

Abstract
The development of microarray technology has had a significant impact on the genetic analysis of human disease. The recently developed single nucleotide polymorphism (SNP) array can be used to measure both DNA polymorphism and dosage changes. Our laboratory has applied SNP microarray analysis to uncover frequent uniparental disomies and sub-microscopic genomic copy number gains and losses in different cancers. This review will focus on the wide range of applications of SNP microarray analysis to cancer research. SNP array genotyping can determine loss of heterozygosity, genomic copy number changes and DNA methylation alterations of cancer cells. The same technology can also be used to investigate allelic association in cancers. Therefore, it can be applied to the identification of cancer predisposition genes, oncogenes and tumor suppressor genes in specific types of tumors. As a consequence, they have potential in cancer risk assessment, diagnosis, prognosis and treatment selection.
Key Words: SNP array, cancer, genome-wide analysis, genotyping, copy number change
INTRODUCTION
Cancer development is accompanied by multiple genetic alterations including chromosomal copy number and structural changes. Identification of all genetic alterations is essential for a full understanding of the etiology of human cancer. Genetic analysis using a genome-wide detection tool is an essential approach to uncover all abnormalities and is also an efficient way to identify key genetic events, such as activation of oncogenes and inactivation of tumor suppressor genes in cancer development and progression. Such an approach can lead to quick discovery of genetic markers for cancer risk assessment, diagnosis and prognosis. Eventually, the full mapping of genotype and genetic alterations may be used for individually stratified medicine.
Karyotyping, the cytogenetic method analyzing the global genetic alterations at the chromosomal level has played an important role in our understanding of human cancers since the 1970’s. Karyotype analysis has led to the identification of tumorigenic fusion genes and tumor suppressor genes and is currently used clinically for the diagnosis and prognosis of haematological malignancies. However, the general application of karyotyping is limited, since it is based on analyzing metaphase cells which are difficult to obtain in many solid tumors. Furthermore the position of chromosome breakpoints needs to be defined by fluorescence in situ hybridization (FISH) analysis.
The development of comparative genomic hybridization (CGH) in 1992 has transformed the genome wide analysis of cancer genetic alterations [1]. CGH can detect DNA copy number changes across the entire genome of a tumor sample in a single experiment by comparing the hybridization signal intensity of a tumor sample against a reference sample along the chromosomes. The initial development of CGH was applied on metaphase chromosomes [1]. Although it has already made a significant impact on cancer research and has been used to identify frequent chromosome region copy number gains and losses in many tumors, particularly solid cancers, the resolution of conventional CGH is low. Changes smaller than 2Mb are undetectable by chromosomal CGH. In 1998, Pinkel et al. [2] applied array technology for high-resolution CGH analysis. In array-CGH, the metaphase chromosomes are replaced by cloned DNA fragments with known genomic locations. The cloned DNA fragments can be genomic DNA or cDNA which are spotted on a glass slide or other supporting materials. As in chromosomal CGH, the DNA copy number changes are shown by the fluorescence ratio between the test and control samples. With array-CGH, sub-microscopic aberrations can be detected.
Tumor suppressor gene plays an important role in tu-morigenesis, and can be detected by deletion and mutation analyses. They can also frequently be indicated by loss-of-heterozygosity (LOH) which can be picked up by genotyping analysis. The development of single nucleotide polymorphism (SNP) arrays [3] enables simultaneous detection of a large number of DNA polymorphic loci in a simple way. Further technical developments make SNP arrays capable of analyzing both signal intensity variations and changes in allelic composition in parallel [4, 5]. SNP arrays can also detect both copy number changes and copy-neutral LOH events [6–8]. Fan et al. have provided a detailed review of the mechanism of different SNP genotyping methods [9]. In this review, we will focus on the stages of SNP array development and its applications in cancer research.
THE DEVELOPMENT OF SNP MICROARRAY TECHNOLOGY
SNP array technology was developed in 1998 for genotyping [3]. Since then, the technique has been improved dramatically and has become one of the most powerful ge-nomic analysis tools. The different development stages of SNP array technology are listed in Table 1.
Table 1.
Historical Stages of the SNP Array Technology and Application Development
| Years | Events |
|---|---|
| 1998 | First test of SNP array analysis (558 SNPs) |
| 2000 | First application of SNP array analysis on cancer |
| 2003 | Affymetrix developed 10K SNP array using single primer amplification of restriction enzyme digested genomic DNA |
| 2004 | SNP array validated for DNA copy number analysis |
| 2004 | SNP array application whole genome amplified DNA samples |
| 2005 | Development of whole-genome genotyping approach which should be scalable for simultaneous analysis of all SNPs in the genome. |
| 2006 | Development of the high resolution karyotyping approach by combined SNP and 24-color FISH analyses |
The first SNP array contained 558 loci, and SNPs present within a sample amplified by multiplex polymerase chain reaction (PCR), in which primer pairs from many different loci were combined in a single reaction [3]. Amplified DNA was then hybridized on the SNP array to detect the genotyping of the 558 SNPs in the sample. A large amount of primers were required to amplify these multiple SNPs in a sample in the multiplex PCR approach for array analysis. However, primer dimer formation limited the number of primer pairs that could be included in a single PCR reaction. Therefore, the sample preparation using this protocol was still labour-extensive for high density SNP array analysis. Therefore, it took two years for this technique to be applied to cancer research. In 2000, two groups separately applied SNP array analysis for multiple detection of LOH and allelic imbalance (AI) in human cancers [10, 11]. The microarray company, Affymetrix, has improved the confidence of SNP array genotyping by interrogating on the array additional offset probes for each SNP locus [11]. The low resolution SNP array has been applied to human tumor samples mainly for LOH and allele imbalance analysis [10, 11]. Several approaches have been used to improve the capacity of multiplex PCR [9]. Among them, the GoldenGate assay [12] is one of the successful highly multiplexed PCR-based SNP genotyping method which the company Illumina has adopted for their commercial SNP array chips. Although these modifications improved the number of SNPs that can be analyzed, the multiplex PCR approach still limits the member of SNPs that can be analyzed.
In 2003, researchers at Affymetrix developed the whole-genome sampling method for SNP genotyping [13, 14]. This approach amplified genomic DNA pre-cut by a restriction enzyme. After digestion of the genomic DNA and ligation of primers, the amplification step was specifically designed to amplify DNA fragments between 400 to 800 base pairs. Using this approach, thousands of SNPs could be analyzed simultaneously. Using XbaI digestion, commercial SNP arrays containing 10,000 SNPs and accompanied by a sample preparation kit were produced. This development made the spot density and the genomic resolution of SNP array analysis higher than that of the 1Mb bacterial artificial chromosome array and cDNA arrays used commonly at that time. Since then, the SNP array has been rapidly applied to many human tumors. The application of whole-genome DNA amplification techniques in combination with SNP array genotyping [15–17] and genomic copy number analysis have been evaluated [4]. The combined genotyping and genomic copy number analysis has made the SNP array a unique technique in cancer genomic research, and revealed many new genetic features in cancer cells such as acquired uniparental disomy (UPD) [7, 8]. In the last couple of years, Affymetrix has improved the coverage of their SNP array chips further into 100K and then 500K by selecting different enzymes to fragment genomic DNA. The 500K SNP array achieved a ge-nomic resolution of average 5Kb per SNP. Combining the 500K SNP array with 24-colour FISH analyses, we developed a high resolution karyotyping approach which can define the majority of chromosome rearrangement breakpoints to 5-50 Kb [18].
