

SUMMARY
The LytTR domain is a DNA-binding motif found within the AlgR/AgrA/LytR family of transcription factors that regulate virulence factor and toxin gene expression in pathogenic bacteria. This previously uncharacterized domain lacks sequence similarity with proteins of known structure. The crystal structure of the DNA-binding domain of Staphylococcus aureus AgrA complexed with a DNA pentadecamer duplex has been determined at 1.6 Å resolution. The structure establishes a 10-stranded β fold for the LytTR domain and reveals a novel mode of interaction with DNA. Residues within loop regions of AgrA contact two successive major grooves and the intervening minor groove on one face of the oligonucleotide duplex, inducing a substantial bend in the DNA. Loss of DNA-binding upon substitution of key interacting residues in AgrA supports the observed binding mode. This novel mode of protein-DNA interacton provides a potential target for future antimicrobial drug design.
INTRODUCTION
Bacterial response regulators of two-component signal transduction systems are used for transcriptional regulation of a wide range of genes in response to cellular and environmental signals. An analysis of 330 bacterial and archaeal genomes identified 5589 DNA binding response regulators (Galperin, 2006). While all characterized response regulators contain a conserved receiver domain necessary for phosphorylation dependent activation and dimerization, 95.3% interact with DNA through a helix-turn-helix DNA-binding domain (HTH) or a variation of this domain (e.g. winged-HTH, helix-ribbon-helix). The remaining 4.7% of proteins annotated as DNA-binding response regulators contain a conserved, uncharacterized DNA-binding domain named the LytTR domain after the Bacillus subtillus LytT and Staphylococcus aureus LytR response regulators (Nikolskaya and Galperin, 2002).
The LytTR domain is a ~105-residue bacterial DNA-binding domain found most commonly as the effector module in response regulators, although it can also be found as a stand-alone domain and in combination with membrane-associated MHYT, ABC transporter, and PAS domains (Nikolskaya and Galperin, 2002). LytTR domains exist mostly in the genomes of γ-proteobacteria and firmicutes where they are encoded in only one or two genes per genome, substantially less prevalent than transcription factors of the more populated response regulator subfamilies. A common function associated with response regulators that contain LytTR domains is the regulation of virulence factor and toxin production. Examples of such proteins include Pseudomonas aeruginosa AlgR, the response regulator that modulates the production of the exopolysaccharide alginate involved in chronic pneumonia (Lizewski et al., 2002; Mohr et al., 1991); Clostridium perfringens VirR, which is required for production of toxins implicated in gas gangrene (Rood, 1998; Shimizu et al., 2002); Streptococcus pneumoniae BlpR, a transcriptional regulator of bacteriocin production and the only response regulator required for growth in that organism (Dawid et al., 2007; de Saizieu et al., 2000); Lactobacillus plantarum PlnC, involved in bacteriocin regulation (Diep et al., 2003; Risøen et al., 2001); and Staphylococcus aureus AgrA, the transcriptional component of a quorum sensing system and global regulator of virulence that upregulates secreted virulence factors and down-regulates cell wall-associated proteins (Abdelnour et al., 1993; Novick et al., 1995; Novick, 2003). The agr locus contains two promoter regions. The first, designated P2, contains two high-affinity LytTR domain binding sites. The second promoter region, P3, is located on the opposite strand, 40 bp downstream of the P2 region and contains both a high-affinity LytTR binding site and a low-affinity binding site (Koenig et al., 2004).
In most systems studied, LytTR domain-containing response regulators dimerize and bind to direct 9-bp repeats, often imperfect, which are separated by 12 bp and are located just upstream of the transcription start site (Cheung and Rood, 2000; de Saizieu, et al., 2000; Diep et al., 1996; Koenig, et al., 2004; Mohr, et al., 1991; Mohr et al., 1992; Risøen et al., 1998; Ween et al., 1999). The number of bp separating the repeats has been reported to be crucial, as reducing the number by a single bp renders the promoter nonfunctional (Knutsen et al., 2004; Risøen, et al., 2001). An exception has been observed in the AlgR system, in which two binding sites separated by 66 bp are located far upstream and a third binding site is located just upstream of the transcriptional start site. This arrangement has been implicated in a DNA looping mechanism required for transcriptional activation (Mohr et al., 1990).
While the roles of LytTR domain transcription factors in regulation of bacterial virulons have been investigated in many pathogenic organisms, structural information is lacking. The absence of sequence similarity to any characterized DNA-binding motif suggests that the LytTR domain might be a unique fold, and little is known about the molecular interaction between LytTR domains and DNA. To address these questions, we have determined the X-ray crystal structure of the C-terminal DNA-binding domain of the Staphylococcus aureus response regulator AgrA in complex with a 15-bp DNA duplex containing a 9-bp consensus binding sequence. The LytTR domain consists of a 10-stranded elongated β–β–β fold that appears to have arisen from duplication of a 5-stranded β motif. Residues within three loops protruding from an edge of the β sheets make base-specific contacts to two adjacent major grooves and the intervening minor groove of the DNA. The AgrA-DNA interface is sterically and electrostatically complementary, with a buried surface of 1500 Å2, and the DNA is significantly bent and distorted as it conforms to the protein surface. This novel protein-DNA interaction provides a unique target in pathogenic bacteria that might be exploited for antimicrobial drug design.
RESULTS AND DISCUSSION
Crystallization of an AgrAC–DNA Complex
Initial attempts to produce quantities of full-length Staphylococcus aureus AgrA suitable for structure determination were unsuccessful due to insolubility. To circumvent this problem, the C-terminal DNA-binding domain, termed AgrAC (residues 137–238 with an initiator methionine), was expressed and purified. We were unsuccessful in crystallizing AgrAC alone, and so pursued investigation of AgrAC–DNA complexes.
We first determined the minimum sequence necessary to obtain a stable interaction between AgrAC and DNA oligonucleotides. Since we were investigating the isolated C-terminal domain, which migrates as an apparent monomer during gel filtration chromatography (data not shown), a single 9-bp repeat was used as the basis for construction of a set of oligonucleotides for DNA-binding studies. Beginning with a 19-bp oligonucleotide that contained the 9-bp consensus binding sequence flanked by 5 bp on each side, corresponding to the sequence of the upstream binding site in the P2 promoter region of the agr locus, we systematically shortened the flanking regions 1 bp at a time, while simultaneously adding 1 bp to the opposite flanking region to maintain a constant length of 19 bp, until we could no longer detect a stable interaction by using electrophoretic mobility shift assays. We determined that ~3 bp on either side of the 9-bp consensus binding sequence were necessary for optimal interaction of AgrAC with DNA.
Using this 15-bp minimum sequence as a starting point, oligonucleotides of various lengths, with and without 1-nucleotide overhangs, were designed for crystallization trials. Diffraction quality crystals were obtained by co-crystallizing AgrAC with a 15-bp duplex containing 1-nucleotide overhangs at the 5′ end of each strand, and centered on the 9-bp consensus binding sequence.
