

Abstract
Background:
Whereas deficiency of the essential nutrient choline is associated with DNA damage and apoptosis in cell and rodent models, it has not been shown in humans.
Objective:
The objective was to ascertain whether lymphocytes from choline-deficient humans had greater DNA damage and apoptosis than did those from choline-sufficient humans.
Design:
Fifty-one men and women aged 18–70 y were fed a diet containing the recommended adequate intake of choline (control) for 10 d. They then were fed a choline-deficient diet for up to 42 d before repletion with 138–550 mg choline/d. Blood was collected at the end of each phase, and peripheral lymphocytes were isolated. DNA damage and apoptosis were then assessed by activation of caspase-3, terminal deoxynucleotide transferase–mediated dUTP nick end-labeling, and single-cell gel electrophoresis (COMET) assays.
Results:
All subjects fed the choline-deficient diet had lymphocyte DNA damage, as assessed by COMET assay, twice that found when they were fed the control diet. The subjects who developed organ dysfunction (liver or muscle) when fed the choline-deficient diet had significantly more apoptotic lymphocytes, as assessed by the activated caspase-3 assay, than when fed the control diet.
Conclusions:
A choline-deficient diet increased DNA damage in humans. Subjects in whom these diets induced liver or muscle dys-function also had higher rates of apoptosis in their peripheral lymphocytes than did subjects who did not develop organ dysfunction. Assessment of DNA damage and apoptosis in lymphocytes appears to be a clinically useful measure in humans (such as those receiving parenteral nutrition) in whom choline deficiency is suspected.
Keywords: Choline deficiency, apoptosis, lymphocytes, humans, DNA damage, single-cell gel electrophoresis, COMET assay, terminal deoxynucleotide transferase–mediated dUTP nick end-labeling, TUNEL assay
INTRODUCTION
Choline is a dietary component that is essential to the normal function of all cells (1). It (or its metabolites) assures the structural integrity and signaling functions of cell membranes; in addition, it is the major source of methyl groups in the diet (one of choline's metabolites, betaine, participates in the methylation of homocysteine to form methionine), and it directly affects cholinergic neurotransmission and the transport of lipids from the liver (1). One of the functional consequences of dietary choline deficiency in humans is the development of liver damage (ie, elevated serum aminotransferase concentrations; 2, 3) and muscle damage (ie, elevated serum creatine phosphokinase concentrations; 4). Cultured hepatocytes and myocytes died by apoptosis when placed in choline-deficient medium (4-7), which may explain why liver and muscle cells leaked enzymes into blood in choline-deficient humans.
Choline deficiency has also been associated with DNA damage. In choline-deficient hepatocytes, when compared with cells grown with choline, leakage of reactive oxygen species from mitochondria was greater (8, 9), and these reactive oxygen species react with lipids and DNA. In choline-deficient rats, lipid peroxides accumulated (10-13), and there was more DNA adduct formation and DNA damage in hepatocytes (14) and in lymphocytes (15) than in control rats. This finding has never been shown in humans.
Better biomarkers for assessment of choline status are important for clinical practice in nutrition. Low plasma choline concentrations occur in up to 84% of the patients who require total parenteral nutrition [(TPN) 3, 16-20], as does liver damage (3, 21) and fatty liver (3). In some patients, the hepatic dysfunction associated with TPN was resolved with choline supplementation but then returned when standard TPN was reinstituted (3). Low choline status is also possible in healthy populations. Dietary choline intakes vary enough in healthy women in the United States (from <300 to >500 mg/d) to significantly influence the risk that a healthy woman will have a baby with a birth defect (22). Current assessment of choline status in humans involves assays of choline, betaine, and phosphatidylcholine concentrations in plasma (2), but it is important to note that measurement of these biomarkers in humans was not sufficient to predict which subjects would develop organ dysfunction when fed a choline-deficient diet, because all choline-depleted subjects had low plasma choline and betaine concentrations, yet only some developed liver or muscle damage (4, 23). Although these plasma concentrations decreased when humans ate a choline-deficient diet, they stabilized after a decrease of 30% to 50%, probably as a result of homeostatic mechanisms (24). Thus, plasma measurements likely do not accurately reflect tissue concentrations of choline. An alternative functional marker for choline deficiency is needed in a readily accessible tissue.
In the current study, men and women were fed a diet containing choline for 10 d and then fed a choline-deficient diet for ≤42 d before repletion with choline. We determined whether DNA damage and apoptosis increased during the period of choline depletion and whether these functional effects of choline deficiency were correlated with susceptibility to liver or muscle damage when the subjects were fed a choline-deficient diet.
SUBJECTS AND METHODS
Study design
Healthy men (n = 31) and women (n = 35) were recruited for the study. Inclusion was contingent on a good, age-typical health status, as ascertained by physical examination and standard clinical laboratory tests such as complete blood count, blood chemistries, fasting lipid and liver function tests, and the presence of no known chronic disease. Twenty-two subjects admitted to the study had minor elevations in blood lipids that the study physician deemed not to have clinical significance. Of the originally recruited 66 subjects, 61 completed at least the initial phase and the depletion phase. Of that group, 1 subject was excluded because of a 9-kg weight loss during the study, and 3 subjects were excluded because they did not comply with diet restrictions; baseline measurements of DNA damage and apoptosis were not performed in 6 subjects. Thus, 51 subjects were included in the analyses. These 20 men and 31 women ranged in age from 18 to 70 y and had a body mass index (in kg/m2) between 19 and 33, which they maintained throughout the study. Two of the post-menopausal women were taking hormone replacement therapy. The race-ethnicity distribution of the participants was white (63%), African American (27%), Asian (6%), and Native American (4%), which reflected the local population characteristics of the Raleigh-Durham-Chapel Hill area.
