

Abstract
The availability of sequenced genomes of human and many experimental animals necessitated the development of new technologies and powerful computational tools that are capable of exploiting these genomic data and ask intriguing questions about complex nature of biological processes. This gave impetus for developing whole genome approaches that can produce functional information of genes in the form of expression profiles and unscramble the relationships between variation in gene expression and the resulting physiological outcome. These profiles represent genetic fingerprints or catalogue of genes that characterize the cell or tissue being studied and provide a basis from which to begin an investigation of the underlying biology. Among the most powerful and versatile tools are high-density DNA microarrays to analyze the expression patterns of large numbers of genes across different tissues or within the same tissue under a variety of experimental conditions or even between species. The wide spread use of microarray technologies is generating large sets of data that is stimulating the development of better analytical tools so that functions can be predicted for novel genes. In this review, the authors discuss how these profiles are being used at various stages of the drug discovery process and help in the identification of new drug targets, predict the function of novel genes, and understand individual variability in response to drugs
Key Words: Expression profiling, microarray analysis, toxicogenomics, pharmacogenomics, biomarkers
INTRODUCTION
It is well known that successful introduction of new drugs and vaccines has contributed to increased life expectancy by as much as 30 years during the past century [1]. With an aged population on the rise, however, the incidence of complex and debilitating diseases such as cancer and Alzheimer’s and Parkinson’s disease, is increasing thereby reducing the overall quality of life. This necessitates a better understanding of the complexity of human physiology and age related cellular degeneration affecting various functions and increased susceptibility to major chronic diseases at the biochemical and molecular level.
The collection of genes that are transcribed or expressed from genomic DNA is a major determinant of cellular phenotype and function and is also responsible for variation of cellular responses to environmental stimuli and perturbations. Hence understanding the function of genes and knowing when, where and to what extent a gene is expressed helps us to understand the biological roles of encoded proteins. In addition, the knowledge gained from these studies, in the context of human health and disease, help us to determine the causes and consequences of diseases that in turn facilitate an understanding of what gene products might have therapeutic uses or may be appropriate as targets for therapeutic manipulation. Over the last few years, expression profiling methodologies have been emerging to monitor and catalog changes in the expression of genes.
With the availability of entire genomic sequences of a large number of prokaryotes and a rapidly growing number of eukaryotes [2] the challenge is to identify the gene products and understand their function. A large number of technologies that allow analysis of multiple DNA sequences rapidly and efficiently have been developed to address this challenge, but the most versatile tools that have gained widest application for this purpose are high-density arrays of oligonucleotides and complementary DNAs [3, 4]. DNA microarrays are made up of high density nucleic acid spots immobilized in an orderly arrangement on a solid substrate. The technology was introduced in the early 1990s, and has since undergone several adaptations and refinements to achieve the current status as the platform of choice for this purpose. In contrast to the gene-by gene approach used to identify novel genes involved in various cellular processes, analysis of gene expression using arrays is a more powerful approach in determining mRNA abundance. As a result, data sets containing massive lists of expressed or repressed genes are generated, thereby providing a starting point from which to begin the investigation of various biological processes [5, 6]. Therefore, in a conventional sense, the experiments involving microarrays are generally question-driven rather than hypothesis driven.
Microarray data analysis and mining has fundamentally changed the way in which biological systems are studied. These studies have generated catalogs of genes and their functions, and this will help to understand how these genes work together in a cellular environment [7]. When the microarray analysis is carried out with emphasis on comparative gene expression of control (e.g. normal) and experimental (e.g. diseased) samples, the results have shed light on several areas of drug discovery and development, particularly in identifying novel targets, biomarkers and delineating mechanisms of action of therapeutic leads [8]. Because the arrays can be designed and made on the basis of partial sequence information also, it is possible to include genes in a survey that are completely uncharacterized. The validity of these approaches has been well documented and proven to be useful in understanding basic biological processes and identifying genes responsible for disease conditions [9]. In this review, the authors highlight some of the applications of expression profiling using microarray analysis in drug development.
TISSUE EXPRESSION PROFILING
Transcriptional or expression profiling analysis of whole and fractionated tissues is an important part of the drug development process. The utilization of information obtained from transcriptional profiling studies has a dramatic effect on multiple areas of the drug discovery including target identification, validation, compound selection, pharmacogenomics, biomarker development, clinical trial evaluation and toxicology.
Over the last decade, pharmaceutical companies have committed tremendous resources to establish extremely large databases of transcriptional profiling data from multiple research species as well as humans [10]. To obtain the maximum benefit for their committed resources, a single array platform is chosen and all tissue profiling studies are conducted on arrays from a single provider using standardized methodologies. This allows the creation of enormous species-specific databases that facilitate and allow confidence in comparison of data sets from different experiments because all the arrays are analyzed using the same quality control standards. This will also permit integration of specific databases that allow for array data from single or multiple orthologous genes from two different species to be rapidly compared and evaluated. Such computational power is very important when comparing gene expression data from human/mouse/rat in all areas of drug development, such as determining whether a specific gene of interest is expressed in the same tissues in all three species at roughly the same intensity.
Within the context of drug discovery, transcriptional profiling is commonly performed on whole tissue or smaller defined regions of tissue, such as purified cells that are unique to the particular tissue of interest [11, 12], small morphologically distinct regions isolated via physical microdissection [13, 14] or laser capture microdissection [15]. The following paragraphs will describe some of the uses of data generated by the transcriptional profiling of whole and fractionated tissues using examples from the literature.
In the early stages of initiating a transcriptional profiling effort on a novel platform, companies will begin by profiling whole tissues. The goal is to increase the power of the profiling databases as rapidly as possible and this is best done by having multiple tissues represented as compared to fewer tissues that have been fractionated prior to profiling. Tissue and RNA from non-regulated research species are often collected and evaluated on arrays “in house” as it is more cost effective and allows more control over the quality of the samples. RNA for the profiling of regulated species (canine, primate, human) is often purchased directly from vendors who specialize in providing uncontaminated and high quality RNA from multiple species.
Alternatively, some companies may purchase transcriptional profiling data, and for them this may represent their total investment in profiling. For many large companies their databases are constructed from a combination of commercially purchased transcriptional profiling data as well as data acquired “in house”. In such cases it is necessary to ensure that the methodologies and quality controls used by both companies are either identical or significantly similar to allow for comparison of the commercial data with the data generated in house.
Large scale, whole tissue transcriptional profiling has multiple uses in the area of drug development. For example, in the area of target identification it is often important to identify genes that have a highly restricted or tissue-specific expression profile. For example, in the authors’ research it is crucial to identify transcripts expressed preferentially in only one tissue, such as the epididymis. To this end, profiling was carried out on whole mouse epididymi using the Affymetrix MOE430AB array set and compared these data to transcriptional profiling data from 23 other normal whole mouse tissues to identify probe sets that were expressed uniquely in the epididymis [13]. Thirty seven probe sets were identified that were specific to the epididymis (Fig. 1). A second example is in the area of toxicology, where transcriptional profiling of an organ during its gestational development [16] or postnatal development [12] can elucidate dramatic changes in gene expression that occur during development and, coupled with insight into the specific activities of the compound under development, provide important information as to where and by what mechanisms toxicological effects are more likely to occur (unpublished observations).