However, the Affymetrix enzyme digestion and the single primer amplification approach still has its limitations. While the selection of enzyme digested DNA fragments reduces the genomic complexity, the selected DNA only represents SNPs in a proportion of the human genome. The PCR amplification of digested DNA also limited the application of these SNP arrays in analyzing degraded DNA. The majority of human clinical cancer tissues are stored as formalin-fixed paraffin-embedded (FFPE) blocks which are invaluable for retrospective studies. Although in certain FFPE blocks, DNA is well preserved so that the fragments are large enough for whole-genome sampling analysis, DNA from the majority of tissues kept as FFPE materials is degraded. Many of the DNA fragments are not long enough to be cut twice by the enzyme, and are therefore subsequently excluded from PCR amplification. Although the application of Affymetrix 10K array analysis on FFPE samples has been reported [19], in our experience, only less than 10% of FFPE samples can be successfully analyzed by the Affymetrix 10K or 100K SNP arrays. In this respect, samples prepared without restriction enzyme digestion worked better for SNP array analysis [20, 21].
Recently, another high throughput genotyping method was been developed by hybridizing the whole genomic DNA to an array of locus-specific capture probes [22, 23]. The SNP genotypes are scored using enzymatic allelic discrimination. Although the SNP array chips currently available for this type of SNP genotyping are limited to 550,000 loci, this principle of SNP array analysis makes the number of SNPs in one chip scalable. Using this approach, theoretically all SNPs existing in the genome can be analyzed in one experiment, and in principle it is applicable to degraded DNA such as that extracted from FFPE materials. Illumina has developed this approach, making BeadChips for high density SNP array analysis.
Accompanying the technical development of microarray arrays, many SNP array analysis tools have been developed. Lin et al. [24] have reported software that can pool replicates to make LOH calls and visualize SNP and LOH data along chromosomes. Using this software, statistical inference to identify shared LOH regions and sample clustering analysis is available. With the validation of high density SNP array for DNA copy number analysis, both robust algorithms and statistical methods to detect gained and/or lost chromosome regions using SNP array hybridization data were also designed [25, 26]. Laframboise et al. further developed software for probe-level allele-specific quantitation (PLASQ) [27, 28]. This procedure analyzes allelic specific copy number changes which can determine not just the region but also the haplotype of amplifications and deletions. Using this software, they found that amplification in lung cancer is essentially monoallelic either due to germ line or somatic variation [28]. Affymetrix has also produced similar SNP array data analyzing software for allelic specific DNA copy number changes and allelic genotyping called Copy Number Analysis With Regression and Tree (CARAT) [29]. A web-accessible SNP array analysis tool, SNPscan, was established for multiple types, combined or separated genotyping and genomic dosage analyses [30]. In our laboratory, we have developed software, GOLF, for parallel detailed analysis of LOH and DNA copy number changes along the chromosomes. Using this software, gene expression data from Affymetrix gene expression or exon arrays can also be analyzed along the chromosomes in parallel.
APPLICATION OF SNP ARRAY IN CANCER RESEARCH
Due to the complexity of genetic alterations in cancer cells, high density SNP array analysis is a very demanding technique in the field of cancer research. Here we describe the main areas of its application, and the tumor types analyzed by SNP array technology are summarized in Table 2.
Table 2.
Tumor Types have been Analyzed by SNP Array
| Analyses | Tumor types |
|---|---|
| LOH | bladder cancer (4), lung cancer (2), prostate cancer (2), breast cancer (2), colorectal cancer (2), oral squamous cell carcinoma (2), tongue squamous cell carcinoma (1), renal cancer (1), acute lymphoblastic leukemia (1), pheochromocytoma (1), phyllodes tumor (1), fibroadenoma of the breast (1), osteosarcoma (1), neuroblastoma (1) |
| CGH | lung cancer (4), prostate cancer (3), colorectal cancer (1), ovarian cancer (1), melanoma (1) |
| Combined approach | acute myeloid leukemia (3), breast cancer (1), colorectal cancer (1), ovarian cancer (1), pancreatic cancer (1), hepatocellular carcinoma (1), oral cancer (1), basal cell carcinoma (1), myeloma (1), lymphoma (1), testicular germ cell tumor (1) |
| Linkage | colorectal cancer (2), hereditary mixed polyposis (colorectal cancer) (1), prostate cancer (1), chronic lymphocytic leukemia (1), esophageal cancer (1), pituitary adenoma (1) |
| DNA methylation | leukemia (1), Wilm’s tumor (1), lung cancer (1) |
Notes: The numbers in parentheses show the numbers of reports in the literature.
AI and LOH Analysis
The initial purpose of SNP array development was to genotype multiple SNPs simultaneously. Therefore they were first used in cancer research for LOH and AI analyses which are important for tumor suppressor gene identification. Before the development of the single primer whole-genome sampling analysis approach and the release of the Affymetrix 10K arrays, SNP array analysis was only used to study AI in cancer cells. In 2000, Lindblad-Toh et al. first applied SNPs array in a LOH study of human cancers. Analyzing small-cell lung cancer and control DNA samples, they found that the SNP arrays detected the same patterns of LOH as simple sequence length polymorphism (SSLPs or micro-satellites) analysis [10]. Subsequently, Primdahl et al. applied SNP arrays on bladder cancer and found that AI occurred more frequently in T2-4 than T1 tumors. A new AI area was found on 6p [31]. Using the same approach, a LOH region on 6p was also found in prostate cancer [32]. Hoque et al., investigating bladder cancer, proved that SNP array analysis picked up LOH patterns consistent with those detected from microsatellite allelotype analysis, but with additional information [33]. They applied this technique further in bladder cancer detection by analyzing urine sediment [34]. In 2004, Janne et al. demonstrated that small-cell lung cancer could be separated from non-small-cell lung cancer by LOH analysis using the 1500 low density SNP arrays and they further validated the efficiency and reliability of the 10K array for LOH analysis [35]. In breast cancer, SNP array analysis detected LOH patterns associated with tumor subgroups which were also partially defined by gene expression patterns [36]. Huang et al. [37] demonstrated that using the contiguous stretches of homozygous markers, LOH can be detected in tumor samples without analyzing the matched normal control samples. This SNP array LOH analysis using unpaired tumor samples was further validated by other research teams [38, 39]. Smoker and non-smoker oral cavity squamous cell carcinomas have been analyzed for AI using SNP array, but no difference was found [40]. In 2005, Koed et al. demonstrated the clinical potential using 10K SNP array for AI analysis in bladder cancer. In agreement with the results from Primdahl and colleagues, they found that AI was strongly stage-dependent. In addition, they found that cancers from different locations were associated with different frequencies of allelic imbalance. Seven out of eight tumors involving the upper urinary tract were genomically stable [41]. Irving et al. demonstrated that high density SNP arrays could detect LOH associated with relapse and bad prognosis in childhood lymphoblastic leukaemia [42]. Dahia et al., using SNP array analysis, identified high frequency of LOH in the pheochromocytoma susceptibility loci. This analysis also further defined the locus to <2 cM [43]. Gaasenbeek et al. combined array-CGH with SNP array analyses to demonstrate the multiple forms of chromosomal instability in colo-rectal cancers [44]. Wang et al. applied the SNP array for LOH analysis in phyllodes tumor and fibroadenoma of breast. LOH was frequently found in phyllodes tumors and primary tumors shared common regions of LOH with paired recurrences from the same patient. However, LOH was rare in fibroademomas [45]. LOH analysis using SNP arrays was also conducted in renal cell cancer [46].