The subjects were admitted to the General Clinical Research Center at the University of North Carolina at Chapel Hill, where they remained under the supervision of study staff for the duration of the study. The diets administered to the subjects, which were composed of high-biologic-value protein (0.8 g/kg), fat (30% of kcal), and carbohydrate (70% of kcal), were prepared in-house to protocol specifications; they are described in detail elsewhere (25). Total food intake was adjusted to be isocaloric and to provide adequate intakes of macronutrients and micronutrients.
Initially, all participants received a diet of normal foods containing 550 mg choline · 70 kg body wt−1 · d−1, which is the presumed adequate intake (26), and 400 dietary folate equivalents (DFE)/d. The dietary choline content was confirmed as described previously (25, 27), and the folate content was calculated by using the US Department of Agriculture Nutrient Database for Standard Reference (release 16; http://www.ars.usda.gov/ba/bhnrc/ndl) and PRONUTRA software (version 3.1.0.13; ProNutra, Princeton, NJ). After 10 d, the subjects were randomly assigned to 1 of 2 groups—dietary folate only or dietary folate supplemented with 400 μg folic acid/d—and then were fed a diet in which the choline content was reduced to <50 mg/d, as confirmed by analysis of duplicate food portions (27, 28). For the rest of the study, all diets offered to the folate-only group contained 100 DFE/d, whereas the folic acid–supplemented group received an additional 668 DFE/d (Figure 1). Plasma folate concentrations were measured by using a microbiological assay with interassay and intraassay CVs of 3.9% and 3.1%, respectively (29). Periodic measurments of urinary choline and betaine concentrations were used to confirm compliance with the dietary restrictions. Briefly, urine was collected (pooled for 24 h) at the end of the baseline, depletion, and repletion phases, as well as on days 13 and 31 of the choline-deficient diet. Aliquots were stored at −80 °C until they were analyzed for choline and betaine by the method of Koc et al (28).
FIGURE 1.
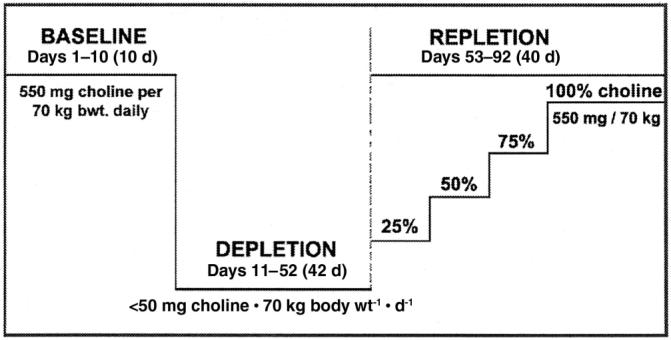
Overview of study design. The choline content of the baseline, depletion, and repletion phases is indicated. Subjects who developed organ dysfunction when fed the choline-deficient diet underwent repletion with graded increasing concentrations of choline for 10 d each until signs of organ dysfunction resolved [137.5 (25%), 275 (50%), 412.5 (75%), and finally 550 mg choline · 70 kg−1 · d−1 (100%)]. The baseline diet provided 400 dietary folate equivalents (DFE)/d. The choline-depletion and -repletion diets provided 100 DFE/d. On day 11, subjects were randomly assigned to receive placebo (100 DFE/d) or 400 μg folic acid supplement/d for the remainder of the study.
Subjects continued to be fed this choline-depletion diet until they developed organ dysfunction associated with choline deficiency or for 42 d if they did not develop such dysfunction. Humans were deemed to have organ dysfunction associated with choline deficiency if their serum creatine phosphokinase (CPK) activity rose >5-fold from baseline (4) or if liver fat content increased by >28% during the choline-depletion diet, and if this greater CPK activity or higher liver fat content resolved when choline was restored to the diet. After the depletion study, subjects with no dysfunction were maintained on a diet containing 550 mg choline/d for ≥3 d before being discharged from the study. Those with organ dysfunction underwent choline repletion by receiving 137.5 mg choline · 70 kg−1 · d−1 (25% adequate intake) for 10 d and then 275, 412.5, and 550 mg choline · 70 kg−1 · d−1 in successive 10-d periods. At the point when the clinical symptoms were resolved, the subjects were fed >550 mg choline · 70 kg−1 · d−1 ≥3d.
Written informed consent was obtained from all participants. The study protocol was approved by the Institutional Review Board at the University of North Carolina at Chapel Hill.
Assessment of fatty liver
Liver fat was measured at the end of the baseline diet, on days 21 and 42 of the choline-deficient diet, and at the end of the repletion period (for subjects with fatty liver when fed the choline-deficient diet). Change in liver fat content was estimated by magnetic resonance imaging with a Siemens Vision 41.5T clinical MR system (Siemens Medical Solutions, Malvern, PA) using a modified “In and Out of Phase” procedure (23, 30). This approach uses the differences in transverse magnetization intensity after a fast low-angle shot [(FLASH) echo time = 2.2 and 4.5 msec, with a flip angle of 80°, and repetition time = 140 msec]. Processing of successive FLASH magnetic resonance images with the software from Siemens Medical Solutions was used to estimate fat content. Organ content was derived from measurements across 3–5 liver slices per subject and standardized by relating the results to similarly measured slices of spleen.
Assessment of liver and muscle cell damage
Fasting blood samples were taken every 3–4 d for blood chemistry analyses (including CPK analysis). For the lymphocyte studies, they were also obtained after 10 d of the baseline diet, at the end of the choline-deficient diet (depletion phase), and at the end of the choline-repletion phase. Absolute lymphocyte counts were performed on an Advia analyzer (Bayer, Tarrytown, NY). Serum was analyzed by using a dry-slide colorimetric method for CPK activity [McClendon Clinical Laboratories, University of North Carolina Hospitals, Chapel Hill, NC, which are accredited by both the Clinical Laboratory Improvement Act (CLIA) and the College of American Pathologists].
