

Summary
The Reelin-signaling pathway regulates nervous system function after birth, in addition to its known role in regulating neuronal positioning during embryogenesis. The receptor-dependent, Reelin-induced tyrosine phosphorylation of the Dab1 docking protein is an established prerequisite for biological responses to this ligand. Here we show that the inactivation of a conditional Dab1 allele reduces process complexity in correctly positioned neurons, in the CA1 region of the mouse hippocampus after birth. Reelin stimulation of cultured hippocampal neurons enhances dendritogenesis by approximately twofold, in a manner dependent on Src family kinases. This enhancement is blocked by reducing expression of Crk family proteins, adaptor molecules that interact with Dab1 in a tyrosine phosphorylation-dependent manner. Retrovirally-expressed inhibitory RNAs used to reduce Crk and CrkL expression did not block BDNF-enhanced dendritogenesis or influence axonogenesis. Together this demonstrates that the Crk family proteins are important downstream components of the Reelin-signaling pathway in the regulation of postnatal hippocampal dendritogenesis.
Keywords: Dab1, Crk, CrkL, dendritogenesis, hippocampus
Introduction
The Reelin gene encodes a signaling protein that has been shown to regulate neuronal positioning in the cerebral cortex, cerebellum, hippocampus and spinal cord during development (D’Arcangelo, 2005; Lambert de Rouvroit and Goffinet, 1998). More recently Reelin has been shown to regulate hippocampal dendritogenesis and long-term potentiation (LTP; Beffert et al., 2005; Niu et al., 2004). To understand how Reelin signaling regulates these diverse biological properties, we have sought to elucidate the downstream molecular components of the signaling pathway. One of its first identified components, Dab1, is a neuronally expressed docking protein that is tyrosine phosphorylated in response to Reelin stimulation. It is now known that upon phosphorylation, Dab1 forms complexes with a number of additional signaling proteins. However, the function of these complexes in the biological response to Reelin remains to be determined. Using inhibitory RNAs to compromise expression, we have demonstrated a role for Dab1 binding proteins in Reelin-enhanced dendritogenesis. This general approach can be used to examine the biological role of other candidate Reelin pathway components.
Reelin binds to the extracellular domains of the ApoER2 and VLDLR receptors (D’Arcangelo et al., 1999; Hiesberger et al., 1999), which in turn bind Dab1 on the cytoplasmic side (Trommsdorff et al., 1999). The augmentation in Dab1 tyrosine phosphorylation that results from Reelin binding to its receptors is dependent upon the activity of Src family kinases (Arnaud et al., 2003; Bock and Herz, 2003; Howell et al., 1999). In support of the relevance of these interactions to the biological function of Reelin, the loss-of-function phenotypes of the genes encoding them share similarities with the Reelin mutant phenotype, observed in the Reeler mouse (Arnaud et al., 2003; D’Arcangelo et al., 1995; Kuo et al., 2005; Sheldon et al., 1997; Trommsdorff et al., 1999). Furthermore, mice that only express Dab1 molecules lacking the tyrosine phosphorylation sites have a similar phenotype to the Dab1 null suggesting that Reelin-induced Dab1 phosphorylation is required for the biological response to the Reelin signal (Howell et al., 2000).
Phosphotyrosine dependent Dab1 binding proteins have been identified by a number of biochemical assays and they include, the adaptors Nckβ, Crk, CrkL, p85 and Lis1 (Assadi et al., 2003; Ballif et al., 2004; Bock et al., 2003; Chen et al., 2004; Huang et al., 2004; Pramatarova et al., 2003). These molecules are known to operate on a number of signaling pathways, making assessment of their role in Reelin signaling more complex. A CrkL mutant has recently been shown to have a partial similarity to the Reeler phenotype in the spinal cord, but it does not have Reeler like phenotypes in other areas of the CNS (Guris et al., 2001; Yip et al., 2007). This may be due to compensation by highly similar adaptor molecules encoded by the Crk gene, CrkI and CrkII. Like CrkL, CrkII has an N-terminal SH2 domain that binds upstream tyrosine phosphorylated molecules, followed by two SH3 domains that interact with downstream effectors (Feller, 2001). CrkI lacks the most C-terminal SH3 domain. Loss of Crk gene function leads to a severe phenotype late in embryonic development with no obvious similarities to the Reeler phenotype, suggesting that Crk plays essential roles in other signaling pathways (Park et al., 2006). It remains to be determined if Crk and CrkL regulate Reelin-dependent cellular events.
Here we demonstrate a role for Dab1 in postnatal hippocampal dendritogenesis in vivo by inactivating a conditional Dab1 allele after birth and examining hippocampal neurons several weeks later by Golgi staining. Having established that postnatal hippocampal neuritogenesis requires Dab1 we employed an in vitro Reelin-induced dendritogenesis model to examine the role of Dab1-binding proteins in the process. We show that reduction of both Crk and CrkL inhibits Reelin induced dendritogenesis but does not affect axonal outgrowth. Reduced expression of Crk and CrkL does not affect BDNF-induced neurite extension, suggesting that Crk and CrkL are not required for the general machinery that drives dendritogenesis. This indicates a role for the Crk family proteins in a direct, quantitative assay for a Reelin-regulated biological activity and provides a model to explore the role of other Dab1 binding proteins in Reelin-regulated dendritogenesis.
Results
Inactivation of a conditional Dab1 allele in neonates leads to a reduction in dendrite complexity in the CA1 region of the hippocampus
It has previously been demonstrated that animals with mutations in the Dab1 and Reelin genes have a reduction in the dendrite complexity of hippocampal neurons (Niu et al., 2004). In cultured neurons, Reelin has been shown to either promote dendrite extension when added to tissue culture media or suppress dendrite outgrowth when it is adsorbed to the culture dishes (Hoareau et al., 2006; Jossin and Goffinet, 2007; Niu et al., 2004). It is therefore important to clarify the in vivo role of Dab1 and other components of the Reelin signaling pathway in dendritogenesis in animals with correctly positioned neurons. In addition, the developmental timing for Dab1 or other components of the Reelin-signaling pathway in dendrite extension is not known. To inactivate the Dab1 gene in the postnatal period we generated mice that are homozygous for a conditional Dab1 allele (Dab1 cKIneo; Pramatarova et al., 2008) and also express an inducible Cre-recombinase fusion. The conditional allele consists of a floxed Dab1 expression cassette in the Dab1 genetic locus. A downstream splice acceptor directs expression of β-galactosidase upon excision of the expression cassette. Germline excision of the Dab1 cKIneo allele by Cre-mediated recombination produces a Dab1-null like phenotype in homozygous animals showing that the Dab1 cKIneo allele is inactivated by excision of the expression cassette (Pramatarova et al., 2008). Mice that are homozygous for the Dab1 cKIneo allele express approximately 15% of the normal level of Dab1 and have mild phenotypes in the cerebellum and neocortex (Pramatarova et al., 2008). The majority of neurons in the hippocampus are correctly positioned in the homozygous Dab1 cKIneo mutants, providing a good background in which to analyze the effects of lost Dab1 expression on dendrite morphology in correctly positioned neurons (Figure 1A).
