (PRKG1) (Entrez Gene 5592) is expressed in hippocampal and other neurons.49,50 Nitric oxide dramatically modulates brain cGMP systems; PRKG1 thus provides a major target for the products of nitric oxide synthases. Mnemonic and addictive functions can each be altered by changes in cGMP-dependent protein kinase and/or nitric oxide synthases.51–53
The genes for triple functional domain/PTPRF interacting protein (Entrez Gene 7204), Cub and Sushi multiple domains 1 (Entrez Gene 64478), sarcoglycan zeta (Entrez Gene 137868), and receptor protein tyrosine phosphatase D (Entrez Gene 5789) are other cell adhesion genes that appear to contain variants that influence both vulnerability to addiction and success in quitting. Although the functions of none of these genes is well understood, each of these genes is highly expressed in the hippocampus (http://www.brain-map.org).
Several of the genes identified by both quit-success and addiction vulnerability GWA approaches are likely to alter protein disposition and half-lives. Peptidase activities are regulated by inhibitory proteins that include the products of serpin peptidase inhibitor A1 (Entrez Gene 5265) and A2 (Entrez Gene 390502) genes, which are identified by results from addiction vulnerability and smoking cessation GWA data. These genes are expressed in the cerebral cortex, hippocampus, and other brain regions of interest. Cellular protein trafficking is likely to be altered by variants in the SorCS1 sortilin-related Vps10p domain containing receptor 1, which is expressed in interesting brain regions that include the cortex and hippocampus.
Signaling within neurons is likely to be altered by variants in a number of the genes that are identified for both quit success and addiction vulnerability. Variants in the A kinase anchor protein 13 gene (AKAP13) (Entrez Gene 11214) are likely to alter cyclic adenosine monophosphate–related signaling pathways, whereas variants in the chimerin 2 gene (CHN2) (Entrez Gene 1124) are also likely to alter signaling. Both of these genes exhibit abundant brain expression in interesting regions.
The neuronal PAS domain protein 3 gene (NPAS3) (Entrez Gene 64067) is expressed in developing and adult brains in ways that suggest that its variants could play important roles.
The gene for ataxin 2–binding protein 1 (A2BP1) (Entrez Gene 54715) is highly expressed in neurons in brain regions that include the hippocampus (http://www.brain-map.org). The A2BP1 gene binds to a UGCAUG splicing enhancer element that borders a substantial number of neuron-specific exons and thus acts to regulate splicing processes that form mature messenger RNAs.54 The A2BP1 gene itself contains a number of splicing variants that are likely to alter its functions.
In addition to these genes that are identified in repeated comparisons between successful vs unsuccessful abstainers and in repeated comparisons between substance-dependent vs control individuals, we have also identified other interesting genes (and intragenic regions) in at least 2 samples of successful vs unsuccessful abstainers that are not consistently identified in comparisons of substance-dependent vs control individuals. These genes represent a large fraction of the total number of genes identified herein, consistent with classic genetic data that suggest modest overlap between the genetics of developing a dependence on tobacco and the genetics of successful cessation. Although detailed discussion of each of these genes is beyond the scope of the present report, the gene for calsyntenin 2 (CLSTN2) (Entrez Gene 64084) falls into this group and was also identified by recent association and linkage studies of individual differences in memory and executive function.30,55 The CLSTN2 gene is heavily expressed in brain regions that include the hippocampus (http://www.brain-map.org) and appears to function as a single trans-membrane domain cell adhesion molecule.
It is important to consider several limitations for these convergent, replicated genome-wide data for smoking cessation. The sample sizes available for this work provide moderate power to detect gene variants. False-negative results are likely, because we require positive data from each of 2 samples and also require positive results from several SNPs that cluster within small chromosomal regions. We focus only on data from autosomal regions herein, allowing us to combine data from male and female smokers in ways that will miss potentially important contributions from genes on sex chromosomes. These subjects volunteered for demanding clinical protocols, potentially rendering them not totally representative of all smokers or of all smokers who seek to quit.56 We used a preplanned analytic approach that represents only 1 of many current approaches to analyzing GWA data (the analysis and technical limits of GWA studies are discussed in the supplemental text available online and at http://www.nhlbi.nih.gov/resources/listserv/number35.htm [RFA-HL-07-010]).33,57 Despite these limitations, the replicated positive results obtained herein and the failure of control experiments to support alternative hypotheses provide substantial confidence in roles for most of the genes reported.