Structure Determination and Analysis
The X-ray crystal structure of the AgrAC–DNA complex was determined at 1.6 Å resolution using SAD phasing with oligonucleotides containing two 5-bromodeoxyuridine bases at positions 14 on strand A and 15 on strand B (Figure 1A). The crystals belonged to space group P41 with one AgrAC–DNA complex per asymmetric unit and a solvent content of 52%. The final electron density maps were of excellent quality and allowed for unambiguous construction of a model containing residues 136–238 of AgrAC as well as the complete DNA molecule. The crystallographic R and Rfree were 0.195 and 0.222, respectively, and the final model exhibited excellent geometry (Table 1). Within the crystal lattice, the DNA forms a pseudo-continuous helix along the crystallographic c axis through a reverse Hoogsteen interaction between complementary overhangs with symmetry-related molecules. Other packing interactions occur between strand β8 of one AgrAC and β5 of an adjacent molecule and between the extended N-terminal 6 residues of AgrAC and the DNA major groove of a symmetry-related complex.
Figure 1. Structure of the AgrAC–DNA Complex.
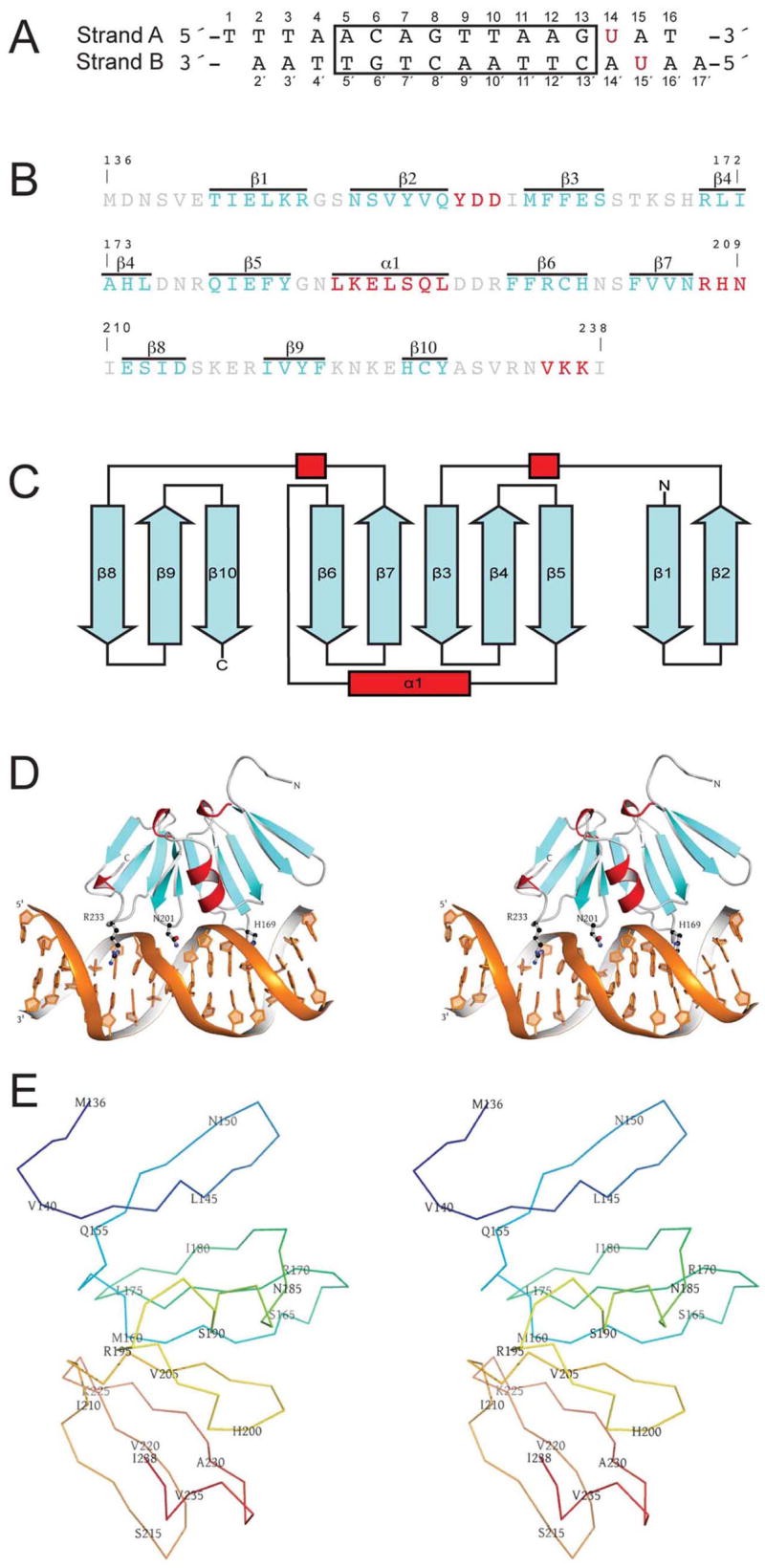
(A) Sequence of the DNA oligonucleotide crystallized in complex with AgrAC. The highly conserved LytTR consensus sequence is boxed. A red U indicates each position substituted with 5′-bromouracil. The coding strand is labeled as Strand A and the non-coding strand as Strand B.
(B) Sequence of the C-terminal DNA-binding domain of AgrA (residues 137–238 and initiator methionine). Loop regions are colored gray, helices are colored red, and strands are colored cyan. The secondary structure elements are indicated above each region.
(C) Topology diagram of AgrAC. Secondary structure elements are colored as described in (B).
(D) A stereo ribbon representation of the AgrAC–DNA complex. AgrAC is shown in an orientation and color scheme similar to that in (B). Residues that make base-specific contacts with the DNA are shown in ball-and-stick representation.
(E) Stereo view of an alpha carbon trace of AgrAC. The chain is color ramped from blue at the N terminus to red at the C terminus with residue numbers indicated at every fifth C α.
Table 1.
Data Collection and Refinement Statistics
| Data collection | |
| Space group | P41 |
| Cell dimensions | |
| a, b, c (Å) | 47.9, 47.9. 100.1 |
| α, β, γ (°) | 90, 90, 90 |
| Resolution (Å) | 17.3−1.60 (1.66−1.60)a |
| Rmergeb (%) | 1.7 (15.5) |
| I/σI | 41.7 (5.7) |
| Completeness (%) | 98.1 (97.3) |
| Redundancy | 3.8 (3.7) |
|
| |
| Refinement | |
|
| |
| Resolution (Å) | 17.3−1.6 |
| No. reflections | 27,747 |
| Rworkc/Rfreed (%) | 19.5/22.2 |
| No. atoms | |
| Protein | 872 |
| DNA | 650 |
| Water | 243 |
| Ions | 2 |
| Average B-factors (Å2) | |
| Protein | 16.5 |
| DNA | 19.6 |
| Water | 21.9 |
| Ions | 19.9 |
| Rmsde | |
| Bond lengths (Å) | 0.007 |
| Bond angles (°) | 1.38 |
Values in parentheses are for the highest-resolution shell.
Rmerge = (ΣhΣi|<I(h)> − I(h)i|)/ΣhΣi I(h)i, where I(h)i is the ith observation of reflection h, and <I(h)> is the mean intensity of all observations of reflection h.