Figure 1. The complexity of pyramidal neurons in the CA1 region is reduced by excision of a Dab1 expression cassette in postnatal mice.
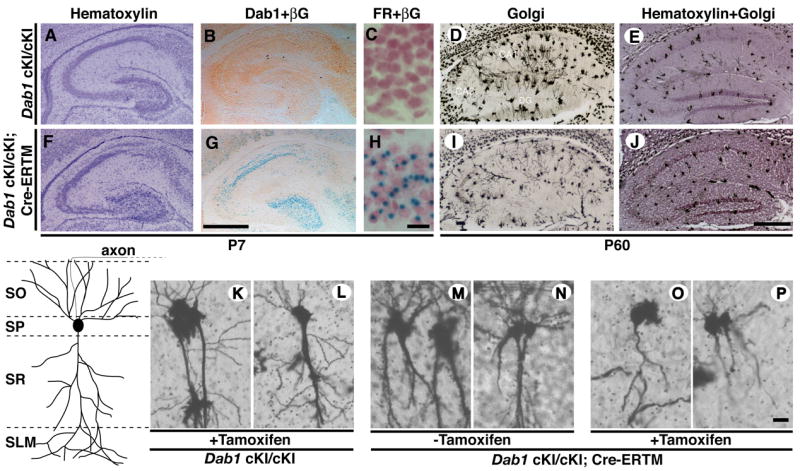
Mice that were homozygous for the Dab1 cKI allele (A-E) or homozygous for the Dab1 cKI allele and carried a tamoxifen inducible Cre transgene (Cre-ERTM; F-J) were injected with tamoxifen at P3 and analyzed for Dab1 and β-galactosidase expression (βG) (B,G), nuclear fast red (FR) and β-galactosidase activity (C,H) at P7, or analyzed by Golgi staining at P60 (D,I). Hematoxylin staining of the hippocampus shows that neuronal cell body position is relatively normal in Dab1 cKI homozygous animals in the absence or presence of the Cre-ERTM transgene at P7 (A,F, respectively) and P60 (E,J, respectively) in tamoxifen treated animals. Pyramidal neurons from the CA1 region of the hippocampus of adult animals have the basic morphology characterized in the diagram. Golgi stained neurons from the CA1 region of the hippocampus were analyzed in two control groups of mice that didn’t carry the Cre-ERTM gene but were treated with tamoxifen (K,L), and mice that carried the Cre-ERTM gene but were not treated with tamoxifen (M,N), as well as one group of experimental animals that carried the Cre-ERTM transgene and were treated with tamoxifen (O,P). The neuronal morphologies displayed are representative of those observed in several sections of at least three brains from each treatment group. SO, stratum oriens; SP, stratum pyramidale; SR, stratum radiatum; SLM, stratum lacunosum moleculare. The scale bar is 500 μm in G, J and 20 μm in H, P.
To examine the role of Dab1 in the regulation of postnatal dendritogenesis we inactivated the conditional Dab1 gene at postnatal day 3 (P3) using an inducible version of the Cre-recombinase. The fusion between Cre and a mutant form of the estrogen receptor (ERTM), which is activated upon tamoxifen exposure, is retained inactive in the cytoplasm until exposure to the ligand (Feil et al., 1996). The ERTM ligand-binding domain is insensitive to endogenous levels of estrogen making it useful for in vivo studies. To investigate the effects of inactivation of the conditional Dab1 allele we treated mice that were homozygous for Dab1 cKIneo and carried the Cre-ERTM transgene (Hayashi and McMahon, 2002) with tamoxifen at P3. These animals were compared to genetic control animals that lacked the Cre-ERTM transgene. Cre-mediated recombination was evaluated by assaying for β-galactosidase expression in parallel with immunohistochemistry to detect Dab1 protein expression in treated animals at P7.
Tamoxifen treatment of the conditional mutants with the Cre-ERTM transgene reduced Dab1 expression to barely detectable levels and activated β-galactosidase expression (Figure 1G). This was in contrast to the genetic control animals where Dab1 expression was apparent and β-galactosidase was not. In Cre-ERTM expressing cells, we detected β-galactosidase signal in greater than 80% of the pyramidal neurons in the CA1 region of the hippocampus and in the granule cells of the dentate gyrus indicating that at least one Dab1 locus was rearranged in these cells (compare Figure 1C with H). Fewer neurons expressed β-galactosidase in the CA3 region in tamoxifen treated animals, but Dab1 expression was lost in this region. This suggests that expression of the β-galactosidase reporter may be less efficient in this region.
To analyze the complexity of neuronal processes after Dab1 gene inactivation, we stained hippocampal brain sections by the Golgi technique, which labels a small percentage of neurons, allowing the visualization of individual processes. We compared hippocampal pyramidal cell in the CA1 region since the excision of the Dab1 expression cassette was apparent in a high percentage of neurons in this region. Eight weeks after tamoxifen treatment, animals that were homozygous for the Dab1 cKIneo allele and carried the Cre-ERTM transgene had less robust dendritic processes than the genetic control animals lacking the Cre-ERTM transgene (compare Figure 1D, K, L to I, O, P). The Cre-ERTM protein was not inappropriately activated in the absence of tamoxifen, since untreated animals that were homozygous for the conditional Dab1 allele and were positive for the Cre-ERTM transgene had similar process complexity to Dab1 cKIneo animals lacking the transgene (compare Figure 1M, N to K, L). Together this suggests that Dab1 is required for hippocampal process development after birth and in correctly positioned neurons, in vivo. Therefore monitoring Reelin-pathway dependent hippocampal neuron dendritogenesis is a realistic model to assess Reelin pathway activity.