Assessment of lymphocyte DNA damage and apoptosis
Peripheral lymphocytes were isolated from blood by using Ficoll-Hypaque gradient in evacuated tubes with sodium citrate (Vacutainer CPT; Becton Dickinson, Franklin Lakes, NJ) according to the manufacturer's instructions (31, 32). DNA was measured by using the method of Labarca and Paigen (33). The cells were then analyzed by using the ApoTarget Caspase-3/CPP32 Colorimetric Protease Assay (BioSource International, Camarillo, CA). Briefly, activated caspase-3 cleaves the 4-nitroaniline peptide substrate DEVD-pNA, and this activity is quantified at 405-nm absorbance by using ultraviolet spectrometry and expressed as integrated optical density/mg DNA. Activated caspase-3 was also measured in a subset of cells by labeling them with carboxyfluorescein (FAM)-labeled peptide fluoromethyl ketone (FMK) caspase inhibitor (FAM-DEVD-FMK, CaspaTag kit; Intergen, Purchase, NY), which specifically binds to activated caspase-3. Nuclei were then stained with 4′-6-diamidino-2-phenylindole, and activated caspase-3 was detected by using a band-pass filter (excitation wavelength: 490 nm; emission wavelength: 520 nm) on a microscope. We used an ultraviolet filter (excitation wavelength: 365 nm; emission wavelength: 4600 nm) to visualize the nuclear staining, and the National Institutes of Health–based SCION IMAGE software (version 4.03; Scion Corporation, Frederick, MD) to quantify the activated caspase-3–integrated absorption.
We measured DNA fragmentation in lymphocytes by using terminal deoxynucleotidyl transferase–mediated dUTP nick end-labeling [(TUNEL) ApopTag Plus in Situ Apoptosis Detection Kit; Chemicon International, Temecula, CA]. Lymphocytes were fixed in 2% paraformaldehyde and suspended on a silanized microscope slide by cytospin. They were labeled and stained with diaminobenzidine-peroxidase, and the nuclei were counterstained with 0.5% methyl green. The slides were mounted, and the TUNEL-positive cells were counted by light microscopy. At least 300 total cells were counted for each data point, and the amount is expressed as a percentage.
We assessed DNA strand breaks by using single-cell gel electrophoresis (COMET assay; 34). Lymphocytes were sandwiched between 0.5% agarose and 0.5% low-melting-point (37 °C) agarose (Fisher, Fair Lawn, NJ). The resulting slides were placed into cold, freshly made lysis solution [10 mmol Tris/L (pH 10), 2.5 mol NaCl/L, 100 mmol EDTA/L, 1% sodium sarcosinate, 10% DMSO, and 1% Triton X-100] at 4 °C for ≥1 h and then treated for 20 min in electrophoresis buffer [300 mmol NaOH/L, 1 mmol EDTA/L (pH 13)]. After electrophoresis was performed at 25 V and 300 mA for 20 min, slides were incubated 3 times for 5 min in neutralization buffer [0.4 mol Tris/L (pH 7.5)], washed with methanol, and stained with 20 mg ethidium bromide/mL. COMET tail length (the distance of DNA migration from the body of the nuclear core) was visualized by using a fluorescence microscope (typically, 100 cells/sample) and SCION IMAGE software. The COMET tail moment (defined as the integrated density in the COMET tail multiplied by the distance from the center of the nucleus to the center of mass of the tail) was calculated by using the NIH IMAGE ANALYSIS MACRO language software (http://dir.nhlbi.nih.gov/labs/ldn/macroanalysis.asp).
Statistical analysis
Significant differences in urinary data between subjects after they consumed the baseline (550 mg choline/d) diet and the same subjects after they consumed the choline-deficient and choline-repletion diets were determined by analysis of variance (ANOVA) and Dunnett's test. A 2-factor analysis with interaction based on the differences between the activated caspase-3, TUNEL, and COMET measurements; the lymphocyte counts; and plasma folate concentrations at the end of the baseline and choline-deficient diets was used to determine significant interactions between organ dysfunction and folate status. The interactions were not significant, so a 2-factor ANOVA was then used to compare these groups. A 2-sample t test based on the differences between the lymphocyte counts and activated caspase-3, TUNEL, COMET, and plasma folate measurements at the end of the baseline and choline-repletion diets for the folate-only and the folic acid–supplemented groups was performed to determine whether folate had an effect on these measurements. If folate had no effect, paired t tests were then used to compare measurements between the baseline and repletion diets. Statistical analysis was performed by using JMP software (version 3.2; SAS Institute Inc, Cary, NC).
RESULTS
Organ dysfunction
Thirty-three (65%) of 51 subjects developed organ dysfunction when fed the choline-deficient diet. Of this group, 26 (8 men and 18 women) had liver dysfunction, 1 (a man) had only muscle dysfunction, and 6 (5 men and 1 woman) had both liver and muscle dysfunction. All subjects returned to normal with choline repletion. Urinary betaine and choline concentrations dropped to 60% of baseline after subjects consumed the choline-deficient diet for 13 d (Figure 2) and were <50% of baseline at the end of the depletion period (P < 0.01, ANOVA and Dunnett's test). When choline was restored to the diet (for 3 d in those with no clinical symptoms and for >3 d in those with symptoms), urinary concentrations of both metabolites increased to >80% of baseline (Figure 2).
FIGURE 2.
