

Abstract
The new BODIPY systems 1 and 2 were prepared then used as substrates to explore SNAr and F–B displacement reactions. Chloride was easily displaced from 1 by a piperidine/ester, methyl magnesium bromide selectively displaced fluoride, and cyanide could attack both sites. System 2 readily added soft nucleophiles to the electrophilic carbon atoms, providing a new method for bioconjugation of BODIPYs to proteins while also introducing a 19F probe.
BODIPY dyes are some of the most widely used fluorescent probes in biotechnology.1,2 They were first prepared in 1969,3 but developments relating to their syntheses and functionalization reactions are far from mature. In that regard, two of the most striking recent innovations in the literature are: (i) SNAr reactions of 3-chloro or 3,5-dichloro systems;4,5 and, (ii) nucleophilic displacement of fluoride atoms from boron.6–13 These studies, reported from other labs, typically feature compounds such as A and B. Hard nucleophiles were not explored for the SNAr substrates A, cyanide was not studied for either system, and no attempts have been made to use nucleophilic amino acid side-chains on proteins for the functionalization reactions.

Work from our group involving BODIPY dyes has exposed different facets of both reaction types mentioned above. This communication focuses on BODIPYs 1 and 2 to illustrate those new issues. Compound 1 was used to introduce groups functionalized for bioconjugation, and to study cases in which nucleophilic displacement of F and SNAr reactions can compete. Compound 2 was used to illustrate how SNAr reactions in particular can be used to conjugate dyes to proteins while simultaneously changing the BODIPY core structure, its fluorescent properties, and enhancing its water solubility.

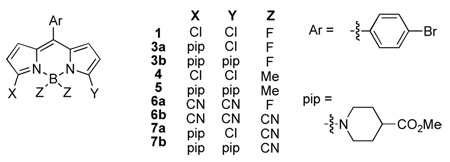
Scheme 1 shows the synthesis developed for the 3,5-dichloroBODIPY 1. Academically, this follows a procedure similar to that already developed for the similar compound A14 but practically the physical properties of the materials and/or minor variations of the procedure made it possible to scale-up the syntheses to 5 grams with minimal chromatography.
Scheme 1.

Synthesis of the 3,5-dichloroBODIPY 1.
Displacement of the first of the two chlorines in 3,5- dichloroBODIPY 1 with a piperidine derivative occurs rapidly (Scheme 2). The second chlorine can be displaced using extended reaction times at elevated temperatures. This particular piperidine derivative was of interest because hydrolysis of the methyl ester would unmask two carboxylic acids that could be used for conjugation of this material to biomolecules.
Scheme 2.

Mono- and bis-substitution on BODIPY 1.
The transformations shown in Scheme 2 were predictable for this substrate in combination with a soft nucleophile. However, reactions of 1 with a harder nucleophile like methylmagnesium bromide could conceivably occur at either of the electrophilic sites. It transpired that displacement of the B–F bonds occurred most rapidly, allowing relatively clean production of the B-dimethylated compound 4. This could then be reacted with a piperidine derivative to give the SNAr product 5.
Cyanide is a softer anion than methylmagnesium bromide, but is harder than amines. The reaction of 3,5-dichloroBODIPY 1 with this nucleophile reflects this intermediate character. Displacement was achieved using Lewis acidic activation of trimethylsilyl cyanide (reactions of solubilized cyanide ions were less satisfactory). The reaction of 1 in the presence of tin tetrachloride occurred selectively at the carbon to give compound 6a, but boron trifluoride promoted both types of displacement reactions to give the tetracyanide 6b. Cyanide has been used in SNAr reactions occurring at the meso (or 8-position) of non-halogenated-BODIPY dyes,15–17 but this is the first time that either mode of reactivity shown in Scheme 3 has been reported.
Scheme 3.

Syntheses of compounds 6 having cyanide substituents.
The Lewis-acid-mediated conditions described above were also applied to the aminated products 3. This led to the corresponding B-dicyanated compounds 7 (Scheme 4).
Scheme 4.

