

Abstract
Vasoconstrictors that bind to phospholipase C–coupled receptors elevate inositol-1,4,5-trisphosphate (IP3). IP3 is generally considered to elevate intracellular Ca2+ concentration ([Ca2+]i) in arterial myocytes and induce vasoconstriction via a single mechanism: by activating sarcoplasmic reticulum (SR)-localized IP3 receptors, leading to intracellular Ca2+ release. We show that IP3 also stimulates vasoconstriction via a SR Ca2+ release–independent mechanism. In isolated cerebral artery myocytes and arteries in which SR Ca2+ was depleted to abolish Ca2+ release (measured using D1ER, a fluorescence resonance energy transfer–based SR Ca2+ indicator), IP3 activated 15 pS sarcolemmal cation channels, generated a whole-cell cation current (ICat) caused by Na+ influx, induced membrane depolarization, elevated [Ca2+]i, and stimulated vasoconstriction. The IP3-induced ICat and [Ca2+]i elevation were attenuated by cation channel (Gd3+, 2-APB) and IP3 receptor (xestospongin C, heparin, 2-APB) blockers. TRPC3 (canonical transient receptor potential 3) channel knockdown with short hairpin RNA and diltiazem and nimodipine, voltage-dependent Ca2+ channel blockers, reduced the SR Ca2+ release–independent, IP3-induced [Ca2+]i elevation and vasoconstriction. In pressurized arteries, SR Ca2+ depletion did not alter IP3-induced constriction at 20 mm Hg but reduced IP3-induced constriction by ≈39% at 60 mm Hg. [Ca2+]i elevations and constrictions induced by endothelin-1, a phospholipase C–coupled receptor agonist, were both attenuated by TRPC3 knockdown and xestospongin C in SR Ca2+-depleted arteries. In summary, we describe a novel mechanism of IP3-induced vasoconstriction that does not occur as a result of SR Ca2+ release but because of IP3 receptor–dependent ICat activation that requires TRPC3 channels. The resulting membrane depolarization activates voltage-dependent Ca2+ channels, leading to a myocyte [Ca2+]i elevation, and vasoconstriction.
Keywords: vascular smooth muscle, voltage-dependent calcium channels, TRPC channels, endothelin-1
Many vasoconstrictors bind to phospholipase (PL)C-coupled receptors, leading to an elevation in intracellular diacylglycerol (DAG) and inositol-1,4,5,-trisphosphate (IP3). DAG directly modulates ion channels, including transient receptor potential (TRP) channels, and activates protein kinase (PK)C, which can phosphorylate a wide variety of proteins that regulate arterial myocyte contractility, including ion channels.1,2 In contrast, IP3 binds to IP3 receptors (IP3Rs) located on the sarcoplasmic reticulum (SR), leading to SR Ca2+ release and an increase in myocyte [Ca2+]i.1 The resulting activation of Ca2+/calmodulin-dependent myosin light chain kinase stimulates vasoconstriction.
The mechanism by which IP3 regulates arterial contractility is generally well accepted.1 Indeed, IP3-induced SR Ca2+ release is considered to be the only mechanism by which this second messenger regulates arterial diameter. However, the physiological mechanisms by which IP3 regulates intracellular Ca2+ signaling and arterial diameter are poorly understood, and few studies have directly tested the accepted view. Arterial contractility regulation by IP3 has primarily been studied by using vasoconstrictors that activate PLC. Because PLC activation elevates both DAG and IP3 and reduces PIP2, mechanisms by which IP3 specifically modulates arterial [Ca2+]i signaling and diameter require additional study.
Here, we investigated IP3 regulation of ion channel activity, intracellular Ca2+ signaling, and contractility in cerebral artery myocytes and pressurized arteries. We show that IP3 activates a nonselective cation current (ICat) in myocytes and induces vasoconstriction via a mechanism that does not require the release of SR Ca2+ but involves IP3R and TRPC3 (canonical transient receptor potential 3) channel activation. IP3-induced Na+ influx produces membrane depolarization, voltage-dependent Ca2+ channel activation, an [Ca2+]i elevation, and vasoconstriction. We also show that TRPC3 channel activation is required for the [Ca2+]i elevation and vasoconstriction induced by endothelin (ET)-1, a PLC-coupled receptor agonist, and that IP3R activation contributes to these responses in SR Ca2+-depleted arteries. These data indicate that IP3R activation can stimulate vascular contraction through a mechanism that is independent of SR Ca2+ release.
Materials and Methods
Tissue Preparation
Animal procedures used were approved by the Animal Care and Use Committee at the University of Tennessee. Sprague–Dawley rats (200 to 250g) of either sex were euthanized, and the brains were removed. Posterior cerebral, cerebellar, and middle cerebral arteries were harvested and used for all measurements. Myocytes were isolated as previously described,3 maintained at 4°C, and used for experimentation within 8 hours.
Adenovirus Construction
cDNA encoding D1ER, a fluorescence resonance energy transfer–based Ca2+ indicator protein that locates to the endoplasmic reticulum lumen, was kindly provided by Dr R.Y. Tsien (University of California, San Diego).4 A recombinant adenovirus expressing D1ER (adenD1ER) was constructed, as previously described.5 DMEM containing adenD1ER was inserted into the lumen of isolated endothelium-denuded cerebral artery segments that were placed in serum-free DMEM supplemented with 1% penicillin/streptomycin and incubated at 37°C (95% O2, 5% CO2) for 4 days before experimentation.
Cytosolic and SR Ca2+ Imaging
Cytosolic [Ca2+]i was measured in isolated myocytes and endothelium-denuded arterial segments using fura-2. Myocytes were imaged using a charge-coupled device camera (Dage-MTI), and arteries were measured using a photomultiplier tube and Ionwizard software (Ionoptix, Milton, Mass), as previously described.6,7 D1ER fluorescence was measured in myocytes of endothelium-denuded cerebral artery segments using a CoolSNAPfx charge-coupled device camera (Photometrics) attached to a Nikon TE300 microscope. D1ER was excited with 436±10 nm light. Emitted light was alternately collected at 480±20 nm (donor, cyan fluorescent protein) and 535±15 nm (acceptor, yellow fluorescent protein), and yellow fluorescent protein/cyan fluorescent protein ratios were calculated every 20 seconds using MetaMorph 6.1 (Universal Imaging Corp).
Patch-Clamp Electrophysiology
Membrane currents were measured using the patch-clamp technique (Axopatch 200B, Clampex 8.2). For perforated-patch recordings, pipette solution contained (in mmol/L): 140 CsCl, 3 MgCl2, 0.1 EGTA, 10 Hepes, and 10 glucose (pH 7.2). Bath solution for perforated-patch and conventional whole-cell experiments contained (in mmol/L): 140 NaCl, 1.8 CaCl2, 1.2 MgCl2, 10 Hepes, and 10 glucose (pH 7.4). For conventional whole-cell, pipette solution contained (in mmol/L): 140 CsCl, 10 Hepes, 10 glucose, 5 Mg-ATP, and 5 EGTA (pH 7.2). Caged IP3 was introduced via the pipette solution, and IP3 was photoreleased using six 1-ms UV flashes, each applied 3 seconds apart, that were transmitted through the microscope objective (Cairn Research). For inside-out cation channel recordings, the conventional whole-cell bath solution was used as the pipette solution and the whole-cell pipette solution was used as the bath solution (compositions above). For inside-out KCa channel recordings, bath solution contained (in mmol/L): 140 KCl, 1.6 hydroxyethylethylenediaminetriacetic acid, 1 EGTA, 2 MgCl2, 10 Hepes, and free [Ca2+] adjusted to 3 μmol/L as previously described (pH 7.2).8 For cell-attached experiments, the pipette solution was the whole-cell bath solution described above and the bath solution contained (in mmol/L): 140 KCl, 1.8 CaCl2, 1.2 MgCl2, 10 Hepes, and 10 glucose (pH 7.4). Where appropriate, NaCl was substituted with equimolar N-methyl-D-glucamine-Cl. Whole-cell currents were measured by applying 940-ms voltage ramps between −120 and +20 mV. Single channel currents were measured at a steady voltage of −60 mV. Single channel activity (NPo) for each condition in each experiment was calculated by analyzing 5 seconds of continuous data by using pClamp 9 (Axon Instruments).