Fig. (1).
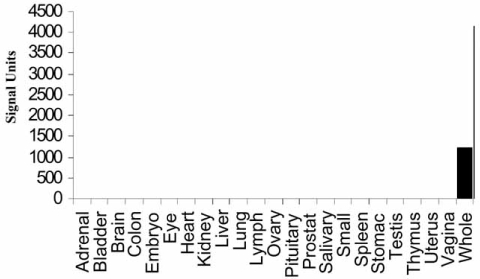
Electronic tissue distribution demonstrates epididymis-specific expression of mRNA levels for mouse EST AW455861 “Whole” refers to entire epididymis [13].
Once a company has established a large set of whole organ transcriptional profiling data, individual research programs will begin to start fractionating the organ into component fractions. Methods for subdividing organs include but are not limited to (1) physical micro dissection of an entire organ into morphological or developmental components [13], (2) laser capture micro dissection of defined regions of a tissue [16], enzymatic dispersion of cells that comprise whole organ followed by purification of defined cell types [12], and physical micro-dissection of individual cell types [11]. Microarray analysis on these tissue or organ fractions provide dramatically increased resolution of the transcriptome of the organ. Expanding the transcriptome in this manner has a powerful and profound effect on target identification, especially when a restricted tissue expression profile is required. For example, transcriptional profiling of the 10 individual segments that comprise the mouse epididymis increased the number of profiling sets identified compared to whole epididymis by 44% (11863 probe sets from total tissue compared to 17096 probe sets from individual segments) [13]. When the transcriptome identified from segmented epididymis was compared to the transcriptional profiling database of 23 whole tissues, the number of probe sets specific to the epididymis increased by approximately 100% (37 probe sets from whole tissue transcriptome, 75 from for segmented epididymal transcriptome). This dramatic increase in potential targets identified justified the extra costs required to conduct the analysis of the segmented transcriptome.
In addition to enhancing target identification, there are many important pieces of information that can be obtained from complete micro-dissection of a tissue into precise morphological or developmental regions, or from isolating defined portions of a tissue that represent a specific developmental event or from isolating and profiling multiple cell types along a developmental process [11, 12]. First, this profiling strategy allows for the identification of a large number of transcripts whose expression is differentially regulated over the series of samples [10, 14]. It is generally assumed that non-regulated genes are more likely to represent “housekeeping” genes and are less likely to transcribe proteins important to the specific biological process under investigation. Second, the increased resolution obtained from fractionation provides important information on the localization of gene expression (and presumably protein expression) within the tissue. Information generated through this process is extremely valuable when trying to isolate native protein translated from a novel gene or to identify a specific region to focus on with a natural or targeted deletion of a gene of interest. Third, when combined with appropriate bioinformatics tools, fractionation provides an enormously powerful mechanism to study the precise relative expression pattern along a developmental progression of single genes or gene families associated with specific biological mechanisms and pathways [13, 14] (Fig. 2). Data from analyses such as these can assist greatly in directing future research by elucidating families and pathways that may have been previously not identified as being important to the biological processes of interest. Fourth, having data on fractionated tissues from multiple species allows for the determination as to whether families of genes or genes whose products are involved in the same biological pathways are conserved across species. When families of genes or individual genes involved in the same pathway are conserved across species in a developmental program, there is a general acceptance that those genes and those programs are important to the biological process and likely to be conserved in humans as well.
Fig. (2).
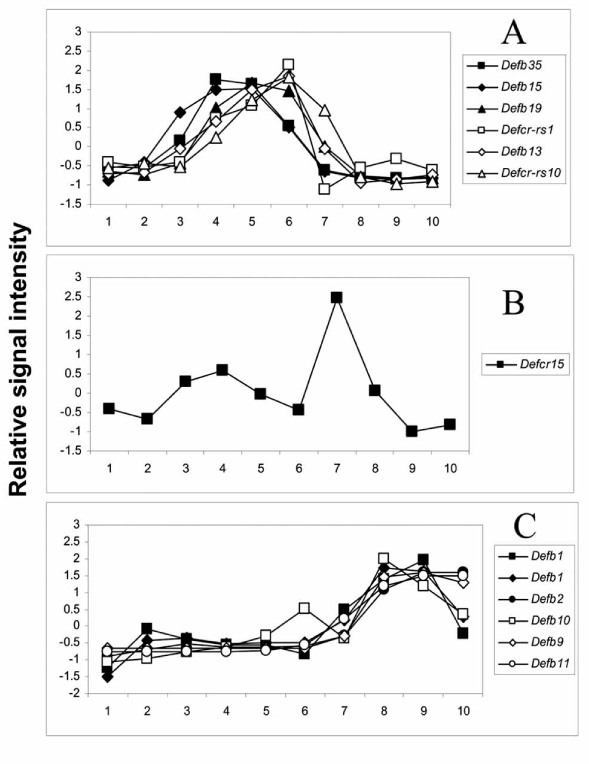
Segmental expression of defensin genes in the mouse epididymis. Gene Ontology annotation was used to select defensin genes as described [13]. Relative gene expressions are displayed for 12 defensin genes represented by 13 probe sets. The values are Z-scored normalized average intensity. The relative expression pattern of the 13 probe sets occurs in three distinct groups. A) One group consists of Defb35, Defb15, Defb19, Defcr-rs1, Defb13 and Defcr-rs10. B) A second group consists of Defcr15. C) A third group consists of Defb1 (two probe sets), Defb2, Defb10, Defb9 and Defb11 [13].
SIDE EFFECT PROFILING
The field of toxicogenomics has emerged from the combination of classical toxicology and gene expression profiling. Understanding the potential toxicological properties of a chemical entity at the transcriptome level of a target organ or cell, could be termed toxicogenomics or side-effect profiling. For the purpose of this review, our discussion of the field will be limited to the literature from the field of DNA microarray based expression profiling for toxicological studies. Use of other techniques such as differential display, subtractive hybridization, SAGE, MPSS, and proteomics in toxicogenomics has been reviewed elsewhere [17]. Although pharmacogenomics is useful throughout the drug development, its greatest impact currently is in designing of clinical trials [18]. It is helping scientists to identify groups of patients that are most likely to benefit from the drug as well as groups of patients that can potentially experience the worst side effects / toxicity outcomes.