Clearly, SNP array analysis, detecting multiple polymorphism loci in one experiment, even at low density has advantages over the traditional SSLP analysis. Comparison studies of SSLP and SNP array analysis showed that SNP array results generally agree with the SSLP analysis and the accuracy of SNP array analysis is even higher than SSLP [10]. With the development of high density SNP arrays, the advantage of SNP array analysis over the traditional SSLP technique for LOH analysis is more significant. Hundreds of thousands of loci can be detected simultaneously for AI, making the genome-wide screen of tumor suppressor genes possible.
DNA Copy Number Aberration Analysis
With the development of the high-density SNP arrays, genomic resolution higher than any other array platforms has been achieved. Due to the high resolution of SNP arrays, their use in DNA copy number analysis had been explored soon after Affymetrix released its 10K array analysis system [4, 47]. Since then, SNP arrays have been applied to many tumor types for genomic copy number changes (Table 2). Rubin et al. used SNP array analysis to detect the copy number gain of the 8q region, where the prostate cancer overex-pressed androgen regulated gene, TPD52 is located [48]. Zhao et al. detected recurrent homozygous deletion and chromosome amplification in lung cancer using the 10K array [49]. Analyzing a large number of lung cancer cell lines using SNP array technology, Sato et al. detected more homozygous deletions and confirmed the most frequent deletion regions on 9p using PCR analysis of an even larger series of cell lines [50]. Garraway et al., combining high density SNP array analysis with gene expression signatures, identified a novel melanoma amplicon and its target gene, MITF. MITF was associated with metastatic disease and was negatively correlated with patient survival [51]. Koochek-pour et al. [52], used SNP arrays to detect amplification of the PSAP gene which played a role in prostate carcinogene-sis. Park et al. used SNP array analysis to identify, in ovarian cancer, an amplicon at 19p13.12, and further gene expression analysis identified Notch3 as the target gene [53]. Recently, the possibility of using SNP array to generate DNA copy number changes for tumor classification has been evaluated [54]. The SNP array has also been used to define the deleted region on 9p in mesothelioma cell lines [55] and chromosomal alterations associated with cancer cell platinum resistance [56]. In pancreatic cancer, many new homozygous deletions have been detected [39]. The application of 100K SNP array in prostate cancer genetic analysis revealed small size novel alterations [57].
The currently available 500K SNP array has an average coverage of 5Kb per SNP. We have applied the 500K SNP array in leukemia and prostate cancer studies and detected many sub-microscopic deletions and amplifications that were not previously identified using lower resolution methods (Fig. 1). Many of these sub-microscopic deletions are homozygous. The finding of sub-microscopic deletions in cytoge-netically normal karyotyped leukemia cells is invaluable in understanding the aetiology of this malignancy.
Fig. (1).
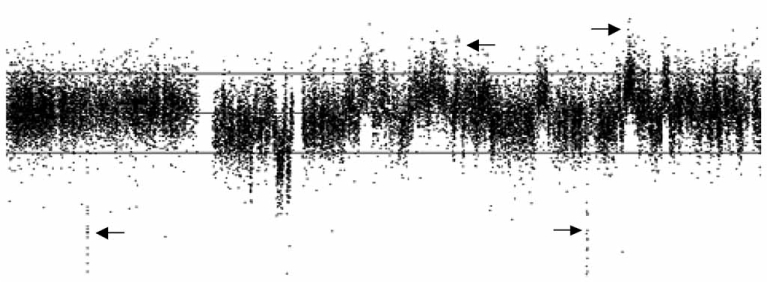
An example of complex chromosome copy number alterations detected by 500K SNP array analysis. This image shows multiple small gains and losses on chromosome 5 including sub-microscopic chromosomal amplifications and deletions (arrows) around 100 Kb in the PC3 prostate cancer cell line. The middle horizontal grey line represents log2 ratio of 0 compared to normal controls; the bottom and upper lines represent log2 ratios of −1 and 1.
Combined LOH and DNA Copy Number Analysis
As SNP array analysis generates information on both LOH and DNA copy number changes, it provides a unique platform for combined genotyping and DNA copy number alteration analysis. Actually, this combined analysis is the most rewarding area of SNP array application in cancer research. Soon after the development of the 10K SNP arrays, in 2004, four groups separately validated the feasibility of using SNP data for both genotyping and DNA copy number analysis [4, 5, 15, 37]. Whole genome amplified DNA can also be used reliably for both type of analyses [5], and Huang et al. [37] demonstrated that both LOH and DNA copy number analyses could tolerate certain levels of normal contamination in tumor samples. In cancer, LOH may not always accompany DNA copy number loss, either through mitotic recombination, chromosome non-disjunction and loss of one parental chromosome and gain of opposite parental material. Analyzing both the LOH and DNA copy number changes, our laboratory pioneered the application of SNP analysis in the study of LOH without loss of DNA copy number in human malignancies [6–8]. In acute myeloid leukemia, we found a high frequency (20%) of UPD, and UPD regions on chromosome 19 coincided with a previously identified homozygous mutation in the CEBPA gene [7]. Further mutation screening identified mutations at four distinct loci, WT1, FLT3, CEBPA, and RUNX1, within the UPD regions in 7 out of 13 cases of acute myeloid leukemia [58]. FLT3-internal tandem duplication was also detected both at diagnosis and relapse of a case of acute myeloid leukemia with chromosome 13 UPD [59]. The high frequency of UPDs in acute myeloid leukemia has been confirmed by other research groups [60]. In basal cell carcinoma, LOH at 9q21-31 was found in 13 of 14 samples and 5 of the LOH are caused by UPD. Patched 1 gene (PTCH) in this region was found mutated in more than half of cases with LOH [8]. These studies demonstrated that mitotic recombination can act as a “second hit”, similar to chromosome deletion, and responsible for removal of the remaining wild-type allele of those tumor suppressor-like genes. Using 10K SNP arrays, we also found a high frequency of large-scale homozygous chromosome regions (HCR) frequently extending to the whole chromosome without loss of chromosome copy number in the nonseminomatous subtype of male germ cell tumors. The frequency of these large-scale HCRs without chromosome copy number losses is significantly higher than the seminomatous subtype of germ cell tumor and the other tumor types, indicating its specific relationship with non-seminoma development [6]. Fig. (2) and (3) present our combined analysis approach and the high frequency of large-scale HCRs without chromosome losses. Since then, UPD has been found using SNP array analysis by other research groups in malignant lymphoma [61], myeloma [62], hepato-cellular carcinoma [63] and colorectal cancers [64]. UPD is prevalent in myeloma [62] and half of the LOH regions in colorectal cancers are present as UPD and predominantly involve 8q, 13q and 20q [64]. In the colorectal cancer study, UPD did not change the expression level of genes in the relevant regions [64]. The correlation between DNA content and gene expression in the same type of cancer was also demonstrated by Tsafrir and colleagues [65] and in hepato-cellular carcinoma by Midorikawa et al. [63].
Fig. (2).

Parallel genotyping and DNA copy number change analysis of testicular germ cell tumor. A). SNP genotype patterns along the chromosomes of two cases of non-seminomas with paired normal controls. From top to bottom, the cases presented are 5T (tumor), 5N (normal), 7T and 7N. It shows acquired large homozygous chromosome regions (HCRs, chromosome regions with 98% or greater homozy-gosities in a minimum region of 50 contiguous SNPs) on chromosome 2q, 5, 10, 11, 13 and 14 in case 5T and chromosome 4, 8q and 13 in case 7T. For each chromosome, two blue lines represent either AA or BB homozygous calls. The red line represents AB heterozygous calls and the grey line on the bottom shows SNPs not called. B). DNA copy number changes along the chromosomes of the same two tumors. The signal intensity of the tumor samples is normalized against the normal controls. The top case is 5T and the bottom case is 7T.