Pressurized Artery Membrane Potential Measurements
The membrane potential of pressurized artery segments was measured as previously described.7
Pressurized Artery Diameter Measurements
Pressurized artery diameter was measured using an arteriograph, as previously described.7
Short Hairpin RNA Silencing Vector Construction
Using pRNA-U6.1/Neo as a template, silencing vectors were constructed to express short hairpin (sh)RNA (GenScript Corp, Piscataway, NJ). shRNA expressed by each vector is processed by Dicer to generate siRNA that specifically targets exon 6 in TRPC3 (TRPC3shV) or a scrambled sequence (TRPC3scrm).9 Expressed DNA sequences were as follows: for TRPC3shV, GTTCATACTTTACTCCTACTA; and for TRPC3scrm, TGAACATCAGTGCTAGGTTAC.
Reverse Permeabilization
Silencing vectors were inserted intracellularly into cerebral artery segments using a reverse permeabilization procedure previously described.10,11 Arteries were placed into serum-free DMEM supplemented with 1% penicillin/streptomycin and incubated at 37°C (95% O2, 5% CO2) for 4 days.
Western Immunoblot Analysis
Experiments were performed and data quantified as previously described.7,8 TRPC3 and TRPC6 channel protein bands were first normalized to actin and relative change in expression calculated as: (TRPC3shV/actin)/(TRPC3scrm/actin).
Statistical Analysis
Data are presented as means±SE. Statistical significance was calculated by using Student’s t tests for paired or unpaired data or ANOVA, followed by Student–Newman–Keuls test for multiple comparisons. P<0.05 was considered significant.
An expanded Materials and Methods section is available in the online data supplement at http://circres.ahajournals.org.
Results
IP3 Elevates [Ca2+]i in Myocytes With Intact and Depleted SR Ca2+
In isolated myocytes, from a resting [Ca2+]i of 114±7 nmol/L, Bt-IP3 (10 μmol/L), a membrane-permeant IP3 analog, caused a sustained, mean [Ca2+]i elevation of ≈111 nmol/L (Figure 1A and 1B). Bt-IP3–induced [Ca2+]i elevations were attenuated by Gd3+ (30 μmol/L), a nonselective cation channel blocker; 2-APB (100 μmol/L), a nonselective cation channel and IP3R inhibitor; and diltiazem (50 μmol/L), a voltage-dependent Ca2+ channel blocker (Figure 1B). A combination of 2-APB (100 μmol/L) and diltiazem (50 μmol/L) blocked Bt-IP3–induced [Ca2+]i elevations (Figure 1B). These data suggest that both SR Ca2+ release and sarcolemmal Ca2+ influx contribute to IP3-induced [Ca2+]i elevations in arterial myocytes.
Figure 1.
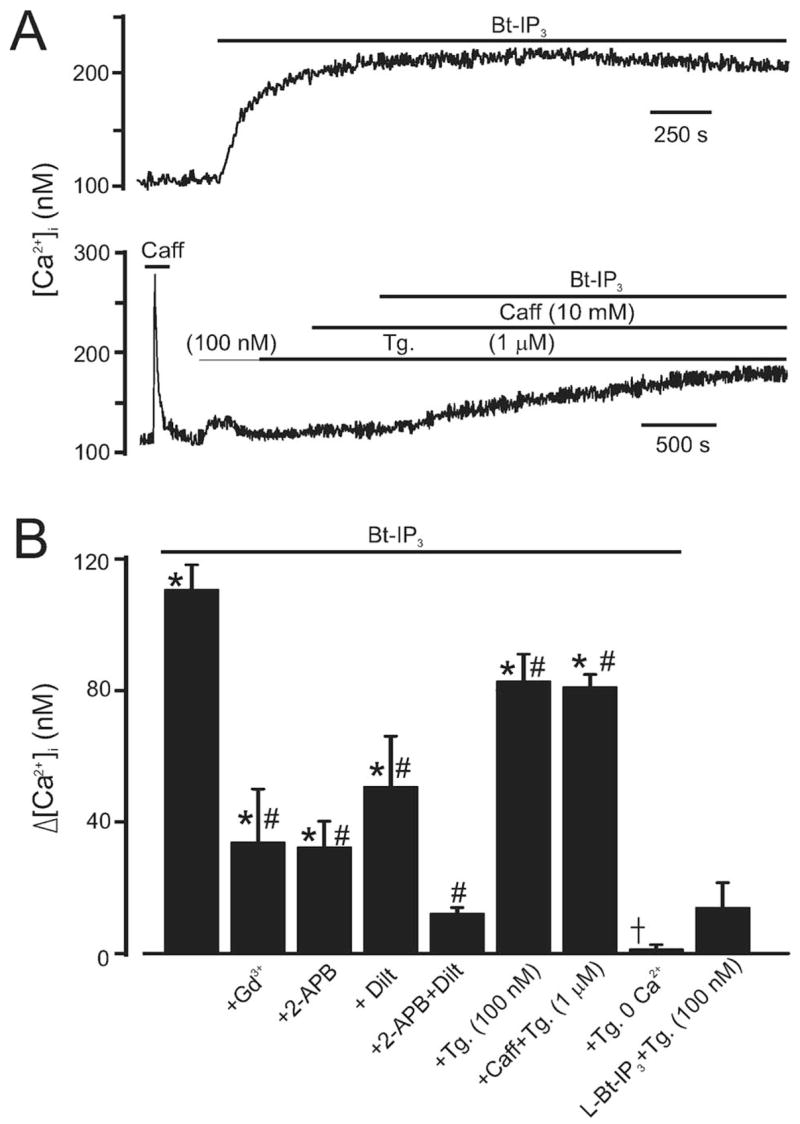
Bt-IP3 elevates [Ca2+]i in arterial myocytes with intact and depleted SR Ca2+. A, Original traces illustrating [Ca2+]i regulation by Bt-IP3 (10 μmol/L) in myocytes with intact (top) and depleted (bottom) SR Ca2+. B, Mean data. Control [Ca2+]i was 114±7 nmol/L (n=53). *P<0.05 compared with control in the same cell, #P<0.05 compared with Bt-IP3, †P<0.05 compared with Bt-IP3+thapsigargin (Tg) (100 nmol/L). The experimental numbers for bars from left to right were 6, 5, 4, 5, 5, 7, 9, 5, and 4, respectively.
Because IP3 is generally considered to elevate myocyte [Ca2+]i by mobilizing SR Ca2+, we studied the contribution of SR Ca2+ release to IP3-induced [Ca ]i elevations. Thapsigargin (100 nmol/L), a SR Ca2+-ATPase blocker, or a combination of thapsigargin (1 μmol/L) plus caffeine (10 mmol/L), a ryanodine receptor channel activator, reduced Bt-IP3–induced [Ca2+]i elevations by ≈25% (Figure 1A). In contrast, Bt-IP3 (10 μmol/L) did not change [Ca2+]i when applied in the absence of extracellular Ca2+ in thapsigargin-pretreated (100 nmol/L; >15 minutes) myocytes (Figure 1B). In the presence of Bt-IP3 (10 μmol/L) plus thapsigargin (100 nmol/L), CCCP (1 μmol/L), a protonophore that depolarizes mitochondria,6,12 further increased [Ca2+]i by 45±6 nmol/L (n=7, P<0.05), indicating that mitochondria attenuate rather than contribute to the IP3-induced [Ca2+]i elevation. L-Bt-IP3, a membrane permeant weak IP3R agonist, did not alter [Ca2+]i (Figure 1B). These data suggest that IP3 elevates [Ca ]i in arterial myocytes through a SR Ca2+ release–independent mechanism that requires plasma membrane Ca2+ influx.
Thapsigargin (100 nmol/L) caused a transient [Ca2+]i elevation of 45±5 nmol/L, which declined to a steady-state elevation of 6±3 nmol/L over prethapsigargin [Ca2+]i in 14.0±1.5 minutes (n=7, Figure 1B, P<0.05 for each). An elevation in thapsigargin concentration from 100 nmol/L to 1 μmol/L did not further change [Ca2+]i, consistent with 100 nmol/L thapsigargin causing SR Ca2+ depletion (Figure 1A, n=7, P<0.05). In agreement, thapsigargin (100 nmol/L to 1 μmol/L) abolished caffeine-induced (10 mmol/L) [Ca2+]i transients (Figure 1A and Figure I in the online data supplement).