The Dab1 binding proteins Crk and CrkL are required for Reelin-induced dendritogenesis in cultured neurons
To employ Reelin-enhanced dendritogenesis as a model to investigate downstream components of the pathway, we developed inhibitory short hairpin RNAs (shRNAs) against the Crk family adaptors. We employed lentiviruses to express shRNAs against Crk and CrkL RNAs since lentiviruses efficiently infect and are well tolerated by hippocampal neurons. We identified shRNA expressing viruses that reduced expression of CrkI, CrkII and CrkL in B16 melanoma cells from tests on several candidates (Figure 2, and data not shown). The shRNA that proved to be most effective against the Crk isoforms, CrkI and CrkII, also reduced CrkL expression in B16 cells (Figure 2A, B). This virus significantly reduced CrkL expression in cortical neurons (Figure 2A, B). This shRNA is directed to a region of high homology between Crk and CrkL and because it is effective at reducing expression of both Crk and CrkL in neurons we refer to it as a Crk&CrkL shRNA. The most effective CrkL shRNA reduced expression of CrkL and did not affect the expression CrkII (Figure 2A, C). In some experiments we observed a reduction in CrkI expression; however, this is unlikely to compromise Crk gene function since CrkII encompasses all the domain structure in CrkI plus an additional SH3 domain. Control vectors with three point mutations in the stem-loops of Crk&CrkL and CrkL shRNAs did not reduce expression of Crk or CrkL (Figure 2).
Figure 2. Inhibitory RNAs directed against Crk and CrkL effectively reduce expression of their respective targets.

(A) The protein expression of Crk and CrkL was compared between cultures of B16 mouse melanoma cells (lanes 1–4) or primary neurons (lanes 5–7) in the absence of infection (lane 1), after infection with control virus (lanes 2, 5), virus targeting Crk&CrkL (lanes 3, 6) or a mutant version of the shRNA (mut-shRNA; lanes 4, 7). Similarly, the effects of the CrkL shRNA lentivirus on Crk and CrkL expression were compared in uninfected (lane 8), and control infected (lane 9) B16 cells, and in those infected with CrkL shRNA virus (lane 10) and mutant CrkL shRNA virus (lane 11) by Western blotting. (B-D) The intensity of bands on Western blots was averaged between three independent experiments. (B, C) The Crk&CrkL shRNA reduced expression of CrkI and CrkII in B16 cells, and in neurons a significant reduction of CrkL was also observed. (D) The CrkL shRNA reduced expression of CrkL and CrkI in B16 cells but had no effect on CrkII.
We examined whether the lentiviral system could be used to investigate Reelin-enhanced dendritogenesis in hippocampal neurons. A CMV promoter driven GFP marker facilitated the identification of infected neurons and determination of their process lengths in culture. The lengths of Map2-positive dendrites in hippocampal neurons infected with GFP control virus were increased in response to stimulation with Reelin conditioned media (CM) as compared to neurobasal alone or control CM treatments after five days (Compare Figure 3C with A, B). Map2 positive dendrites were approximately twofold longer in Reelin treated samples than controls (Figure 3J). This establishes that dendritogenesis is enhanced by Reelin treatment of wild-type hippocampal neurons and is not altered by infection with control lentiviruses expressing GFP. This is consistent with a previous study demonstrating Reelin-enhanced dendritogenesis in Reeler mutant neurons (Niu et al., 2004).
Figure 3. Reduction of Crk and CrkL prevents Reelin-enhanced dendrite extension in hippocampal neurons.

Primary hippocampal neurons harvested from E17 mouse brains were infected with lentiviruses. Two days later neurons were replated in neurobasal-B27 growth media (A, D, G), control conditioned media (CM) (B, E, H) or Reelin CM (C, F, I). MAP2 positive dendrites of GFP control virus infected neurons, measured after 5 days of growth, were longer when grown in Reelin CM (C) than control CM (B) or neurobasal media (A). Reelin CM treatment also enhanced dendritogenesis neurons infected with CrkL shRNA virus (F) as compared to control CM (E) or neurobasal media (D) media treatment of the same population of neurons. In contrast Reelin CM treatment did not promote extension of Crk&CrkL shRNA virus infected neurons (I) relative to Control CM (H) or neurobasal media(G) treatments. The scale bar is 50 μm. Quantification of these and similar experiments demonstrated Reelin CM promoted an approximately twofold increase in dendrite length that was blocked by Crk&CrkL shRNA virus infection, but not by Crk&CrkL mutant (mt) shRNA, or GFP-expressing control viruses (J). SFK inhibitors PP1 and PP2 also prevented Reelin CM enhanced dendritogenesis. The combined effect of SFK inhibitors and Crk&CrkL shRNA viruses did not cause further reductions in process lengths over that induced by either agent alone (J). The CrkL shRNA virus did not significantly inhibit Reelin CM enhanced dendritogenesis (J). The number of neurons measured is indicated at the base of each bar. * P<0.001. Error bars indicate s.e.m.
To determine if Crk and/or CrkL are required for Reelin-enhanced dendritogenesis we infected neurons with the lentiviruses characterized above to reduce expression of these adaptor molecules. We observed that the Crk&CrkL shRNA, but not the CrkL shRNA reduced the process extension of Reelin CM treated cultures down to the baseline length observed in untreated and control CM treated cultures (compare Figure 3I with F, C). The defective Crk&CrkL shRNA virus, which did not compromise Crk or CrkL expression, did not prevent dendrite augmentation in Reelin containing cultures (Figure 3J). The Src family kinase (SFK) inhibitors PP1 and PP2 prevented Reelin-induced process extension as demonstrated previously (Niu et al., 2004). These inhibitors have been used extensively to block Reelin-induced Dab1 tyrosine phosphorylation and thereby block downstream signaling (Arnaud et al., 2003; Bock and Herz, 2003). The combination of SFK inhibitors and the Crk&CrkL lentivirus did not lead to a further reduction in dendrite length than either agent alone (Figure 3J). This is consistent with the idea that Crk, CrkL and SFKs work on a common linear pathway downstream of Reelin.