Rwork = (Σ||Fo| − |Fc||)/Σ|Fo|, where |Fo| and |Fc| are observed and calculated structure factor amplitudes, respectively.
Rfree was calculated for 5% of the randomly selected reflections of data sets that were not used in the refinement.
Rmsd, root-mean-square deviation.
The LytTR Domain has an Elongated β-β-β Fold
The structure of AgrAC reveals a novel topology, having ten β strands arranged into three antiparallel β sheets and a small two-turn α helix that is not involved in DNA binding (Figures 1B–1E). The sheets are arranged roughly parallel to each other in an elongated β-β-β sandwich. A hydrophobic five-stranded β sheet (sheet 2: β3–β7) is at the center of the domain with two smaller amphipathic β sheets (sheet 1: β1–β2 and sheet 3: β8–β10) positioned on either side (Figures 1C and 1D).
The α helix is located between strands β5 and β6 and is packed along the edges of sheet 1 and sheet 2. Each sheet is linked to the next by a 310 turn followed by a buried isoleucine that anchors the turn. A salt bridge interaction between residues D157 and H208, each located in a turn of 310 helix act to further stabilize the connections between the sheets. Salt-bridges are also observed between residues R195, located in the loop between helix 1 and strand β6, and residue E141 located in the beginning of strand β1 and D157. Salt bridges between residues D176 and K223 and between H174 and E226 also stabilize the interaction between sheets 2 and 3 (Figure 2). A single turn of 310 helix at the C terminus packs between sheets 2 and 3. The 6 residues at the N terminus of AgrAC adopt an extended conformation that is likely to be different in the context of the full-length protein (Figures 1D and 1E).
Figure 2. Salt Bridge Interactions in AgrAC.
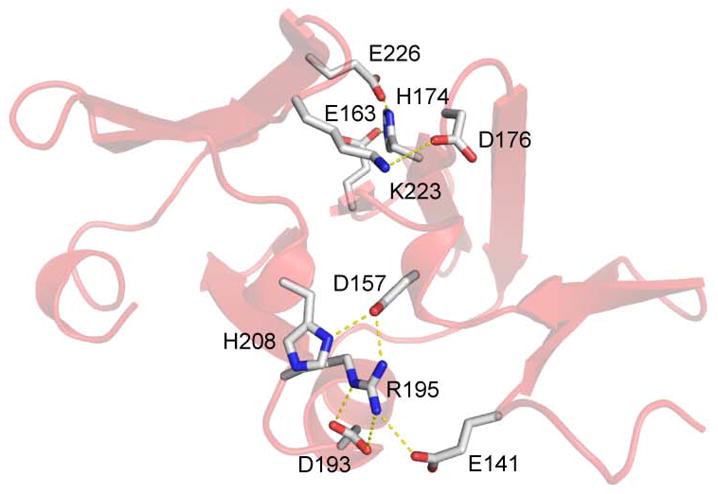
Ribbon representation of AgrAC displaying salt bridges that stabilize the fold. Sidechains of residues involved in salt bridge interactions are shown in stick representation with carbon in white, oxygen in red, and nitrogen in blue. The salt bridges are concentrated on the surface opposite of the surface that contacts DNA.
An interesting feature of this fold is the two-fold symmetry between strands β1–β5 and β6–β10. A least-squares alignment of residues 136–193 and 194–238 shows that the connectivity and spatial positioning of the strands is remarkably similar, suggesting that this fold is derived from duplication of a smaller domain (Figure 1C).
Structural homology between AgrAC and Sac7d
DALI (Holm and Sander, 1995) and VAST (Gibrat et al., 1996) structure homology searches revealed that AgrAC is similar to the well-characterized 66-residue archaeal DNA-binding protein Sac7d from Sulfolobus acidocaldarius (Sso7d in Sulfolobus solfataricus) and a 74-residue protein of unknown function from Pseudomonas aeruginosa (Lin et al., 2006). Sac7d/Sso7d are basic, highly abundant 7-kDa proteins that bind nonspecifically to DNA conferring thermo, chemical, and acid stability. The structure of Sac7d/Sso7d is comprised of five antiparallel β strands arranged into a two-stranded β sheet connected to a three-stranded β sheet by way of a single turn of 310 helix, remarkably similar to strands 1–5 and 6–10 of AgrAC (Figure 3A) (McCrary et al., 1996; Robinson et al., 1998). Sac7d/Sso7d contact DNA with the β sheet of Sac7d/Sso7d interacting in the minor groove by intercalating two hydrophobic residues between the DNA bases (Figure 3B) (Peters et al., 2004; Robinson, et al., 1998). This interaction causes significant unwinding and severely kinks the DNA by 72°.
Figure 3. Structural Comparison of AgrAC and Sac7d.
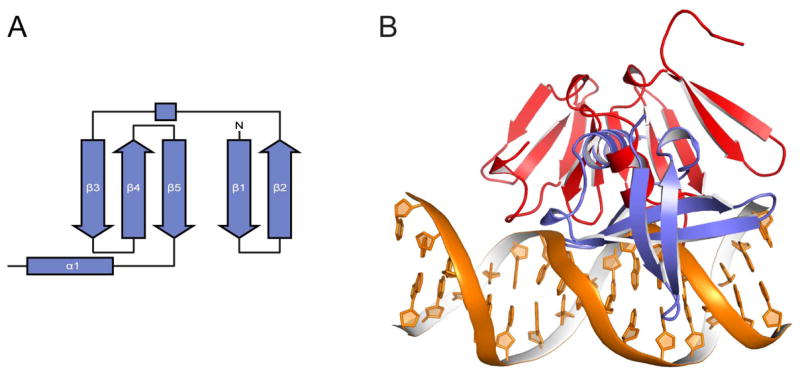
(A) Toplogy diagram of Sulfolobus acidocaldarius Sac7d.
(B) Different DNA-binding orientations of AgrAC and Sac7d. Structures of the DNA molecules within the AgrAC–DNA and Sac7d–DNA (Robinson, et al., 1998) complexes were superimposed using the program SuperPose (Maiti et al., 2004). For clarity in illustrating the different orientations of the two proteins when interacting with DNA, only a single DNA duplex, that of the AgrAC–DNA complex, is shown.
The mode of interaction of Sac7d/Sso7d with DNA is distinctly different than that of AgrAC, which binds specifically to a conserved DNA sequence through loops that act as fingers, inserting themselves into successive major grooves and gently bending the DNA (Figure 3B). Although Sac7d/Sso7d share identical secondary structure topology and nearly identical tertiary structure with the subdomains of AgrAC, these proteins have evolved completely different mechanisms for interacting with DNA.