To determine whether IP3 elevated [Ca2+]i by releasing a Ca2+ store that was not depleted by SR Ca2+-ATPase inhibition or ryanodine receptor channel activation, we measured [Ca2+] within the entire SR using D1ER, a fluorescence resonance energy transfer–based Ca2+indicator that targets to the endoplasmic reticulum lumen.4 This methodology also provides a time course of SR Ca2+ depletion. Thapsigargin (100 nmol/L) reduced D1ER mean fluorescence ratio to a plateau of 0.87±0.04 within 15 minutes, consistent with a reduction in SR Ca2+ concentration ([Ca2+]SR, n=6, P<0.05, Figure 2). Coapplication of Bt-IP3 (10 μmol/L) with thapsigargin (100 nmol/L), or an elevation in thapsigargin concentration to 5 μmol/L, did not further change D1ER ratio (to 0.88±0.04 and 0.85±0.04, respectively, P<0.05 for each, n=6, Figure 2). Collectively, these data indicate that, in cerebral artery myocytes, a 15-minute application of 100 nmol/L thapsigargin depletes Ca2+ in the entire SR; that IP3 does not further reduce [Ca2+]SR when applied in the presence of 100 nmol/L thapsigargin; and that IP3 elevates [Ca2+]i in arterial myocytes via a SR Ca2+ release-independent mechanism.
Figure 2.
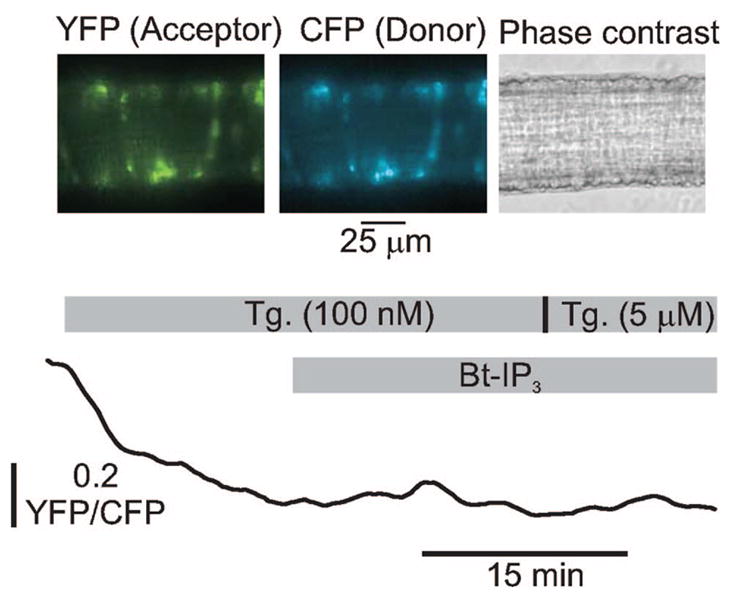
Thapsigargin (100 nmol/L to 5 μmol/L) depletes myocyte SR Ca2+. Images illustrate D1ER fluorescence in myocytes of a cerebral artery segment. Original trace is shown illustrating the time course of normalized D1ER yellow fluorescent protein (YFP)/cyan fluorescent protein (CFP) ratio change in the same artery in response to thapsigargin and Bt-IP3 (10 μmol/L). Data are representative of 6 separate experiments.
IP3 Activates a ICat in Myocytes With Intact and Depleted SR Ca2+
To investigate mechanisms by which IP3 elevates [Ca2+]i in myocytes, we measured plasma membrane ICat. When using the perforated patch-clamp configuration, Bt-IP3 (10 μmol/L) increased mean whole-cell ICat amplitude from ≈28 to 120 pA, or ≈4.3-fold at −120 mV (Figure 3B). Bt-IP3 similarly activated ICat when applied to myocytes pretreated with thapsigargin (Figure 3A and 3B). ICat regulation by IP3 or photolytic release of intracellular caged IP3 (both introduced via the pipette solution) was also studied in thapsigargin-treated myocytes by using the conventional whole-cell configuration, with [Ca2+]i strongly buffered using EGTA. IP3 and release of caged IP3 increased ICat≈2.6- to 3.4-fold (Figure 3B). Gd3+ reversed ICat activation by Bt-IP3 and caged IP3 (Figure 3B). In contrast, thapsigargin (100 nmol/L to 5 μmol/L) did not alter ICat (Figure 3B). These data indicate that IP3 activates a ICat in myocytes with either intact or depleted SR Ca2+.
Figure 3.
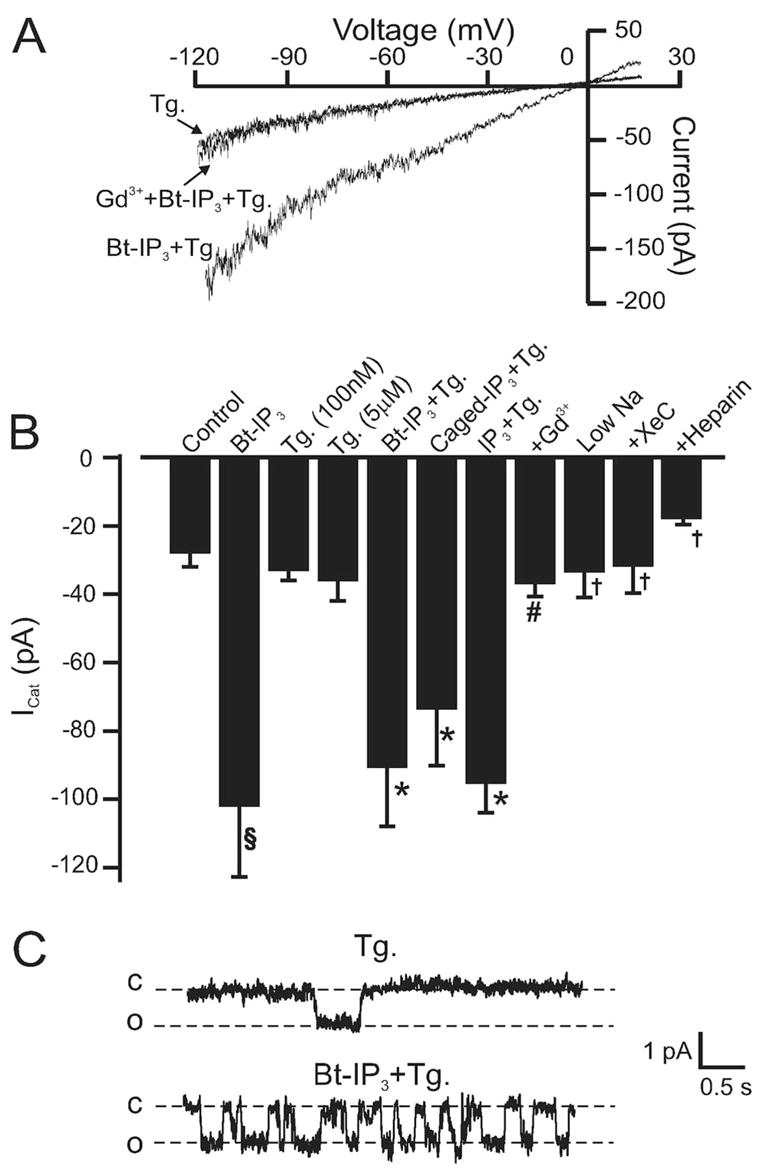
IP3 activates a Na+-permeant ICat as a result of IP3R activation in myocytes with intact and depleted SR Ca2+. A, Original recordings obtained using the perforated-patch configuration illustrating that Bt-IP3 (10 μmol/L) activates a Gd3+-sensitive (30 μmol/L) whole-cell ICat in a myocyte with depleted SR Ca2+. B, Mean data: control perforated-patch (p-p), n=9; Bt-IP3 (10 μmol/L) p-p, n=9; thapsigargin (Tg) (100 nmol/L; p-p [n=12] and conventional whole-cell [w-c] [n=9] were statistically similar and pooled, giving n=21; Tg [5 μmol/L] p-p, n=4; Bt-IP3+Tg [100 nmol/L] p-p, n=5; caged IP3 [2 μmol/L]+ Tg [100 nmol/L] w-c, n=14; IP3 [20 μmol/L]+ Tg [100 nmol/L] w-c, n=17; +Gd3+ [30 μmol/L] [n=5] represents Bt-IP3 [10 μmol/L, n=2] and caged IP3 [2 μmol/L, n=3], which were statistically similar and pooled; IP3 [20 μmol/L]+ Tg [100 nmol/L] with 20 mmol/L bath Na+ w-c, n=5; IP3 [20 μmol/L]+ Tg [100 nmol/L]+ XeC [20 μmol/L, via pipette], w-c, n=6; IP3 [20 μmol/L]+ Tg [100 nmol/L]+ heparin [1 mg/mL via pipette], w-c, n=7). Bt-IP3 shifted the reversal potential (Erev) from −9±1 to −5±0.4 mV. C, Bt-IP3 (10 μmol/L) activates single channel currents in a myocyte (cell-attached configuration) with depleted SR Ca2+ (Tg 100 nmol/L) at −60 mV. c indicates closed; o, open. §P<0.05 compared with control, *P<0.05 compared with Tg, #P<0.05 compared with Bt-IP3 and IP3, †P<0.05 compared with IP3.