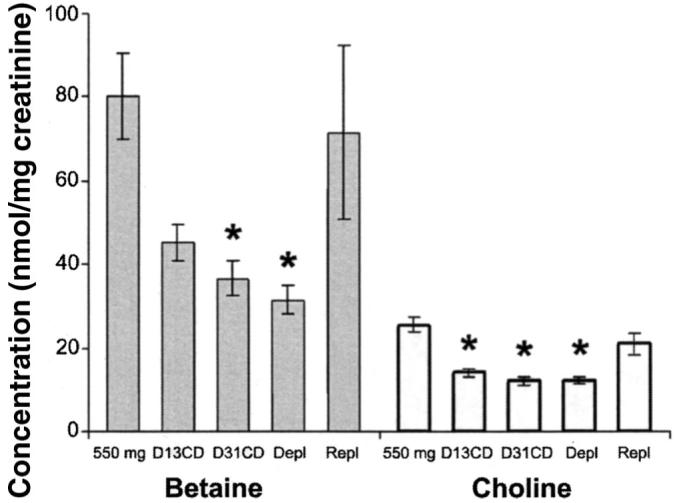
Mean (±SE) urinary betaine and choline concentrations (n = 32–45/time point), from which dietary compliance was assessed. Subjects were treated as described in the legend for Figure 1. Urine samples (24-h) were collected at the end of the baseline (550 mg), depletion (Depl), and repletion (Repl) phases and on days 13 and 31 of the choline-deficient diet (D13CD and D31CD, respectively), and aliquots were analyzed for betaine ( ) and choline (□) concentrations nmol/mg creatinine as described in Methods. *Significantly different from baseline, P < 0.01 (one-factor ANOVA and Dunnett's test).
) and choline (□) concentrations nmol/mg creatinine as described in Methods. *Significantly different from baseline, P < 0.01 (one-factor ANOVA and Dunnett's test).
Lymphocyte counts
Mean total lymphocyte counts at the end of the choline-depletion period (2.3±0.1×106/mL) did not differ significantly from baseline values, regardless of organ dysfunction or folate status (Table 1). Lymphocyte counts in the subjects at the end of the repletion diets (2.2 ±0.1 ×106/mL) also did not differ significantly from baseline values (P = 0.479, paired t test).
TABLE 1.
Markers of apoptosis and DNA damage in the lymphocytes of humans after a diet with 550 mg choline/d and after a choline-deficient diet1
| Activated caspase-3 | TUNEL | COMET | Lymphocyte count | Plasma folate | |
|---|---|---|---|---|---|
|
integrated OD/mg DNA |
% cells positive |
tail moment | × 106/mL | nmol/L | |
| Baseline diet (n = 51) | 11.5 ± 1.2 | 1.6 ± 0.2 | 1.0 ± 0.1 | 2.3 ± 0.1 | 26.0 ± 0.9 |
| Low-choline diet | |||||
| Organ dysfunction, 100 DFE (n = 17) | 21.5 ± 3.92 | 4.2 ± 0.92 | 2.2 ± 0.2 | 2.1 ± 0.2 | 20.7 ± 1.62 |
| Organ dysfunction, 768 DFE (n = 16) | 15.6 ± 4.02 | 2.0 ± 0.4 | 2.1 ± 0.2 | 2.2 ± 0.1 | 28.2 ± 1.7 |
| No organ dysfunction, 100 DFE (n = 8) | 5.2 ± 1.0 | 3.3 ± 1.32 | 1.7 ± 0.2 | 2.4 ± 0.2 | 23.0 ± 2.42 |
| No organ dysfunction, 768 DFE (n = 10) | 11.0 ± 2.7 | 2.4 ± 0.7 | 1.8 ± 0.2 | 2.2 ± 0.3 | 27.1 ± 2.0 |
All values are x̄ ±SE. OD, optical density; TUNEL, terminal deoxynucleotidyl transferase–mediated dUTP nick end-labeling; COMET, single-cell gel electrophoresis; DFE, dietary folate equivalents. Subjects were fed a diet providing 550 mg choline/70 kg body wt daily (baseline) for 10 d and were then fed a low-choline diet diet for up to 42 d. They were randomly assigned to receive only dietary folate (100 DFE/d) or dietary folate plus a daily folic acid supplement (combined intake: 768 DFE/d) while consuming a choline-deficient diet. The organ dysfunction×folate interaction was not significant; thus, a 2-wayANOVA based on the differences between measurements at the end of the baseline and choline-deficient periods was used to compare the groups.
Significantly different from all other choline-deficient groups, P < 0.05.
Activated caspase-3 assay
The amount of activated caspase-3 in the lymphocytes of subjects who developed organ dysfunction after they were fed a low choline-diet was significantly greater than their baseline values (P = 0.018 by 2-factor ANOVA; Table 1). Subjects who did not develop organ dysfunction had no increase in mean activated caspase-3 in their lymphocytes. The effect of the choline-deficient diet was not changed by supplementation with folic acid (Table 1). When choline was restored to the diet of the subjects with organ dysfunction, activated caspase-3 returned to 104 ± 21% of baseline (P = 0.842 for difference from baseline, paired t test; n = 27).
TUNEL assay
There were more TUNEL-positive lymphocytes in subjects at the end of the choline-depletion period than when they were fed the baseline diet (Table 1). There was no significant interaction between liver-muscle dysfunction and folate status, but subjects who were not supplemented with folic acid had significantly (P = 0.026) more TUNEL-positive cells than did subjects receiving the supplement. When choline was restored to the diet of the subjects with organ dysfunction, there was a partial reduction in the proportion of TUNEL-positive lymphocytes (to 2.4 ± 0.4%; P = 0.034 for difference from baseline values, paired t test; n = 23); there was no effect of folate status on the postrepletion TUNEL assay values (P = 0.940, t test).