Binding of AgrAC Significantly Distorts DNA
The majority of the DNA within the AgrAC–DNA complex adopts the B form, although local distortions demonstrate characteristics of both C and D forms. The DNA adopts a global bend of ~38° as it conforms to the concave DNA-binding surface of AgrAC (Figure 1D). Previously reported footprinting experiments performed with full-length AgrA bound to the P2 region demonstrated an increased sensitivity to DNase I along a single face of the DNA, indicating that AgrA binds along a single face of the DNA (Koenig, et al., 2004), consistent with the structure presented here. The binding interaction causes a compression of the two major grooves and the minor groove between them. Strong distortion is observed for bp A7-T7′, located just after the specific interaction with residue R233 in the major groove, and A12-T12′, located just before the specific H169 interaction in the succeeding major groove (Figures 4A and 4B). In both bp there is a high degree of stagger, buckle, and opening as well as base-step distortions in the tilt and roll. The bp T9-A9 and T10-A10, absolutely conserved in all LytTR domain consensus binding sequences, demonstrate a high degree of buckle and tilt as well as a large degree of twist, thus acting as a hinge that enables the DNA to conform to the protein surface.
Figure 4. Interactions between AgrAC and the LytTR Consensus Sequence.
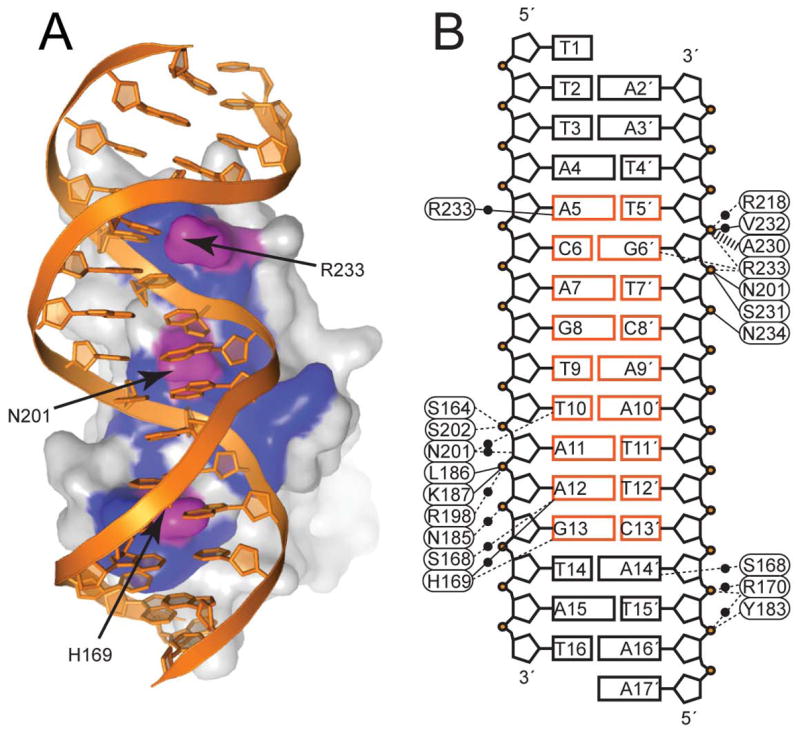
(A) Surface representation of AgrAC bound to DNA showing the DNA contact region of AgrAC. The DNA, shown in a ribbon representation, is colored orange. The surface of AgrAC is shown in white with residues that make base-specific contacts colored magenta and those involved in non-specific contacts colored blue.
(B) Schematic diagram of AgrAC–DNA interactions. Amino acid residues of AgrAC are identified as ovals and bases are identified as rectangles with the consensus DNA-binding sequence colored orange. Solid lines indicate contacts between backbone atoms of amino acid residues and DNA. Dashed lines indicate contacts between sidechain atoms and DNA. Black circles indicate water-mediated interactions. The wide dashed line indicates a van der Waals interaction.
AgrAC Contacts Successive Major Grooves
Unlike most other major groove DNA-binding proteins, AgrAC does not interact through a recognition helix. Instead, it sits on the DNA and inserts long loops into successive major grooves, making single direct base contacts in each major groove (Figure 1D). AgrAC binds to a single face of the DNA with its long axis running along the DNA backbone, covering nearly 16 bp or a length of ~50 Å, with a buried surface area of 1516 Å2. The N terminus of the domain is positioned at the edge of the β sheet that lies opposite to the edge contacting DNA. Thus the N-terminal regulatory domain attached to this terminus would presumably be positioned in a location sterically compatible with DNA binding by the LytTR domain. The C terminus of the domain also lacks contacts with DNA. This is interesting in regard to mutations at the C terminus of AgrA that have been identified in both laboratory and clinical strains of non-haemolytic S. aureus (Traber and Novick, 2006). A frameshift mutation that lengthens AgrA by 3 residues results in partially defective Agr-mediated transcription, as evidenced by delayed and lower production of RNAIII, while a mutation that adds 21 amino acids results in complete loss of RNAIII production. The structure provides no explanation for how these altered C-terminal residues would directly interfere with DNA binding. Thus it seems likely that the transcriptional defects in these variant AgrA proteins result from either decreased protein stability or loss of other intra- or intermolecular interactions such as contacts with the regulatory domain, polymerase, or other transcription factors.
There are a large number of direct and water-mediated nonspecific stabilizing contacts between the protein and the DNA (Figure 4). Indeed, the ratio of nonspecific DNA backbone interactions (10) to direct base contacts (2) made by AgrAC is similar to that observed for nonspecific DNA-binding proteins (Luscombe and Thornton, 2002). However, binding of AgrA is specific for the highly conserved 9-bp consensus sequence. As many of the bp within this consensus make neither direct nor water-mediated contacts to AgrAC, it is likely that the strict DNA sequence conservation reflects a role other than protein–DNA interactions such as allowing for specific DNA conformation. The regions flanking the 9-bp consensus binding sequence are involved in nonspecific DNA backbone interactions with AgrAC and appear to contribute primarily to stability rather than specificity.
The protein has a calculated pI of 7.8 and an asymmetric distribution of surface charge. An electrostatic representation shows that AgrAC contains the highest concentration of positively charged residues along the DNA-binding surface while the remainder of the protein is predominately negatively charged (data not shown). The shape and charge complementarity between AgrAC and DNA indicate a snug interaction within the complex.
H169 and R233 Make Specific DNA Contacts
Surprisingly, only two amino acid sidechains are involved in direct base-specific interactions within the AgrAC–DNA complex. Residue H169, located in the loop between β4 and β5, interacts with G13 on strand A, forming a Nε2-O6 hydrogen bond interaction. The backbone amide of H169 also forms a water-mediated hydrogen bond with A12 N7 (Figure 5A). In the succeeding major groove, residue R233, located in the loop between β10 and the C-terminal 310 helix, forms a bidentate NH1-N7 and NH2-O6 hydrogen bond interaction with G6 on strand B, as well as a water-mediated interaction between NH2 and N6 of A5 (Figure 5A). These two specific interactions involve G-C bp separated by 6 intervening bp, providing sequence specificity. In the minor groove, N201 makes sidechain water-mediated contacts with T10 and the ribose of A11.
Figure 5. AgrAC Residues Required for Specific Interaction with DNA.