Reduction of Crk and CrkL does not affect BDNF regulated dendritogenesis
Other tyrosine kinase based signaling pathways, such as BDNF-TrkB signaling, are known to enhance process outgrowth (Niu et al., 2004). Unlike the Reelin receptors, the BDNF receptor TrkB has an intrinsic tyrosine kinase activity that is stimulated by ligand binding resulting in autophosphorylation, binding of adaptor molecules and downstream events such as Src kinase activation (Reichardt, 2006). We investigated a role for Crk and CrkL in BDNF-treated hippocampal neurons to determine if the Crk family adaptors are generally required for enhancement of dendritogenesis downstream of tyrosine kinases based signaling. We found that inclusion of BDNF in the culture media of primary neurons enhanced dendrite outgrowth approximately twofold in control virus infected cells (Figure 4). This effect was blocked by treatment with the inhibitors PP1 and PP2 analogous to what we observed for Reelin. Infection with the Crk&CrkL shRNA virus did not diminish the BDNF enhancement of dendrite outgrowth, however (Figure 4). This suggests that in hippocampal neurons Crk is not required for BDNF to promote dendritogenesis. This highlights the role for Crk as a component of Reelin signaling and not as a general mediator of dendritogenesis or signaling-enhanced dendritogenesis.
Figure 4. BDNF enhanced hippocampal dendrite outgrowth is blocked by Src inhibitors but not by the Crk&CrkL shRNA virus.
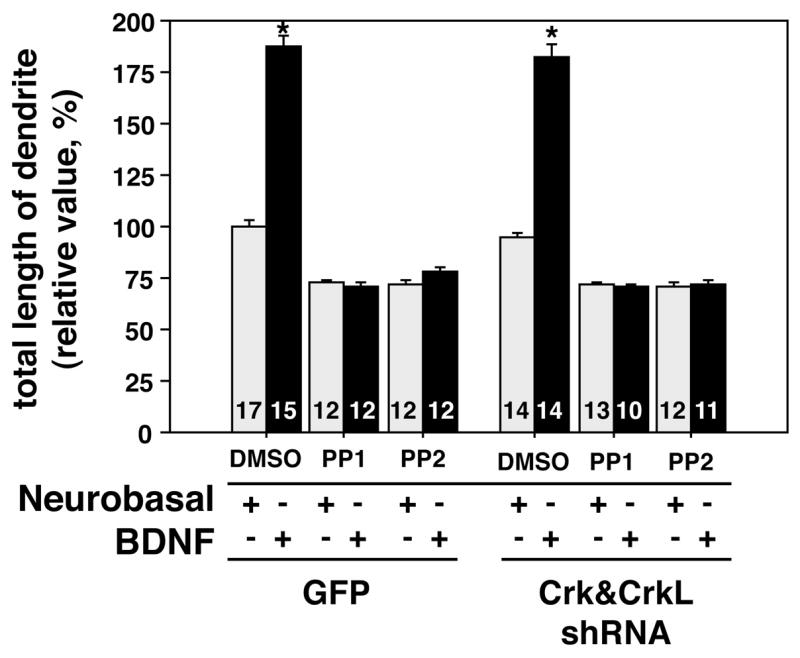
BDNF enhances the growth of MAP 2 positive hippocampal dendrites approximately 1.8 fold after 5 days of treatment as compared to growth in neurobasal-B27 media alone. Inclusion of the SFK family kinase inhibitors PP1 and PP2 during incubation prevented the BDNF-induced augmentation of process extension. However reduction of Crk and CrkL expression did not significantly block the BDNF effect on dendrites. The number of neurons measured is indicated at the base of each bar. * P<0.001. Error bars indicate SEM values.
Reduction in Crk and CrkL does not affect axonogenesis
Since neurons are polarized cells and some signals that regulate axonogenesis differ from those that regulate dendrite growth and branching we investigated the effects of Reelin and the requirements for Crk and CrkL in this process (Solecki et al., 2006). Hippocampal axons extend much greater distances and much more quickly than dendrites. We therefore examined the lengths of these processes after day 3, sooner than in experiments designed to examine dendritogenesis, to facilitate analysis. We observed no significant differences between axon lengths of neurons grown in Reelin CM, control CM or neurobasal-B27 media (Figure 5). This is consistent with previous reports that suggested Reelin affects only dendrite extension (Jossin and Goffinet, 2001; Niu et al., 2004).
Figure 5. Axon lengths of hippocampal neurons is not altered by Reelin treatment or reduction in Crk levels.
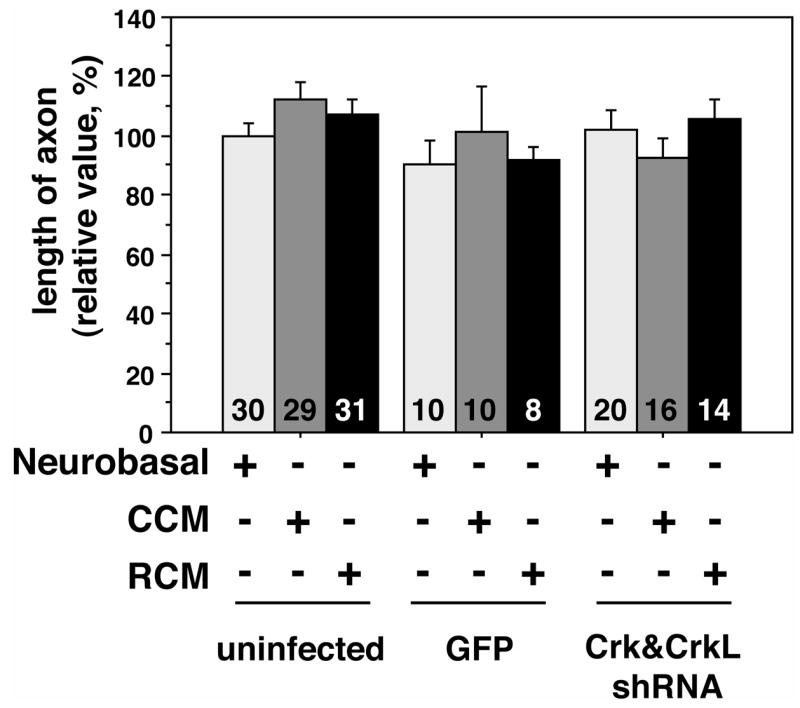
Tau-positive axons lengths were measured in hippocampal neurons 3 days after viral infection and 1 day after replating in the presence of Reelin CM, control CM or neurobasal-B27 media. Reelin CM treatment did not significantly alter process lengths under these conditions. Similarly, infection of neurons with the Crk&CrkL shRNA virus had no effect on the extension of axons in hippocampal neurons. The number of neurons measured is indicated at the base of each bar.