The cell-attached and excised inside-out patch-clamp configurations were used to study IP3-activated single channel currents in myocytes with depleted SR Ca2+ (100 nmol/L thapsigargin; >15 minutes). In 8 of 9 cell-attached patches at −60 mV, Bt-IP3 increased the mean activity (NPo) of 0.91±0.13 pA single channels from 0.10±0.02 to 0.75±0.22 (Figure 3C). In inside-out patch-clamp experiments, we ensured that the internal membrane surface was accessible to the bath solution by confirming that KCa channels observed in a K+-containing bath solution were inhibited on switching to the CsCl-containing bath solution used to measure cation channels. In inside-out membrane patches at −60 mV, IP3 (20 μmol/L, n=27), oleoyl acetyl glycerol (OAG) (10 μmol/L, n=8), or IP3 (20 μmol/L) plus OAG (10 μmol/L, n=8) did not activate single channels. Therefore, these data suggest that an intact myocyte is required for IP3-induced cation channel activation.
We next investigated mechanisms mediating IP3-induced ICat activation. Xestospongin C (XeC), a membrane permeant IP3R blocker, and heparin, an impermeant IP3R inhibitor (1 mg/mL), reduced mean IP3-induced ICat in SR Ca2+-depleted myocytes from ≈95 to ≈32 and 18 pA, respectively (Figure 3B). A reduction in extracellular Na+ from 140 to 20 mmol/L also reduced mean IP3-induced ICat to ≈33 pA (Figure 3B). These data indicate that IP3R activation is required for IP3-induced ICat activation and that Na+ is the principal cation generating ICat.
IP3 Depolarizes and Constricts Pressurized
Arteries With Intact and Depleted SR Ca2+ Physiological functions of IP3-induced ICat activation were studied by measuring membrane potential and diameter regulation of pressurized endothelium-denuded arteries. Arteries were studied primarily at low intravascular pressure (20 mm Hg) to: (1) improve success of sustained microelectrode impalement by reducing vasomotion; (2) maintain a relatively negative membrane potential, which creates a larger driving force for cation influx; and (3) reduce negative-feedback regulation of membrane potential and diameter by Ca2+ sparks and KCa channels.3,13,14 At 20 mm Hg, Bt-IP3 depolarized SR Ca2+-depleted arteries by ≈16 mV (Figure 4A and 4B). At 20 mm Hg, Bt-IP3 also reduced the diameter of pressurized arteries with intact SR Ca2+ by ≈12 μm, from a mean diameter of 142±9 μm (Figure 5C). At 20 mm Hg, thapsigargin (100 nmol/L to 3 μmol/L) did not change diameter and did not reduce Bt-IP3–induced vasoconstriction (Figure 5A and 5C). Nimodipine, a voltage-dependent Ca2+ channel blocker, reduced Bt-IP3–induced constriction by 83% (Figure 5B and 5C). At 60 mm Hg, Bt-IP3 reduced the diameter of arteries with intact SR Ca2+ by ≈11 μm (Figure 5D). At 60 mm Hg, thapsigargin (100 nmol/L) reduced diameter by ±12 μm and attenuated the Bt-IP3–induced constriction by ±39% (Figure 5D). These data indicate that SR Ca2+ release is not required for IP3-induced vasoconstriction, IP3 constricts via voltage-dependent Ca2+ channel activation, and diameter regulation by IP3-induced SR Ca2+ release and SR Ca2+ depletion are both pressure-dependent.
Figure 4.
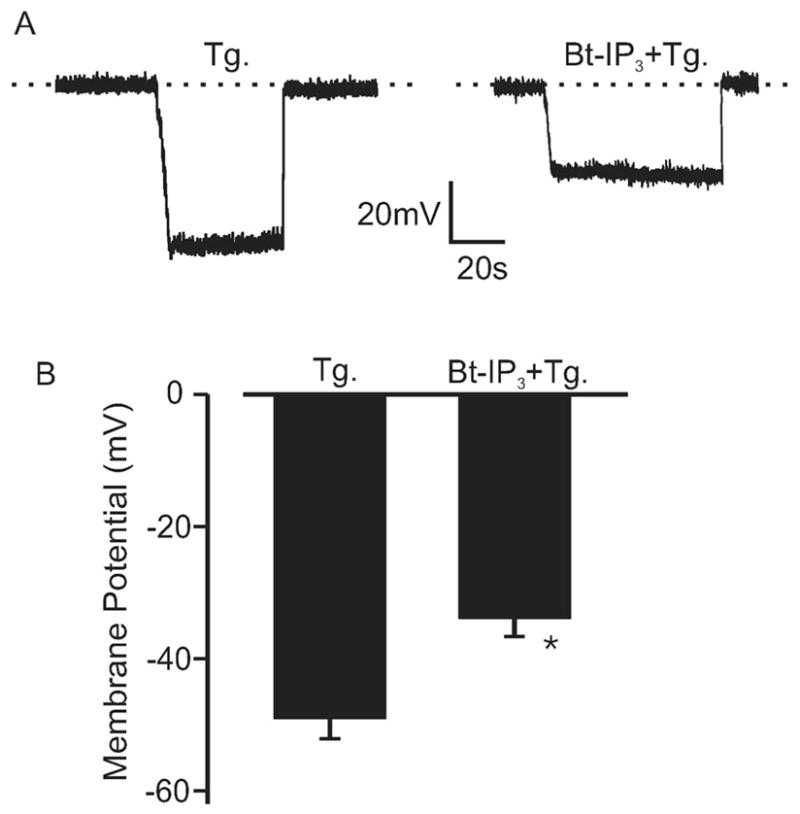
IP3 depolarizes pressurized arteries with depleted SR Ca2+. A, Original traces illustrating membrane depolarization induced by Bt-IP3 (1 μmol/L) in the same SR Ca2+ depleted (100 nmol/L thapsigargin [Tg]; >15 minutes) pressurized (20 mm Hg) artery. Negative deflections from 0 voltage (dotted line) illustrate intracellular microelectrode impalement to measure membrane potential, followed by subsequent removal. B, Mean data (Tg, n=5; Bt-IP3+Tg, n=9). *P<0.05 compared with Tg.
Figure 5.
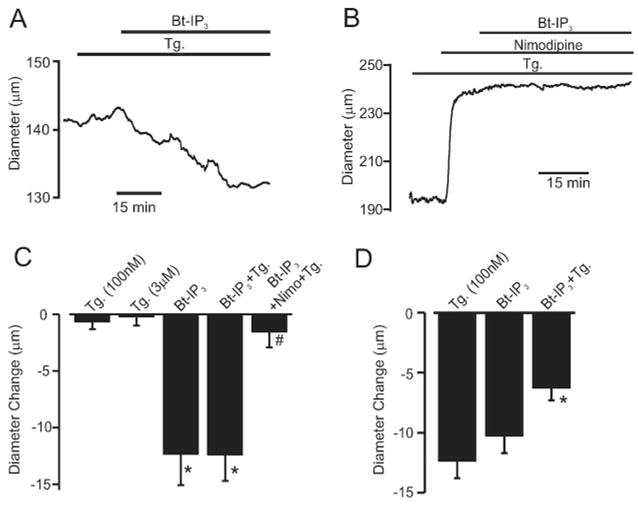
Bt-IP3 constricts pressurized arteries with intact and depleted [Ca2+]SR. A, Bt-IP3 (10 nmol/L) constricts an artery at 20 mm Hg with depleted SR Ca2+ (thapsigargin [Tg],100 nmol/L; >15 minutes). B, At 20 mm Hg, nimodipine (1 μmol/L) caused a large vasodilation and attenuated Bt-IP3–induced vasoconstriction. C, Mean data illustrating Bt-IP3–induced vasoconstriction at 20 mm Hg in arteries with intact (n=8) and depleted (n=7) [Ca2+]SR and inhibition by nimodipine (n=5). D, Mean data illustrating vasoregulation at 60 mm Hg by thapsigargin (n=10), Bt-IP3 (n=7), and Bt-IP3+thapsigargin (n=7). Mean myogenic tone was (in %): 20 mm Hg, 24±2 (n=25); 60 mm Hg, 42.1±10.7 (n=7). *P<0.05 compared with respective control or thapsigargin, #P<0.05 compared with Bt-IP3+thapsigargin.