Fig. (3).
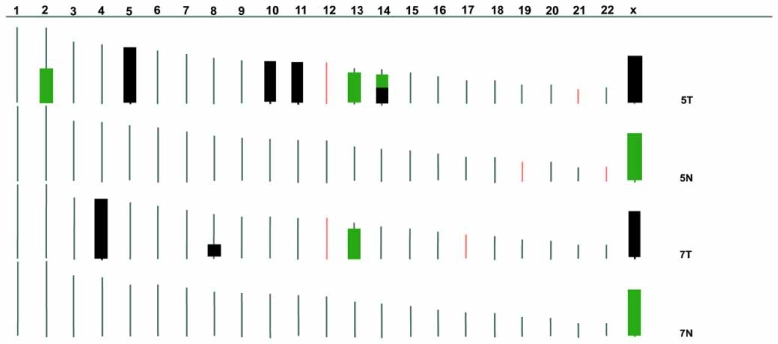
A summary of the HCRs on each chromosome for the two pairs of samples analyzed by SNP arrays. Black bars are HCRs without significant chromosome copy number changes (signal intensity ratio compared to case average between 0.75-1.25); green bars are HCRs with relative chromosome copy number loss (signal intensity ratio < 0.75).
Linkage Analysis for Cancer Susceptibility Loci
As SNP arrays can analyze genome-wide constitutional DNA polymorphisms in individuals, it is applicable for linkage analysis to identify cancer predisposition genes. In 2004, using the 10K array, Schaid et al. first validated the application of SNP array in linkage screen for cancer-susceptibility loci. Although they found that SNP array analysis can inflate LOD scores due to the presence of linkage disequilibrium, clearly, they demonstrated that SNP array analysis largely increased the linkage information content [66]. Causing inflated nonparametric linkage by the presence of high linkage disequilibrium was confirmed by Sellick and colleagues in a separate SNP array linkage analysis of chronic lymphocytic leukemia [67]. Despite this, Sellick et al. found some susceptibility loci such as 11p11, 5q22-23, 6p22 and 10q25 in chronic lymphocytic leukaemia. In colorectal cancer patients, Kemp et al. identified a disease susceptibility region of 3q21-24 which linked to the majority of families they analyzed [68]. Using 10K SNP arrays, Hu et al. [69] identified 37 SNPs associated with esophageal cancer and applied these 37 SNPs using principle components analysis to predict patients and normal individuals at more than 90% accuracy. Cao et al. [70] using SNP array analysis identified a 7 cM region on 10q23 with linkage to hereditary mixed poly-posis syndrome, a disease which eventually leads to colorec-tal cancer. Subsequently, in one family they identified an 11 bp deletion in the bone morphogenesis protein receptor 1A (BMPR1A) gene. In pituitary adenoma, by screening a family using SNP array technology, Vierimaa et al. mapped a susceptibility locus to a previously reported chromosome region, 11q12–11q13. Further expression array analysis led to the selection of AIP gene for mutation analysis and identification of this low-penetrance tumor-susceptibility gene [71]. SNP array analysis has also been used to analyze the polymorphism of genes in xenobiotic metabolism and some SNPs in those genes have been associated with increased or decreased risk of colorectal cancers [72].
Other Applications of SNP Array Analysis in Cancer Research
Although SNP arrays were originally developed for ge-nomic analysis, they were also adopted for epigenetic and gene expression analysis. As the current principle of Af-fymetrix array analysis is to apply selected restriction enzyme digested DNA samples for hybridization, it can be easily adjusted for DNA methylation studies by comparing the presence and absence of DNA sequences between the methylation sensitive and non-sensitive enzymes. Yuan and colleagues validated this application in decitabine treated leukemia and tumor/normal comparison of Wilm’s tumor using bisulfite restriction analysis and bisulfite PCR as confirmation tools [73]. At the same time, application of the Golden-Gate SNP array system for methylation analysis was validated at Illumina with the ability to distinguish lung cancer samples from controls [74]. DNA methylation can lead to differential expression of genes between paternal and maternal alleles, which is referred to as DNA imprinting. Other factors such as DNA polymorphisms in gene regulatory factors can also cause imbalanced allelic expression [75]. Using cDNA instead of genomic DNA as starting material, allelic specific gene expression can also be detected by SNP array analysis [76, 77]. Allelic specific gene expression may be important in tumorigenesis. However, allelic specific gene expression pattern analysis directly using SNP arrays has not been explored in the field of cancer research.
ADVANTAGE AND LIMITATION OF SNP ARRAY ANALYSIS
The advantage of SNP array in high throughput DNA genotyping over PCR based individual microsatellite marker polymorphism analysis is obvious and has been demonstrated in papers summarized above. Compared with other array technology, the high density resolution of SNP arrays is clearly an advantage. It is estimated that there are over 20 million SNPs in the entire human genome, on average every 150 base pair per SNP. The whole-genome genotyping SNP array technology under current development has the potential to comprise almost all existing SNPs into a single experiment. The resolution of genetic abnormalities picked up by high density SNP array analysis will be more than sufficient for direct PCR and sequencing analysis. The unique feature of SNP arrays, and the concurrent analysis of both genotype and DNA copy number changes, make SNP arrays irreplaceable by any other high throughput technology.
However, every technique has its limitations. Similar to the other DNA array-based methods, SNP array analysis cannot detect balanced chromosomal translocations, inversions and whole-genome ploidy changes without combination with other techniques. Therefore, SNP array analysis of DNA extracted from a cell population cannot indicate the heterogeneity within the sample. In this respect, combination of data with those from individual cell analysis methods such as FISH or 24-color FISH karyotyping is necessary. Although SNP arrays can be used for allelic specific gene expression analysis, they are principally designed for genomic study. More specific expression arrays such as oligonucleo-tide and exon array are required for transcription level study. Further functional studies should also be correlated with ge-nomic and expression alterations. In the case of evaluating known genes or established genetic markers in a large series of samples, PCR and tissue array analyses will be more efficient.
CONCLUSION
SNP array technology is a powerful genomic analysis tool. It concurrently generates data identifying genome-wide genotyping and DNA dosage alterations. The application of SNP arrays in combination with PCR, tissue array and functional analysis technologies will eventually lead to the full revelation of the complex genetic alterations in human cancers.
ACKNOWLEDGEMENTS
Supported by Orchid Cancer Appeal and Cancer Research UK. We thank Dr. Sharon James for critical reading of this review.
