

Abstract
A variety of chroman spiroketals are synthesized via inverse-demand [4 + 2] cycloaddition of enol ethers and ortho-quinone methides (o-QMs). Low temperature o-QM generation in situ allows for the kinetic, diastereoselective construction of these motifs, providing entry to a number of unusual chroman spiroketal natural products.
Aliphatic spiroketals are a common substructure of natural products isolated in a large variety from both marine and terrestrial sources.1 Studies aimed at understanding the origins of their conformational preference have resulted in the general assumption that the spiroketal carbon corresponds to the thermodynamically most stable isomer as determined by stereoelectronic influences. Hence, the biosynthesis of these fluxional natural products usually results in the construction of the most stable diastereomer or a mixture that reflects energy differences between respective diastereomers.
For some time, we have speculated that a rare subset consisting of chroman spiroketals (1,2 2,3 and 3,4 Figure 1) may possess features that refute this basic tenet and require a kinetic assembly. These uniquely robust chroman spiroketal scaffolds have caused us to consider new methods for their construction.
Figure 1.

Natural products that contain a chroman spiroketal.
Some time ago, we developed a method that enables the controlled, low-temperature generation of o-quinone methides (o-QMs) (Figure 2, I).5 The process, which is driven by the relative stability of sequential anions formed along the cascade, begins by formation of an alkoxide. This species intercepts a neighboring carbonyl of a phenolic carbonate and thus liberates a phenoxide that subsequently undergoes β-elimination of a carboxylate and thereby forms a reactive o-QM species, which captures the first nucleophile that it subsequently encounters.
Figure 2.

(I) Cascade leading to o-QMs. (II) Preferred endo transition state for [4 + 2] cycloaddition with chiral vinyl ether derivatives. (III) Proposal to examine the effects of R1, R2, R3 on the reactions of methylene dihydrofurans.
This is a useful method for o-QM generation because, since the reactive o-QM intermediate is generated at low temperature in low concentrations, it subsequently participates in very precise, kinetically controlled reactions.6 Using this procedure, we demonstrated the first examples of diastereoselective o-QM cycloadditions and found that the inverse demand [4 + 2] significantly favors an endo transition state with electron-rich alkenes.7 In further experiments, we showed that chiral enol ethers containing remote stereocenters, such as those derived from (1S,2R)-2-phenyl-cyclohexanol, will participate in diastereoselective cycloadditions to yield the corresponding chroman acetals with an R configuration (Figure 2, II).8 On the basis of these findings, we began to speculate that an assortment of chiral 2-methylene tetrahydrofurans with R1, R2, and R3 substituents should also participate in cycloadditions with similar diastereoselectivity (Figure 2, III). Detailed analysis of our proposed transition state allowed us to further speculate that substituents R1 and R3 would have a pronounced effect on the stereochemical outcome, whereas the R2 residue would not, allowing it to be tolerated on the same face as the reaction. Therefore, we began to examine model systems that might eventually be developed into strategies leading to the construction of the chroman spiroketal natural products 1–3.
Our investigation began with the preparation of known enol ethers 4,9 5,10 6,11 and 7,12 as well as the unknown enol ether 8, which we prepared from the known acid 1113 by sequential acetalization with formaldehyde and methylenation with Tebbe’s reagent (Scheme 1).
Scheme 1.

Known and Readily Available Five-Membered Ring Enol Ethers Containing Exocyclic Olefins
We also prepared enol ethers 9 and 10 in a short sequence. We began by subjecting the unsaturated lactone 13 (2.4 M in THF) to lithiated acetonitrile 14, which results in exclusive 1,4-addition. Use of the latter as the nucleophile avoids the problem of an undesirable proton transfer during the course of the reaction. Thus, the intermediate enolate undergoes subsequent C-methylation with methyl iodide and affords the trans-substituted lactone 15 in 73% yield (>10:1 dr). Freshly prepared Tebbe reagent14 fails to provide the desired product and returns 15 unchanged. However, if 15 (0.1 M in THF) is refluxed with Petasis’ reagent (2.5 equiv, 4 h) and sequentially treated to a methanol/water workup, the enol ether 10 arises whereby the nitrile functionality has also been transformed into a methyl ketone. While investigation of the crude product mixture by 1H NMR showed compound 10 as the only identifiable product, bulb-to-bulb distillation with base washed glassware provided only a 20–30% yield. To the best of our knowledge, the conversion of a nitrile into a methyl ketone by the Petasis reagent has never before been reported.15 Doxsee has observed a related transformation with Tebbe’s reagent in the presence of DMAP and trimethyl phosphine.16 If the amount of THF is limited, however, to the minimal amount required for solubility (toluene/THF, 3:2), the reaction affords the enol ether 9 (4:1 ratio of 9/10) in a 40–50% yield after flash chromatography and distillation.
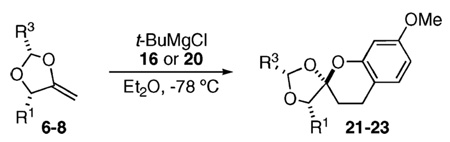
With these enol ethers in hand, we began examining their reactivity with o-QMs. When the known benzyl alcohol 1617 (0.1 M in toluene, −78 °C, 2–4 equiv of the 1,3-dihy-droisobenzofuran enol ether 4) is treated with t-BuMgCl, an endo-selective cycloaddition proceeds as the reaction mixture slowly warms to room temperature (Scheme 2). The reaction cleanly produces the chroman spiroketal 17 in 60% isolated yield (>8:1 dr by crude 1H NMR). This transformation, therefore, serves as a practical model for an eventual synthetic strategy aimed toward (+)-paecilospirone (3).
Scheme 2.