COMET assay
At the end of the choline-depletion period, we observed that all subjects, regardless of whether they became sufficiently choline-depleted to develop liver or muscle dysfunction, had lymphocyte DNA damage (assessed by the COMET assay) twice that seen when they were fed the control diet (Table 1). This increase in tail moment occurred regardless of whether subjects also developed other signs of organ dysfunction when fed the choline-deficient diet and was independent of dietary folate intake. At the end of the choline-repletion period, when all choline deficiency–associated organ dysfunction had resolved, the average COMET tail moment in the lymphocytes of subjects was 1.2 ± 0.1 (P = 0.428 for difference from baseline, paired t test; n = 27).
Folate assay
At the end of the baseline diet period, mean plasma folate concentrations in the subjects were 26.0 ± 0.9 nmol/L (Table 1), which was within the range of values reported for humans consuming 427 μg folate/d (35). When subjects were fed the choline-deficient diet supplemented with folic acid, the mean plasma folate concentration was 27.8 ± 1.3 nmol/L (P = 0.038 for difference from baseline, paired t test; n = 26). When these subjects underwent repletion with added choline, the mean plasma folate concentration did not change (28.2 ± 1.9 nmol/L). When subjects were fed the choline-deficient diet and did not receive folic acid (100 DFE/d from diet alone), mean plasma folate concentrations decreased to 21.4 ± 1.3 nmol/L (P = 0.0003 for difference from the supplemented group, 2-factor ANOVA). This value is still well above that which indicates folate deficiency (7 nmol/L; 36). In these subjects at the end of the choline-repletion period, the mean plasma folate concentration (26.2 ± 1.6 nmol/L) did not differ from baseline.
DISCUSSION
We found that choline deficiency in humans was associated with significant damage to DNA and with apoptosis in peripheral lymphocytes. This association had not previously been shown in humans.
Caspase-3 is a critical effector protein in apoptosis, and it exists within the cytosol as an inactive dimer (37). Cleavage within a linker segment is required for activation, which occurs only during the terminal execution cascade of signals mediating apoptosis (37). Thus, activated caspase-3 is an extremely specific marker for apoptosis. We observed that activated caspase-3 increased in the lymphocytes of subjects who developed organ dysfunction when fed a choline-deficient diet (Table 1). TUNEL labeling is often cited as a specific biomarker for apoptosis because the technique identifies cells with single-strand nicks in DNA. However, the assay also detects nonapoptotic events that result in DNA damage (38, 39). Our data are consistent with this finding, because our TUNEL assay values appear to be intermediate between activated caspase-3 values, which increased only in subjects who developed organ dysfunction when fed a choline-deficient diet, and COMET tail moment values, which increased in all subjects fed a choline-deficient diet. The COMET assay measures double-strand breaks in DNA (34) as well as DNA modifications such as abasic sites (AP sites) (40). This assay is different from both the TUNEL assay, which detects single- and double-strand breaks in DNA, and the activated caspase-3 assay, which detects specific activation of the apoptotic signaling cascade that is responsible for cell execution (41). Whether DNA lesions detected by the COMET assay go on to become gene mutations depends on whether they are correctly repaired without resulting in permanent genetic alterations (40).
As noted earlier, in animal models, choline deficiency increased leakage of reactive oxygen species from mitochondria (8, 9) and increased DNA adduct formation and DNA damage in liver (14, 15). In the current study, we observed that all subjects, as compared with the values when they were fed the control diet, had twice as much lymphocyte DNA damage (COMET assay) when they were fed the choline-deficient diet, even if choline was not sufficiently depleted to lead to liver or muscle dysfunction (Table 1). No evidence exists of a mechanism for choline interactions with DNA that is particular to lymphocytes, rather than to other tissues, and thus it is likely that damage to DNA is not organ specific. Perhaps damage to DNA is the earliest functional effect of choline deficiency and occurs before intracellular concentrations of choline or phosphatidylcholine decrease enough to induce apoptosis and leakage of enzymes from liver and muscle or fatty liver. If so, increased DNA damage as assessed by COMET assays could be the earliest marker for identification of a tissue as choline-deficient.
We suggest that choline deficiency–induced apoptosis shares a mechanism with other measures of organ dysfunction associated with choline deficiency—ie, diminished synthesis of phosphatidylcholine. Fatty liver occurs in choline deficiency because phosphatidylcholine is not available for secretion of VLDL from liver (42). Muscle dysfunction in choline deficiency likely occurs because lower membrane phosphatidylcholine concentrations make myocytes fragile (4). We previously reported that membrane phosphatidylcholine concentrations may be the critical variable for choline deficiency–mediated induction of apoptotic cascades (9, 43).
Choline and folate metabolisms are interrelated (1), and it has been suggested that folate status can modify susceptibility to choline deficiency (44). In the current study, the changes in plasma folate were modest, although they did occur in the direction we expected. It is possible that the experimental period in the current study—which was as short as 2 wk and as long as 6 wk—was not long enough for the dietary changes (from 400 to 100 or 768 DFE) to reach a plateau in plasma folate; it has been reported that 6–14 wk is required for that outcome (45). It could have been predicted that humans ingesting a choline-deficient diet containing 100 DFE/d would have decreased plasma folate concentrations because this folate intake is less than that estimated for humans eating an ad libitum diet (≈200 DFE/d from naturally occurring food folate, in addition to the contribution from foods fortified with folic acid) (46). We present folate data to show that our subjects did not become frankly folate deficient (plasma folate concentrations were well above those indicative of folate deficiency; 36). Supplementary folic acid mitigated some but not all of the effects of the choline-deficient diet on increasing TUNEL labeling but did not mitigate choline deficiency–associated changes in activated caspase-3 or in COMET tail moment. The restoration to the diet of choline, without restoration of folate, returned COMET and activated caspase-3 values to normal. Thus, choline deficiency and not diminished folate status was responsible for the changes that we report here.