ABBREVIATIONS
- SNP
Single nucleotide polymorphism
- FISH
Fluorescence in situ hybridization
- CGH
Comparative genomic hybridization
- LOH
Loss of heterozygosity
- PCR
Polymerase chain reaction
- AI
Allelic imbalance
- SSLP
Simple sequence length polymorphism
- UPD
Uniparental disomy
- HCR
Homozygous chromosome region
REFERENCES
- 1.Kallioniemi A, Kallioniemi OP, Sudar D, Rutovitz D, Gray JW, Waldman F, Pinkel D. Comparative genomic hybridization for molecular cytogenetic analysis of solid tumors. Science. 1992;258:818–821. doi: 10.1126/science.1359641. [DOI] [PubMed] [Google Scholar]
- 2.Pinkel D, Segraves R, Sudar D, Clark S, Poole I, Kowbel D, Collins C, Kuo WL, Chen C, Zhai Y, Dairkee SH, Ljung BM, Gray JW, Albertson DG. High resolution analysis of DNA copy number variation using comparative genomic hybridization to microarrays. Nat Genet. 1998;20:207–211. doi: 10.1038/2524. [DOI] [PubMed] [Google Scholar]
- 3.Wang DG, Fan JB, Siao CJ, Berno A, Young P, Sapolsky R, Ghandour G, Perkins N, Winchester E, Spencer J, Kruglyak L, Stein L, Hsie L, Topaloglou T, Hubbell E, Robinson E, Mittmann M, Morris MS, Shen N, Kilburn D, Rioux J, Nusbaum C, Rozen S, Hudson TJ, Lipshutz R, Chee M, Lander ES. Large-scale identification, mapping, and genotyping of single-nucleotide polymorphisms in the human genome. Science. 1998;280:1077–1082. doi: 10.1126/science.280.5366.1077. [DOI] [PubMed] [Google Scholar]
- 4.Bignell GR, Huang J, Greshock J, Watt S, Butler A, West S, Grigorova M, Jones KW, Wei W, Stratton MR, Futreal PA, Weber B, Shapero MH, Wooster R. High-resolution analysis of DNA copy number using oligonucleotide microarrays. Genome Res. 2004;14:287–295. doi: 10.1101/gr.2012304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhou X, Mok SC, Chen Z, Li Y, Wong DT. Concurrent analysis of loss of heterozygosity (LOH) and copy number abnormality (CNA) for oral premalignancy progression using the Affymetrix 10K SNP mapping array. Hum Genet. 2004;115:327–330. doi: 10.1007/s00439-004-1163-1. [DOI] [PubMed] [Google Scholar]
- 6.Lu YJ, Yang J, Noel E, Skoulakis S, Chaplin T, Raghavan M, Purkis T, McIntyre A, Kudahetti SC, Naase M, Berney D, Shipley J, Oliver RT, Young BD. Association between large-scale genomic homozygosity without chromosomal loss and nonseminomatous germ cell tumor development. Cancer Res. 2005;65:9137–9141. doi: 10.1158/0008-5472.CAN-05-1697. [DOI] [PubMed] [Google Scholar]
- 7.Raghavan M, Lillington DM, Skoulakis S, Debernardi S, Chaplin T, Foot NJ, Lister TA, Young BD. Genome-wide single nucleotide polymorphism analysis reveals frequent partial uniparental disomy due to somatic recombination in acute myeloid leukemias. Cancer Res. 2005;65:375–378. [PubMed] [Google Scholar]
- 8.Teh MT, Blaydon D, Chaplin T, Foot NJ, Skoulakis S, Raghavan M, Harwood CA, Proby CM, Philpott MP, Young BD, Kelsell DP. Genomewide single nucleotide poly-morphism microarray mapping in basal cell carcinomas unveils uniparental disomy as a key somatic event. Cancer Res. 2005;65:8597–8603. doi: 10.1158/0008-5472.CAN-05-0842. [DOI] [PubMed] [Google Scholar]
- 9.Fan JB, Chee MS, Gunderson KL. Highly parallel genomic assays. Nat Rev Genet. 2006;7:632–644. doi: 10.1038/nrg1901. [DOI] [PubMed] [Google Scholar]
- 10.Lindblad-Toh K, Tanenbaum DM, Daly MJ, Winchester E, Lui WO, Villapakkam A, Stanton SE, Larsson C, Hudson TJ, Johnson BE, Lander ES, Meyerson M. Loss-of-heterozygosity analysis of small-cell lung carcinomas using single-nucleotide polymorphism arrays. Nat Biotechnol. 2000;18:1001–1005. doi: 10.1038/79269. [DOI] [PubMed] [Google Scholar]
- 11.Mei R, Galipeau PC, Prass C, Berno A, Ghandour G, Patil N, Wolff RK, Chee MS, Reid BJ, Lockhart DJ. Genome-wide detection of allelic imbalance using human SNPs and high-density DNA arrays. Genome Res. 2000;10:1126–1137. doi: 10.1101/gr.10.8.1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fan JB, Oliphant A, Shen R, Kermani BG, Garcia F, Gunderson KL, Hansen M, Steemers F, Butler SL, Deloukas P, Galver L, Hunt S, McBride C, Bibikova M, Rubano T, Chen J, Wickham E, Doucet D, Chang W, Campbell D, Zhang B, Kruglyak S, Bentley D, Haas J, Rigault P, Zhou L, Stuelpnagel J, Chee MS. Highly parallel SNP genotyping. Cold Spring Harb Symp Quant Biol. 2003;68:69–78. doi: 10.1101/sqb.2003.68.69. [DOI] [PubMed] [Google Scholar]
- 13.Kennedy GC, Matsuzaki H, Dong S, Liu WM, Huang J, Liu G, Su X, Cao M, Chen W, Zhang J, Liu W, Yang G, Di X, Ryder T, He Z, Surti U, Phillips MS, Boyce-Jacino MT, Fodor SP, Jones KW. Large-scale genotyping of complex DNA. Nat Biotechnol. 2003;21:1233–1237. doi: 10.1038/nbt869. [DOI] [PubMed] [Google Scholar]
- 14.Matsuzaki H, Loi H, Dong S, Tsai YY, Fang J, Law J, Di X, Liu WM, Yang G, Liu G, Huang J, Kennedy GC, Ryder TB, Marcus GA, Walsh PS, Shriver MD, Puck JM, Jones KW, Mei R. Parallel genotyping of over 10,000 SNPs using a one-primer assay on a high-density oligonucleotide array. Genome Res. 2004;14:414–425. doi: 10.1101/gr.2014904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Paez JG, Lin M, Beroukhim R, Lee JC, Zhao X, Richter DJ, Gabriel S, Herman P, Sasaki H, Altshuler D, Li C, Meyerson M, Sellers WR. Genome coverage and sequence fidelity of phi29 polymerase-based multiple strand displacement whole genome amplification. Nucleic Acids Res. 2004;32:e71. doi: 10.1093/nar/gnh069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wong KK, Tsang YT, Shen J, Cheng RS, Chang YM, Man TK, Lau CC. Allelic imbalance analysis by high-density single-nucleotide polymorphic allele (SNP) array with whole genome amplified DNA. Nucleic Acids Res. 2004;32:e69. doi: 10.1093/nar/gnh072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhou X, Temam S, Chen Z, Ye H, Mao L, Wong DT. Allelic imbalance analysis of oral tongue squamous cell carcinoma by high-density single nucleotide polymorphism arrays using whole-genome amplified DNA. Hum Genet. 2005;118:504–507. doi: 10.1007/s00439-005-0069-x. [DOI] [PubMed] [Google Scholar]
- 18.Mao X, James SY, Yanez-Munoz RJ, Chaplin T, Molloy G, Oliver RTD, Young BD, Lu YJ. Rapid high-resolution karyo-typing with precise identification of chromosome breakpoints. Gene Chromosome Cancer. 2007 doi: 10.1002/gcc.20452. In press. [DOI] [PubMed] [Google Scholar]