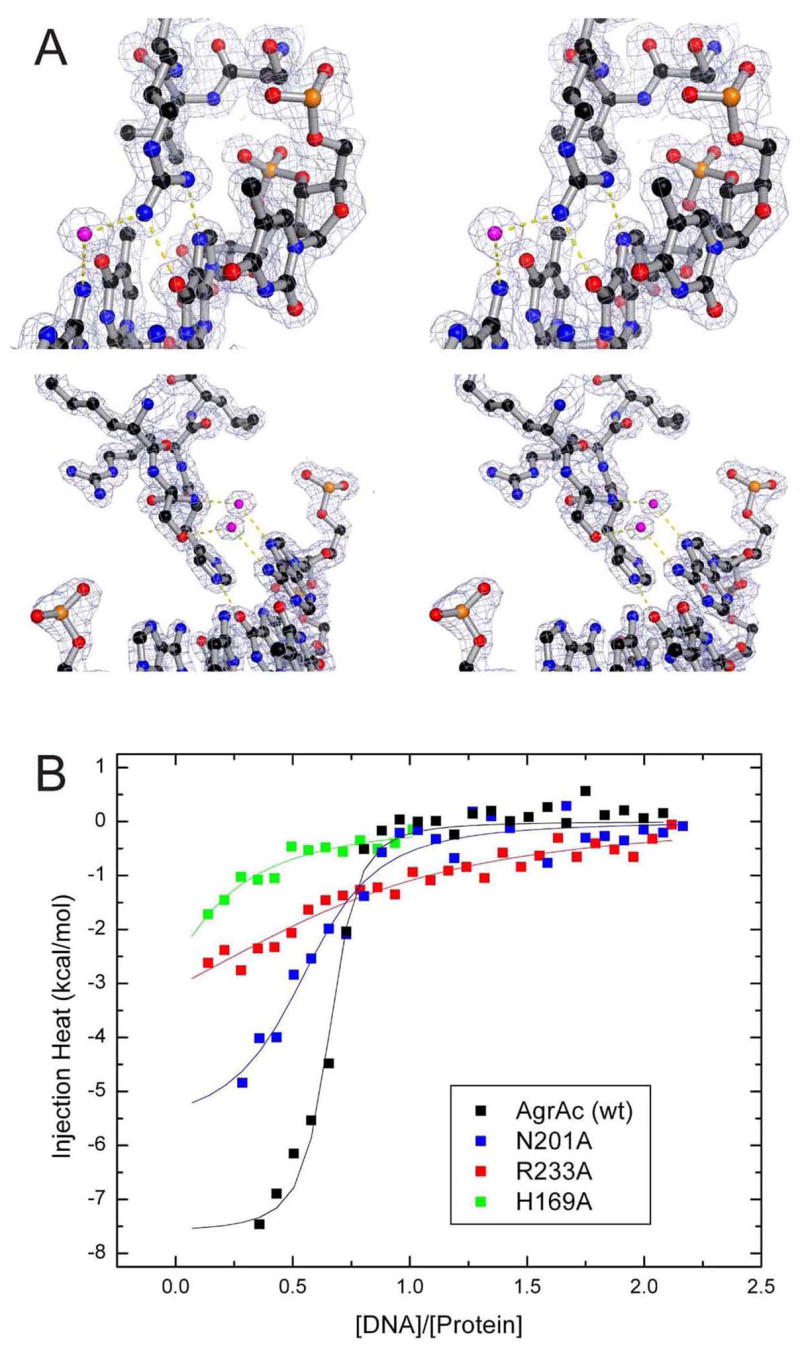
(A) Stereo views of 2FO − FC electron density contoured at 1.5σ (gray mesh) for the residues of AgrAC that make base-specific contacts with DNA. Residue R233 (top) and H169 (bottom) are shown in ball-and-stick representation with carbon in black, oxygen in red, nitrogen in blue, and phosphorus in gold.
(B) Isothermal titration calorimetry analyses of wild-type and mutant AgrAC proteins binding to DNA. Titrations were performed as described in Experimental Procedures. Data were fit with a model for one binding site and yielded Kd values of 0.08, 3, 0.7, and 7 μM for wild-type, H169A, N201A, and R233A AgrAC, respectively.
Although the LytTR domain DNA consensus binding sequence is highly conserved, the residues that make base-specific contacts in AgrA (H169, N201 and R233) are not strictly conserved in the LytTR family. However, assessment of conservation is hindered by several factors. A relatively low level of sequence identity between family members, short β strands with poorly defined endpoints and register, and a relatively large percentage of residues in variable loop regions preclude unambiguous sequence alignments. Furthermore, it is the loop regions of the LytTR domain, rather than repetitive secondary structure elements, that contact DNA. It is likely that many residues within loops will not superimpose in different three-dimensional structures, thus similar functional roles might be provided by residues with slightly different positions in primary sequence. This might be especially true for residues corresponding to H169 and R233, which lie within relatively large loops. Additionally, the relatively large range of geometries available to residues within loops and the added versatility of water-mediated contacts would potentially allow for similar protein-DNA contacts to be mediated by different amino acid sidechains.
H169 and R233 are Essential for DNA Binding
In order to assess the importance of sidechains that were observed to make base-specific contacts in the AgrAC–DNA complex, site-specific substitutions of alanine were introduced into AgrAC at positions H169, N201, and R233. The DNA-binding affinities of the wild-type and alanine-subsituted AgrAC proteins were compared by isothermal titration calorimetry. Wild-type AgrAC binds specifically to a 19-bp duplex containing the 9-bp consensus binding sequence with a Kd of ~80 nM (Figure 5B).
When H169 or R233, residues that make direct contacts to DNA bases, re substituted with alanines, the Kd values are ~40 to 90-fold higher (Figure 5B). These data are consistent with results reported for VirR, the Clostridium perfringens LytTR domain-containing response regulator, which showed that mutating a residue corresponding to R233 in AgrAC abolished DNA-binding activity (McGowan et al., 2003). Moreover, an N201A substitution in AgrAC results in a significant decrease in binding efficiency, with almost a 10-fold increase in Kd, suggesting that this interaction might be important for stabilizing the AgrAC–DNA interaction at the point where the greatest bending of the DNA occurs. This region in Clostridium perfringens VirR was previously investigated by site-specific substitution, and consistent with the results observed with AgrAC, alterations of this residue caused diminished DNA binding (McGowan et al., 2002).
The DNA-binding affinity observed for AgrAC and a DNA duplex containing a single binding site is significantly weaker than affinities previously reported for full-length AgrA. Using electromobility shift assays and the dimeric binding site within the P2 promoter, Kd values of 3.8 nM and 0.16 nM were determined for unphosphorylated and phosphorylated full-length AgrA, respectively (Koenig, et al., 2004). The 500-fold higher binding affinity of phosphorylated AgrA relative to that of the isolated DNA-binding domain likely reflects either the enhanced affinity of a dimeric binding site, contributions of the N-terminal domain to binding, or a combination of both.