TRPC3 Channels Are Required for IP3-Induced [Ca2+]i Elevation and Vasoconstriction
Silencing vectors were constructed that express either shRNA targeting TRPC3 exon 6 (TRPC3shV) or scrambled shRNA (TRPC3scrm). Western blot analysis indicated that TRPC3shV reduced TRPC3 channel protein to ≈34% of TRPC3scrm (Figure 6A and 6B). In contrast, TRPC3shV did not alter TRPC6 channel expression in the same arteries (Figure 6A and 6B).
Figure 6.
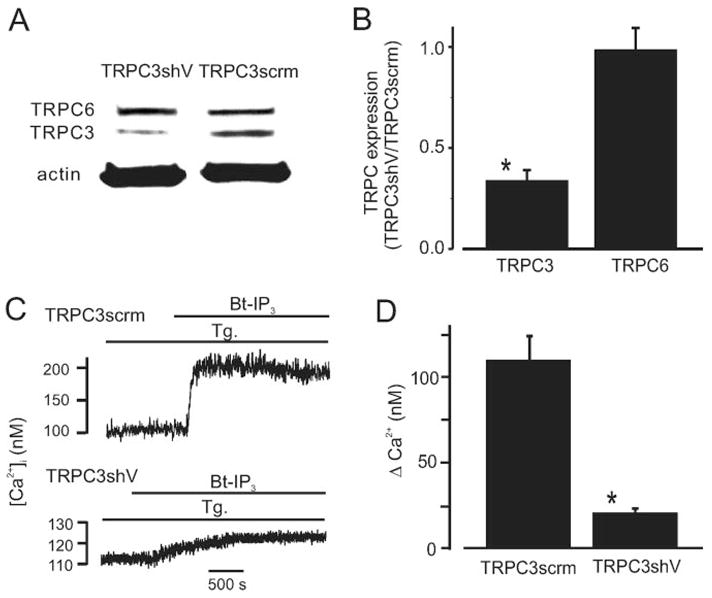
TRPC3 knockdown attenuates Bt-IP3–induced [Ca2+]i elevations in arterial myocytes with depleted SR Ca2+. A, Original Western blot data illustrating that TRPC3shV reduces TRPC3 protein but does not change TRPC6 in the same arteries. Lanes and bands shown were obtained from the same original Western blot that was reprobed with antibodies for TRPC3, TRPC6, and actin and combined for presentation. For clarity, an unrelated lane that was between the 2 illustrated lanes was not shown. B, Mean data (n=5). C, TRPC3 knockdown attenuates Bt-IP3–induced (10 μmol/L) [Ca2+]i elevations in SR Ca2+-depleted myocytes (thapsigargin [Tg], 100 nmol/L; >15 minutes). D, Mean data (TRPC3scrm, n=14; TRPC3shV, n=19). *P<0.05 when compared with TRPC3scrm.
[Ca2+]i was measured in SR Ca2+-depleted myocytes that were isolated from arteries treated with TRPC3shV or TRPC3scrm vectors. TRPC3 knockdown did not alter resting [Ca2+]i (TRPC3scrm, 113≈4 nmol/L, n=14; TRPC3shV, 116≈3 nmol/L, n=19, P<0.05). However, mean Bt-IP3–induced [Ca2+]i elevations in TRPC3shV-treated myocytes were ≈18% of those in TRPC3scrm-treated myocytes (Figure 6C and 6D). In addition, Bt-IP3–induced constrictions in SR Ca2+-depleted, pressurized (20 mm Hg), endothelium-denuded, TRPC3shV-treated arteries were ≈29% of those in TRPC3scrm-treated arteries (Figure 7A through 7C). Collectively, these data indicate that TRPC3 channel activation contributes to the SR Ca2+ release–independent [Ca2+]i elevation and vasoconstriction induced by IP3.
Figure 7.
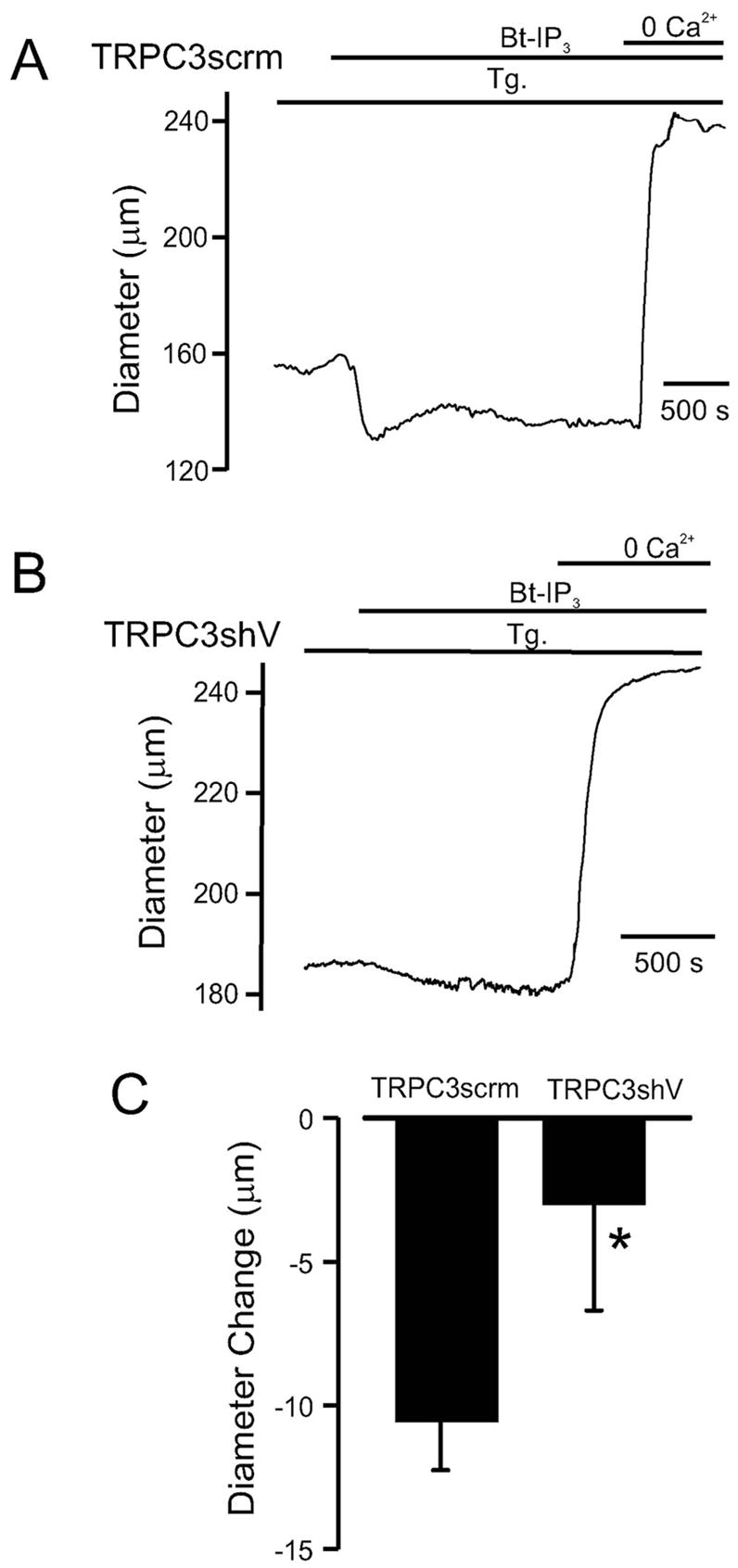
TRPC3 knockdown reduces vasoconstriction induced by Bt-IP3. Original traces illustrating Bt-IP3–induced (10 nmol/L) diameter responses in pressurized (20 mm Hg) arteries treated with TRPC3scrm (A) or TRPC3shV (B). C, Mean Bt-IP3–induced vasoconstriction in arteries treated with TRPC3scrm (n=7) or TRPC3shV (n-6). *P<0.05 compared with TRPC3scrm.