With the rising costs of drug development, there is a need to conduct toxicity studies at earlier stages and on as many potential drug candidates as possible. With the increases in the size of chemical libraries, there has been an exponential increase in compounds selected for toxicity testing. Since later stages of development are immensely expensive, it is important to identify the most promising drug candidates, with highest safety margins, early on in the drug development process. Gene expression studies have been used to determine the mechanism of toxicity of drug candidates as well as in a predictive mode to identify potential safety liabilities [19]. Thus, toxicogenomics could play a major role in prioritizing lead compounds that could be advanced for further development. Datasets from toxicogenomic studies have increasingly become part of recent submissions to various regulatory agencies [20].
Understanding the molecular mechanism of toxicity is critical for monitoring, managing and determining the acceptability levels of side effects for each therapeutic indication [21]. Compared to the classical methods of toxicology based on organ pathology and survival endpoints, toxicogenomics has the added power to detect dysregulation of cellular biological pathways, which could lead to the elucidation of the mechanism of toxicity [22]. How critical and where in the drug discovery pipeline a particular compound is, and availability of backup compounds with similar efficacies but with better liability profiles based on the gene expression datasets etc., will determine whether the mechanism of toxicity will be fully explored. If several backup compounds with similar pharmacological profile are available at an early stage of the drug discovery program, it may not be worthwhile investigating the mechanism of toxicity of a single compound [19].
Changes in a small number of signature gene sets have been used to match or differentiate the unknown mechanism of toxicity of new chemical entities to compounds with known safety profiles and mechanisms of actions [17, 23–26]. Investigators at Abbott laboratories have used this method to identify the mechanism of hepatotoxicity of a novel NFkB antagonist (A-277249) using its signature gene expression pattern similar to that of Aroclor 1254 and 3-methylcholanthrene, two activators of the aryl hydrocarbon receptor (AhR). In this toxicogenomic study, the authors established that the toxicity of A-277249 was through the activation of AhR [27–29].
Genomic data have great predictive value but may not correlate directly to toxicity outcomes and hence, when viewed in isolation, may lead to more questions than answers [19, 30, 31]. Dysregulation of genes associated with a new chemical entity may reflect complex pharmacological, biochemical and physiological processes interacting with one another resulting in a multitude of toxicological or beneficial endpoints [19, 32]. It is therefore very important to separate the direct effect of the drug on the toxicity being investigated from any secondary effects [19]. As an emerging tool in drug development, side-effect profiling databases need to reach a critical threshold with datasets from numerous reference compounds, to deliver the promised predictive potential of the technique. Ideal toxicogenomic databases should consist of many known pharmaceutical compounds, toxins and control compounds, at multiple doses and time points, with biological replicates for each condition [21, 27, 30]. Such libraries would permit hierarchical clustering of the new chemical entity with known compounds to understand potential toxicity pathways, risks and benefits.
Assessment of reproductive toxicity is an essential component of the drug development process [21]. Prominent morphological changes associated with testicular toxicity may not be manifested at early stages of most studies. Several groups have efficiently used gene expression profiling to understand and potentially predict the mechanism of testicular toxicity [33–36]. For example, expression of several genes such as HSP70.2, SP22 etc., with known functions in spermatogenesis were shown to be dysregulated in mice treated with bromochloroacetic acid [36]. Genes involved in Sertoli cell-germ cell interactions were also significantly affected in the mice treated with this toxicant. Dysregulation of the Fas – Fas ligand system that leads to germ cell death was demonstrated as the leading mechanism of testicular toxicity in gene expression studies using several toxicants [33]. Such studies are powerful in predicting potential reproductive safety liabilities faster and more efficiently.
Toxicogenomics to screen compounds at early stages of drug development, applied to in vitro cultured cells or tissues, has the potential to significantly increase throughput and cut costs to the industry [29]. Primary hepatocytes in short-term cultures have been shown to retain their drug metabolic pathways [37–40]. Human primary hepatocytes are difficult to obtain and exhibit significant differences in gene expression profiles depending on the lifestyle of the donor [19]. Primary rat hepatocytes have been used in gene expression profiling to understand the mechanism of hepatotoxicity [26, 41]. Several hepatic cell lines have also been used in toxicogenomic studies [42] and the toxicogenomics of several nuclear receptor agonists were reviewed recently [43].
Expression analysis of the cytochrome P450 family of genes that are involved in drug metabolism is an important focus in toxicogenomics [18]. Variations in CYP2D6, one of the enzymes in the P450 super family, accounts for large variations in drug tolerability among patients [18, 44]. Also, genetic variations in the enzyme UGT1A1 have been shown to be responsible for the toxicity associated with chemotherapeutic drug Irinotecan [45, 46]. FDA has recently approved the use of a new gene chip called the AmpliChip to test the genetic variations in two drug metabolizing enzymes CYP2D6 and CYP2C19 [47].
In a toxicogenomic study, Kier et.al, treated primary human hepatocytes with three thiazolidinediones (TZDs) used in type II diabetes treatments [48]. One of the drugs, troglita-zone, results in hepatotoxicity in a small percentage of patients and has been pulled from the market. Microarray analysis of the treated hepatocytes demonstrated that troglitazone, unlike rosiglitazone or pioglitazone, leads to the dys-regulation of a number of genes in the hepatocytes [48]. These examples highlight the power of toxicogenomics in preclinical drug safety evaluation using in vitro systems.
Gene expression changes may occur outside the drug target tissue and these surrogate tissues could be used to detect toxicological biomarkers. Changes that occur in the transcriptome of PBMCs (peripheral blood mononuclear cells) or in mucosal epithelial cells have been used in toxicogenomics [19]. PBMCs are especially useful in studying allergic reactions to drugs by looking at the expression of cytokines and toxicity biomarkers [19]. Gene expression analysis is also broadening the understanding of immunological processes that occur upon immunotoxicant exposure. Recent developments in the use of toxicogenomics for the assessment of immunotoxicity was reviewed by Baken and coworkers [49].
One of the most challenging aspects of realizing the potential of toxicogenomics involves establishing a flexible and comprehensive knowledge base of gene expression and its integration to classical ADME, histopathology, clinical chemistry, and toxicity data [30, 31]. Several toxicogenomics databases have recently been established both in the private and public domains. These databases have utilized the standard of Minimum Information About a Microarray Experiment (MIAME) as a guide [30]. Commercial databases such as Gene Logic (http://www.genelogic.com), and Iconix Pharmaceuticals (http://www.iconixpharm.com) have gained popularity and are widely used by pharmaceutical companies. A comprehensive list of databases is listed in a recent review [30]. Recently, the architecture and construction of DrugMatrix, the Iconix database was described. DrugMatrix is a comprehensive toxicogenomic database based on short term repeat-dose rat studies for marketed and withdrawn drugs, toxicants and reference compounds [50]. Recent release of the DrugMatrix contains toxicogenomic data on over 600 compounds [50]. This database combines standardized toxicology data with gene expression profiling from seven different tissues (bone marrow, heart, intestine, liver, kidney, spleen and thigh muscle) and rat primary hepatocytes.