To determine if Crk and CrkL are required for axonogenesis we infected neurons with the control or Crk&CrkL lentiviruses (Figure 5). Similar to what we observed for dendrite growth, control virus infection did not alter axon extension. The lentivirus expressing the Crk&CrkL inhibitory RNA (RNAi) also did not alter axonogenesis (Figure 5). This suggests that the Crk family of adaptor proteins is not required for hippocampal axonogenesis, downstream of Reelin or other signaling ligands.
Discussion
In addition to its known developmental role in neuronal positioning, Reelin signaling also regulates events in the postnatal brain in a Dab1-dependent manner. We demonstrate that the inactivation of the Dab1 gene after birth leads to a reduction in the complexity of pyramidal neurons that were appropriately placed in the CA1 region of the hippocampus. This validates the use of in vitro dendritogenesis assays to examine the molecular consequences of Reelin signaling. Here we used shRNAs that target the Crk family adaptors to examine a role for these molecules in Reelin signaling. Reducing the expression of Crk and CrkL prevented Reelin-enhanced dendritogenesis but not BDNF-regulated dendritogenesis or axonogenesis. This demonstrates a biological role for Crk family proteins on the Reelin-signaling pathway. This assay can be adapted to examine the role of other molecules thought to act downstream of Reelin.
Previous studies have put forth conflicting ideas regarding the effects of Reelin signaling on dendritogenesis. Studying Reeler and Dab1 mutants and Reelin stimulated neurons in culture, Nui et al concluded that Reelin promotes dendritogenesis in a Dab1 and Src dependent manner (Niu et al., 2004). In a confounding study, Dab1-mutant neurons were shown to have the same dendritic length and complexity as wild-type neurons if cultured for 20 days in vitro (MacLaurin et al., 2007). In another study, coating tissue culture dishes with Reelin inhibited the process extension of hippocampal neurons (Hoareau et al., 2006). The in vivo experiments presented here show that the inactivation of the Dab1 gene after birth dramatically reduces the complexity of pyramidal neuron at 2 months of age, suggesting that Dab1 is required for the appropriate development and/or maintenance of dendritic processes.
The reduced expression of Dab1 that we previously documented in Dab1 cKIneo homozygous animals (Pramatarova et al., 2008) is sufficient to support the normal positioning of hippocampal neurons (Figure 1A-E). Cre-mediated excision of the Dab1-expression cassette at P3 reduced Dab1 protein expression and activated β-galactosidase reporter expression in approximately 80% of neurons in the CA1 region of the hippocampus by P7 (Figure 1G, H). At P60 the pyramidal neurons in this region were qualitatively less complex than neurons in control animals as judged by Golgi staining (Figure 1, compare panels O, P to K-N). In addition to the suspected attenuation of the development of hippocampal processes it is possible that secondary effects, such as degeneration contribute to this phenotype. Reduced dendrite length and complexity have also been observed in cortical neurons, where Dab1 expression was suppressed by a Dab1 specific RNAi beginning at E16 (Olson et al., 2006). Neuronal ectopia was also observed in those animals. We did not observe significant neuronal ectopia in the CA1 region of the hippocampus, where we compared process complexity of pyramidal neurons by Golgi staining. This demonstrates that appropriately positioned hippocampal neurons rely on Dab1 for normal process development and/or maintenance in postnatal animals.
It has previously been demonstrated that Reelin promotes the formation of Dab1-Crk and Dab1-CrkL complexes in a phosphorylation dependent manner (Ballif et al., 2004; Chen et al., 2004; Huang et al., 2004). In addition Reelin signaling activates the tyrosine phosphorylation of C3G, a Rap activator known to bind Crk (Ballif et al., 2004). Tyrosine phosphorylated Dab1 also indirectly forms complexes with Dock1 (Dock180), an exchange factor for the small G protein Rac and a Crk binding protein (Chen et al., 2004). Evidence from an exogenous model system suggests that tyrosine phosphorylated Dab1 can also influence cellular responses through Dock1 relatives (Chen et al., 2004). However, definitive evidence that Crk family proteins play a physiological role in neurons in response to Reelin has been lacking. We have demonstrated that reduced expression of Crk family members blocked Reelin-enhanced dendritogenesis. In contrast the Crk&CrkL shRNA did not block BDNF-enhanced dendritogenesis or axon propagation, demonstrating that the effect was specific and did not compromise the health of the neurons or prevent their maturation. The combined effect of SFK inhibition and reduced Crk and CrkL expression was not additive, consistent with the idea that both reagents substantially block the same pathway.
Reelin-enhanced dendritogenesis also requires mammalian target of rapamycin, (mTor) (Jossin and Goffinet, 2007). This kinase is activated downstream of phosphatidylinositol 3-kinase (PI3K) and AKT in Reelin-stimulated neurons. The p85 subunit of PI3K and the Crk and CrkL adaptors bind to tyrosine phosphorylated Dab1 independently and therefore represent a bifurcation in the signal propagation (Ballif et al., 2004; Bock et al., 2003; Chen et al., 2004). Interestingly, inhibition of mTor by rapamycin does not cause Reeler or Dab1 mutant-like defects in the positioning of neurons in slice culture assays (Jossin and Goffinet, 2007). This suggests that different components of the Reelin-Dab1 signaling pathway play specific roles in the various biological pathways regulated by these molecules. It remains to be determined if Crk and CrkL are required for neuronal positioning in the neocortex downstream of Dab1 tyrosine phosphorylation. The CrkL mutant has some similarities to Reeler-Dab1 phenotypes in the spinal cord, but not in the neocortex (Guris et al., 2001; Yip et al., 2007). The Crk-null animals die around E16 with cardiac and craniofacial defects precluding examination for a Reeler-like phenotype (Park et al., 2006). It will therefore require analysis of neurons with reductions in both Crk and CrkL in the developing brain to determine the extent to which these molecules are required for Reelin-regulated neuronal positioning. This study provides evidence that the Crk family adaptors act in a dominant manner to regulate Reelin-enhanced dendritogenesis. It will now be interesting to determine which signaling molecules downstream of Crk family proteins are required and how these signals are integrated with mTor to enhance dendrite but not axon growth.