Comparing data from successful vs unsuccessful abstainers from those treated with bupropion, NRT, or placebo provides samples that allow us to ask whether molecular genetic results are consistent with the idea that each gene variant identified in this work provides identical influences on responses to NRT and bupropion. There is a pharmacological rationale for differential effects because bupropion’s relatively high affinity as a dopamine transporter blocker contrasts with nicotine’s relatively high affinity for nicotinic acetylcholine receptors58 (but bupropion also has moderate affinities for nicotinic acetylcholine receptors59). The results reported herein provide initial evidence that the genetics underlying successful smoking cessation with bupropion are not identical to the genetics of successful smoking cessation with NRT. There should be caution because the NRT- and bupropion-selective genes are identified in a single analysis that is not undergirded by twin study data. Nevertheless, these results (1) provide sets of clustered, nominally positive SNPs that converge with the SNPs where allele frequencies distinguish nondependent smokers from dependent smokers (Monte Carlo P < .001),23 and (2) identify the CYP2B7P1/CYP2B6 gene cluster in which cluster products are important for generating bupropion’s pharmacologically active metabolite hydroxybupropion.58 Replication of these data will allow use of such SNPs to individualize therapeutic approaches by selecting better candidates for NRT vs bupropion therapies and allow us to consider whether each gene’s effect is additive or might depend on allelic variants at other loci (epistasis).
The data reported herein provide molecular genetic support for the idea that a smoker’s ability to abstain from nicotine has polygenic genetic components. The data support modest overlap between these heritable components and the genes that contribute to vulnerability to dependence on addictive substances. Several of the genes identified in this work are consistent with emerging views of the importance of mnemonic mechanisms for the development of and remission from addictions.3 The convergent, replicated results presented herein are supplemented by initial data that nominate differential genetic influences on quit success depending on which smoking cessation treatment is used. Taken together, the current data provide promise that we may soon use molecular genetics to match the type and/or intensity of antismoking treatments with the smokers most likely to benefit from them.
Supplementary Material
Additional Information: The eTables and supplemental text are available at http://www.archgenpsychiatry.com.
Acknowledgments
Funding: This study was supported by the National Institutes of Health (NIH)–Intramural Research Program, National Institute on Drug Abuse, Department of Health and Social Services (Dr Uhl); NIH grants P50CA/DA84718 and RO1CA 63562 (Dr Lerman); NIH grants HL32318, DA08511, and P50CA84719 (Dr Niaura) and NIH grant 1K08 DA14276-05 (Dr David); the Pennsylvania Department of Health (Dr Lerman); and GlaxoSmithKline, Inc.
Footnotes
Financial Disclosure: Drs Uhl and Rose report possible proprietary interests of several authors in a provisional patent filed by Duke University related to the use of genetic markers to predict smoking cessation success. The patent application was filed after this work was accepted for publication. Dr Lerman reports no financial interest.
Disclaimer: The Pennsylvania Department of Health takes no responsibility for any of the analyses, interpretations, or conclusions of this study.
Additional Contributions: Leonard Epstein, PhD, Larry Hawk, PhD, Peter Shields, MD, Freda Patterson, PhD, Angela Pinto, BA, Margaret Rukstalis, MD, Wade Berrettini, MD, Rick Brown, PhD, Elizabeth Richardson, PhD, Frederique M. Behm, MD, Prity Kukovich, BA, Eric C. Westman, MD, and Greg Samsa, PhD, contributed to the design, execution, and/or analyses of the clinical trials that underpin this work.