In recent years, the FDA and pharmaceutical companies have recognized the importance of developing a framework for the submission of toxicogenomic datasets for regulatory review. Several mock submissions were made recently, containing microarray data along with corresponding classical toxicology data in order to familiarize the Nonclinical Pharmacogenomic subcommittee (NPSC) which is comprised of CDER pharmacology and toxicology reviewers and researchers [20]. Proposed submission guidelines and quality control including MIAME/MINTox (minimum information about a microarray experiment / minimum information needed for a toxicology experiment) were discussed recently in a report [20]. In addition to these ongoing efforts to unify the regulatory submission guidelines, pharmaceutical companies have been voluntarily providing toxicogenomic datasets as part of IND and NDA submissions [19, 20, 30].
One of the mock submissions included studies to assess the data quality for a single agent used to inhibit HMG-CoA reductase. Female rats were dosed orally at three daily dose levels for 1 month and the data generated included classical toxicological endpoints combined with RatGenome U34 Microarray (Affymetrix) gene expression analysis of liver samples collected 1, 7, and 30 days of treatment. This submission included controls for efficiency of RNA preparation and processing as well as sensitivity of hybridization. Quality control metrics used in the studies and the need for simplified SOPs were discussed [20]. A second mock submission assessed the effects of several compounds in a class on a single tissue. Marketed PPARalpha agonists, fenofibrate, ciprofibrate, used to treat dyslipidemia and a proprietary pan-PPAR agonist were administered to cynomologus monkeys. Liver samples from the animals were studied using HGU95Av2 arrays. One of the assumptions that the sponsor made was that the human based probe sets will hybridize with the correct homologous monkey sequences. In this study aimed to understand the toxicity associated with the PPARpan compound, by principal component analysis of the array data, PPARpan effects appeared to cluster closely with higher dose levels of fenofibrate. Technical challenges associated with sample quality, species and genetic variability etc were also addressed in this study.
The third mock submission, potential toxicity of a selective serotonin reuptake inhibitor (SSRI) was assessed in male rats treated daily for 5 days with vehicle or two dose levels of the test compound. Higher dose was based on the MTD for decrease in body weight gain. The data was compared to the microarray data for 600 known compounds. The data found that the SSRI was non-hepatotoxic. The sponsors were able to identify drug signature of the SSRI compound that delineated it from other compounds that predicted the effects confirmed by conventional toxicological methods. This mode of comparative expression analysis may be very valuable and effective in future IND/NDA submissions. The NPSC anticipates increased use of contextual databases as a source for the drug signatures that would become validated with future biomarkers of toxicity [20].
PHARMACOGENOMICS
It is generally recognized that the failure rate for drugs in clinical development is unacceptably high and is a major factor contributing to the perceived negative attitudes towards the pharmaceutical industry’s recent low productivity. Although termination of drug development can occur at any stage, because of the large expense associated with performing Phase III studies, these are the most expensive. Close to half of all drugs entering Phase III fail due to either lack of efficacy or toxicity issues. Drugs that pass this hurdle and make it to the market, where they are prescribed to a larger patient pool, occasionally show the emergence of unanticipated variations in efficacy and safety. This variability in drug response can include such extremes as a complete absence of efficacy or the manifestation of a fatal adverse drug reaction (ADR). The frequency of ADRs from prescribed drugs has been estimated at over 2 million cases per year, resulting in over 100,000 fatalities [51]. Failures in efficacy are evidenced by the treatment of depression with SSRIs, where only 60% of patients exhibit an adequate therapeutic response [52]. These “kinks” in the drug discovery process have prompted an expedited maturation of the field of pharmacogenomics (PGx), mediated by collaboration between the drug makers and the FDA.
PGx in its broadest sense describes the profiling of inter-individual differences in response to drug treatment via the use of a comprehensive set of profiling technologies. This can include the well-established oligonucleotide microarrays [53], which query mRNA levels, as well as other profiling methods that examine DNA single nucleotide polymorphisms (SNPs) or differences between individuals, commonly referred to as pharmocogenetic (PGt) analysis. A more inclusive definition of PGx would encompass the analysis of other features of mRNA (i.e. processing, microRNA levels etc.) and DNA (i.e. insertions, deletions, rearrangements, copy number etc.). Additional layers of comparative analysis, which can serve to enhance the interpretation of the drug-mediated response but are not within the scope of PGx include the measurement of protein levels (pharmacoproteomics) and metabolite levels (pharmacome-tabonomics). In combination, the information from all these profiling methods would provide a more comprehensive picture of the consequences of drug treatment at the molecular level. In practice, however, only the oligonucleotide microar- ray and SNP profiling technologies have advanced to the point where large scale analysis of multiple samples is economically and technically feasible.
PGx is an area where improvements in gene expression profiling (GEP) have made a tremendous impact on the quantity and quality of data generated. Some notable advances include the advent of higher density chips, more accurate annotation and the ability to work with smaller quantities of experimental samples. These advances have been expedited by large scale sequencing projects that have led to the elucidation of several mammalian genomes. Despite the progress in annotating the sequences tiled on the chips there still remain gaps in our knowledge concerning the encoded function of many of these sequences. This information will become valuable for interpreting the significance of co-regulated gene networks and thereby elucidating new signaling pathways associated with a pathophysiology or a drug response.
The ultimate goal of PGx is to provide predictive information, in the form of a biomarker(s), concerning the efficacy and safety of a pharmacological agent, for a specific individual. The road leading to this destination now begins very early in the drug discovery process, contains numerous twists and turns and frequent dead ends. The challenges associated with this journey are predetermined by several factors, including complexity of the diseased state, degree of target resolution, and accessibility of patient samples and availability of relevant preclinical models. Nevertheless, strategic applications of PGx can serve to streamline drug discovery, from lead compound selection through effective clinical trial execution and beyond. Examples of successful applications of PGx at different stages of drug discovery will be highlighted to reveal the potential of this promising technology.
For each pharmacological treatment of a particular cell type it is possible to derive a characteristic signature, represented by a collection of differentially expressed genes, which may be predictive of responsiveness or outcome. An elegant marriage of PGx and biological state was described recently [54] and will pave the way for more extensive studies aimed at establishing predictive databases for drug classes. A similar approach was used for establishing a predictive expression profile fingerprint for discriminating between different psychoactive drug classes [55]. Continued expansion of these types of studies will enhance our ability to make predictions about drug action at the cellular level, thereby serving to triage and catalogue lead compounds prior to testing against in vivo models.
Applications of PGx in a preclinical model of allergic asthma serve to illustrate a suitable approach for identifying biomarkers, defining the pathways involved and identifying additional targets [56]. These investigators were able to show an IL-13-induced gene profile that overlapped with genes induced by ovalbumin (OVA). Furthermore, co-treatment with a soluble form of the IL-13 receptor significantly reduced a subset of the OVA-induced genes. The challenge for all biomarkers identified in either in vitro or preclinical in vivo models is demonstrating relevance in the clinical setting.