ET-1 Elevates [Ca2+]i and Induces Vasoconstriction Independently of SR Ca2+ Release and via IP3R and TRPC3 Channel Activation
To investigate functional modulation of cerebral artery Ca2+ signaling and diameter by IP3Rs, we studied responses to ET-1, a vasoconstrictor that binds to myocyte PLC-coupled receptors, in endothelium-denuded arteries.15 ET-1 similarly elevated [Ca2+]i when applied to nonpressurized control arteries or arteries with depleted SR Ca2+ (Figure 8B). In SR Ca2+-depleted arteries, XeC did not alter [Ca2+]i when applied alone (3±6 nmol/L change, n=5, P<0.05) but reduced mean ET-1–induced [Ca2+]i elevation by ≈73% (Figure 8A and 8B). TRPC3 knockdown also reduced mean ET-1–induced [Ca2+]i elevation to ≈32% of that in TRPC3scrm-treated arteries (Figure 8B). In agreement with the [Ca2+]i responses, ET-1 similarly constricted pressurized (20 mm Hg) arteries with intact or depleted SR Ca2+ (Figure 8C and 8D). XeC also reduced ET-1–induced vasoconstriction in arteries with depleted SR Ca2+ by ≈53% (Figure 8C and 8D). In addition, TRPC3 knockdown reduced mean ET-1–induced constriction in pressurized (20 mm Hg) arteries to ≈55% of that in TRPC3scrm-treated arteries (Figure 8D). These data indicate that TRPC3 channel activation is necessary for ET-1 to elevate [Ca2+]i and induce vasoconstriction in cerebral arteries and that IP3R activation contributes to the SR Ca2+ release–independent responses.
Figure 8.
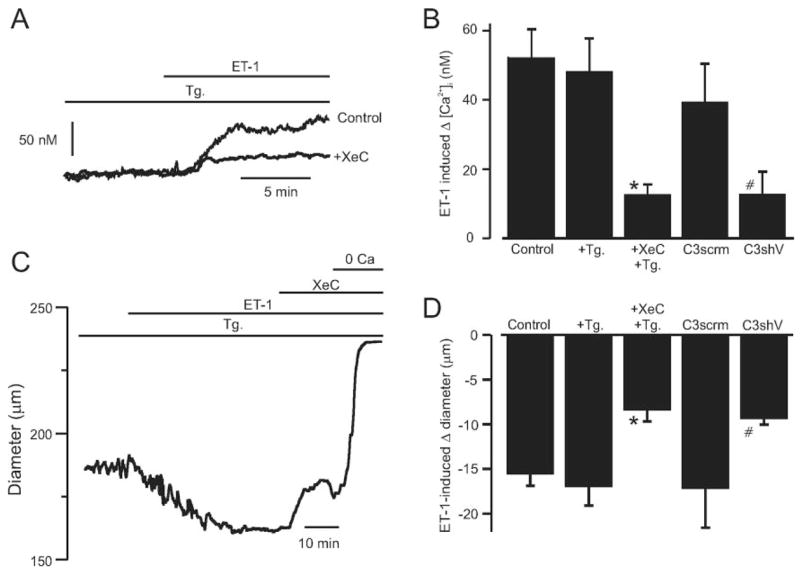
ET-1–induced [Ca2+]i elevation and vasoconstriction requires TRPC3 channels and is attenuated by IP3R inhibition in SR Ca2+-depleted arteries. A, Original recordings illustrating ET-1–induced (300 pmol/L) [Ca2+]i elevations and inhibition by XeC (3 μmol/L) in SR Ca2+-depleted (thapsigargin [Tg], 100 nmol/L; >15 minutes) arteries. B, Mean data for ET-1–induced [Ca2+]i elevations in arteries in control (n=6), the presence of Tg (100 nmol/L; >15 minutes, n=16), and Tg (100 nmol/L)+ XeC (3 μmol/L, n=5) or in arteries treated with TRPC3scrm (n=4) or TRPC3shV (n=4). C, XeC (3 μmol/L) partially reverses an ET-1–induced (300 pmol/L) vasoconstriction in a pressurized (20 mm Hg) artery with depleted SR Ca2+. D, Mean data for ET-1–induced vasoconstriction in arteries in control (n=5), in the presence of Tg (100 nmol/L; >15 minutes, n=6), or Tg+XeC (n=7) or in arteries treated with TRPC3scrm (n=5) or TRPC3shV (n=4). *P<0.05 compared with ET-1+Tg, #P<0.05 compared with TRPC3scrm.
Discussion
It is generally well accepted that IP3 contracts arterial myocytes by stimulating SR Ca2+ release. Here, we provide an additional mechanism by which IP3 regulates arterial contractility. In arterial myocytes, a transient or sustained IP3 elevation activates a ICat that occurs primarily because of Na+ influx. Importantly, activation of this contractile mechanism release. We by IP3 does not require the stimulation of SR Ca2+ also show that the SR Ca2+ release–independent, IP3-induced [Ca2+]i elevation and vasoconstriction require IP3R, TRPC3 channel, and voltage-dependent Ca2+ channel activation. These data indicate that IP3R activation stimulates a ICat to which TRPC3 channels contribute, leading to membrane depolarization, voltage-dependent Ca2+ channel activation, an [Ca2+]i elevation, and vasoconstriction. These findings reveal a novel physiological signaling pathway for IP3 in the vasculature.
To study IP3 regulation of myocyte ICat, [Ca2+]i, and contractility through SR Ca2+ release–independent mechanisms, SR Ca2+ was depleted using thapsigargin. A concentration of 100 nmol/L thapsigargin abolished caffeine-induced [Ca2+]i transients, indicating that SR containing ryanodine receptor channels was Ca2+-depleted. We also used D1ER to measure [Ca2+] in the entire SR and to obtain a time course of the thapsigargin-induced reduction in [Ca2+]SR. Data indicated that a 15-minute application of 100 nmol/L thapsigargin depleted SR Ca2+. D1ER measurements also indicated that when IP3 was applied with thapsigargin, the ICat activation, [Ca2+]i elevation, and vasoconstriction did not occur as a result of a further change in [Ca2+]SR. In addition, in conventional whole-cell experiments, IP3 activated a ICat in myocytes that were exposed to thapsigargin and internally perfused with a pipette solution containing 5 mmol/L EGTA, which would deplete Ca2+ in all SR compartments. Collectively, these data indicate that IP3 activates a ICat via a SR Ca2+ release–independent mechanism.
Store-operated cation currents have been reported in myocytes from several vessels, including mesenteric artery, portal vein, and aorta.16 The regulation, ion selectivity, and conductance of myocyte store-operated cation currents vary depending on the anatomic origin of the vasculature.16 In the present study, thapsigargin (100 nmol/L to 5 μmol/L) did not activate a myocyte ICat and did not alter the diameter of arteries pressurized to 20 mm Hg but constricted arteries pressurized to 60 mm Hg. These data indicate that cerebral artery diameter regulation by SR Ca2+ depletion is pressure-dependent. These data are also consistent with previous evidence that SR Ca2+ depletion constricts cerebral arteries by inhibiting Ca2+ sparks and KCa channels, which are activated by an elevation in intravascular pressure.3,13,17–19 In nonpressurized rabbit and rat cerebral arteries, store depletion also did not induce depolarization and caused an [Ca2+]i elevation that did not stimulate contraction.20,21 Here, thapsigargin caused a myocyte [Ca2+]i transient that declined to a steady-state [Ca2+]i elevation of ≈6 nmol/L. Consistent with these findings, SR Ca2+-ATPase inhibition also likely elevates cytosolic [Ca2+]i by removing a SR Ca2+ sink. In contrast, in myocytes of rabbit pial arteriole fragments and murine cerebral artery myocytes, SR Ca2+ depletion activated an inward current.22,23 Different findings of these studies may be attributable to experimental approach, species, vessel size, and anatomic origin. However, it is unclear why in the present study the thapsigargin-induced [Ca2+]i transient did not cause an associated transient vasoconstriction in pressurized arteries. Conceivably, this may occur because of Ca2+ compartmentalization in a noncontractile location that can activate CREB, as previously proposed.20,24
At 20 mm Hg, SR Ca2+ depletion did not alter IP3 or ET-1–induced vasoconstriction. In contrast, at 60 mm Hg, SR Ca2+ depletion reduced IP3-induced vasoconstriction by ≈39%. These data indicate that vasoregulation by IP3 is pressure-dependent. In addition, findings indicate that over this pressure range, IP3R activation promotes vasoconstriction primarily by activating a ICat, which is contrary to the conventional view of IP3-induced vasoconstriction. The increasing contribution of SR Ca2+ release to the IP3-induced vasoconstriction likely occurs because pressure-induced arterial depolarization activates voltage-dependent Ca2+ channels, which elevates cytosolic [Ca2+]i and [Ca2+]SR. Thus, data suggest that arterial depolarization increases the contribution of SR Ca2+ release to the IP3-induced [Ca2+]i elevation and vasoconstriction. In isolated myocytes, SR Ca2+ depletion reduced the IP3-induced [Ca2+]i elevation by ≈25%. An explanation for this result is that isolated myocytes are more depolarized than myocytes in arteries at low pressure. Thus, isolated myocytes have a higher [Ca2+]SR and IP3-induced SR Ca2+ release contributes to the [Ca2+]i elevation.