References
- 1.Mokdad AH, Marks JS, Stroup DF, Gerberding JL. Actual causes of death in the United States, 2000. JAMA. 2004;291(10):1238–1245. doi: 10.1001/jama.291.10.1238. [DOI] [PubMed] [Google Scholar]
- 2.Warner KE, Burns DM. Hardening and the hard-core smoker: concepts, evidence, and implications. Nicotine Tob Res. 2003;5(1):37–48. doi: 10.1080/1462220021000060428. [DOI] [PubMed] [Google Scholar]
- 3.Uhl GR. Molecular genetics of addiction vulnerability. NeuroRx. 2006;3(3):295–301. doi: 10.1016/j.nurx.2006.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xian H, Scherrer JF, Madden PA, Lyons MJ, Tsuang M, True WR, Eisen SA. The heritability of failed smoking cessation and nicotine withdrawal in twins who smoked and attempted to quit. Nicotine Tob Res. 2003;5(2):245–254. [PubMed] [Google Scholar]
- 5.Karkowski LM, Prescott CA, Kendler KS. Multivariate assessment of factors influencing illicit substance use in twins from female-female pairs. Am J Med Genet. 2000;96(5):665–670. [PubMed] [Google Scholar]
- 6.Tsuang MT, Lyons MJ, Meyer JM, Doyle T, Eisen SA, Goldberg J, True W, Lin N, Toomey R, Eaves L. Co-occurrence of abuse of different drugs in men: the role of drug-specific and shared vulnerabilities. Arch Gen Psychiatry. 1998;55(11):967–972. doi: 10.1001/archpsyc.55.11.967. [DOI] [PubMed] [Google Scholar]
- 7.Uhl GR, Elmer GI, Labuda MC, Pickens RW. Genetic influences in drug abuse. In: Gloom FE, Kupfer DJ, editors. Psychopharmacology: The Fourth Generation of Progress. New York, NY: Raven Press; 1995. pp. 1793–2783. [Google Scholar]
- 8.True WR, Heath AC, Scherrer JF, Xian H, Lin N, Eisen SA, Lyons MJ, Goldberg J, Tsuang MT. Interrelationship of genetic and environmental influences on conduct disorder and alcohol and marijuana dependence symptoms. Am J Med Genet. 1999;88(4):391–397. doi: 10.1002/(sici)1096-8628(19990820)88:4<391::aid-ajmg17>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 9.Schnoll RA, Lerman C. Current and emerging pharmacotherapies for treating tobacco dependence. Expert Opin Emerg Drugs. 2006;11(3):429–444. doi: 10.1517/14728214.11.3.429. [DOI] [PubMed] [Google Scholar]
- 10.Cofta-Woerpel L, Wright KL, Wetter DW. Smoking cessation 1: pharmacological treatments. Behav Med. 2006;32(2):47–56. doi: 10.3200/BMED.32.2.47-56. [DOI] [PubMed] [Google Scholar]
- 11.Munafò MR, Shields AE, Berrettini WH, Patterson F, Lerman C. Pharmacogenetics and nicotine addiction treatment. Pharmacogenomics. 2005;6(3):211–223. doi: 10.1517/14622416.6.3.211. [DOI] [PubMed] [Google Scholar]
- 12.Munafò MR, Elliot KM, Murphy MF, Walton RT, Johnstone EC. Association of the muopioid receptor gene with smoking cessation. Pharmacogenomics J. 2007;7(5):353–361. doi: 10.1038/sj.tpj.6500432. [DOI] [PubMed] [Google Scholar]
- 13.Johnstone EC, Yudkin PL, Hey K, Roberts SJ, Welch SJ, Murphy MF, Griffiths SE, Walton RT. Genetic variation in dopaminergic pathways and short-term effectiveness of the nicotine patch. Pharmacogenetics. 2004;14(2):83–90. doi: 10.1097/00008571-200402000-00002. [DOI] [PubMed] [Google Scholar]