The successful application of PGx for predicting outcomes of efficacy, in a clinical setting, is evidenced by its use with oncology therapeutics. Gene expression profiling was successfully applied to identify predictive gene signatures for drug response with renal cell carcinoma [57] and multiple myeloma [58]. This success is attributed, in part, to the nature of the disease, which provides ready access to clinical samples for profiling studies. Other disease states are not as permissive in terms of providing access to affected tissue. As such, the identification of a relevant biomarker from a pre-clinical model can be a challenging endeavor. A potential solution to this dilemma is provided by the use of peripheral blood mononuclear cells as a source for the identification of surrogate markers [59].
The availability of an early surrogate marker associated with disease progression or remission would allow for earlier decisions about compounds, especially in cases where the compound appears destined to fail. In this particular situation the decision to terminate a study will have to be guided by the degree of confidence with the prognostic marker and how accurately it associates/correlates with disease progression. Beyond the science, economic factors will ultimately determine the degree to which PGx will be used for personalized medicine, based on cost-effectiveness [60].
BIOMARKERS
As previously discussed, the cost of developing new drugs is increasing at an alarming rate and most of those increases have been attributed to increases in clinical development (phase I, II and III). This increased clinical cost is reflected both by expenses specifically associated with the clinical trials but also with the high rate of drug failure in phase II. In response to all of these pressures, the industry has been taking a closer look at the way they are doing business, and a significant area of focus has been on improving the success rate of drug development in the clinical phases. This can, in theory, be done by two approaches. First, targets/therapeutics in preclinical development need to be assessed with additional stringency so that decisions with regard to project viability in clinical trials can be made sooner rather than later. This would ensure that only those targets/therapeutics with the greatest possibility of success be put into phase I. Second, once targets/therapeutics are in phase I, there needs to be strategies in place to determine whether these targets/therapeutics can move forward to phase II and IIa (i.e., safety, efficacy, proof of concept).
One approach to address these issues both at the preclinical and clinical development stages is to develop strategies to assess, as early as possible, whether a therapeutic agent is having a desired profile with respect to both safety and modulating its target and altering the disease state in a fashion that would predict an ultimate successful therapeutic outcome. Although there are plenty of examples where success in preclinical animal models does not translate to humans, the increasing use of biomarkers and diagnostic markers in drug development will help, in theory, to reduce this problem. Biomarkers have been defined as either physical endpoints (e.g., blood pressure; heart rate; pupil dilation) or laboratory measurements (e.g., cholesterol) capable of being detected in association with both the normal biological process and a particular pathological process that could have putative diagnostic and/or prognostic utility [61].
In the drug development process biomarkers are objective measurements used to assess whether a particular therapeutic is modulating the target for which it was developed, and is either treating the symptom or the progress of disease. In addition, biomarkers can be used to assess pharmacokinetic, pharmacodynamic and toxicological characteristics of a particular therapeutic entity. The identification and development of relevant biomarkers for these purposes can, and will, aid in the internal decision making process regarding future preclinical or clinical go/no go decision points, and, in theory, should reduce drug development costs since decisions to abort programs will be made early.
The fields of genomics and proteomics have contributed to the identification and development of biomarkers that are of diverse molecular nature, ranging from simple electrolytes and amino acids to proteins and other biomolecules. The use of these disciplines to screen normal and diseased tissues/fluids to identify differences in gene expression can ultimately yield specific fingerprints that may be associated with critical pathways leading to the diseased states. Likewise, comparison of tissues/fluids from normal and drug-treated individuals can help to identify biomarkers that reflect drug interactions with targets, as well as drug toxicity. Although genomics and proteomics holds great promise for the development of biomarkers it must be emphasized that the establishment of a validated biomarker represents a significant effort since specificity and resolution are key foundations of success.
CONCLUSIONS
The breadth of array based observations in high probability guarantees surprising findings. In addition, since arrays often contain probes for genes of unknown function, gene profiling analysis not only sheds light on new genes involved in a pathway but also produces potential drug targets or bio-markers that can be used in a predictive or diagnostic fashion. Mining and compiling expression databases generate gene expression patterns in human diseases and identify gene expression signatures that correlate with specific clinical outcomes. The knowledge of these signatures could be translated into either full-fledged clinical diagnostic tests, or novel targets to develop therapeutics.
Despite the wide use of array technology, doubts still exist regarding the reproducibility and variability of microarray data, and the compatibility of results on different platforms. Some of these issues are arising due to inter-laboratory variations in experimental design and sample preparation as well as methods for data acquisition, statistical analysis and data interpretation. To this end, the microarray community and FDA have formed a consortium, the microarray quality control (MAQC) project, to develop a set of criteria to assure data quality, identify factors affecting quality, and standardize microarray procedures. Once the recommendations from this project are finalized and implemented, it is expected that quality control metrics and thresholds for objective assessment of the achievable performances by different microarray platforms and evaluation of merits and limitations of various data analysis methods can be established. This would ensure that the biological interpretation and decision making is based on reliable and reproducible data.
By embracing the power of expression profiling and transcriptome analysis, drug developers will be able to increase the chance of significant advances in efficacy of the novel drugs in addition to having the potential for reduced adverse events. This improved safety profile will play an important role in bringing forward next generation medicines for a variety of ailments.