- 19.Thompson ER, Herbert SC, Forrest SM, Campbell IG. Whole genome SNP arrays using DNA derived from formalin-fixed, paraffin-embedded ovarian tumor tissue. Hum Mutat. 2005;26:384–389. doi: 10.1002/humu.20220. [DOI] [PubMed] [Google Scholar]
- 20.Lips EH, Dierssen JW, van Eijk R, Oosting J, Eilers PH, Tollenaar RA, de Graaf EJ, van’t Slot R, Wijmenga C, Morreau H, van Wezel T. Reliable high-throughput genotyping and loss-of-heterozygosity detection in formalin-fixed, paraffin-embedded tumors using single nucleotide polymorphism arrays. Cancer Res. 2005;65:10188–10191. doi: 10.1158/0008-5472.CAN-05-2486. [DOI] [PubMed] [Google Scholar]
- 21.Schubert EL, Hsu L, Cousens LA, Glogovac J, Self S, Reid BJ, Rabinovitch PS, Porter PL. Single nucleotide polymor-phism array analysis of flow-sorted epithelial cells from frozen versus fixed tissues for whole genome analysis of allelic loss in breast cancer. Am J Pathol. 2002;160:73–79. doi: 10.1016/S0002-9440(10)64351-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gunderson KL, Steemers FJ, Lee G, Mendoza LG, Chee MS. A genome-wide scalable SNP genotyping assay using microarray technology. Nat Genet. 2005;37:549–554. doi: 10.1038/ng1547. [DOI] [PubMed] [Google Scholar]
- 23.Steemers FJ, Chang W, Lee G, Barker DL, Shen R, Gunderson KL. Whole-genome genotyping with the single-base extension assay. Nat Methods. 2006;3:31–33. doi: 10.1038/nmeth842. [DOI] [PubMed] [Google Scholar]
- 24.Lin M, Wei LJ, Sellers WR, Lieberfarb M, Wong WH, Li C. dChipSNP: significance curve and clustering of SNP-array-based loss-of-heterozygosity data. Bioinformatics. 2004;20:1233–1240. doi: 10.1093/bioinformatics/bth069. [DOI] [PubMed] [Google Scholar]
- 25.Nannya Y, Sanada M, Nakazaki K, Hosoya N, Wang L, Hangaishi A, Kurokawa M, Chiba S, Bailey DK, Kennedy GC, Ogawa S. A robust algorithm for copy number detection using high-density oligonucleotide single nucleotide polymorphism genotyping arrays. Cancer Res. 2005;65:6071–6079. doi: 10.1158/0008-5472.CAN-05-0465. [DOI] [PubMed] [Google Scholar]
- 26.Lai Y, Zhao H. A statistical method to detect chromosomal regions with DNA copy number alterations using SNP-array-based CGH data. Comput Biol Chem. 2005;29:47–54. doi: 10.1016/j.compbiolchem.2004.12.004. [DOI] [PubMed] [Google Scholar]
- 27.Laframboise T, Harrington D, Weir BA. PLASQ: A Generalized Linear Model-Based Procedure to Determine Allelic Dosage in Cancer Cells from SNP Array Data. Biostatistics. 2006 doi: 10.1093/biostatistics/kxl012. [DOI] [PubMed] [Google Scholar]
- 28.LaFramboise T, Weir BA, Zhao X, Beroukhim R, Li C, Harrington D, Sellers WR, Meyerson M. Allele-specific amplification in cancer revealed by SNP array analysis. PLoS Comput Biol. 2005;1:e65. doi: 10.1371/journal.pcbi.0010065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huang J, Wei W, Chen J, Zhang J, Liu G, Di X, Mei R, Ishikawa S, Aburatani H, Jones KW, Shapero MH. CARAT: a novel method for allelic detection of DNA copy number changes using high density oligonucleotide arrays. BMC Bioinformatics. 2006;7:83. doi: 10.1186/1471-2105-7-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ting JC, Ye Y, Thomas GH, Ruczinski I, Pevsner J. Analysis and visualization of chromosomal abnormalities in SNP data with SNPscan. BMC Bioinformatics. 2006;7:25. doi: 10.1186/1471-2105-7-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Primdahl H, Wikman FP, von der Maase H, Zhou XG, Wolf H, Orntoft TF. Allelic imbalances in human bladder cancer: genome-wide detection with high-density single-nucleotide polymorphism arrays. J Natl Cancer Inst. 2002;94:216–223. doi: 10.1093/jnci/94.3.216. [DOI] [PubMed] [Google Scholar]
- 32.Dumur CI, Dechsukhum C, Ware JL, Cofield SS, Best AM, Wilkinson DS, Garrett CT, Ferreira-Gonzalez A. Genome-wide detection of LOH in prostate cancer using human SNP microarray technology. Genomics. 2003;81:260–269. doi: 10.1016/s0888-7543(03)00020-x. [DOI] [PubMed] [Google Scholar]
- 33.Hoque MO, Lee CC, Cairns P, Schoenberg M, Sidransky D. Genome-wide genetic characterization of bladder cancer: a comparison of high-density single-nucleotide polymorphism arrays and PCR-based microsatellite analysis. Cancer Res. 2003;63:2216–2222. [PubMed] [Google Scholar]
- 34.Hoque MO, Lee J, Begum S, Yamashita K, Engles JM, Schoenberg M, Westra WH, Sidransky D. High-throughput molecular analysis of urine sediment for the detection of bladder cancer by high-density single-nucleotide polymorphism array. Cancer Res. 2003;63:5723–5726. [PubMed] [Google Scholar]
- 35.Janne PA, Li C, Zhao X, Girard L, Chen TH, Minna J, Christiani DC, Johnson BE, Meyerson M. High-resolution single-nucleotide polymorphism array and clustering analysis of loss of heterozygosity in human lung cancer cell lines. Oncogene. 2004;23:2716–2726. doi: 10.1038/sj.onc.1207329. [DOI] [PubMed] [Google Scholar]
- 36.Wang ZC, Lin M, Wei LJ, Li C, Miron A, Lodeiro G, Harris L, Ramaswamy S, Tanenbaum DM, Meyerson M, Iglehart JD, Richardson A. Loss of heterozygosity and its correlation with expression profiles in subclasses of invasive breast cancers. Cancer Res. 2004;64:64–71. doi: 10.1158/0008-5472.can-03-2570. [DOI] [PubMed] [Google Scholar]
- 37.Huang J, Wei W, Zhang J, Liu G, Bignell GR, Stratton MR, Futreal PA, Wooster R, Jones KW, Shapero MH. Whole genome DNA copy number changes identified by high density oligonucleotide arrays. Hum Genomics. 2004;1:287–299. doi: 10.1186/1479-7364-1-4-287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Beroukhim R, Lin M, Park Y, Hao K, Zhao X, Garraway LA, Fox EA, Hochberg EP, Mellinghoff IK, Hofer MD, Descazeaud A, Rubin MA, Meyerson M, Wong WH, Sellers WR, Li C. Inferring loss-of-heterozygosity from unpaired tumors using high-density oligonucleotide SNP arrays. PLoS Comput Biol. 2006;2:e41. doi: 10.1371/journal.pcbi.0020041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Calhoun ES, Hucl T, Gallmeier E, West KM, Arking DE, Maitra A, Iacobuzio-Donahue CA, Chakravarti A, Hruban RH, Kern SE. Identifying Allelic Loss and Homozygous Deletions in Pancreatic Cancer without Matched Normals Using High-Density Single-Nucleotide Polymorphism Arrays. Cancer Res. 2006;66:7920–7928. doi: 10.1158/0008-5472.CAN-06-0721. [DOI] [PubMed] [Google Scholar]