Insights from a Model of the AgrAC Dimer
The characteristics of DNA binding sites for LytTR domain transcription factors have been studied for Staphylococcus aureus AgrA (Koenig, et al., 2004), Clostridium perfringens VirR (Cheung and Rood, 2000), Lactobacillus plantarum PlnC and PlnD (Risøen, et al., 1998; Risøen, et al., 2001; Straume et al., 2006), Streptococcus pneumoniae BlpR and ComE (Knutsen, et al., 2004), and Pseudomonas aeruginosa AlgR (Kato and Chakrabarty, 1991; Mohr, et al., 1990; Mohr, et al., 1991; Mohr, et al., 1992). In all cases except the AlgR promoter, which consists of two tandem repeats and another repeat located far upstream of the transcriptional start site, the LytTR domain binding region consists of two direct repeats separated by exactly 12 bp. The exact spacing has been found to be important for binding, but the molecular basis of this requirement has not been determined.
To investigate this issue, we constructed a model of AgrAC domains bound to two direct repeats with the intervening 12-bp spacer modeled as B-form DNA (Figure 6). When the AgrAC–DNA complex is positioned in a tandem orientation with the 12-bp intervening sequence, the two AgrAC molecules sit along the same face of the DNA. The distance between the two protein molecules (10.1 Å at their closest point of contact) suggests that they do not interact, and that the dimeric interaction must be mediated solely by the N-terminal domains. In this scenario, the stringent spacing between the DNA binding sites would propagate from the orientation and spacing of the N-terminal domains within the dimer through rigid interfaces between the N-terminal and C-terminal domains of each protomer. This model implies that the N-terminal domains dimerize with translational rather than rotational symmetry, paralleling the symmetry of the direct repeat DNA binding sites.
Figure 6. A Model of the AgrAC Dimer Bound to DNA.
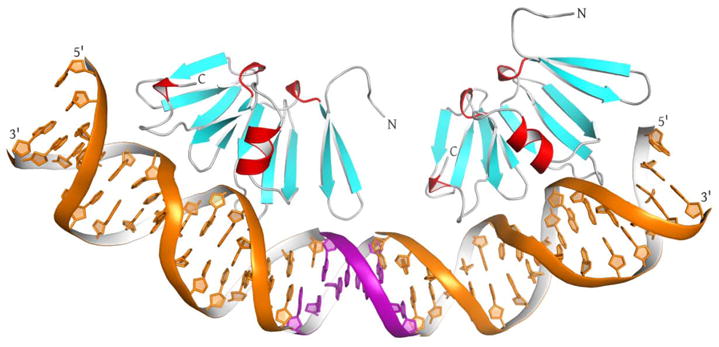
An AgrAC dimer bound to direct repeat recognition elements was modeled by aligning two AgrAC–DNA complexes together with 3 bp of B-form DNA (purple) in order to achieve the proper 12-bp spacer between the consensus binding sequences. The molecules were manually aligned in PyMol (Delano, 2002) using the termini of the DNA strands as guides.
EXPERIMENTAL PROCEDURES
Plasmid Construction
A T7-based inducible expression vector for the C-terminal domain of S. aureus AgrA (AgrAC, residues 137–238) was constructed by PCR amplification of the coding region using plasmid pJR1 DNA (Koenig, et al., 2004) as template. Forward and reverse primers, 5′-GATCCATATGGATAATAGCGTTGAAACGATTGAATTAAAACG-3′ and 5′-GATCAAGCTTTTATATTTTTTTAACGTTTCTCACCGATG-3′, respectively, introduced a unique NdeI restriction site and initiator methionine codon immediately prior to the codon for Asp137 and a unique BamHI restriction site immediately following the stop codon. The PCR product was digested with NdeI and BamHI and ligated into pET9a (Novagen), generating plasmid pDS3. The nucleotide sequence of the coding region of pDS3 was determined by automated sequencing and verified (genbank 78172216). Expression vectors for AgrAC mutant proteins (H169A, N201A, R233A, and H169A/R233A) were produced from plasmid pDS3 using the Quick-change Mutagenesis kit (Stratagene), and sequences of the coding region were determined.
Expression, Purification, and Crystallization
Recombinant AgrAC was expressed from pDS3 in Escherichia coli BL21(DE3) (Novagen). Cells were grown with shaking in 6 l Terrific Broth with 30 mg/l kanamycin at 37°C to an optical density at 600 nm of ~ 0.5, then cooled to 18°C. Protein expression was induced by addition of 0.3 mM isopropyl-β-D-thiogalactopyranoside and incubation was continued with shaking at 18°C for 24 h. Cells were collected by centrifugation at 6400 × g for 10 min and frozen at −20°C. Frozen cells were resuspended in 20 mM sodium sodium phosphate (pH 7.0), 0.1 M NaCl, 5 mM phenylmethylsulfonyl fluoride at a ratio of 3 ml per g cells and then lysed by sonication at 0°C. All subsequent steps were performed at 4°C, unless indicated otherwise. All protein solutions were filtered through 0.2 μm filter units prior to loading onto columns. Unbroken cells, cell debris and membranes were removed by centrifugation at 100,000 × g for 1 h. (NH4)2SO4, 51.6 g per 100 ml lysate, was added with constant stirring on ice for >30 min and precipitated protein was collected by centrifugation at 26,900 × g for 20 min. The pellet was resuspended in 20 mM sodium potassium phosphate (pH 7.0), 0.1 M NaCl, dialyzed against the same buffer, and applied to 2 × 5-ml HiTrap SP HP columns (GE Healthcare) equilibrated with 20 mM sodium potassium phosphate (pH 7.0), 0.1 M NaCl. Bound AgrAC was eluted using a 10-column volume 0.3–0.6 M NaCl gradient in 20 mM sodium potassium phosphate. Fractions containing AgrAC were pooled, 4.0 M (NH4)2SO4 was added to a final concentration of 1.0 M, and then applied to a HiLoad 16/10 Phenyl Sepharose HP column (GE Healthcare) equilibrated with 20 mM sodium postassium phosphate (pH 7.0), 1.0 M (NH4)2SO4. AgrAC eluted in the column flow-through. Fractions containing AgrAC were pooled, concentrated to ~4 ml using an Amicon Ultra-15 (Ultracel10) centrifugal filter unit (Millipore), and applied to a HiLoad 26/60 Superdex 75 gel filtration column (GE Healthcare) equilibrated with 20 mM Bis-Tris (pH 6.0), 0.1 M NaCl. AgrAC migrated as a single peak with a mobility corresponding to ~12 kDa. The identity of the protein was confirmed by N-terminal amino acid sequence analysis. Yields of soluble AgrAC were typically 5–10 mg/l cells. Alanine-substituted AgrAC proteins were purified by similar procedures.
Oligonucleotides used in crystallization, 5′-TTTAACAGTTAAGTAT-3′ (designated strand A, nucleotides 1–16) and 5′-AATACTTAACTGTTAA-3′ (designated strand B, nucleotides 17′-2′), were purchased from Integrated DNA Technologies with standard desalting. Duplex DNA was formed by resuspending the single-stranded oligonucleotides in annealing buffer (10 mM Tris-Cl (pH 7.5), 150 mM NaCl, 5 mM MgCl2), mixing equimolar amounts of each strand and heating to 70°C for 10 min followed by slow cooling to 20°C overnight. Modified DNA containing two covalently bound bromines (5′-bromouracil in place of thymine at nucleotides 14 and 15′) was used for crystallographic phasing. The AgrAC–DNA complex was formed by mixing 1.0 mM AgrAC with 1.2 mM DNA, incubated for 30 min, and passed through a 0.2 μM filter. Crystals of the AgrAC–DNA complex were grown at 20°C using the hanging drop vapor diffusion method. Crystals were obtained in 2 days using 40% PEG 400, 0.1 M Bis-Tris (pH 5.5) as the reservoir solution and equal volumes of reservoir and AgrAC–DNA solution in the drop. No additions were required for cryo-protection.