Some [4 + 2] Cycloadditions of Benzofurans and an o-QM
We next examined a similar cycloaddition using the enol ether 5. The 2-methylbenzofuran 18 is produced along with a small amount of the desired chroman spiroketal 19 in 10% yield. Presumably, the fragile 2,3-dihydrobenzofuran enol ether 5 succumbs to isomerization under these conditions.18 Therefore, due to the low yield for this transformation, we sought an alternative strategy for the construction of 1 by investigating o-QM cycloadditions with the enol ethers 6–8.
The starting benzyl alcohol 16 (0.1 M in Et2O at −78 °C, 2 equiv of 6) was subjected again to t-BuMgCl (Table 1). A cycloaddition occurs as the mixture slowly warms to room temperature, affording a single diastereomer as determined by crude 1H NMR (45% isolated yield of 21). On the basis of our extensive experience with these cycloadditions, we speculate that the product 21 has the stereochemistry shown, which reflects a reaction proceeding through an endo orientation on the face opposite of phenyl residues at R1 and R3. Next, we examined the corresponding reaction of enol ether 7 and benzyl alcohol 2019 using similar conditions. Again, 1H NMR revealed formation of 22 as a single diastereomer (>20:1). This result suggested to us that the presence of an R1 substituent was sufficient to control the diastereoselectivity of the reaction. Thus, we were not altogether surprised that use of the enol ether 8, which expresses a substituted aryl ring, undergoes a diastereoselective reaction with the o-QM derived from 16 in comparable ratio and yield.
Table 1.
Cycloadditions of Methylene-1,3-dioxolanes
 | ||||||
|---|---|---|---|---|---|---|
| enol ether | benzyl alcohol | product | yield | dr | ||
| 6 | 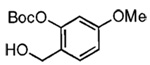 |
16 | 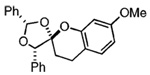 |
21 | 45% | >20:1 |
| 7 | 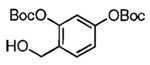 |
20 | 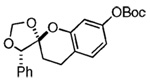 |
22 | 52% | >20:1 |
| 8 | 16 | 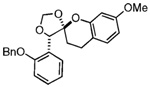 |
23 | 45% | >20:1 | |
With the chroman spiroketal 23 in hand, we paused to investigate its reformulation into the corresponding benzo-furan spiro-adduct (Scheme 3). The benzyl ether in 23 (0.5 M in 95% EtOH) was cleaved in nearly quantitative yield by exposure of the heterogeneous mixture containing 5% Pd/C to a hydrogen atmosphere to provide phenol 24. Subsequent treatment of 24 with various proton sources, as well as a cadre of Lewis acids, failed to afford the desired spiro-adduct 26 (via 25). In most instances, the starting material remained unchanged or suffered decomposition.
Scheme 3.