That choline deficiency induces significant DNA damage in humans is an important observation. In rodent models, prolonged (ie, 1 y) choline deficiency results in the development of liver cancers (47) and in greater sensitivity to chemical carcinogens (48). These conditions could be the result of mutations due to DNA damage. Similarly, choline deficiency–induced apoptosis is of clinical significance, because liver damage is a common side effect of prolonged parenteral nutrition (16). At present, there is no definitive clinical test that can be used to identify persons who are choline deficient. Plasma choline, betaine, and phosphatidylcholine concentrations decreased in humans fed a choline-deficient diet, but they plateau after falling 30%–50% (2, 24). Urinary betaine and choline concentrations also decreased in humans consuming a choline-deficient diet, and we used that measure as a compliance marker in this study. Lower concentrations of choline metabolites are necessary but not sufficient to predict who will develop organ dysfunction (4, 23). In the current study, we identified a number of biomarkers that can be used in conjunction with plasma choline metabolites to make a diagnosis of choline deficiency in humans. COMET tail moment may be the most sensitive measure, and dose-response studies should be performed to ascertain the exact dietary intake at which COMET values become abnormal. Activated caspase-3 measurements in lymphocytes correlated best with liver and muscle dysfunction that was associated with choline deficiency. Thus, this assessment has clinical value for physicians who might use it as an indicator for ordering studies of liver or muscle function in patients suspected of being choline deficient. The TUNEL assay was influenced by folate status as well as by choline status, and thus it is not as specific a biomarker for choline deficiency as are the other assays.
The human dietary requirement for an essential nutrient is defined as the amount that prevents organ dysfunction. The Institute of Medicine Dietary Reference Intake Panel defined the human requirement for choline according to the amount of dietary intake needed to prevent liver dysfunction (ie, elevated serum transaminase concentrations; 26). Because apoptosis and DNA damage in lymphocytes are measures of significantly abnormal cell function, we suggest that these measures in a clinically accessible tissue could be used to define the human dietary requirement for choline.
Acknowledgments
We thank Mihaela Badea for help with the caspase assays and Lester Kwock for assistance with MRI measurements.
KD participated in the design of the studies, supervised blood collection and processing and the caspase assays, participated in statistical analyses and data interpretation, and participated in writing the manuscript. MDN performed the TUNEL analyses, and CC performed the COMET assays; both participated in writing the manuscript. LMF participated in the supervision of the human study. SHZ was responsible for the conceptualization, implementation, and design of the human study; participated in statistical analyses and data interpretation; and participated in writing the manuscript. SHZ receives grant support from Mead Johnson for studies other than those described here and serves on an advisory board for the Solae Company.
Footnotes
Some of the data presented here were presented in poster form at the Experimental Biology Meeting in San Diego, CA in 2003 and published in FASEB J 2003;17:A1157.
Supported by grant no. DK55865 from the National Institutes of Health and by grants from the NIH to the UNC Clinical Nutrition Research Unit (DK56350), the UNC General Clinical Research Center (RR00046), the Center for Gastrointestinal Biology and Disease (DK34987), and the Center for Environmental Health and Susceptibility (ES10126). The Solae Company provided the lecithin used to formulate the diets.
None of the authors had a personal or financial conflict of interest.
REFERENCES
- 1.Zeisel SH, Blusztajn JK. Choline and human nutrition. Annu Rev Nutr. 1994;14:269–96. doi: 10.1146/annurev.nu.14.070194.001413. [DOI] [PubMed] [Google Scholar]
- 2.Zeisel SH, da Costa K-A, Franklin PD, et al. Choline, an essential nutrient for humans. FASEB J. 1991;5:2093–8. [PubMed] [Google Scholar]
- 3.Buchman AL, Ament ME, Sohel M, et al. Choline deficiency causes reversible hepatic abnormalities in patients receiving parenteral nutrition: proof of a human choline requirement: a placebo-controlled trial. JPEN J Parenter Enteral Nutr. 2001;25:260–8. doi: 10.1177/0148607101025005260. [DOI] [PubMed] [Google Scholar]
- 4.da Costa KA, Badea M, Fischer LM, Zeisel SH. Elevated serum creatine phosphokinase in choline-deficient humans: mechanistic studies in C2C12 mouse myoblasts. Am J Clin Nutr. 2004;80:163–70. doi: 10.1093/ajcn/80.1.163. [DOI] [PubMed] [Google Scholar]
- 5.Albright CD, Lui R, Bethea TC, da Costa K-A, Salganik RI, Zeisel SH. Choline deficiency induces apoptosis in SV40-immortalized CWSV-1 rat hepatocytes in culture. FASEB J. 1996;10:510–6. doi: 10.1096/fasebj.10.4.8647350. [DOI] [PubMed] [Google Scholar]