B–F displacements using cyanide anion in: a mono-; and, b diaminated BODIPY dyes.
Table 1 shows photophysical data for molecules 1 and 3 – 7. Several trends can be observed using the starting material 1 as a basis for comparison. Monoamination and diamination cause red-shifts in the fluorescence, reduced quantum yields, and peak broadening. The Stokes’ shift for 3a is high because the absorption maximum for this material is blue-shifted. The B-dimethylated compound 4 is spectroscopically similar to the parent 1 in all respects, and, just like 1 amination to give 5 blue-shifts the absorption maximum. Replacement of the 3,5-dichloro-substituents in 1 with cyanides gave the BF2 compound 6a and the B(CN)2-compound 6b. These changes did not significantly affect the wavelengths for the absorption or fluorescence maxima, but they did increase the quantum yields by a factor of 4 – 5. Again, monoamination and diamination caused red-shifts in the fluorescence, peak broadening, and reduced quantum yields.
Table 1.
Spectroscopic data for BODIPYs 1, and 3–7.
 | |||||
|---|---|---|---|---|---|
| dye | λabsa (nm) | εa (M−1 cm−1) | λemissa (nm) | fwhma (nm) | Φ a |
| 1 | 512 | 77600 | 523 | 27 | 0.150±01b |
| 3a | 482 | 68400 | 562 | 78 | 0.003c |
| 3b | 573 | 39900 | 614 | 52 | 0.01d |
| 4 | 503 | 134300 | 516 | 30 | 0.13±0.01c |
| 5 | 483 | 61800 | 564 | 84 | 0.002c |
| 6a | 514 | 127400 | 526 | 25 | 0.66±0.01b |
| 6b | 510 | 36500 | 523 | 25 | 0.80±0.02b |
| 7a | 485 | 33700 | 560 | 75 | 0.006c |
| 7b | 517 | 26100 | 613 | 61 | 0.008b |
In MeOH.
Rhodamine 6G was used as a standard (Φ = 0.94 in EtOH).
Fluorescein was used as a standard (Φ = 0.92 in 0.1 N NaOHaq).
Rhodamine 101 was used as a standard (Φ = 1.00 in EtOH).
The probes containing piperidine were prepared because the carboxylic acid might be used to conjugate them to biomolecules. However, this was not pursued further since the quantum yields for these derivatives were low. Further, hydrolysis of the esters gave carboxylic acids that, at least in the protonated form, tended to destabilize the structure, perhaps via a pH effect. Consequently, an alternative approach was adopted; this involved derivatives of system 2.
The BODIPY dye 2 was targeted because it might have several attributes as a biochemical probe. First, the electron withdrawing trifluoromethyl group would make it more reactive in SNAr reactions, and we anticipated that these reactions could be used to directly link the probe to proteins. Second, the probe is small. Third, the trifluoromethyl group might be useful as an NMR marker. Finally, the trifluoromethyl substituent makes the carbon that is destined to become the meso-carbon more electrophilic, and this facilitates synthesis of the probe. In the event, a convenient synthesis was developed, in which the trifluoromethyl groups stabilized dipyrromethane and dipyrromethene intermediates 8 and 9 so that these were isolable (Scheme 5).18
Scheme 5.

Synthesis of CF3-dichloroBODIPY 2.
Scheme 6 shows how compound 2 was reacted with avidin (chosen as a model protein). We assume this gave the product of displacement of one chlorine atom since excess of the dye was used, and because of the experiments depicted in Scheme 6 featuring reactions with simpler substrates. Indeed, it was necessary to prepare compounds like 10 and 12 to estimate extinction coefficients for the dyes on proteins, and therefore dye-to-protein ratios.19 Thus the calculated dye-to-protein ratio was 10:1 for 2-avidin, and 3:1 for 11-avidin.
Scheme 6.

Syntheses of dye avidin conjugates, and model compounds.
The quantum yields measured for the model compounds 10 and 12 were higher than similar derivatives of 1 (Table 2). This may be attributed to removal of a pathway for non-radiative decay when the freely rotating meso-aryl group is replaced by CF3, and/or change in oxidative potential of the BODIPY core leading to reduced photoinduced electron transfer. Absorbance and fluorescence spectra of 2- and 11- avidin are close to the control compounds 10 and 12 (Figure 1 and Table 2); this supports the assertion that the substitution pattern is predominantly that shown in Scheme 6. Finally, CD spectra recorded for avidin and 2-avidin showed no significant differences in the 190 – 320 nm region, indicating the labeling event did not cause a radical change in the protein tertiary structure.
Table 2.
Photophysics studies on Compounds 2, 10, 11 and 12.
| dye | λabsa(nm) | ε a (M−1 cm−1) | λemissa (nm) | Φ a |
|---|---|---|---|---|
| 2b | 548 | 86861 | 554 | 1.00±0.02 c |
| 2-avidin | 481 | - | 546 | - |
| 10 | 469 | 17066 | 542 | 0.740±02 d |
| 11 | 569 | 57032 | 584 | 0.950±02 e |
| 12 | 477 | 41810 | 584 | 0.70±0.01 d |
| 11-avidin | 492 | - | 592 | - |
In pH = 7.4 buffer (0.1 N Lithium Phosphate).
In CH2Cl2.
Rhodamine B was used as a standard (Φ = 0.73 in EtOH).
Fluorescein was used as a standard (Φ = 0.92 in 0.1 N NaOHaq).
Rhodamine 101 was used as a standard (Φ = 1.00 in EtOH).
Figure 1.