Structure Determination
A single-wavelength anomalous diffraction dataset was collected at 100 K using an ADSC Q4R CCD detector at the National Synchrotron Light Source beamline X4A. The peak wavelength was determined to be 0.9204 Å by a fluorescence scan. Data were collected using the inverse beam method with an oscillation angle of 1.5° per frame. Diffraction intensities were integrated and scaled using HKL2000 (Table 1) (Otwinowski and Minor, 1997). The asymmetric unit contains one AgrAC–DNA complex, with a solvent content of 52%.
The positions of the two Br atoms and the phases were calculated and refined using SHELX (Schneider and Sheldrick, 2002). Following solvent flattening with DM (Cowtan, 1994) the correct hand was interpretable and an initial model was built into electron density using Coot (Emsley and Cowtan, 2004). Phases were further improved by positional refinement using REFMAC (Murshudov et al., 1997) with data to 1.8 Å, followed by multiple cycles of manual rebuilding and positional refinement and refinement of atomic B-factors using data to 1.6 Å resolution. Water molecules were added where supported by both chemistry and geometry and the difference electron density was >3σ and the 2|Fobs|−|Fcalc| density was >1σ. The quality and stereochemistry of the model were evaluated using PROCHECK (Laskowski et al., 1993). Refinement statistics are show in Table 1. Structural images were generated using PyMol (Delano, 2002).
Analysis of DNA Binding by Isothermal Titration Calorimetry
Isothermal titration calorimetry (ITC) measurements were conducted at 25°C on a MicroCal VP-ITC (MicroCal, Inc., Northampton, MA). The solution conditions for all ITC measurements were 10 mM potassium phosphate (pH 7.0), 0.1 M NaCl. In each experiment, 10-μl aliquots of a 150-μM solution of 19-mer duplex DNA (ATTTAACAGTTAAGTATTT and complement) were sequentially injected from a 300- μl rotating syringe (300 RPM) into an isothermal sample chamber containing 1.42 ml of 15 M protein. The duration of each injection was 10 sec, with a 60-sec initial delay prior to μthe first injection and 300-sec delays between injections. Each DNA–protein experiment was accompanied by the corresponding control experiment, in which the 19-mer duplex DNA was injected into a solution of buffer alone. Each injection generated a heat burst curve (μcal/s versus s), the area under which was determined by integration [using Origin version 7.0 software (MicroCal, Inc., Northampton, MA)], to obtain a measure of the heat associated with that injection. The measure of the heat associated with each DNA-buffer injection was subtracted from that of the corresponding heat associated with each DNA-protein injection to yield the heat of DNA binding for that injection. The buffer-corrected ITC profiles for the binding of each protein to 19-mer duplex DNA were fit with a model for one set of binding sites.
Acknowledgments
We thank Robin Koenig and Barry Hurlburt for plasmid pJR1 and Randy Abramowitz and John Schwanof at the NSLS X4 beamlines for technical assistance during data collection. We thank Michael Galperin for valuable discussions and insight regarding LytTR domains. This work was supported in part by U.S. National Institutes of Health grant 2R37GM47958. A.M.S. is an investigator of the Howard Hughes Medical Institute. The authors declare no competing financial interests.
Footnotes
Accession Numbers
The atomic coordinates and structure factors for AgrAC have been deposited in the Protein Data Bank (accession code 3BS1).
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abdelnour A, Arvidson S, Bremell T, Ryden C, Tarkowski A. The accessory gene regulator (agr) controls Staphylococcus aureus virulence in a murine arthritis model. Infect Immun. 1993;61:3879–3885. doi: 10.1128/iai.61.9.3879-3885.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung JK, Rood JI. The VirR response regulator from Clostridium perfringens binds independently to two imperfect direct repeats located upstream of the pfoA promoter. J Bacteriol. 2000;182:57–66. doi: 10.1128/jb.182.1.57-66.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowtan K. ‘DM’: An automated procedure for phase improvement by density modification. Joint CCP4 and ESF-EACBM Newsletter on Protein. Crystallography. 1994;31:34–38. [Google Scholar]
- Dawid S, Roche AM, Weiser JN. The blp bacteriocins of Streptococcus pneumoniae mediate intraspecies competition both in vitro and in vivo. Infect Immun. 2007;75:443–451. doi: 10.1128/IAI.01775-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Saizieu A, Gardes C, Flint N, Wagner C, Kamber M, Mitchell TJ, Keck W, Amrein KE, Lange R. Microarray-based identification of a novel Streptococcus pneumoniae regulon controlled by an autoinduced peptide. J Bacteriol. 2000;182:4696–4703. doi: 10.1128/jb.182.17.4696-4703.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delano WL. The Pymol Molecular Graphics System. DeLano Scientific; San Carlos, CA, USA: 2002. [Google Scholar]
- Diep DB, Havarstein LS, Nes IF. Characterization of the locus responsible for the bacteriocin production in Lactobacillus plantarum C11. J Bacteriol. 1996;178:4472–4483. doi: 10.1128/jb.178.15.4472-4483.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diep DB, Myhre R, Johnsborg O, Aakra A, Nes IF. Inducible bacteriocin production in Lactobacillus is regulated by differential expression of the pln operons and by two antagonizing response regulators, the activity of which is enhanced upon phosphorylation. Mol Microbiol. 2003;47:483–494. doi: 10.1046/j.1365-2958.2003.03310.x. [DOI] [PubMed] [Google Scholar]
- Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Galperin MY. Structural classification of bacterial response regulators: diversity of output domains and domain combinations. J Bacteriol. 2006;188:4169–4182. doi: 10.1128/JB.01887-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibrat JF, Madej T, Bryant SH. Surprising similarities in structure comparison. Curr Opin Struct Biol. 1996;6:377–385. doi: 10.1016/s0959-440x(96)80058-3. [DOI] [PubMed] [Google Scholar]
- Holm L, Sander C. Dali: a network tool for protein structure comparison. Trends Biochem Sci. 1995;20:478–480. doi: 10.1016/s0968-0004(00)89105-7. [DOI] [PubMed] [Google Scholar]