- 40.Tong BC, Dhir K, Ha PK, Westra WH, Alter BP, Sidransky D, Koch WM, Califano JA. Use of single nucleotide polymorphism arrays to identify a novel region of loss on chromosome 6q in squamous cell carcinomas of the oral cavity. Head Neck. 2004;26:345–352. doi: 10.1002/hed.10391. [DOI] [PubMed] [Google Scholar]
- 41.Koed K, Wiuf C, Christensen LL, Wikman FP, Zieger K, Moller K, von der Maase H, Orntoft TF. High-density single nucleotide polymorphism array defines novel stage and location-dependent allelic imbalances in human bladder tumors. Cancer Res. 2005;65:34–45. [PubMed] [Google Scholar]
- 42.Irving JA, Bloodworth L, Bown NP, Case MC, Hogarth LA, Hall AG. Loss of heterozygosity in childhood acute lym-phoblastic leukemia detected by genome-wide microarray single nucleotide polymorphism analysis. Cancer Res. 2005;65:3053–3058. doi: 10.1158/0008-5472.CAN-04-2604. [DOI] [PubMed] [Google Scholar]
- 43.Dahia PL, Hao K, Rogus J, Colin C, Pujana MA, Ross K, Magoffin D, Aronin N, Cascon A, Hayashida CY, Li C, Toledo SP, Stiles CD. Novel pheochromocytoma susceptibility loci identified by integrative genomics. Cancer Res. 2005;65:9651–9658. doi: 10.1158/0008-5472.CAN-05-1427. [DOI] [PubMed] [Google Scholar]
- 44.Gaasenbeek M, Howarth K, Rowan AJ, Gorman PA, Jones A, Chaplin T, Liu Y, Bicknell D, Davison EJ, Fiegler H, Carter NP, Roylance RR, Tomlinson IP. Combined array-comparative genomic hybridization and single-nucleotide polymorphism-loss of heterozygosity analysis reveals complex changes and multiple forms of chromosomal instability in colorectal cancers. Cancer Res. 2006;66:3471–3479. doi: 10.1158/0008-5472.CAN-05-3285. [DOI] [PubMed] [Google Scholar]
- 45.Wang ZC, Buraimoh A, Iglehart JD, Richardson AL. Genome-wide analysis for loss of heterozygosity in primary and re-current phyllodes tumor and fibroadenoma of breast using single nucleotide polymorphism arrays. Breast Cancer Res Treat. 2006;97:301–309. doi: 10.1007/s10549-005-9124-5. [DOI] [PubMed] [Google Scholar]
- 46.Lam CW, To KF, Tong SF. Genome-wide detection of allelic imbalance in renal cell carcinoma using high-density single-nucleotide polymorphism microarrays. Clin Biochem. 2006;39:187–190. doi: 10.1016/j.clinbiochem.2006.01.012. [DOI] [PubMed] [Google Scholar]
- 47.Rauch A, Ruschendorf F, Huang J, Trautmann U, Becker C, Thiel C, Jones KW, Reis A, Nurnberg P. Molecular karyotyping using an SNP array for genomewide genotyping. J Med Genet. 2004;41:916–922. doi: 10.1136/jmg.2004.022855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rubin MA, Varambally S, Beroukhim R, Tomlins SA, Rhodes DR, Paris PL, Hofer MD, Storz-Schweizer M, Kuefer R, Fletcher JA, Hsi BL, Byrne JA, Pienta KJ, Collins C, Sellers WR, Chinnaiyan AM. Overexpression, amplification, and androgen regulation of TPD52 in prostate cancer. Cancer Res. 2004;64:3814–3822. doi: 10.1158/0008-5472.CAN-03-3881. [DOI] [PubMed] [Google Scholar]
- 49.Zhao X, Weir BA, LaFramboise T, Lin M, Beroukhim R, Garraway L, Beheshti J, Lee JC, Naoki K, Richards WG, Sugarbaker D, Chen F, Rubin MA, Janne PA, Girard L, Minna J, Christiani D, Li C, Sellers WR, Meyerson M. Homozygous deletions and chromosome amplifications in human lung carcinomas revealed by single nucleotide polymorphism array analysis. Cancer Res. 2005;65:5561–5570. doi: 10.1158/0008-5472.CAN-04-4603. [DOI] [PubMed] [Google Scholar]
- 50.Sato M, Takahashi K, Nagayama K, Arai Y, Ito N, Okada M, Minna JD, Yokota J, Kohno T. Identification of chromosome arm 9p as the most frequent target of homozygous deletions in lung cancer. Genes Chromosomes Cancer. 2005;44:405–414. doi: 10.1002/gcc.20253. [DOI] [PubMed] [Google Scholar]
- 51.Garraway LA, Widlund HR, Rubin MA, Getz G, Berger AJ, Ramaswamy S, Beroukhim R, Milner DA, Granter SR, Du J, Lee C, Wagner SN, Li C, Golub TR, Rimm DL, Meyerson ML, Fisher DE, Sellers WR. Integrative genomic analyses identify MITF as a lineage survival oncogene amplified in malignant melanoma. Nature. 2005;436:117–122. doi: 10.1038/nature03664. [DOI] [PubMed] [Google Scholar]
- 52.Koochekpour S, Zhuang YJ, Beroukhim R, Hsieh CL, Hofer MD, Zhau HE, Hiraiwa M, Pattan DY, Ware JL, Luftig RB, Sandhoff K, Sawyers CL, Pienta KJ, Rubin MA, Vessella RL, Sellers WR, Sartor O. Amplification and overexpression of prosaposin in prostate cancer. Genes Chromosomes Cancer. 2005;44:351–364. doi: 10.1002/gcc.20249. [DOI] [PubMed] [Google Scholar]
- 53.Park JT, Li M, Nakayama K, Mao TL, Davidson B, Zhang Z, Kurman RJ, Eberhart CG, Shih Ie M, Wang TL. Notch3 gene amplification in ovarian cancer. Cancer Res. 2006;66:6312–6318. doi: 10.1158/0008-5472.CAN-05-3610. [DOI] [PubMed] [Google Scholar]
- 54.Wang Y, Makedon F, Pearlman J. Tumor classification based on DNA copy number aberrations determined using SNP arrays. Oncol Rep. 2006;15:1057–1059. doi: 10.3892/or.15.4.1057. [DOI] [PubMed] [Google Scholar]
- 55.Pei J, Kruger WD, Testa JR. High-resolution analysis of 9p loss in human cancer cells using single nucleotide polymorphism-based mapping arrays. Cancer Genet Cytogenet. 2006;170:65–68. doi: 10.1016/j.cancergencyto.2006.05.002. [DOI] [PubMed] [Google Scholar]
- 56.Stordal B, Peters G, Davey R. Similar chromosomal changes in cisplatin and oxaliplatin-resistant sublines of the H69 SCLC cell line are not associated with platinum resistance. Genes Chromosomes Cancer. 2006;45:1094–1105. doi: 10.1002/gcc.20373. [DOI] [PubMed] [Google Scholar]
- 57.Liu W, Chang B, Sauvageot J, Dimitrov L, Gielzak M, Li T, Yan G, Sun J, Sun J, Adams TS, Turner AR, Kim JW, Meyers DA, Zheng SL, Isaacs WB, Xu J. Comprehensive assessment of DNA copy number alterations in human prostate cancers using Affymetrix 100K SNP mapping array. Genes Chromosomes Cancer. 2006;45:1018–1032. doi: 10.1002/gcc.20369. [DOI] [PubMed] [Google Scholar]
- 58.Fitzgibbon J, Smith LL, Raghavan M, Smith ML, Debernardi S, Skoulakis S, Lillington D, Lister TA, Young BD. Association between acquired uniparental disomy and homozygous gene mutation in acute myeloid leukemias. Cancer Res. 2005;65:9152–9154. doi: 10.1158/0008-5472.CAN-05-2017. [DOI] [PubMed] [Google Scholar]