Failed Attempts to Effect Trans-Ketalization
The resiliency of the chroman spiroketal 24 and related structures is appreciated through an understanding of resonance effects. The lone pair of electrons on the chroman oxygen is delocalized throughout the adjoining aromatic ring. Therefore, it is difficult for the lone pair of electrons to assist in the departure of the aliphatic ether, which results in the formation of the oxonium 25. Conversely, the lone pair of electrons belonging to the chroman oxygen is also difficult to protonate, which would lead to the cleavage of the corresponding aryl ether. These facts also account for the unusual stability of MOM-aryl ethers as compared to the corresponding aliphatic variety. On the basis of these results, we abandoned an o-QM strategy for 1 and devised an alternative method for the construction of naphthoquinone spiroketals.20
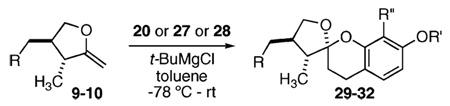
Our attention now turned toward the chroman spiroketal framework found in the natural product 2. Similar treatment of 27 and 9 with t-BuMgCl affords 29 along with its diastereomer in a 61% yield as an inseparable 3:1 ratio (Table 2). We assumed that the R1 substituent (−Me), which is closer to the site of bond formation, exerted greater stereo-control than the R2 substituent (−CH2CN), as the latter is further removed from the site of reaction. Nevertheless, the ratio was disappointing and we decided to examine the effects of electronics within the o-QM on the stereoselectivity for the reaction. Upon exchange of the −OBn for a more electron deficient −OBoc residue, the reaction of the o-QM derived from 20 exhibits a small increase in selectivity (3.5:1) favoring the chroman spiroketal 30. Unfortunately, the diastereomers again prove inseparable by chromatography (77% combined yield). Introduction of a bromine atom results in a higher yield and better selectivity (4.5:1) for 31.21 Moreover, the bromine atom assists in the subsequent separation of diastereomers by chromatography. Similar findings were observed for the ketone 32 that arises from the reaction of 28 and the enol ether 10, which provides further evidence for the mildness of this method and its compatibility with a variety of functional groups. The lower diastereoselectivity compared with that in Table 1 may indicate that the steric bulk of R1 (Figure 2) plays a substantial role in the stereochemical outcome of the reaction.
Table 2.
Cycloadditions of Some Exocyclic Furan Enol Ethers
 | ||||||
|---|---|---|---|---|---|---|
| enol ether | benzyl alcohol | product | yield | dr | ||
| 9 | 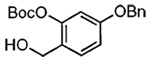 |
27 | 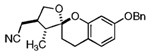 |
29 | 61% | 3:1 |
| 9 | 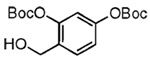 |
20 | 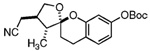 |
30 | 77% | 3.5:1 |
| 9 | 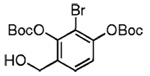 |
28 | 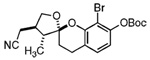 |
31 | 72% | 4.5:1 |
| 10 | 28 | 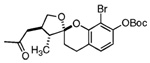 |
32 | 72% | 4.5:1 | |
While we are confident of our structural assignments based upon analysis of coupling data and NOE effects, we wished to unequivocally establish the relative stereochemistry of adducts 17, 21–23, and 29–32. Unfortunately, our attempts to crystallize these materials failed. We therefore decided to cleave the Boc group from compound 31, in hope that the phenol 33 would crystallize. This task was accomplished by short exposure to 1 N LiOH. Crystallization from CH2-Cl2/MeOH (1:1) provided X-ray quality crystals of 33 (Figure 3). The resulting structure determination proved 31 arises from reaction of the enol ether in endo orientation on the face opposite the R1 substituent and on the same side as the R3 substituent.
Figure 3.