- Kato J, Chakrabarty AM. Purification of the regulatory protein AlgR1 and its binding in the far upstream region of the algD promoter in Pseudomonas aeruginosa. Proc Natl Acad Sci USA. 1991;88:1760–1764. doi: 10.1073/pnas.88.5.1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knutsen E, Ween O, Havarstein LS. Two separate quorum-sensing systems upregulate transcription of the same ABC transporter in Streptococcus pneumoniae. J Bacteriol. 2004;186:3078–3085. doi: 10.1128/JB.186.10.3078-3085.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koenig RL, Ray JL, Maleki SJ, Smeltzer MS, Hurlburt BK. Staphylococcus aureus AgrA binding to the RNAIII-agr regulatory region. J Bacteriol. 2004;186:7549–7555. doi: 10.1128/JB.186.22.7549-7555.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski RA, McArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallogr. 1993;26:282–291. [Google Scholar]
- Lin YC, Liu G, Shen Y, Bertonati C, Yee A, Honig B, Arrowsmith CH, Szyperski T. NMR structure of protein PA2021 from Pseudomonas aeruginosa. Proteins. 2006;65:767–770. doi: 10.1002/prot.21098. [DOI] [PubMed] [Google Scholar]
- Lizewski SE, Lundberg DS, Schurr MJ. The transcriptional regulator AlgR is essential for Pseudomonas aeruginosa pathogenesis. Infect Immun. 2002;70:6083–6093. doi: 10.1128/IAI.70.11.6083-6093.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luscombe NM, Thornton JM. Protein-DNA interactions: amino acid conservation and the effects of mutations on binding specificity. J Mol Biol. 2002;320:991–1009. doi: 10.1016/s0022-2836(02)00571-5. [DOI] [PubMed] [Google Scholar]
- Maiti R, Van Domselaar GH, Zhang H, Wishart DS. SuperPose: a simple server for sophisticated structural superposition. Nucleic Acids Res. 2004;32:W590–W594. doi: 10.1093/nar/gkh477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCrary BS, Edmondson SP, Shriver JW. Hyperthermophile protein folding thermodynamics: differential scanning calorimetry and chemical denaturation of Sac7d. J Mol Biol. 1996;264:784–805. doi: 10.1006/jmbi.1996.0677. [DOI] [PubMed] [Google Scholar]
- McGowan S, Lucet IS, Cheung JK, Awad MM, Whisstock JC, Rood JI. The FxRxHrS motif: a conserved region essential for DNA binding of the VirR response regulator from Clostridium perfringens. J Mol Biol. 2002;322:997–1011. doi: 10.1016/s0022-2836(02)00850-1. [DOI] [PubMed] [Google Scholar]
- McGowan S, O’Connor JR, Cheung JK, Rood JI. The SKHR motif is required for biological function of the VirR response regulator from Clostridium perfringens. J Bacteriol. 2003;185:6205–6208. doi: 10.1128/JB.185.20.6205-6208.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohr CD, Martin DW, Konyecsni WM, Govan JR, Lory S, Deretic V. Role of the far-upstream sites of the algD promoter and the algR and rpoN genes in environmental modulation of mucoidy in Pseudomonas aeruginosa. J Bacteriol. 1990;172:6576–6580. doi: 10.1128/jb.172.11.6576-6580.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohr CD, Hibler NS, Deretic V. AlgR, a response regulator controlling mucoidy in Pseudomonas aeruginosa, binds to the FUS sites of the algD promoter located unusually far upstream from the mRNA start site. J Bacteriol. 1991;173:5136–5143. doi: 10.1128/jb.173.16.5136-5143.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohr CD, Leveau JH, Krieg DP, Hibler NS, Deretic V. AlgR-binding sites within the algD promoter make up a set of inverted repeats separated by a large intervening segment of DNA. J Bacteriol. 1992;174:6624–6633. doi: 10.1128/jb.174.20.6624-6633.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- Nikolskaya AN, Galperin MY. A novel type of conserved DNA-binding domain in the transcriptional regulators of the AlgR/AgrA/LytR family. Nucleic Acids Res. 2002;30:2453–2459. doi: 10.1093/nar/30.11.2453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novick RP, Projan SJ, Kornblum J, Ross HF, Ji G, Kreiswirth B, Vandenesch F, Moghazeh S. The agr P2 operon: an autocatalytic sensory transduction system in Staphylococcus aureus. Mol Gen Genet. 1995;248:446–458. doi: 10.1007/BF02191645. [DOI] [PubMed] [Google Scholar]
- Novick RP. Autoinduction and signal transduction in the regulation of staphylococcal virulence. Mol Microbiol. 2003;48:1429–1449. doi: 10.1046/j.1365-2958.2003.03526.x. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- Peters WB, Edmondson SP, Shriver JW. Thermodynamics of DNA binding and distortion by the hyperthermophile chromatin protein Sac7d. J Mol Biol. 2004;343:339–360. doi: 10.1016/j.jmb.2004.08.042. [DOI] [PubMed] [Google Scholar]
- Risøen PA, Håvarstein LS, Diep DB, Nes IF. Identification of the DNA-binding sites for two response regulators involved in control of bacteriocin synthesis in Lactobacillus plantarum C11. Mol Gen Genet. 1998;259:224–232. doi: 10.1007/pl00008627. [DOI] [PubMed] [Google Scholar]
- Risøen PA, Johnsborg O, Diep DB, Hamoen L, Venema G, Nes IF. Regulation of bacteriocin production in Lactobacillus plantarum depends on a conserved promoter arrangement with consensus binding sequence. Mol Genet Genomics. 2001;265:198–206. doi: 10.1007/s004380000397. [DOI] [PubMed] [Google Scholar]
- Robinson H, Gao YG, McCrary BS, Edmondson SP, Shriver JW, Wang AH. The hyperthermophile chromosomal protein Sac7d sharply kinks DNA. Nature. 1998;392:202–205. doi: 10.1038/32455. [DOI] [PubMed] [Google Scholar]
- Rood JI. Virulence genes of Clostridium perfringens. Annu Rev Microbiol. 1998;52:333–360. doi: 10.1146/annurev.micro.52.1.333. [DOI] [PubMed] [Google Scholar]
- Schneider TR, Sheldrick GM. Substructure solution with SHELXD. Acta Crystallogr D Biol Crystallogr. 2002;58:1772–1779. doi: 10.1107/s0907444902011678. [DOI] [PubMed] [Google Scholar]
- Shimizu T, Shima K, Yoshino K, Yonezawa K, Shimizu T, Hayashi H. Proteome and transcriptome analysis of the virulence genes regulated by the VirR/VirS system in Clostridium perfringens. J Bacteriol. 2002;184:2587–2594. doi: 10.1128/JB.184.10.2587-2594.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straume D, Axelsson L, Nes IF, Diep DB. Improved expression and purification of the correctly folded response regulator PlnC from lactobacilli. J Microbiol Methods. 2006;67:193–201. doi: 10.1016/j.mimet.2006.03.022. [DOI] [PubMed] [Google Scholar]
- Traber K, Novick R. A slipped-mispairing mutation in AgrA of laboratory strains and clinical isolates results in delayed activation of agr and failure to translate delta- and alpha-haemolysins. Mol Microbiol. 2006;59:1519–1530. doi: 10.1111/j.1365-2958.2006.04986.x. [DOI] [PubMed] [Google Scholar]
- Ween O, Gaustad P, Havarstein LS. Identification of DNA binding sites for ComE, a key regulator of natural competence in Streptococcus pneumoniae. Mol Microbiol. 1999;33:817–827. doi: 10.1046/j.1365-2958.1999.01528.x. [DOI] [PubMed] [Google Scholar]