- 14.Yudkin P, Munafò M, Hey K, Roberts S, Welch S, Johnstone E, Murphy M, Griffiths S, Walton R. Effectiveness of nicotine patches in relation to genotype in women versus men: randomised controlled trial. BMJ. 2004;328(7446):989–990. doi: 10.1136/bmj.38050.674826.AE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Swan GE, Valdes AM, Ring HZ, Khroyan TV, Jack LM, Ton CC, Curry SJ, McAfee T. Dopamine receptor DRD2 genotype and smoking cessation outcome following treatment with bupropion SR. Pharmacogenomics J. 2005;5(1):21–29. doi: 10.1038/sj.tpj.6500281. [DOI] [PubMed] [Google Scholar]
- 16.Lerman C, Jepson C, Wileyto EP, Epstein LH, Rukstalis M, Patterson F, Kaufmann V, Restine S, Hawk L, Niaura R, Berrettini W. Role of functional genetic variation in the dopamine D2 receptor (DRD2) in response to bupropion and nicotine replacement therapy for tobacco dependence: results of two randomized clinical trials. Neuropsychopharmacology. 2006;31(1):231–242. doi: 10.1038/sj.npp.1300861. [DOI] [PubMed] [Google Scholar]
- 17.Lerman C, Wileyto EP, Patterson F, Rukstalis M, Audrain-McGovern J, Restine S, Shields PG, Kaufmann V, Redden D, Benowitz N, Berrettini WH. The functional mu opioid receptor (OPRM1) Asn40Asp variant predicts short-term response to nicotine replacement therapy in a clinical trial. Pharmacogenomics J. 2004;4(3):184–192. doi: 10.1038/sj.tpj.6500238. [DOI] [PubMed] [Google Scholar]
- 18.Liu QR, Drgon T, Johnson C, Walther D, Hess J, Uhl GR. Addiction molecular genetics: 639,401 SNP whole genome association identifies many “cell adhesion” genes. Am J Med Genet B Neuropsychiatr Genet. 2006;141(8):918–925. doi: 10.1002/ajmg.b.30436. [DOI] [PubMed] [Google Scholar]
- 19.Uhl GR, Liu QR, Drgon T, Johnson C, Walther D, Rose JE. Molecular genetics of nicotine dependence and abstinence: whole genome association using 520,000 SNPs. BMC Genet. 2007;8(1):10. doi: 10.1186/1471-2156-8-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Uhl GR, Liu QR, Walther D, Hess J, Naiman D. Polysubstance abuse-vulnerability genes: genome scans for association, using 1004 subjects and 1494 single-nucleotide polymorphisms. Am J Hum Genet. 2001;69(6):1290–1300. doi: 10.1086/324467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu QR, Drgon T, Walther D, Johnson C, Poleskaya O, Hess J, Uhl GR. Pooled association genome scanning: validation and use to identify addiction vulnerability loci in two samples. Proc Natl Acad Sci U S A. 2005;102(33):11864–11869. doi: 10.1073/pnas.0500329102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Johnson C, Drgon T, Liu QR, Walther D, Edenberg H, Rice J, Foroud T, Uhl GR. Pooled association genome scanning for alcohol dependence using 104,268 SNPs: validation and use to identify alcoholism vulnerability loci in unrelated individuals from the collaborative study on the genetics of alcoholism. Am J Med Genet B Neuropsychiatr Genet. 2006;141(8):844–853. doi: 10.1002/ajmg.b.30346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bierut LJ, Madden PA, Breslau N, Johnson EO, Hatsukami D, Pomerleau OF, Swan GE, Rutter J, Bertelsen S, Fox L, Fugman D, Goate AM, Hinrichs AL, Konvicka K, Martin NG, Montgomery GW, Saccone NL, Saccone SF, Wang JC, Chase GA, Rice JP, Ballinger DG. Novel genes identified in a high-density genome wide association study for nicotine dependence. Hum Mol Genet. 