- 59.Bungaro S, Raghavan M, Dell’Oro MG, Paolucci P, Young BD, Biondi A, Cazzaniga G. Assessment of submicroscopic genetic lesions by single nucleotide polymorphism arrays in a child with acute myeloid leukemia and FLT3-internal tandem duplication. Haematologica. 2006;91:998–1000. [PubMed] [Google Scholar]
- 60.Gorletta TA, Gasparini P, D’Elios MM, Trubia M, Pelicci PG, Di Fiore PP. Frequent loss of heterozygosity without loss of genetic material in acute myeloid leukemia with a normal karyo-type. Genes Chromosomes Cancer. 2005;44:334–337. doi: 10.1002/gcc.20234. [DOI] [PubMed] [Google Scholar]
- 61.Nielaender I, Martin-Subero JI, Wagner F, Martinez-Climent JA, Siebert R. Partial uniparental disomy: a recurrent genetic mechanism alternative to chromosomal deletion in malignant lym-phoma. Leukemia. 2006;20:904–905. doi: 10.1038/sj.leu.2404173. [DOI] [PubMed] [Google Scholar]
- 62.Walker BA, Leone PE, Jenner MW, Li C, Gonzalez D, Johnson DC, Ross FM, Davies FE, Morgan GJ. Integration of global SNP-based mapping and expression arrays reveals key regions, mechanisms, and genes important in the pathogenesis of multiple myeloma. Blood. 2006;108:1733–1743. doi: 10.1182/blood-2006-02-005496. [DOI] [PubMed] [Google Scholar]
- 63.Midorikawa Y, Yamamoto S, Ishikawa S, Kamimura N, Igarashi H, Sugimura H, Makuuchi M, Aburatani H. Molecular karyotyping of human hepatocellular carcinoma using single-nucleotide polymorphism arrays. Oncogene. 2006;25:5581–5590. doi: 10.1038/sj.onc.1209537. [DOI] [PubMed] [Google Scholar]
- 64.Andersen CL, Wiuf C, Kruhoffer M, Korsgaard M, Laurberg S, Orntoft TF. Frequent occurrence of uniparental disomy in colorectal cancer. Carcinogenesis. 2006 doi: 10.1093/carcin/bgl086. [DOI] [PubMed] [Google Scholar]
- 65.Tsafrir D, Bacolod M, Selvanayagam Z, Tsafrir I, Shia J, Zeng Z, Liu H, Krier C, Stengel RF, Barany F, Gerald WL, Paty PB, Domany E, Notterman DA. Relationship of gene expression and chromosomal abnormalities in colorectal cancer. Cancer Res. 2006;66:2129–2137. doi: 10.1158/0008-5472.CAN-05-2569. [DOI] [PubMed] [Google Scholar]
- 66.Schaid DJ, Guenther JC, Christensen GB, Hebbring S, Rosenow C, Hilker CA, McDonnell SK, Cunningham JM, Slager SL, Blute ML, Thibodeau SN. Comparison of micro-satellites versus single-nucleotide polymorphisms in a genome linkage screen for prostate cancer-susceptibility Loci. Am J Hum Genet. 2004;75:948–965. doi: 10.1086/425870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sellick GS, Webb EL, Allinson R, Matutes E, Dyer MJ, Jonsson V, Langerak AW, Mauro FR, Fuller S, Wiley J, Lyttelton M, Callea V, Yuille M, Catovsky D, Houlston RS. A high-density SNP genomewide linkage scan for chronic lympho-cytic leukemia-susceptibility loci. Am J Hum Genet. 2005;77:420–429. doi: 10.1086/444472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kemp Z, Carvajal-Carmona L, Spain S, Barclay E, Gorman M, Martin L, Jaeger E, Brooks N, Bishop DT, Thomas H, Tomlinson I, Papaemmanuil E, Webb E, Sellick GS, Wood W, Evans G, Lucassen A, Maher ER, Houlston RS. Evidence for a colorectal cancer susceptibility locus on chromosome 3q21-q24 from a high-density SNP genome-wide linkage scan. Hum Mol Genet. 2006;15:2903–2910. doi: 10.1093/hmg/ddl231. [DOI] [PubMed] [Google Scholar]
- 69.Hu N, Wang C, Hu Y, Yang HH, Giffen C, Tang ZZ, Han XY, Goldstein AM, Emmert-Buck MR, Buetow KH, Taylor PR, Lee MP. Genome-wide association study in esophageal cancer using GeneChip mapping 10K array. Cancer Res. 2005;65:2542–2546. doi: 10.1158/0008-5472.CAN-04-3247. [DOI] [PubMed] [Google Scholar]
- 70.Cao X, Eu KW, Kumarasinghe MP, Li HH, Loi C, Cheah PY. Mapping of hereditary mixed polyposis syndrome (HMPS) to chromosome 10q23 by genomewide high-density single nucleotide polymorphism (SNP) scan and identification of BMPR1A loss of function. J Med Genet. 2006;43:e13. doi: 10.1136/jmg.2005.034827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Vierimaa O, Georgitsi M, Lehtonen R, Vahteristo P, Kokko A, Raitila A, Tuppurainen K, Ebeling TM, Salmela PI, Paschke R, Gundogdu S, De Menis E, Makinen MJ, Launonen V, Karhu A, Aaltonen LA. Pituitary adenoma pre-disposition caused by germline mutations in the AIP gene. Science. 2006;312:1228–1230. doi: 10.1126/science.1126100. [DOI] [PubMed] [Google Scholar]
- 72.Landi S, Gemignani F, Moreno V, Gioia-Patricola L, Chabrier A, Guino E, Navarro M, de Oca J, Capella G, Canzian F. A comprehensive analysis of phase I and phase II metabolism gene polymorphisms and risk of colorectal cancer. Pharmacogenet Genomics. 2005;15:535–546. doi: 10.1097/01.fpc.0000165904.48994.3d. [DOI] [PubMed] [Google Scholar]
- 73.Yuan E, Haghighi F, White S, Costa R, McMinn J, Chun K, Minden M, Tycko B. A single nucleotide polymorphism chip-based method for combined genetic and epigenetic profiling: validation in decitabine therapy and tumor/normal comparisons. Cancer Res. 2006;66:3443–3451. doi: 10.1158/0008-5472.CAN-05-3739. [DOI] [PubMed] [Google Scholar]
- 74.Bibikova M, Lin Z, Zhou L, Chudin E, Garcia EW, Wu B, Doucet D, Thomas NJ, Wang Y, Vollmer E, Goldmann T, Seifart C, Jiang W, Barker DL, Chee MS, Floros J, Fan JB. High-throughput DNA methylation profiling using universal bead arrays. Genome Res. 2006;16:383–393. doi: 10.1101/gr.4410706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Pastinen T, Ge B, Gurd S, Gaudin T, Dore C, Lemire M, Lepage P, Harmsen E, Hudson TJ. Mapping common regulatory variants to human haplotypes. Hum Mol Genet. 2005;14:3963–3971. doi: 10.1093/hmg/ddi420. [DOI] [PubMed] [Google Scholar]
- 76.Lo HS, Wang Z, Hu Y, Yang HH, Gere S, Buetow KH, Lee MP. Allelic variation in gene expression is common in the human genome. Genome Res. 2003;13:1855–1862. doi: 10.1101/gr.1006603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Pant PV, Tao H, Beilharz EJ, Ballinger DG, Cox DR, Frazer KA. Analysis of allelic differential expression in human white blood cells. Genome Res. 2006;16:331–339. doi: 10.1101/gr.4559106. [DOI] [PMC free article] [PubMed] [Google Scholar]