IP3, photoreleased IP3, and membrane permeant IP3 all activated a ICat in arterial myocytes via a mechanism that did not require the stimulation of SR Ca2+ release. ICat activation by IP3 was blocked by IP3R inhibitors, cation channel blockers, and a reduction in extracellular Na+. In intact myocytes, IP3 activated 15 pS (0.91 pA at −60 mV) single channels. Our data suggest that, in cerebral artery myocytes, IP3 does not directly activate cation channels, and that DAG does not act as a cofactor for activation. In intact cerebral artery myocytes, DAG stimulates cation channels via PKC activation.25 The lack of effect of OAG in inside-out patches suggests that activated PKC translocates from the cytosol to the membrane to stimulate cation channels. These data indicate that IP3R activation mediates IP3-induced ICat activation. In contrast, in rabbit portal vein myocyte excised patches, IP3 activated 2 pS Ca2+-selective channels.26 In rabbit coronary artery myocytes, IP3 only activated Ca2+-selective channels if patches were also exposed to OAG.27 Variability in ICat regulation by PLC products or SR Ca2+ depletion in myocytes of anatomically diverse arteries may occur because of cell type–specific TRP channel expression or heteromultimer formation.16,28 Vascular myocytes express at least 4 TRP families: TRPC (canonical TRP), TRPM (melastatin TRP), TRPV (vanilloid TRP), and TRPP (polycystin TRP).29–32 Rat cerebral artery myocytes express several TRP channels, including TRPC1, -3, -4, and -6 and TRPM4 and TRPV4.10,21,29,33,34 Although TRPC3 channels are Na+- and Ca2+-permeant (PNa:Pca = 1:1.5), Na+ influx would be predominant with physiological extracellular cation gradients, consistent with observations made here.2,35 The IP3-activated channels described here are also similar in conductance to TRPC3 channels.28 We show that TRPC3 channel knockdown attenuates both the ET-1– and IP3–induced [Ca2+]i elevation in myocytes and vasoconstriction in pressurized arteries. Whether the IP3R-activated channels in myocytes are formed from homomultimers or heteromultimers containing TRPC3 remains to be resolved, particularly because vascular myocyte heteromultimeric TRPC channels have been described.36 In cerebral artery myocytes, UTP, another PLC-coupled receptor agonist, activates TRPC3 channels but the mechanisms mediating this effect are unclear.34 Here, XeC reduced both the ET-1–induced [Ca2+]i elevation and the constriction in SR Ca2+-depleted arteries. Voltage-dependent Ca2+ channel blockers also robustly reduced the IP3-induced [Ca2+]i elevation and vasoconstriction. Thus, data suggest that IP3 is one second messenger mediating vasoconstrictor-induced ICat activation, IP3R activation mediates this response, and IP3-induced Ca2+ influx occurs primarily through depolarization-induced, voltage-dependent Ca2+ channel activation.
A major finding of the present study is that IP3 constricted arteries not by stimulation of SR Ca2+ release but by a mechanism which required IP3R and TRPC3 channel activation. Although the molecular mechanism mediating IP3-induced ICat activation was not determined, conformational coupling between IP3Rs and TRP channels regulates plasma membrane cation influx.37,38 Indeed, IP3Rs interact molecularly with many TRPC channel isoforms, including TRPC3.28 In HEK293 cells, IP3-induced TRPC3 channel activation required IP3Rs but did not require endoplasmic reticulum Ca2+ release.39 Similarly, in DT40 cells IP3 recognition by nonconducting IP3Rs activated plasma membrane cation influx.40 Thus, physical coupling between IP3Rs and TRP channels may mediate vasoregulation by IP3.
In summary, we describe a novel mechanism of IP3-induced vasoconstriction that requires IP3R activation but not the stimulation of SR Ca2+ release. We show that IP3R activation by IP3 stimulates a ICat. The resulting Na+ influx induces membrane depolarization, leading to voltage-dependent Ca2+ channel activation, an [Ca2+]i elevation, and vasoconstriction. We also show that TRPC3 channel activation contributes to the IP3-induced [Ca2+]i elevation and vasoconstriction. These findings supplement the conventional view that IP3 regulates arterial diameter only by stimulating SR Ca2+ release.
Acknowledgments
Sources of Funding
This study was supported by NIH grants HL077678 and HL67061 (to J.H.J.); HL064981 (to C.M.W.); and HL063886 and HL072902 (to A.H.). Q.X. and A.A. are recipients of American Heart Association Postdoctoral Fellowships (Southeast Affiliate).
Footnotes
Circulation Research is available at http://circres.ahajournals.org
Disclosures
None.
References
- 1.Davis MJ, Hill MA. Signaling mechanisms underlying the vascular myogenic response. Physiol Rev. 1999;79:387–423. doi: 10.1152/physrev.1999.79.2.387. [DOI] [PubMed] [Google Scholar]
- 2.Clapham DE, Julius D, Montell C, Schultz G. International Union of Pharmacology. XLIX. Nomenclature and structure-function relationships of transient receptor potential channels. Pharmacol Rev. 2005;57:427–450. doi: 10.1124/pr.57.4.6. [DOI] [PubMed] [Google Scholar]
- 3.Jaggar JH. Intravascular pressure regulates local and global Ca2+ signaling in cerebral artery smooth muscle cells. Am J Physiol. 2001;281:C439–C448. doi: 10.1152/ajpcell.2001.281.2.C439. [DOI] [PubMed] [Google Scholar]
- 4.Palmer AE, Jin C, Reed JC, Tsien RY. Bcl-2-mediated alterations in endoplasmic reticulum Ca2+ analyzed with an improved genetically encoded fluorescent sensor. Proc Natl Acad Sci U S A. 2004;101:17404–17409. doi: 10.1073/pnas.0408030101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dixit M, Zhuang D, Ceacareanu B, Hassid A. Treatment with insulin uncovers the motogenic capacity of nitric oxide in aortic smooth muscle cells: dependence on Gab1 and Gab1-SHP2 association. Circ Res. 2003;93:e113–e123. doi: 10.1161/01.RES.0000100391.98425.BB. [DOI] [PubMed] [Google Scholar]
- 6.Cheranov SY, Jaggar JH. Mitochondrial modulation of Ca2+ sparks and transient KCa currents in smooth muscle cells of rat cerebral arteries. J Physiol. 2004;556:755–771. doi: 10.1113/jphysiol.2003.059568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Adebiyi A, Zhao G, Cheranov SY, Ahmed A, Jaggar JH. Caveolin-1 abolishment attenuates the myogenic response in murine cerebral arteries. Am J Physiol. 2007;292:H1584–H1592. doi: 10.1152/ajpheart.00584.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xi Q, Tcheranova D, Parfenova H, Horowitz B, Leffler CW, Jaggar JH. Carbon monoxide activates KCa channels in newborn cerebral arteriole smooth muscle cells by increasing the apparent Ca2+-sensitivity of α-subunits. Am J Physiol. 2004;286:H610–H618. doi: 10.1152/ajpheart.00782.2003. [DOI] [PubMed] [Google Scholar]
- 9.Yu JY, DeRuiter SL, Turner DL. RNA interference by expression of short-interfering RNAs and hairpin RNAs in mammalian cells. Proc Natl Acad Sci U S A. 2002;99:6047–6052. doi: 10.1073/pnas.092143499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Welsh DG, Morielli AD, Nelson MT, Brayden JE. Transient receptor potential channels regulate myogenic tone of resistance arteries. Circ Res. 2002;90:248–250. doi: 10.1161/hh0302.105662. [DOI] [PubMed] [Google Scholar]