2007;16(1):24–35. doi: 10.1093/hmg/ddl441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.David SP, Brown RA, Papandonatos GD, Kahler CW, Lloyd-Richardson EE, Munafò MR, Shields PG, Lerman C, Strong D, McCaffery J, Niaura R. Pharmacogenetic clinical trial of sustained-release bupropion for smoking cessation. Nicotine Tob Res. 2007;9(8):821–833. doi: 10.1080/14622200701382033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brown RA, Burgess ES, Sales SD, Whiteley JA, Evans DM, Miller IW. Reliability and validity of a smoking timeline follow-back interview. Psychol Addict Behav. 1998;12(12):101–112. [Google Scholar]
- 26.Rose JE, Behm FM, Westman EC. Nicotine-mecamylamine treatment for smoking cessation: the role of pre-cessation therapy. Exp Clin Psychopharmacol. 1998;6(3):331–343. doi: 10.1037//1064-1297.6.3.331. [DOI] [PubMed] [Google Scholar]
- 27.Bang-Ce Y, Peng Z, Bincheng Y, Songyang L. Estimation of relative allele frequencies of single-nucleotide polymorphisms in different populations by micro-array hybridization of pooled DNA. Anal Biochem. 2004;333(1):72–78. doi: 10.1016/j.ab.2004.05.016. [DOI] [PubMed] [Google Scholar]
- 28.Sham P, Bader JS, Craig I, O’Donovan M, Owen M. DNA pooling: a tool for large-scale association studies. Nat Rev Genet. 2002;3(11):862–871. doi: 10.1038/nrg930. [DOI] [PubMed] [Google Scholar]
- 29.Hinds DA, Seymour AB, Durham LK, Banerjee P, Ballinger DG, Milos PM, Cox DR, Thompson JF, Frazer KA. Application of pooled genotyping to scan candidate regions for association with HDL cholesterol levels. Hum Genomics. 2004;1(6):421–434. doi: 10.1186/1479-7364-1-6-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu F, Arias-Vásquez A, Sleegers K, Aulchenko YS, Kayser M, Sanchez-Juan P, Feng B-J, Bertoli-Avella AM, van Swieten J, Axenovich TI, Heutink P, van Broeckhoven C, Oostra BA, van Duijn CM. A genomewide screen for late-onset Alzheimer disease in a genetically isolated Dutch population. Am J Hum Genet. 2007;81(1):17–31. doi: 10.1086/518720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Papassotiropoulos A, Stephan DA, Huentelman MJ, Hoerndli FJ, Craig DW, Pearson JV, Huynh KD, Brunner F, Corneveaux J, Osborne D, Wollmer MA, Aerni A, Coluccia D, Hanggi J, Mondadori CR, Buchmann A, Reiman EM, Caselli RJ, Henke K, de Quervain DJ. Common Kibra alleles are associated with human memory performance. Science. 2006;314(5798):475–478. doi: 10.1126/science.1129837. [DOI] [PubMed] [Google Scholar]
- 32.Dupont WD, Plummer WD., Jr Power and sample size calculations: a review and computer program. Control Clin Trials. 1990;11(2):116–128. doi: 10.1016/0197-2456(90)90005-m. [DOI] [PubMed] [Google Scholar]
- 33.Wellcome Trust Case Control Consortium. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447(7145):661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Swan GE, Carmelli D, Rosenman RH, Fabsitz RR, Christian JC. Smoking and alcohol consumption in adult male twins: genetic heritability and shared environmental influences. J Subst Abuse. 1990;2(1):39–50. doi: 10.1016/s0899-3289(05)80044-6. [DOI] [PubMed] [Google Scholar]
- 35.Broms U, Silventoinen K, Madden PA, Heath AC, Kaprio J. Genetic architecture of smoking behavior: a study of Finnish adult twins. Twin Res Hum Genet. 2006;9(1):64–72. doi: 10.1375/183242706776403046. [DOI] [PubMed] [Google Scholar]