REFERENCES
- 1.Oeppen J, Vaupel JW. DEMOGRAPHY: Enhanced: Broken Limits to Life Expectancy. Science. 2002;296:1029–1031. doi: 10.1126/science.1069675. [DOI] [PubMed] [Google Scholar]
- 2.Janssen P, Audit B, Cases I, Darzenta N, Goldovsky L, Kunin V, Nuria LG, Manuel PAJBPLJ, Tsoka S, Ouzounis C. Beyond 100 Genomes. Genome Biol. 2003;4:402–505. doi: 10.1186/gb-2003-4-5-402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kane MD, Jatkoe TA, Stumpf CR, Lu J, Thomas JD, Madore SJ. Assessment of the sensitivity and specificity of oligonucleotide (50mer) microarrays. Nucl Acids Res. 2000;28:4552–4557. doi: 10.1093/nar/28.22.4552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beaucage SL. Strategies in the Preparation of DNA Oligonucleotide Arrays for Diagnostic Applications. Curr Med Chem. 2001;8:1213–1244. doi: 10.2174/0929867013372463. [DOI] [PubMed] [Google Scholar]
- 5.Ganter B, Tugendreich S, Pearson CI, Ayanoglu E, Baumhueter S, Bostian KA, Brady L, Browne LJ, Calvin JT, Day GJ, Breckenridge N, Dunlea S, Eynon BP, Furness LM, Ferng J, Fielden MR, Fujimoto SY, Gong L, Hu C, Idury R, Judo MSB, Kolaja KL, Lee MD, McSorley C, Minor JM, Nair RV, Natsoulis G, Nguyen P, Nicholson SM, Pham H, Roter AH, Sun D, Tan S, Thode S, Tolley AM, Vladimirova A, Yang J, Zhou Z, Jarnagin K. Development of a large-scale chemogenomics database to improve drug candidate selection and to understand mechanisms of chemical toxicity and action. J Biotechnol. 2005;119:219–244. doi: 10.1016/j.jbiotec.2005.03.022. [DOI] [PubMed] [Google Scholar]
- 6.Fielden MR, Eynon BP, Natsoulis G, Jarnagin K, Banas D, Kolaja KL. A Gene Expression Signature that Predicts the Future Onset of Drug-Induced Renal Tubular Toxicity. Toxicol Pathol. 2005;33:675–683. doi: 10.1080/01926230500321213. [DOI] [PubMed] [Google Scholar]
- 7.Lamb J, Crawford ED, Peck D, Modell JW, Blat IC, Wrobel MJ, Lerner J, Brunet JP, Subramanian A, Ross KN, Reich M, Hieronymus H, Wei G, Armstrong SA, Haggarty SJ, Clemons PA, Wei R, Carr SA, Lander ES, Golub TR. The Connectivity Map: Using Gene-Expression Signatures to Connect Small Molecules, Genes, and Disease. Science. 2006;313:1929–1935. doi: 10.1126/science.1132939. [DOI] [PubMed] [Google Scholar]
- 8.Shaw KJ, Morrow BJ. Transcriptional profiling and drug discovery. Cur Opi Pharmacol. 2003;3:508–512. doi: 10.1016/s1471-4892(03)00110-3. [DOI] [PubMed] [Google Scholar]
- 9.Korkola JE, Blaveri E, DeVries S, Moore DHI, Hwang ES, Yunn-Yi C, Estep ALH, Chew KL, Jensen RH, Waldman FM. Identification of a Robust Gene Signature that Predicts Breast Cancer Outcome in Independent Data Sets. BMC Cancer. 2007;11:61–74. doi: 10.1186/1471-2407-7-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Johnston DS, Bai Y, Kopf GS. Turner TT, Hinton BT. Epididymis III: The third international conference on the epididymis. Charlottesville: The Van Doren Company; 2002. Utilization of Human Genome Databases in Strategies for Contraceptive Development; pp. 256–270. [Google Scholar]
- 11.Pan H, O’Brien MJ, Wigglesworth K, Eppig JJ, Schultz RM. Transcript profiling during mouse oocyte development and the effect of gonadotropin priming and development in vitro. Develop Biol. 2005;286:493–506. doi: 10.1016/j.ydbio.2005.08.023. [DOI] [PubMed] [Google Scholar]
- 12.Shima JE, McLean DJ, McCarrey JR, Griswold MD. The Murine Testicular Transcriptome: Characterizing Gene Expression in the Testis During the Progression of Spermatogenesis. Biol Reprod. 2004;71:319–330. doi: 10.1095/biolreprod.103.026880. [DOI] [PubMed] [Google Scholar]
- 13.Johnston DS, Jelinsky SA, Bang HJ, DiCandeloro P, Wilson E, Kopf GS, Turner TT. The Mouse Epididymal Transcriptome: Transcriptional Profiling of Segmental Gene Expression in the Epididymis. Biol Reprod. 2005;73:404–413. doi: 10.1095/biolreprod.105.039719. [DOI] [PubMed] [Google Scholar]
- 14.Jelinsky SA, Turner TT, Bang HJ, Finger JN, Solarz MK, Wilson E, Brown EL, Kopf GS, Johnston DS. The Rat Epididymal Transcriptome: Comparison of Segmental Gene Expression in the Rat and Mouse Epididymides. Biol Reprod. 2007;76:561–570. doi: 10.1095/biolreprod.106.057323. [DOI] [PubMed] [Google Scholar]
- 15.Luzzi VI, Holtschlag V, Watson MA. Gene expression profiling of primary tumor cell populations using laser capture microdis-section, RNA transcript amplification, and GeneChip microarrays. Meth Mol Biol. 2005;293:187–207. doi: 10.1385/1-59259-853-6:187. [DOI] [PubMed] [Google Scholar]
- 16.Liang WS, Dunckley T, Beach TG, Grover A, Mastroeni D, Walker DG, Caselli RJ, Kukull WA, McKeel D, Morris JC, Hulette C, Schmechel D, Alexander GE, Reiman EM, Rogers J, Stephan DA. Gene expression profiles in anatomically and functionally distinct regions of the normal aged human brain. Physiol Genomics. 2007;28:311–322. doi: 10.1152/physiolgenomics.00208.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.de Longueville F, Atienzar FA, Marcq L, Dufrane S, Evrard S, Wouters L, Leroux F, Bertholet V, Gerin B, Whomsley R, Arnould T, Remacle J, Canning M. Use of a low-density microarray for studying gene expression patterns induced by hepato-toxicants on primary cultures of rat hepatocytes. Toxicol Sci. 2003;75:378–392. doi: 10.1093/toxsci/kfg196. [DOI] [PubMed] [Google Scholar]
- 18.Service RF. Pharmacogenomics. Going from genome to pill. Science. 2005;308:1858–1860. doi: 10.1126/science.308.5730.1858. [DOI] [PubMed] [Google Scholar]
- 19.Yang Y, Blomme EA, Waring JF. Toxicogenomics in drug discovery: from preclinical studies to clinical trials. Chem Biol Interact. 2004;150:71–85. doi: 10.1016/j.cbi.2004.09.013. [DOI] [PubMed] [Google Scholar]