Materials and Methods
Antibodies, growth factors and inhibitors
The following antibodies were used for immunohistochemistry or for Western blots: anti-MAP2 (monoclonal HM-2; Sigma, St. Louis, MO), anti- Tau1 (MAB 3420; Chemicon, Temecula, CA) anti-β-Actin (monoclonal AC-15; Sigma), anti-Crk antibody (monoclonal, BD Biosciences, La Jolla, CA) anti-CrkL antibody (rabbit polyclonal, Upstate Biotechnology, Charlottesville, VA), Alexa 488-conjugated anti-GFP antibody (Invitrogen) and Alexa 568-conjugated anti-mouse IgG (Invitrogen). Brain derived neurotrophic factor (BDNF) and Src family protein kinase inhibitors, PP1 and PP2, were purchased from EMD Biosciences (La Jolla, CA)
Mouse genetics
The generation of the Dab1 cKIneo mouse line has been described previously (Pramatarova et al., 2008). Briefly the Dab1-conditional allele consists of a floxed Dab1 cDNA expression cassette under control of the endogenous promoter/enhancer, followed by a splice acceptor β –galactosidase reporter gene. The excision of the Dab1 expression cassette by Cre-mediated recombination results in the expression of β-galactosidase in cells with active Dab1 locus transcription (Figure 1, data not shown). A PGK-neo drug selectable marker is 3′ to the β-galactosidase gene. The tamoxifen inducible Cre-ERTMtransgenic line (Tg(cre/Esr1)5Amc/J) was purchased from the Jackson Laboratory (Hayashi and McMahon, 2002). Tamoxifen (Sigma) was dissolved in corn oil (20mg/mL) and was administered intra-peritoneally (225mg/g body weight) to P3 newborn mice. Four days later mice were sacrificed and brains were processed for immunostaining. For Golgi staining mice were sacrificed at P60. All mice used in this study were handled in accordance with the animal care and use guidelines of the NIH.
RNAi vectors
Complementary short hairpin sequences were cloned into pLentilox 3.7 under control of a U6 promoter and transfected into 293T cells along with the support vectors VSVG, RSV-REV, and pMDLg/pRRE to generate lentiviruses that transcribe short hairpin RNAs (shRNA) (Rubinson et al., 2003). The efficacy of each virus was tested by infecting B16 mouse melanoma cells (MOI 20) and immunoblotting cell lysates for Crk and CrkL six days later (Figure 2). Effective target sequences included the hairpin loops from base pair (bp) 88–106 of CrkL (GGCCAGCGCCATGGCATGT) and (bp) 492–513 of Crk (GCCTGAAGAGCAGTGGTGGAAT). This second target sequence has been used previously to reduce expression of the Crk isoforms, CrkI and CrkII (Iwahara et al., 2004). It also has extensive overlap with CrkL RNA (21 of 22 bp) and it reduced expression of CrkL and we therefore refer to it as a Crk&CrkL shRNA vector (Figure 2A, B, C). Specific control shRNAs were generated by introducing three point mutations into the above sequences to yield the following constructs: GTCCAGCGCAATGGCATAT for CrkL and GCATGAAGATCAGTGGTGGAGT for Crk&CrkL.
Cell culture
Primary hippocampal neurons and astrocyte feeder cells were prepared from embryonic mice at E17. The CA1 region of the hippocampi was dissected into Hanks buffered salt solution (HBSS) on ice. Hippocampi were digested using Papain (Worthington, Lakewood NJ) with 0.1% DNaseI (Roche Diagnostics, Indianapolis, IN) at 37°C for 15 minutes. After stopping the digestion reaction with fetal bovine serum, the tissue was collected by centrifugation and triturated in a solution of 0.1% DNaseI solution and applied to cell strainer (BD Falcon, Bedford, MA). Neurons were plated at 3×105 cells/cm2 in 12-well plates previously coated with 0.1 mg/ml poly-L-lysine and a mixture of entactin, collagen and laminin (E-C-L, Upstate Biotechnology), then grown in Neurobasal-A medium containing 2% B-27 supplement (Invitrogen). For the biochemical studies in Figure 2 primary cortical neurons grow essentially as described previously (Pramatarova et al., 2006).
For dendrite outgrowth assays, neurons were infected with virus (MOI 20) 3 hours after plating, 2 days later they were removed from plates by papain treatment and re-plated onto coverslips at 1×104 cells/cm2. There-plating of the cells allowed us to study the behavior of the neurons after the viruses have integrated into the genome and the mRNA and proteins levels of target genes have reached a new, lower equilibrium. Coverslips were placed on a feeder layer of astrocytes and grown in Neurobasal-A media supplemented with 2 % B-27 and with control CM or Reelin CM where indicated (Niu et al., 2004). After five days cells were fixed with 4% paraformaldehyde in PBS.
In preparation for immunostaining, fixed cells were rinsed with PBS and incubated in a blocking solution containing 20 mM Tris-HCl (pH 7.4), 1% BSA, 5% normal goat serum, and 0.05% sodium azide in the presence of 0.25% Triton X-100 for 10 minutes at 21°C. Coverslips were incubated overnight at 4°C with primary antibodies diluted in the blocking solution, followed by washing and incubation with secondary antibodies for 1.5 hours at ambient temperature. Quantitative analysis of dendrite length was performed by measuring the total length and the number of MAP2-labeled processes from individual neurons manually using the softWoRx measuring tool (Applied Precision, WA) on images collected on a DeltaVision microscope system (Applied Precision). Statistical analysis was conducted using student-t test.
Tissue preparation and histological staining
Animals were perfused with 4% paraformaldehyde and their brains were isolated. These were cryoprotected with 20% sucrose and O.C.T. (Tissue-Tek) in PBS and sectioned (16 μm thick for Hematoxylin staining, LacZ, and nuclear staining and 80 μm thick for Golgi staining) using a cryomicrotome (Leica, Germany). After immunostaining with indicated antibodies or stains, the sections were mounted with Permount (Fisher, Scientific). Golgi staining was performed using a FD Rapid GolgiStain kit following the manufacturer’s recommendations (FD NeuroTechnologies Inc, Ellicott City, MD). Images of stained sections were visualized using an Axiovert 100 M microscope (Zeiss, Germany) or MZFL III stereoscope (Leica, Germany).