- 36.Maes HH, Sullivan PF, Bulik CM, Neale MC, Prescott CA, Eaves LJ, Kendler KS. A twin study of genetic and environmental influences on tobacco initiation, regular tobacco use and nicotine dependence. Psychol Med. 2004;34(7):1251–1261. doi: 10.1017/s0033291704002405. [DOI] [PubMed] [Google Scholar]
- 37.Carmelli D, Swan GE, Robinette D, Fabsitz RR. Heritability of substance use in the NAS-NRC Twin Registry. Acta Genet Med Gemellol (Roma) 1990;39(1):91–98. doi: 10.1017/s0001566000005602. [DOI] [PubMed] [Google Scholar]
- 38.Takeuchi T, Misaki A, Liang SB, Tachibana A, Hayashi N, Sonobe H, Ohtsuki Y. Expression of T-cadherin (CDH13, H-cadherin) in human brain and its characteristics as a negative growth regulator of epidermal growth factor in neuroblastoma cells. J Neurochem. 2000;74(4):1489–1497. doi: 10.1046/j.1471-4159.2000.0741489.x. [DOI] [PubMed] [Google Scholar]
- 39.Fredette BJ, Miller J, Ranscht B. Inhibition of motor axon growth by T-cadherin substrata. Development. 1996;122(10):3163–3171. doi: 10.1242/dev.122.10.3163. [DOI] [PubMed] [Google Scholar]
- 40.Kipmen-Korgun D, Osibow K, Zoratti C, Schraml E, Greilberger J, Kostner GM, Jurgens G, Graier WF. T-cadherin mediates low-density lipoprotein-initiated cell proliferation via the Ca(2+)-tyrosine kinase-Erk1/2 pathway. J Cardiovasc Pharmacol. 2005;45(5):418–430. doi: 10.1097/01.fjc.0000157458.91433.86. [DOI] [PubMed] [Google Scholar]
- 41.Philippova M, Ivanov D, Allenspach R, Takuwa Y, Erne P, Resink T. RhoA and Rac mediate endothelial cell polarization and detachment induced by T-cadherin. FASEB J. 2005;19(6):588–590. doi: 10.1096/fj.04-2430fje. [DOI] [PubMed] [Google Scholar]
- 42.Hug C, Wang J, Ahmad NS, Bogan JS, Tsao TS, Lodish HF. T-cadherin is a receptor for hexameric and high-molecular-weight forms of Acrp30/adiponectin. Proc Natl Acad Sci U S A. 2004;101(28):10308–10313. doi: 10.1073/pnas.0403382101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ivanov DB, Philippova MP, Tkachuk VA. Structure and functions of classical cadherins. Biochemistry (Mosc) 2001;66(10):1174–1186. doi: 10.1023/a:1012445316415. [DOI] [PubMed] [Google Scholar]
- 44.Lachman HM, Fann CS, Bartzis M, Evgrafov OV, Rosenthal RN, Nunes EV, Miner C, Santana M, Gaffney J, Riddick A, Hsu CL, Knowles JA. Genomewide suggestive linkage of opioid dependence to chromosome 14q. Hum Mol Genet. 2007;16(11):1327–1334. doi: 10.1093/hmg/ddm081. [DOI] [PubMed] [Google Scholar]
- 45.Dean C, Scholl FG, Choih J, DeMaria S, Berger J, Isacoff E, Scheiffele P. Neurexin mediates the assembly of presynaptic terminals. Nat Neurosci. 2003;6(7):708–716. doi: 10.1038/nn1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yamakawa K, Huot YK, Haendelt MA, Hubert R, Chen XN, Lyons GE, Korenberg JR. DSCAM: a novel member of the immunoglobulin superfamily maps in a Down syndrome region and is involved in the development of the nervous system. Hum Mol Genet. 1998;7(2):227–237. doi: 10.1093/hmg/7.2.227. [DOI] [PubMed] [Google Scholar]
- 47.Chen BE, Kondo M, Garnier A, Watson FL, Puettmann-Holgado R, Lamar DR, Schmucker D. The molecular diversity of DSCAM is functionally required for neuronal wiring specificity in Drosophila. Cell. 2006;125(3):607–620. doi: 10.1016/j.cell.2006.03.034. [DOI] [PubMed] [Google Scholar]