X-ray crystal structure of 33.
In conclusion, we provide the first example of kinetically controlled, diastereoselective construction of chiral chroman spiroketals using enol ethers and o-QMs. This versatile method, due to its stereocontrol and functional group compatibility, seems applicable for subsequent stereoselective synthetic routes toward chroman spiroketal-containing natural products (Scheme 1). These results will be reported in due course.
Acknowledgment
Support from the University of California, Cancer Research Coordinating Committee for investigations leading to the synthesis of 1 and 2 is greatly appreciated. We applaud Neil Schore (UC Davis) for his oversight of this important program.
Footnotes
Supporting Information Available: Experimental procedures and spectroscopic data (1H NMR, 13C NMR, FT-IR, HRMS) for compounds 8–12, 15, 17, 19, 21–24, 27, and 29–33. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Perron F, Albizati KF. Chem. Rev. 1989;89:1617. [Google Scholar]
- 2.(a) Puder C, Loya S, Hizi A, Zeeck A. Eur. J. Org. Chem. 2000:729. [Google Scholar]; (b) Brockmann H, Lenk W, Schwantje G, Zeeck A. Tetrahedron Lett. 1966;7:3525. doi: 10.1016/s0040-4039(01)82822-7. [DOI] [PubMed] [Google Scholar]; (c) Brockmann H, Lenk W, Schwantje G, Zeeck A. Chem. Ber. 1969;102:126. doi: 10.1002/cber.19691020118. [DOI] [PubMed] [Google Scholar]; (d) Brockmann H, Zeeck A. Chem. Ber. 1970;103:1709. doi: 10.1002/cber.19701030607. [DOI] [PubMed] [Google Scholar]
- 3.Stierle AA, Stierle DB, Kelly K. J. Org. Chem. 2006;71:5357. doi: 10.1021/jo060018d. [DOI] [PubMed] [Google Scholar]
- 4.(a) Namikoshi M, Kobayashi H, Yoshimoto T, Meguro S. Chem. Lett. 2000:308. doi: 10.1248/cpb.48.1452. [DOI] [PubMed] [Google Scholar]; (b) Namikoshi M, Kobayashi H, Yoshimoto T, Meguro S. Chem. Pharm. Bull. 2000:1452. doi: 10.1248/cpb.48.1452. [DOI] [PubMed] [Google Scholar]
- 5.Van de Water RW, Magdziak DJ, Chau JN, Pettus TRR. J. Am. Chem. Soc. 2000;122:6502. [Google Scholar]
- 6.Van de Water RW, Pettus TRR. Tetrahedron. 2002;58:5367. [Google Scholar]
- 7.Jones RM, Selenski C, Pettus TRR. J. Org. Chem. 2002;67:6911. doi: 10.1021/jo020224v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.(a) Selenski C, Pettus TRR. J. Org. Chem. 2004;69:9196. doi: 10.1021/jo048703c. [DOI] [PubMed] [Google Scholar]; (b) Selenski C, Mejorado LH, Pettus TRR. Synlett. 2004;6:1101. [Google Scholar]
- 9.Okazoe T, Takai K, Oshima K, Utimoto K. J. Org. Chem. 1987;52:4410. [Google Scholar]
- 10.Pine SH, Zahler R, Evans DA, Grubbs RH. J. Am. Chem. Soc. 1980;102:3270. [Google Scholar]
- 11.Petasis NA, Lu S. J. Am. Chem. Soc. 1995;117:6394. [Google Scholar]
- 12.Diaz-Ortiz A, Diez-Barra E, de la Hoz A, Prieto P. Synth. Comm. 1993;23:1935. [Google Scholar]
- 13.Baltzer S, Schoentjes B, Van Dorsselaer V. Fr. Demande. 2004:85. [Google Scholar]
- 14.Payack JF, Hughes DL, Cai D, Cottrell IF, Verhoeven TR. Org. Synth. 2002;79:19. [Google Scholar]
- 15.For reactions of the Petasis reagent with nitriles, see: Petasis NA, Fu D-K. Organometallics. 1993;12:3776.Barluenga J, del Poso Lasada C, Olano B. Tetrahedron Lett. 1992;33:7579.(c) Treatment of phenylacetonitrile with Petasis’ reagent smoothly affords 1-phenylacetone in 75% yield. Thus, this mild nitrile-to-ketone transformation appears general and proceeds in better yields with less complicated examples
- 16.(a) Doxsee KM, Farahi JB. J. Am. Chem. Soc. 1988;110:7239. [Google Scholar]; (b) Takeda T. Bull. Chem. Soc. Jpn. 2005;78:195. [Google Scholar]; (c) Doxsee KM, Farahi JB. J. Chem. Soc., Chem. Commun. 1990;20:1452. [Google Scholar]
- 17.(a) Marsini MA, Huang Y, Van De Water RW, Pettus TRR. Org. Lett. 2007;7:3229. doi: 10.1021/ol0710257. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Lindsey CC, Pettus TRR. Tetrahedron Lett. 2006;47:201. doi: 10.1016/j.tetlet.2005.10.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhou G, Zheng D, Da S, Xie Z, Li Y. Tetrahedron Lett. 2006;47:3349. [Google Scholar]
- 19.Hoarau C, Pettus TRR. Org. Lett. 2006;8:2843. doi: 10.1021/ol061000s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.(a) Wu K-L, Wilkinson S, Reich NO, Pettus TRR. Org. Lett. 2007;9:5537. doi: 10.1021/ol702450d. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Lindsey CC, Wu K-L, Pettus TRR. Org. Lett. 2006;8:2365. doi: 10.1021/ol0606886. [DOI] [PubMed] [Google Scholar]
- 21.Mejorado LH, Hoarau C, Pettus TRR. Org. Lett. 2004;6:1535. doi: 10.1021/ol0498592. [DOI] [PMC free article] [PubMed] [Google Scholar]