Immunoblotting
Western blotting was done essentially as previously described (Matsuki et al., 2001). Briefly, cell cultures were lysed in RIPA buffer {20 mM Tris-HCl (pH 7.4), 0.15 M NaCl, 1% Nonidet P-40, 2 mM EDTA, 1% sodium deoxycholate, 0.1% SDS, 5 mM 2-mercaptoethanol, 50 mM Sodium Fluoride, phosphatase inhibitor cocktail 1 (Sigma), 1 mM phenylarsine oxide (Sigma), and protease inhibitors (complete mini, EDTA free; Roche)}. Cell lysates were sonicated and centrifuged at 14,000 rpm (20,000 g) for 10 minutes, and resolved by 4%–12% SDS-PAGE gel electrophoresis. Resolved proteins were transferred onto PVDF membrane. Blots were blocked with 5% skimmed milk in Tris-buffered saline with 0.5% Tween-20 for 1 hour and then incubated with indicated primary antibodies overnight at 4°C followed by incubation with appropriate secondary antibodies conjugated to horseradish peroxidase and signal detection using SuperSignal West Pico solutions (Pierce, Rockford, IL).
Acknowledgments
We would like to thank Kelian Chen for technical assistance, Ellen Carpenter and Patricia Phelps for advice on the histological analysis of the Dab1 cKI mutant mice, Mara Pennuto and Isabella Palazzolo for interesting discussions, Tyler Pierson for comments on the manuscript and Frank Gertler for the pLentiLox system. This work was supported by NINDS intramural funds.
References
- Arnaud L, Ballif BA, Förster E, Cooper JA. Fyn tyrosine kinase is a critical regulator of Disabled-1 during brain development. Curr Biol. 2003;13:9–17. doi: 10.1016/s0960-9822(02)01397-0. [DOI] [PubMed] [Google Scholar]
- Assadi AH, Zhang G, Beffert U, McNeil RS, Renfro AL, Niu S, Quattrocchi CC, Antalffy BA, Sheldon M, Armstrong DD, et al. Interaction of reelin signaling and Lis1 in brain development. Nat Genet. 2003;35:270–6. doi: 10.1038/ng1257. [DOI] [PubMed] [Google Scholar]
- Ballif BA, Arnaud L, Arthur WT, Guris D, Imamoto A, Cooper JA. Activation of a Dab1/CrkL/C3G/Rap1 pathway in Reelin-stimulated neurons. Curr Biol. 2004;14:606–10. doi: 10.1016/j.cub.2004.03.038. [DOI] [PubMed] [Google Scholar]
- Beffert U, Weeber EJ, Durudas A, Qiu S, Masiulis I, Sweatt JD, Li WP, Adelmann G, Frotscher M, Hammer RE, et al. Modulation of synaptic plasticity and memory by reelin involves differential splicing of the lipoprotein receptor apoer2. Neuron. 2005;47:567–79. doi: 10.1016/j.neuron.2005.07.007. [DOI] [PubMed] [Google Scholar]
- Bock HH, Herz J. Reelin activates Src family tyrosine kinases in neurons. Curr Biol. 2003;13:18–26. doi: 10.1016/s0960-9822(02)01403-3. [DOI] [PubMed] [Google Scholar]
- Bock HH, Jossin Y, Liu P, Forster E, May P, Goffinet AM, Herz J. Phosphatidylinositol 3-kinase interacts with the adaptor protein Dab1 in response to Reelin signaling and is required for normal cortical lamination. J Biol Chem. 2003;278:38772–9. doi: 10.1074/jbc.M306416200. [DOI] [PubMed] [Google Scholar]
- Chen K, Ochalski PG, Tran TS, Sahir N, Schubert M, Pramatarova A, Howell BW. Interaction between Dab1 and CrkII is promoted by Reelin signaling. J Cell Sci. 2004;117:4527–36. doi: 10.1242/jcs.01320. [DOI] [PubMed] [Google Scholar]
- D’Arcangelo G. The reeler mouse: anatomy of a mutant. Int Rev Neurobiol. 2005;71:383–417. doi: 10.1016/s0074-7742(05)71016-3. [DOI] [PubMed] [Google Scholar]
- D’Arcangelo G, Homayouni R, Keshvara L, Rice DS, Sheldon M, Curran T. Reelin is a ligand for lipoprotein receptors. Neuron. 1999;24:471–9. doi: 10.1016/s0896-6273(00)80860-0. [DOI] [PubMed] [Google Scholar]
- D’Arcangelo G, Miao GG, Chen SC, Soares HD, Morgan JI, Curran T. A protein related to extracellular matrix proteins deleted in the mouse mutant reeler. Nature. 1995;374:719–23. doi: 10.1038/374719a0. [DOI] [PubMed] [Google Scholar]
- Feil R, Brocard J, Mascrez B, LeMeur M, Metzger D, Chambon P. Ligand-activated site-specific recombination in mice. Proc Natl Acad Sci U S A. 1996;93:10887–90. doi: 10.1073/pnas.93.20.10887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feller SM. Crk family adaptors-signalling complex formation and biological roles. Oncogene. 2001;20:6348–71. doi: 10.1038/sj.onc.1204779. [DOI] [PubMed] [Google Scholar]
- Guris DL, Fantes J, Tara D, Druker BJ, Imamoto A. Mice lacking the homologue of the human 22q11.2 gene CRKL phenocopy neurocristopathies of DiGeorge syndrome. Nat Genet. 2001;27:293–8. doi: 10.1038/85855. [DOI] [PubMed] [Google Scholar]
- Hayashi S, McMahon AP. Efficient recombination in diverse tissues by a tamoxifen-inducible form of Cre: a tool for temporally regulated gene activation/inactivation in the mouse. Dev Biol. 2002;244:305–18. doi: 10.1006/dbio.2002.0597. [DOI] [PubMed] [Google Scholar]
- Hiesberger T, Trommsdorff M, Howell BW, Goffinet A, Mumby MC, Cooper JA, Herz J. Direct binding of Reelin to VLDL receptor and ApoE receptor 2 induces tyrosine phosphorylation of disabled-1 and modulates tau phosphorylation. Neuron. 1999;24:481–9. doi: 10.1016/s0896-6273(00)80861-2. [DOI] [PubMed] [Google Scholar]
- Hoareau C, Borrell V, Soriano E, Krebs MO, Prochiantz A, Allinquant B. APP cytoplasmic domain antagonizes reelin neurite outgrowth inhibition of hippocampal neurons. Neurobiol Aging. 2006 doi: 10.1016/j.neurobiolaging.2006.11.012. in press. [DOI] [PubMed] [Google Scholar]