- 48.Keene AC, Krashes MJ, Leung B, Bernard JA, Waddell S. Drosophila dorsal paired medial neurons provide a general mechanism for memory consolidation. Curr Biol. 2006;16(15):1524–1530. doi: 10.1016/j.cub.2006.06.022. [DOI] [PubMed] [Google Scholar]
- 49.Feil S, Zimmermann P, Knorn A, Brummer S, Schlossmann J, Hofmann F, Feil R. Distribution of cGMP-dependent protein kinase type I and its isoforms in the mouse brain and retina. Neuroscience. 2005;135(3):863–868. doi: 10.1016/j.neuroscience.2005.06.051. [DOI] [PubMed] [Google Scholar]
- 50.Demyanenko GP, Halberstadt AI, Pryzwansky KB, Werner C, Hofmann F, Maness PF. Abnormal neocortical development in mice lacking cGMP-dependent protein kinase I. Brain Res Dev Brain Res. 2005;160(1):1–8. doi: 10.1016/j.devbrainres.2005.07.013. [DOI] [PubMed] [Google Scholar]
- 51.Weitzdoerfer R, Hoeger H, Engidawork E, Engelmann M, Singewald N, Lubec G, Lubec B. Neuronal nitric oxide synthase knock-out mice show impaired cognitive performance. Nitric Oxide. 2004;10(3):130–140. doi: 10.1016/j.niox.2004.03.007. [DOI] [PubMed] [Google Scholar]
- 52.Itzhak Y, Martin JL, Black MD, Huang PL. The role of neuronal nitric oxide synthase in cocaine-induced conditioned place preference. Neuroreport. 1998;9(11):2485–2488. doi: 10.1097/00001756-199808030-00011. [DOI] [PubMed] [Google Scholar]
- 53.Kleppisch T, Wolfsgruber W, Feil S, Allmann R, Wotjak CT, Goebbels S, Nave KA, Hofmann F, Feil R. Hippocampal cGMP-dependent protein kinase I supports an age- and protein synthesis–dependent component of long-term potentiation but is not essential for spatial reference and contextual memory. J Neurosci. 2003;23(14):6005–6012. doi: 10.1523/JNEUROSCI.23-14-06005.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Underwood JG, Boutz PL, Dougherty JD, Stoilov P, Black DL. Homologues of the Caenorhabditis elegans Fox-1 protein are neuronal splicing regulators in mammals. Mol Cell Biol. 2005;25(22):10005–10016. doi: 10.1128/MCB.25.22.10005-10016.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Koob GF. The neurobiology of addiction: a neuroadaptational view relevant for diagnosis. Addiction. 2006;101(suppl 1):23–30. doi: 10.1111/j.1360-0443.2006.01586.x. [DOI] [PubMed] [Google Scholar]
- 56.Hughes JR, Giovino GA, Klevens RM, Fiore MC. Assessing the generalizability of smoking studies. Addiction. 1997;92(4):469–472. [PubMed] [Google Scholar]
- 57.Dudbridge F, Gusnanto A, Koeleman BP. Detecting multiple associations in genome-wide studies. Hum Genomics. 2006;2(5):310–317. doi: 10.1186/1479-7364-2-5-310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Damaj MI, Carroll FI, Eaton JB, Navarro HA, Blough BE, Mirza S, Lukas RJ, Martin BR. Enantioselective effects of hydroxy metabolites of bupropion on behavior and on function of monoamine transporters and nicotinic receptors. Mol Pharmacol. 2004;66(3):675–682. doi: 10.1124/mol.104.001313. [DOI] [PubMed] [Google Scholar]
- 59.Ascher JA, Cole JO, Colin JN, Feighner JP, Ferris RM, Fibiger HC, Golden RN, Martin P, Potter WZ, Richelson E, Sulser F. Bupropion: a review of its mechanism of antidepressant activity. J Clin Psychiatry. 1995;56(9):395–401. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Information: The eTables and supplemental text are available at http://www.archgenpsychiatry.com.