- 20.Leighton JK, Brown P, Ellis A, Harlow P, Harrouk W, Pine PS, Robison T, Rosario L, Thompson K. Workgroup report: Review of genomics data based on experience with mock submissions—view of the CDER Pharmacology Toxicology Nonclinical Pharmacogenomics Subcommittee. Environ Health Perspect. 2006;114:573–578. doi: 10.1289/ehp.8318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Khor TO, Ibrahim S, Kong AN. Toxicogenomics in drug discovery and drug development: potential applications and future challenges. Pharm Res. 2006;23:1659–1664. doi: 10.1007/s11095-006-9003-8. [DOI] [PubMed] [Google Scholar]
- 22.de Longueville F, Bertholet V, Remacle J. DNA microarrays as a tool in toxicogenomics. Comb Chem High Through Screen. 2004;7:207–211. doi: 10.2174/1386207043328841. [DOI] [PubMed] [Google Scholar]
- 23.Hamadeh HK, Bushel PR, Jayadev S, DiSorbo O, Bennett L, Li L, Tennant R, Stoll R, Barrett JC, Paules RS, Blanchard K, Afshari CA. Prediction of compound signature using high density gene expression profiling. Toxicol Sci. 2002;67:232–240. doi: 10.1093/toxsci/67.2.232. [DOI] [PubMed] [Google Scholar]
- 24.Hamadeh HK, Bushel PR, Jayadev S, Martin K, DiSorbo O, Sieber S, Bennett L, Tennant R, Stoll R, Barrett JC, Blanchard K, Paules RS, Afshari CA. Gene expression analysis reveals chemical-specific profiles. Toxicol Sci. 2002;67:219–231. doi: 10.1093/toxsci/67.2.219. [DOI] [PubMed] [Google Scholar]
- 25.Morgan KT. Gene expression analysis reveals chemical-specific profiles. Toxicol Sci. 2002;67:155–156. doi: 10.1093/toxsci/67.2.155. [DOI] [PubMed] [Google Scholar]
- 26.de Longueville F, Surry D, Meneses-Lorente G, Bertholet V, Talbot V, Evrard S, Chandelier N, Pike A, Worboys P, Rasson JP, Le Bourdelles B, Remacle J. Gene expression profiling of drug metabolism and toxicology markers using a low-density DNA microarray. Biochem Pharmacol. 2002;64:137–149. doi: 10.1016/s0006-2952(02)01055-9. [DOI] [PubMed] [Google Scholar]
- 27.Waring JF, Cavet G, Jolly RA, McDowell J, Dai H, Ciurlionis R, Zhang C, Stoughton R, Lum P, Ferguson A, Roberts CJ, Ulrich RG. Development of a DNA microarray for toxicology based on hepatotoxin-regulated sequences. EHP Toxicogenom. 2003;111:53–60. [PubMed] [Google Scholar]
- 28.Waring JF, Gum R, Morfitt D, Jolly RA, Ciurlionis R, Heindel M, Gallenberg L, Buratto B, Ulrich RG. Identifying toxic mechanisms using DNA microarrays: evidence that an experimental inhibitor of cell adhesion molecule expression signals through the aryl hydrocarbon nuclear receptor. Toxicology. 2002;182:537–550. doi: 10.1016/s0300-483x(02)00477-8. [DOI] [PubMed] [Google Scholar]
- 29.Yang Y, Abel SJ, Ciurlionis R, Waring JF. Development of a toxicogenomics in vitro assay for the efficient characterization of compounds. Pharmacogenomics. 2006;7:177–186. doi: 10.2217/14622416.7.2.177. [DOI] [PubMed] [Google Scholar]
- 30.Boverhof DR, Zacharewski TR. Toxicogenomics in risk assessment: applications and needs. Toxicol Sci. 2006;89:352–360. doi: 10.1093/toxsci/kfj018. [DOI] [PubMed] [Google Scholar]
- 31.Lord PG, Nie A, McMillian M. Application of genomics in preclinical drug safety evaluation. Basic Clin Pharmacol Toxicol. 2006;98:537–546. doi: 10.1111/j.1742-7843.2006.pto_444.x. [DOI] [PubMed] [Google Scholar]
- 32.Guerreiro N, Staedtler F, Grenet O, Kehren J, Chibout SD. Toxicogenomics in drug development. Toxicol Pathol. 2003;31:471–479. doi: 10.1080/01926230390224656. [DOI] [PubMed] [Google Scholar]
- 33.Lee J, Richburg JH, Shipp EB, Meistrich ML, Boekelheide K. The Fas system, a regulator of testicular germ cell apoptosis, is differentially up-regulated in Sertoli cell versus germ cell injury of the testis. Endocrinology. 1999;140:852–858. doi: 10.1210/endo.140.2.6479. [DOI] [PubMed] [Google Scholar]
- 34.Richburg JH, Nanez A, Williams LR, Embree ME, Boekel-heide K. Sensitivity of testicular germ cells to toxicant-induced apoptosis in gld mice that express a nonfunctional form of Fas ligand. Endocrinology. 2000;141:787–793. doi: 10.1210/endo.141.2.7325. [DOI] [PubMed] [Google Scholar]
- 35.Mantovani A, Maranghi F. Risk assessment of chemicals potentially affecting male fertility. Contraception. 2005;72:308–313. doi: 10.1016/j.contraception.2005.04.014. [DOI] [PubMed] [Google Scholar]
- 36.Richburg JH, Johnson KJ, Schoenfeld HA, Meistrich ML, Dix DJ. Defining the cellular and molecular mechanisms of toxicant action in the testis. Toxicol Lett. 2002;135:167–183. doi: 10.1016/s0378-4274(02)00254-0. [DOI] [PubMed] [Google Scholar]
- 37.Boess F, Kamber M, Romer S, Gasser R, Muller D, Albertini S, Suter L. Gene expression in two hepatic cell lines, cultured primary hepatocytes, and liver slices compared to the in vivo liver gene expression in rats: possible implications for toxicogenomics use of in vitro systems. Toxicol Sci. 2003;73:386–402. doi: 10.1093/toxsci/kfg064. [DOI] [PubMed] [Google Scholar]
- 38.Ruepp S, Boess F, Suter L, de Vera MC, Steiner G, Steele T, Weiser T, Albertini S. Assessment of hepatotoxic liabilities by transcript profiling. Toxicol Appl Pharmacol. 2005;207:161–170. doi: 10.1016/j.taap.2005.05.008. [DOI] [PubMed] [Google Scholar]
- 39.Li AP, Reith MK, Rasmussen A, Gorski JC, Hall SD, Xu L, Kaminski DL, Cheng LK. Primary human hepatocytes as a tool for the evaluation of structure-activity relationship in cyto-chrome P450 induction potential of xenobiotics: evaluation of ri-fampin, rifapentine and rifabutin. Chem Biol Interact. 1997;107:17–30. doi: 10.1016/s0009-2797(97)00071-9. [DOI] [PubMed] [Google Scholar]
- 40.Ulrich RG, Bacon JA, Cramer CT, Peng GW, Petrella DK, Stryd RP, Sun EL. Cultured hepatocytes as investigational models for hepatic toxicity: practical applications in drug discovery and development. Toxicol Lett. 1995;83:107–115. doi: 10.1016/0378-4274(95)03547-8. [DOI] [PubMed] [Google Scholar]