- 6.Albright CD, da Costa KA, Craciunescu CN, Klem E, Mar MH, Zeisel SH. Regulation of choline deficiency apoptosis by epidermal growth factor in CWSV-1 rat hepatocytes. Cell Physiol Biochem. 2005;15:59–68. doi: 10.1159/000083653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Albright CD, Salganik RI, Kaufmann WK, Vrablic AS, Zeisel SH. A p53-dependent G1 checkpoint function is not required for induction of apoptosis by acute choline deficiency in immortalized rat hepatocytes in culture. J Nutr Biochem. 1998;9:476–81. [Google Scholar]
- 8.Albright CD, Salganik RI, Craciunescu CN, Mar MH, Zeisel SH. Mitochondrial and microsomal derived reactive oxygen species mediate apoptosis induced by transforming growth factor-beta1 in immortalized rat hepatocytes. J Cell Biochem. 2003;89:254–61. doi: 10.1002/jcb.10498. [DOI] [PubMed] [Google Scholar]
- 9.Vrablic AS, Albright CD, Craciunescu CN, Salganik RI, Zeisel SH. Altered mitochondrial function and overgeneration of reactive oxygen species precede the induction of apoptosis by 1-O-octadecyl-2-methyl-rac-glycero-3-phosphocholine in p53-defective hepatocytes. FASEB J. 2001;15:1739–44. doi: 10.1096/fj.00-0300com. [DOI] [PubMed] [Google Scholar]
- 10.Rushmore T, Lim Y, Farber E, Ghoshal A. Rapid lipid peroxidation in the nuclear fraction of rat liver induced by a diet deficient in choline and methionine. Cancer Lett. 1984;24:251–5. doi: 10.1016/0304-3835(84)90020-x. [DOI] [PubMed] [Google Scholar]
- 11.Yoshida LS, Miyazawa T, Fujimoto K, Kaneda T. Liver phosphatidylcholine hydroperoxidation provoked by ethionine-containing choline-deficient diet in mice. Lipids. 1990;25:565–9. doi: 10.1007/BF02537166. [DOI] [PubMed] [Google Scholar]
- 12.Ghoshal AK, Farber E. Liver biochemical pathology of choline deficiency and of methyl group deficiency: a new orientation and assessment. Histol Histopathol. 1995;10:457–62. [PubMed] [Google Scholar]
- 13.Banni S, Corongiu FP, Dessi MA, et al. Free radicals and lipid peroxidation in liver of rats kept on a diet devoid of choline. Free Radic Res Commun. 1989;7:233–40. doi: 10.3109/10715768909087947. [DOI] [PubMed] [Google Scholar]
- 14.Pogribny IP, Basnakian AG, Miller BJ, Lopatina NG, Poirier LA, James SJ. Breaks in genomic DNA and within the p53 gene are associated with hypomethylation in livers of folate/methyl-deficient rats. Cancer Res. 1995;55:1894–901. [PubMed] [Google Scholar]
- 15.James SJ, Yin L. Diet-induced DNA damage and altered nucleotide metabolism in lymphocytes from methyl-donor-deficient rats. Carcino-genesis. 1989;10:1209–14. doi: 10.1093/carcin/10.7.1209. [DOI] [PubMed] [Google Scholar]
- 16.Buchman A, Dubin M, Moukarzel A, et al. Choline deficiency: a cause of hepatic steatosis during parenteral nutrition that can be reversed with intravenous choline supplementation. Hepatology. 1995;22:1399–403. [PubMed] [Google Scholar]
- 17.Buchman AL, Moukarzel A, Jenden DJ, Roch M, Rice K, Ament ME. Low plasma free choline is prevalent in patients receiving long term parenteral nutrition and is associated with hepatic aminotransferase abnormalities. Clin Nutr. 1993;12:33–7. doi: 10.1016/0261-5614(93)90143-r. [DOI] [PubMed] [Google Scholar]
- 18.Burt ME, Hanin I, Brennan MF. Choline deficiency associated with total parenteral nutrition. Lancet. 1980;2:638–9. doi: 10.1016/s0140-6736(80)90301-3. [DOI] [PubMed] [Google Scholar]
- 19.Sheard NF, Tayek JA, Bistrian BR, Blackburn GL, Zeisel SH. Plasma choline concentration in humans fed parenterally. Am J Clin Nutr. 1986;43:219–24. doi: 10.1093/ajcn/43.2.219. [DOI] [PubMed] [Google Scholar]
- 20.Shronts EP. Essential nature of choline with implications for total parenteral nutrition. J Am Diet Assoc. 1997;97:639–46. doi: 10.1016/S0002-8223(97)00161-2. 649. [DOI] [PubMed] [Google Scholar]
- 21.Tayek JA, Bistrian B, Sheard NF, Zeisel SH, Blackburn GL. Abnormal liver function in malnourished patients receiving total parenteral nutrition: a prospective randomized study. J Am Coll Nutr. 1990;9:76–83. doi: 10.1080/07315724.1990.10720353. [DOI] [PubMed] [Google Scholar]
- 22.Shaw GM, Carmichael SL, Yang W, Selvin S, Schaffer DM. Periconceptional dietary intake of choline and betaine and neural tube defects in offspring. Am J Epidemiol. 2004;160:102–9. doi: 10.1093/aje/kwh187. [DOI] [PubMed] [Google Scholar]
- 23.da Costa KA, Gaffney CE, Fischer LM, Zeisel SH. Choline deficiency in mice and humans is associated with increased plasma homocysteine concentration after a methionine load. Am J Clin Nutr. 2005;81:440–4. doi: 10.1093/ajcn.81.2.440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Savendahl L, Mar M-H, Underwood L, Zeisel S. Prolonged fasting results in diminished plasma choline concentration but does not cause liver dysfunction. Am J Clin Nutr. 1997;66:622–5. doi: 10.1093/ajcn/66.3.622. [DOI] [PubMed] [Google Scholar]
- 25.Busby MG, Fischer L, Da Costa KA, Thompson D, Mar MH, Zeisel SH. Choline- and betaine-defined diets for use in clinical research and for the management of trimethylaminuria. J Am Diet Assoc. 2004;104:1836–45. doi: 10.1016/j.jada.2004.09.027. [DOI] [PubMed] [Google Scholar]