- 11.Lesh RE, Somlyo AP, Owens GK, Somlyo AV. Reversible permeabilization. A novel technique for the intracellular introduction of antisense oligodeoxynucleotides into intact smooth muscle. Circ Res. 1995;77:220–230. doi: 10.1161/01.res.77.2.220. [DOI] [PubMed] [Google Scholar]
- 12.Xi Q, Cheranov SY, Jaggar JH. Mitochondria-derived reactive oxygen species dilate cerebral arteries by activating Ca2+ sparks. Circ Res. 2005;14(97):354–362. doi: 10.1161/01.RES.0000177669.29525.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brayden JE, Nelson MT. Regulation of arterial tone by activation of calcium-dependent potassium channels. Science. 1992;256:532–535. doi: 10.1126/science.1373909. [DOI] [PubMed] [Google Scholar]
- 14.Knot HJ, Nelson MT. Regulation of arterial diameter and wall [Ca2+] in cerebral arteries of rat by membrane potential and intravascular pressure. J Physiol. 1998;508:199–209. doi: 10.1111/j.1469-7793.1998.199br.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Masaki T. Historical review: endothelin. Trends Pharmacol Sci. 2004;25:219–224. doi: 10.1016/j.tips.2004.02.008. [DOI] [PubMed] [Google Scholar]
- 16.Albert AP, Saleh SN, Peppiatt-Wildman CM, Large WA. Multiple activation mechanisms of store-operated TRPC channels in smooth muscle cells. J Physiol. 2007;583:25–36. doi: 10.1113/jphysiol.2007.137802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Knot HJ, Standen NB, Nelson MT. Ryanodine receptors regulate arterial diameter and wall [Ca2+] in cerebral arteries of rat via Ca2+-dependent K+ channels. J Physiol. 1998;508:211–221. doi: 10.1111/j.1469-7793.1998.211br.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jaggar JH, Porter VA, Lederer WJ, Nelson MT. Calcium sparks in smooth muscle. Am J Physiol. 2000;278:C235–C256. doi: 10.1152/ajpcell.2000.278.2.C235. [DOI] [PubMed] [Google Scholar]
- 19.Nelson MT, Cheng H, Rubart M, Santana LF, Bonev AD, Knot HJ, Lederer WJ. Relaxation of arterial smooth muscle by calcium sparks. Science. 1995;270:633–637. doi: 10.1126/science.270.5236.633. [DOI] [PubMed] [Google Scholar]
- 20.Flemming R, Cheong A, Dedman AM, Beech DJ. Discrete store-operated calcium influx into an intracellular compartment in rabbit arteriolar smooth muscle. J Physiol. 2002;543:455–464. doi: 10.1113/jphysiol.2002.023366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bergdahl A, Gomez MF, Wihlborg AK, Erlinge D, Eyjolfson A, Xu SZ, Beech DJ, Dreja K, Hellstrand P. Plasticity of TRPC expression in arterial smooth muscle: correlation with store-operated Ca2+ entry. Am J Physiol. 2005;288:C872–C880. doi: 10.1152/ajpcell.00334.2004. [DOI] [PubMed] [Google Scholar]
- 22.Dietrich A, Kalwa H, Storch U, Mederos YS, Salanova B, Pinkenburg O, Dubrovska G, Essin K, Gollasch M, Birnbaumer L, Gudermann T. Pressure-induced and store-operated cation influx in vascular smooth muscle cells is independent of TRPC1. Pflugers Arch. 2007;455:465–477. doi: 10.1007/s00424-007-0314-3. [DOI] [PubMed] [Google Scholar]
- 23.Xu SZ, Boulay G, Flemming R, Beech DJ. E3-targeted anti-TRPC5 antibody inhibits store-operated calcium entry in freshly isolated pial arterioles. Am J Physiol. 2006;291:H2653–H2659. doi: 10.1152/ajpheart.00495.2006. [DOI] [PubMed] [Google Scholar]
- 24.Pulver RA, Rose-Curtis P, Roe MW, Wellman GC, Lounsbury KM. Store-operated Ca2+ entry activates the CREB transcription factor in vascular smooth muscle. Circ Res. 2004;94:1351–1358. doi: 10.1161/01.RES.0000127618.34500.FD. [DOI] [PubMed] [Google Scholar]
- 25.Slish DF, Welsh DG, Brayden JE. Diacylglycerol and protein kinase C activate cation channels involved in myogenic tone. Am J Physiol. 2002;283:H2196–H2201. doi: 10.1152/ajpheart.00605.2002. [DOI] [PubMed] [Google Scholar]
- 26.Liu M, Albert AP, Large WA. Facilitatory effect of Ins 1,4,5 P3 on store-operated Ca2+-permeable cation channels in rabbit portal vein myocytes. J Physiol. 2005;566:161–171. doi: 10.1113/jphysiol.2005.088260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Peppiatt-Wildman CM, Albert AP, Saleh SN, Large WA. Endothelin-1 activates a Ca2+-permeable cation channel with TRPC3 and TRPC7 properties in rabbit coronary artery myocytes. J Physiol. 2007;580:755–764. doi: 10.1113/jphysiol.2006.126656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vazquez G, Wedel BJ, Aziz O, Trebak M, Putney JW., Jr The mammalian TRPC cation channels. Biochim Biophys Acta. 2004;1742:21–36. doi: 10.1016/j.bbamcr.2004.08.015. [DOI] [PubMed] [Google Scholar]
- 29.Earley S, Heppner TJ, Nelson MT, Brayden JE. TRPV4 forms a novel Ca2+ signaling complex with ryanodine receptors and BKCa channels. Circ Res. 2005;97:1270–1279. doi: 10.1161/01.RES.0000194321.60300.d6. [DOI] [PubMed] [Google Scholar]
- 30.Beech DJ, Muraki K, Flemming R. Non-selective cationic channels of smooth muscle and the mammalian homologues of Drosophila TRP. J Physiol. 2004;559:685–706. doi: 10.1113/jphysiol.2004.068734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Walker RL, Hume JR, Horowitz B. Differential expression and alternative splicing of TRP channel genes in smooth muscles. Am J Physiol. 2001;280:C1184–C1192. doi: 10.1152/ajpcell.2001.280.5.C1184. [DOI] [PubMed] [Google Scholar]
- 32.Facemire CS, Mohler PJ, Arendshorst WJ. Expression and relative abundance of short transient receptor potential channels in the rat renal microcirculation. Am J Physiol. 2004;286:F546–F551. doi: 10.1152/ajprenal.00338.2003. [DOI] [PubMed] [Google Scholar]
- 33.Earley S, Waldron BJ, Brayden JE. Critical role for transient receptor potential channel TRPM4 in myogenic constriction of cerebral arteries. Circ Res. 2004;95:922–929. doi: 10.1161/01.RES.0000147311.54833.03. [DOI] [PubMed] [Google Scholar]
- 34.Reading SA, Earley S, Waldron BJ, Welsh DG, Brayden JE. TRPC3 mediates pyrimidine receptor-induced depolarization of cerebral arteries. Am J Physiol. 2005;288:H2055–H2061. doi: 10.1152/ajpheart.00861.2004. [DOI] [PubMed] [Google Scholar]
- 35.Welsh DG, Brayden JE. Mechanisms of coronary artery depolarization by uridine triphosphate. Am J Physiol. 2001;280:H2545–H2553. doi: 10.1152/ajpheart.2001.280.6.H2545. [DOI] [PubMed] [Google Scholar]
- 36.Maruyama Y, Nakanishi Y, Walsh EJ, Wilson DP, Welsh DG, Cole WC. Heteromultimeric TRPC6-TRPC7 channels contribute to arginine vasopressin-induced cation current of A7r5 vascular smooth muscle cells. Circ Res. 2006;98:1520–1527. doi: 10.1161/01.RES.0000226495.34949.28. [DOI] [PubMed] [Google Scholar]
- 37.Berridge M. Conformational coupling: a physiological calcium entry mechanism. Sci STKE. 2004;2004:e33. doi: 10.1126/stke.2432004pe33. [DOI] [PubMed] [Google Scholar]
- 38.Irvine RF. ‘Quantal’ Ca2+ release and the control of Ca2+ entry by inositol phosphates–a possible mechanism. FEBS Lett. 1990;263:5–9. doi: 10.1016/0014-5793(90)80692-c. [DOI] [PubMed] [Google Scholar]
- 39.Kiselyov K, Mignery GA, Zhu MX, Muallem S. The N-terminal domain of the IP3 receptor gates store-operated hTrp3 channels. Mol Cell. 1999;4:423–429. doi: 10.1016/s1097-2765(00)80344-5. [DOI] [PubMed] [Google Scholar]
- 40.van Rossum DB, Patterson RL, Kiselyov K, Boehning D, Barrow RK, Gill DL, Snyder SH. Agonist-induced Ca2+ entry determined by inositol 1,4,5-trisphosphate recognition. Proc Natl Acad Sci U S A. 2004;101:2323–2327. doi: 10.1073/pnas.0308565100. [DOI] [PMC free article] [PubMed] [Google Scholar]