- 41.Waring JF, Ciurlionis R, Jolly RA, Heindel M, Ulrich RG. Microarray analysis of hepatotoxins in vitro reveals a correlation between gene expression profiles and mechanisms of toxicity. Toxicol Lett. 2001;120:359–368. doi: 10.1016/s0378-4274(01)00267-3. [DOI] [PubMed] [Google Scholar]
- 42.Hong Y, Muller UR, Lai F. Discriminating two classes of toxicants through expression analysis of HepG2 cells with DNA arrays. Toxicol In Vitro. 2003;17:85–92. doi: 10.1016/s0887-2333(02)00122-4. [DOI] [PubMed] [Google Scholar]
- 43.Ulrich RG. The toxicogenomics of nuclear receptor agonists. Curr Opin Chem Biol. 2003;7:505–510. doi: 10.1016/s1367-5931(03)00080-2. [DOI] [PubMed] [Google Scholar]
- 44.Cascorbi I. Genetic basis of toxic reactions to drugs and chemicals. Toxicol Lett. 2006;162:16–28. doi: 10.1016/j.toxlet.2005.10.015. [DOI] [PubMed] [Google Scholar]
- 45.Goetz MP, Erlichman C, Windebank AJ, Reid JM, Sloan JA, Atherton P, Adjei AA, Rubin J, Pitot H, Galanis E, Ames MM, Goldberg RM. Phase I and pharmacokinetic study of two different schedules of oxaliplatin, irinotecan, Fluorouracil, and leucovorin in patients with solid tumors. J Clin Oncol. 2003;21:3761–3769. doi: 10.1200/JCO.2003.01.238. [DOI] [PubMed] [Google Scholar]
- 46.Goetz MP, Grothey A. Developments in combination chemotherapy for colorectal cancer. Expert Rev Anticancer Ther. 2004;4:627–637. doi: 10.1586/14737140.4.4.627. [DOI] [PubMed] [Google Scholar]
- 47.Heller T, Kirchheiner J, Armstrong VW, Luthe H, Tzvetkov M, Brockmoller J, Oellerich M. AmpliChip CYP450 GeneChip: a new gene chip that allows rapid and accurate CYP2D6 genotyping. Ther Drug Monit. 2006;28:673–677. doi: 10.1097/01.ftd.0000246764.67129.2a. [DOI] [PubMed] [Google Scholar]
- 48.Kier LD, Neft R, Tang L, Suizu R, Cook T, Onsurez K, Tiegler K, Sakai Y, Ortiz M, Nolan T, Sankar U, Li AP. Applications of microarrays with toxicologically relevant genes (tox genes) for the evaluation of chemical toxicants in Sprague Dawley rats in vivo and human hepatocytes in vitro. Mutat Res. 2004;549:101–113. doi: 10.1016/j.mrfmmm.2003.11.015. [DOI] [PubMed] [Google Scholar]
- 49.Baken KA, Vandebriel RJ, Pennings JL, Kleinjans JC, van Loveren H. Toxicogenomics in the assessment of immunotoxicity. Methods. 2007;41:132–141. doi: 10.1016/j.ymeth.2006.07.010. [DOI] [PubMed] [Google Scholar]
- 50.Ganter B, Snyder RD, Halbert DN, Lee MD. Toxicogenomics in drug discovery and development: mechanistic analysis of compound/class-dependent effects using the DrugMatrix database. Pharmacogenomics. 2006;7:1025–1044. doi: 10.2217/14622416.7.7.1025. [DOI] [PubMed] [Google Scholar]
- 51.Lazarou J, Pomeranz BH, Corey PN. Incidence of adverse drug reactions in hospitalized patients: a meta-analysis of prospective studies. JAMA. 1998;279:1200–1205. doi: 10.1001/jama.279.15.1200. [DOI] [PubMed] [Google Scholar]
- 52.Nelson JC. A review of the efficacy of serotonergic and noradrenergic reuptake inhibitors for treatment of major depression. Biol Psychiatry. 1999;46:1301–1308. doi: 10.1016/s0006-3223(99)00173-0. [DOI] [PubMed] [Google Scholar]
- 53.Lockhart DJ, Dong H, Byrne MC, Follettie MT, Gallo MV, Chee MS, Mittmann M, Wang C, Kobayashi M, Horton H, Brown EL. Expression monitoring by hybridization to high-density oligonucleotide arrays. Nat Biotechnol. 1996;14:1675–1680. doi: 10.1038/nbt1296-1675. [DOI] [PubMed] [Google Scholar]
- 54.Lamb J, Crawford ED, Peck D, Modell JW, Blat IC, Wrobel MJ, Lerner J, Brunet JP, Subramanian A, Ross KN, Reich M, Hieronymus H, Wei G, Armstrong SA, Haggarty SJ, Clemons PA, Wei R, Carr SA, Lander ES, Golub TR. The Connectivity Map: using gene-expression signatures to connect small molecules, genes, and disease. Science. 2006;313:1929–1935. doi: 10.1126/science.1132939. [DOI] [PubMed] [Google Scholar]
- 55.Gunther EC, Stone DJ, Gerwien RW, Bento P, Heyes MP. Prediction of clinical drug efficacy by classification of drug-induced genomic expression profiles in vitro. Proc Natl Acad Sci USA. 2003;100:9608–9613. doi: 10.1073/pnas.1632587100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Follettie MT, Ellis DK, Donaldson DD, Hill AA, Diesl V, DeClercq C, Sypek JP, Dorner AJ, Wills-Karp M. Gene expression analysis in a murine model of allergic asthma reveals overlapping disease and therapy dependent pathways in the lung. Pharmacogenomics J. 2006;6:141–152. doi: 10.1038/sj.tpj.6500357. [DOI] [PubMed] [Google Scholar]
- 57.Burczynski ME, Twine NC, Dukart G, Marshall B, Hidalgo M, Stadler WM, Logan T, Dutcher J, Hudes G, Trepicchio WL, Strahs A, Immermann F, Slonim DK, Dorner AJ. Transcriptional profiles in peripheral blood mononuclear cells prognostic of clinical outcomes in patients with advanced renal cell carcinoma. Clin Cancer Res. 2005;11:1181–1189. [PubMed] [Google Scholar]
- 58.Mulligan G, Mitsiades C, Bryant B, Zhan F, Chng WJ, Roels S, Koenig E, Fergus A, Huang Y, Richardson P, Trepicchio WL, Broyl A, Sonneveld P, Shaughnessy JD, Jr, Bergsagel PL, Schenkein D, Esseltine DL, Boral A, Anderson KC. Gene expression profiling and correlation with outcome in clinical trials of the proteasome inhibitor bortezomib. Blood. 2007;109:3177–3188. doi: 10.1182/blood-2006-09-044974. [DOI] [PubMed] [Google Scholar]
- 59.Burczynski ME, Dorner AJ. Transcriptional profiling of peripheral blood cells in clinical pharmacogenomic studies. Pharmacogenomics. 2006;7:187–202. doi: 10.2217/14622416.7.2.187. [DOI] [PubMed] [Google Scholar]
- 60.Veenstra DL, Higashi MK, Phillips KA. Assessing the cost-effectiveness of pharmacogenomics. AAPS PharmSci. 2000;2:E29. doi: 10.1208/ps020329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Colburn WA. Biomarkers in drug discovery and development: from target identification through drug marketing. J Clin Pharmacol. 2003;43:329–341. doi: 10.1177/0091270003252480. [DOI] [PubMed] [Google Scholar]