- 26.Institute of Medicine, National Academy of Sciences . Dietary reference intakes for folate, thiamin, riboflavin, niacin, vitamin B12, panthothenic acid, biotin, and choline. National Academy Press; Washington, DC: 1998. Choline; pp. 390–422. [PubMed] [Google Scholar]
- 27.Zeisel SH, Mar MH, Howe JC, Holden JM. Concentrations of choline-containing compounds and betaine in common foods. J Nutr. 2003;133:1302–7. doi: 10.1093/jn/133.5.1302. [DOI] [PubMed] [Google Scholar]
- 28.Koc H, Mar MH, Ranasinghe A, Swenberg JA, Zeisel SH. Quantitation of choline and its metabolites in tissues and foods by liquid chromatography/electrospray ionization-isotope dilution mass spectrometry. Anal Chem. 2002;74:4734–40. doi: 10.1021/ac025624x. [DOI] [PubMed] [Google Scholar]
- 29.Horne DW, Patterson D. Lactobacillus casei microbiological assay of folic acid derivatives in 96-well microtiter plates. Clin Chem. 1988;34:2357–9. [PubMed] [Google Scholar]
- 30.Fishbein M, Gardner K, Potter C, Schmalbrock P, Smith M. Introduction of fast MR imaging in the assessment of hepatic steatosis. Magn Reson Imaging. 1997;15:287–93. doi: 10.1016/s0730-725x(96)00224-x. [DOI] [PubMed] [Google Scholar]
- 31.Fotino M, Merson E, Allen F. Micromethod for rapid separation of lymphocytes from peripheral blood. Ann Clin Lab Sci. 1971;1:131–3. [PubMed] [Google Scholar]
- 32.Ting A, Morris P. A technique for lymphocyte preparation from stored heparinized blood. Vox Sang. 1971:561–3. doi: 10.1111/j.1423-0410.1971.tb00469.x. [DOI] [PubMed] [Google Scholar]
- 33.Labarca C, Paigen K. A simple, rapid and sensitive DNA assay procedure. Anal Biochem. 1980;102:344–52. doi: 10.1016/0003-2697(80)90165-7. [DOI] [PubMed] [Google Scholar]
- 34.Sauvaigo S, Serres C, Signorini N, Emonet N, Richard MJ, Cadet J. Use of the single-cell gel electrophoresis assay for the immunofluorescent detection of specific DNA damage. Anal Biochem. 1998;259:1–7. doi: 10.1006/abio.1998.2628. [DOI] [PubMed] [Google Scholar]
- 35.Hertrampf E, Cortes F, Erickson JD, et al. Consumption of folic acid-fortified bread improves folate status in women of reproductive age in Chile. J Nutr. 2003;133:3166–9. doi: 10.1093/jn/133.10.3166. [DOI] [PubMed] [Google Scholar]
- 36.Institute of Medicine, National Academy of Sciences . Dietary reference intakes for folate, thiamin, riboflavin, niacin, vitamin B12, panthothenic acid, biotin, and choline. National Academy Press; Washington, DC: 1998. Folate; pp. 196–305. [PubMed] [Google Scholar]
- 37.Boatright KM, Salvesen GS. Mechanisms of caspase activation. Curr Opin Cell Biol. 2003;15:725–31. doi: 10.1016/j.ceb.2003.10.009. [DOI] [PubMed] [Google Scholar]
- 38.Grasl-Kraupp B, Ruttkay-Nedecky B, Koudelka H, Bukowska K, Bursch W, Schulte-Hermann R. In situ detection of fragmented DNA (TUNEL assay) fails to discriminate among apoptosis, necrosis, and autolytic cell death: a cautionary note. Hepatology. 1995;21:1465–8. doi: 10.1002/hep.1840210534. [DOI] [PubMed] [Google Scholar]
- 39.Cervos-Navarro J, Schubert T. Pitfalls in the evaluation of apoptosis using TUNEL. Brain Pathol. 1996;6:347–8. [PubMed] [Google Scholar]
- 40.Brendler-Schwaab S, Hartmann A, Pfuhler S, Speit G. The in vivo comet assay: use and status in genotoxicity testing. Mutagenesis. 2005;20:245–54. doi: 10.1093/mutage/gei033. [DOI] [PubMed] [Google Scholar]
- 41.Roser S, Pool-Zobel BL, Rechkemmer G. Contribution of apoptosis to responses in the comet assay. Mutat Res. 2001;497:169–75. doi: 10.1016/s1383-5718(01)00255-8. [DOI] [PubMed] [Google Scholar]
- 42.Yao ZM, Vance DE. The active synthesis of phosphatidylcholine is required for very low density lipoprotein secretion from rat hepatocytes. J Biol Chem. 1988;263:2998–3004. [PubMed] [Google Scholar]
- 43.Yen CL, Mar MH, Zeisel SH. Choline deficiency-induced apoptosis in PC12 cells is associated with diminished membrane phosphatidylcholine and sphingomyelin, accumulation of ceramide and diacylglycerol, and activation of a caspase. FASEB J. 1999;13:135–42. [PubMed] [Google Scholar]
- 44.Jacob RA, Jenden DJ, Allman-Farinelli MA, Swendseid ME. Folate nutriture alters choline status of women and men fed low choline diets. J Nutr. 1999;129:712–7. doi: 10.1093/jn/129.3.712. [DOI] [PubMed] [Google Scholar]
- 45.Quinlivan EP, Gregory JF., 3rd Effect of food fortification on folic acid intake in the United States. Am J Clin Nutr. 2003;77:221–5. doi: 10.1093/ajcn/77.1.221. [DOI] [PubMed] [Google Scholar]
- 46.Choumenkovitch SF, Selhub J, Wilson PW, Rader JI, Rosenberg IH, Jacques PF. Folic acid intake from fortification in United States exceeds predictions. J Nutr. 2002;132:2792–8. doi: 10.1093/jn/132.9.2792. [DOI] [PubMed] [Google Scholar]
- 47.Newberne PM, Rogers AE. Labile methyl groups and the promotion of cancer. Annu Rev Nutr. 1986;6:407–32. doi: 10.1146/annurev.nu.06.070186.002203. [DOI] [PubMed] [Google Scholar]
- 48.Rogers AE, Akhtar R, Zeisel SH. Procarbazine carcinogenicity in methotrexate-treated or lipotrope-deficient male rats. Carcinogenesis. 1990;11:1491–5. doi: 10.1093/carcin/11.9.1491. [DOI] [PubMed] [Google Scholar]