- Howell BW, Herrick TM, Cooper JA. Reelin-induced tyrosine phosphorylation of disabled 1 during neuronal positioning. Genes Dev. 1999;13:643–8. doi: 10.1101/gad.13.6.643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howell BW, Herrick TM, Hildebrand JD, Zhang Y, Cooper JA. Dab1 tyrosine phosphorylation sites relay positional signals during mouse brain development. Curr Biol. 2000;10:877–85. doi: 10.1016/s0960-9822(00)00608-4. [DOI] [PubMed] [Google Scholar]
- Huang Y, Magdaleno S, Hopkins R, Slaughter C, Curran T, Keshvara L. Tyrosine phosphorylated Disabled 1 recruits Crk family adapter proteins. Biochem Biophys Res Commun. 2004;318:204–12. doi: 10.1016/j.bbrc.2004.04.023. [DOI] [PubMed] [Google Scholar]
- Iwahara T, Akagi T, Fujitsuka Y, Hanafusa H. CrkII regulates focal adhesion kinase activation by making a complex with Crk-associated substrate, p130Cas. Proc Natl Acad Sci U S A. 2004;101:17693–8. doi: 10.1073/pnas.0408413102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jossin Y, Goffinet AM. Reelin does not directly influence axonal growth. J Neurosci. 2001;21:RC183. doi: 10.1523/JNEUROSCI.21-23-j0001.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jossin Y, Goffinet AM. Reelin signals through PI3K and Akt to control cortical development, and through mTor to regulate dendritic growth. Mol Cell Biol. 2007 doi: 10.1128/MCB.00928-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo G, Arnaud L, Kronstad-O’Brien P, Cooper JA. Absence of Fyn and Src causes a reeler-like phenotype. J Neurosci. 2005;25:8578–86. doi: 10.1523/JNEUROSCI.1656-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert de Rouvroit C, Goffinet AM. The reeler mouse as a model of brain development. Adv Anat Embryol Cell Biol. 1998;150:1–106. [PubMed] [Google Scholar]
- MacLaurin SA, Krucker T, Fish KN. Hippocampal dendritic arbor growth in vitro: regulation by Reelin-Disabled-1 signaling. Brain Res. 2007;1172:1–9. doi: 10.1016/j.brainres.2007.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuki T, Kiyama A, Kawabuchi M, Okada M, Nagai K. A novel protein interacts with a clock-related protein, rPer1. Brain Res. 2001;916:1–10. doi: 10.1016/s0006-8993(01)02857-8. [DOI] [PubMed] [Google Scholar]
- Niu S, Renfro A, Quattrocchi CC, Sheldon M, D’Arcangelo G. Reelin Promotes Hippocampal Dendrite Development through the VLDLR/ApoER2-Dab1 Pathway. Neuron. 2004;41:71–84. doi: 10.1016/s0896-6273(03)00819-5. [DOI] [PubMed] [Google Scholar]
- Olson EC, Kim S, Walsh CA. Impaired neuronal positioning and dendritogenesis in the neocortex after cell-autonomous Dab1 suppression. J Neurosci. 2006;26:1767–75. doi: 10.1523/JNEUROSCI.3000-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park TJ, Boyd K, Curran T. Cardiovascular and craniofacial defects in Crk-null mice. Mol Cell Biol. 2006;26:6272–82. doi: 10.1128/MCB.00472-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pramatarova A, Chen K, Howell B. A genetic interaction between the APP and Dab1 genes influences brain development. Mol Cell Neurosci. 2008;37:178–186. doi: 10.1016/j.mcn.2007.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pramatarova A, Ochalski PG, Chen K, Gropman A, Myers S, Min KT, Howell BW. Nck beta interacts with tyrosine-phosphorylated disabled 1 and redistributes in Reelin-stimulated neurons. Mol Cell Biol. 2003;23:7210–21. doi: 10.1128/MCB.23.20.7210-7221.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pramatarova A, Ochalski PG, Lee CH, Howell BW. Mouse disabled 1 regulates the nuclear position of neurons in a Drosophila eye model. Mol Cell Biol. 2006;26:1510–7. doi: 10.1128/MCB.26.4.1510-1517.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichardt LF. Neurotrophin-regulated signalling pathways. Philos Trans R Soc Lond B Biol Sci. 2006;361:1545–64. doi: 10.1098/rstb.2006.1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubinson DA, Dillon CP, Kwiatkowski AV, Sievers C, Yang L, Kopinja J, Rooney DL, Ihrig MM, McManus MT, Gertler FB, et al. A lentivirus-based system to functionally silence genes in primary mammalian cells, stem cells and transgenic mice by RNA interference. Nat Genet. 2003;33:401–6. doi: 10.1038/ng1117. [DOI] [PubMed] [Google Scholar]
- Sheldon M, Rice DS, D’Arcangelo G, Yoneshima H, Nakajima K, Mikoshiba K, Howell BW, Cooper JA, Goldowitz D, Curran T. Scrambler and yotari disrupt the disabled gene and produce a reeler-like phenotype in mice. Nature. 1997;389:730–3. doi: 10.1038/39601. [DOI] [PubMed] [Google Scholar]
- Solecki DJ, Govek EE, Tomoda T, Hatten ME. Neuronal polarity in CNS development. Genes Dev. 2006;20:2639–47. doi: 10.1101/gad.1462506. [DOI] [PubMed] [Google Scholar]
- Trommsdorff M, Gotthardt M, Hiesberger T, Shelton J, Stockinger W, Nimpf J, Hammer RE, Richardson JA, Herz J. Reeler/Disabled-like disruption of neuronal migraton in knockout mice lacking the VLDL receptor and ApoE receptor-2. Cell. 1999:689–701. doi: 10.1016/s0092-8674(00)80782-5. [DOI] [PubMed] [Google Scholar]
- Yip YP, Kronstadt-O’brien P, Capriotti C, Cooper JA, Yip JW. Migration of sympathetic preganglionic neurons in the spinal cord is regulated by reelin-dependent Dab1 tyrosine phosphorylation and CrkL. J Comp Neurol. 2007;502:635–43. doi: 10.1002/cne.21318. [DOI] [PubMed] [Google Scholar]