a Absorption and b emission spectra for 2-Avidin and its model compound 10; and c absorption and d emission for compound 11-Avidin and its model compound 12. Throughout, spectra were recorded in 0.1 M lithium phosphate buffer pH 7.4 at 10−6 M conc in protein or 10−5 M dye.
In summary, the work described in this paper highlights numerous ways in which the BODIPY core may be modified via nucleophilic substitution reactions. In the particular case of SNAr reactions on compound 2 provides a route to label proteins that breaks with well-used approaches involving activation of pendant carboxylic acid functionalities on the dye.
Supplementary Material
Acknowledgements
We thank the TAMU/LBMS-Applications Laboratory for assistance with mass spectrometry. Support for this work was provided by The National Institutes of Health (HG 01745 and GM72041) and by The Robert A. Welch Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes
- 1.In Molecular Probes. Invitrogen Corporation; 2006. http://probes.invitrogen.com. [Google Scholar]
- 2.Loudet A, Burgess K. Chem. Rev. 2007 doi: 10.1021/cr078381n. ASAP. [DOI] [PubMed] [Google Scholar]
- 3.Treibs A, Kreuzer FH. Justus Liebigs Ann. Chem. 1969;721:116–120. [Google Scholar]
- 4.Rohand T, Baruah M, Qin W, Boens N, Dehaen W. Chem. Commun. 2006:266–268. doi: 10.1039/b512756d. [DOI] [PubMed] [Google Scholar]
- 5.Baruah M, Qin W, Vallee RAL, Beljonne D, Rohand T, Dehaen W, Boens N. Org. Lett. 2005;7:4377–4380. doi: 10.1021/ol051603o. [DOI] [PubMed] [Google Scholar]
- 6.Goze C, Ulrich G, Mallon LJ, Allen BD, Harriman A, Ziessel R. J. Am. Chem. Soc. 2006;128:10231–10239. doi: 10.1021/ja062405a. [DOI] [PubMed] [Google Scholar]
- 7.Ulrich G, Goze C, Guardigli M, Roda A, Ziessel R. Angew. Chem. Int. Ed. 2005;117:3760–3764. doi: 10.1002/anie.200500808. [DOI] [PubMed] [Google Scholar]
- 8.Ziessel R, Goze C, Ulrich G. Synthesis. 2007;6:936–949. [Google Scholar]
- 9.Ziessel R, Ulrich G, Harriman A. New J. Chem. 2007;31:496–501. [Google Scholar]
- 10.Goze C, Ulrich G, Ziessel R. Org. Lett. 2006;8:4445–4448. doi: 10.1021/ol061601j. [DOI] [PubMed] [Google Scholar]
- 11.Ulrich G, Goze C, Goeb S, Retailleau P, Ziessel R. New J. Chem. 2006;30:982–986. [Google Scholar]
- 12.Gabe Y, Ueno T, Urano Y, Kojima H, Nagano T. Anal. Biochem. 2006;386:621–626. doi: 10.1007/s00216-006-0587-y. [DOI] [PubMed] [Google Scholar]
- 13.Tahtaoui C, Thomas C, Rohmer F, Klotz P, Duportail G, Mely Y, Bonnet D, Hibert M. J. Org. Chem. 2007;72:269–272. doi: 10.1021/jo061567m. [DOI] [PubMed] [Google Scholar]
- 14.Baruah M, Qin W, Basaric N, De Borggraeve Wim M, Boens N. J. Org. Chem. 2005;70:4152–4157. doi: 10.1021/jo0503714. [DOI] [PubMed] [Google Scholar]
- 15.Treibs A, Kreuzer F-H. Liebigs Ann. Chem. 1968;718:208–223. doi: 10.1002/jlac.19687180118. [DOI] [PubMed] [Google Scholar]
- 16.Boyer JH, Haag AM, Sathyamoorthi G, Soong ML, Thangaraj K, Pavlopoulos TG. Heteroat. Chem. 1993;4:39–49. [Google Scholar]
- 17.Sathyamoorthi G, Boyer JH, Allik TH, Chandra S. Heteroat. Chem. 1994;5:403–407. [Google Scholar]
- 18.Wijesekera TP. Can. J. Chem. 1996;74:1868–1871. [Google Scholar]
- 19.Majumdar RB, Ernst LA, Majumdar SR, Lewis CJ, Waggoner AS. Bioconjugate Chem. 1993;4:105–111. doi: 10.1021/bc00020a001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.