

Abstract
Marek’s disease (MD) is a highly contagious, lymphoproliferative disease of chickens caused by the cell-associated MD virus (MDV), a member of the alphaherpesvirus subfamily. In a previous study we showed that the absence of the serine-threonine protein kinase (pUS3) encoded in the MDV unique-short region resulted in accumulation of primarily enveloped virions in the perinuclear space and significant impairment of virus growth in vitro. It was also shown that pUS3 is involved in actin stress fiber breakdown (Schumacher et al., 2005). Here, we constructed a recombinant virus to test the importance of pUS3 kinase activity for MDV replication and its functions in actin rearrangement. Disruption of the kinase active site was achieved by substituting a lysine at position 220 with an alanine (K220A). Titers of a kinase-negative MDV mutant, 20US3*K220A, were reduced when compared to parental virus in a fashion similar to that of the US3 deletion mutant. We were also able to demonstrate complete absence of phosphorylation of MDV-specific phosphoprotein pp38 in cells infected with the kinase-deficient virus, indicating that pp38 phosphorylation depends entirely on the kinase activity of pUS3. Expression of pUS3*K220A was, however, still capable of mediating breakdown of the actin cytoskeleton in transfection studies, and this activity was indistinguishable from that of wild-type pUS3*. Furthermore, we demonstrated that pUS3 possesses anti-apoptotic activity, which is dependent on its kinase activity. Taken together, our results demonstrate that pUS3 and MDV-specific phosphoprotein pp38 represent a kinase-substrate pair and that growth impairment in the absence of pUS3 is caused by the absence of kinase activity. The unaltered disruption of F-actin by the K220A pUS3 mutant suggests that F-actin disassembly is unrelated to MDV growth restrictions in the absence of the unique-short protein kinase.
Introduction
Marek’s disease (MD) is a lymphomatous and neuropathic disease of chickens (Marek, 1907). The causative agent of MD was discovered 60 years after the first description of the clinical disease, when a cell-associated herpesvirus, Marek’s disease virus (MDV), was found in MD-specific lesions (Churchill and Biggs, 1967; Nazerian et al., 1968). Currently, MDV is classified as a member of the Alphaherpesvirinae subfamily, along with various other animal viruses, such as pseudorabies virus (PRV) of pigs, bovine herpesvirus type 1 (BHV-1), as well as the human varicella zoster virus (VZV) and herpes simplex virus type 1 (HSV-1) (Fukuchi et al., 1984; Lee et al., 2000; Tulman et al., 2000). It is further classified within the genus Mardivirus, which consists of Gallid Herpesvirus 2 (GaHV-2, MDV) and its close relatives, Gallid Herpesvirus 3 (GaHV-3) and herpesvirus of turkeys (Meleagrid Herpesvirus 1, HVT) (Kingham et al., 2001; Osterrieder and Vautherot, 2004). Only MDV can cause MD, while GaHV-3 and HVT are naturally occurring viruses that are a- or only weakly pathogenic and not oncogenic in chickens (Osterrieder et al., 2006)
MDV is highly infectious in vivo where it lytically infects epithelia and predominantly B lymphocytes (Calnek and Hirai, 2001; Shek et al., 1983). In certain lymphocyte subsets, particularly CD4+ T cells, latent infection is established and lymphocytes harboring latent MDV can be malignantly transformed (Calnek, 1986; Ichijo et al., 1981).. Transformed cells ultimately establish visceral lymphomas and cause neural lesions that are characterized by massive infiltration of lymphoma cells into nerve cords, particularly the sciatic and pectoral nerves (Baigent et al., 1998). MDV reactivates from latently infected T-cells and is shed to contact animals from the feather follicle epithelium (FFE), the only place where fully infectious virus is assembled (Calnek et al., 1970; Osterrieder et al., 2006).
The MDV genome is approximately 177 kbp in size and encodes for over 100 proteins (Lee et al., 2000; Tulman et al., 2000). Most MDV genes share similarities with those present in other members of the Alphaherpesvirinae, and their functions have largely been deduced based on analogy to more closely studied members of the alphaherpesviruses or by comparison to GaHV-3 and HVT (Kingham et al., 2001). Our understanding of the detailed functions of many MDV proteins and their requirement for pathogenesis is incomplete, partly caused by the highly cell-associated nature of the virus and the difficulties associated with this property with respect to the generation of virus mutants. Another complication is the fact that MDV only grows efficiently in primary chicken and duck cells (Osterrieder et al., 2006), which makes the generation of complementing cell lines and the analysis of essential open reading frames almost impossible (Osterrieder et al., 2006; Schumacher et al., 2000).
The MDV US3 orthologue encodes a serine/threonine protein kinase, which is highly conserved among all alphaherpesviruses. Although nonessential for growth in vitro in the case of HSV-1, MDV, and PRV, many different functions have been attributed to pUS3 (de Wind, Domen, and Berns, 1992; Purves et al., 1987; Schumacher et al., 2005). Besides phosphorylating the cellular proteins HDAC1, HDAC2 and PKA (Poon et al., 2006; Poon and Roizman, 2007), it also phosphorylates the viral proteins encoded by the UL31 and UL34 orthologues. Phosphorylation of both proteins is required for their even distribution along the nuclear rim and proper primary envelopment of newly synthesized nucleocapsids (Reynolds et al., 2001; Ryckman and Roller, 2004). Lately, pUS3 has also been proven to phosphorylate lamin A/C, thereby altering its localization and simultaneously regulating nuclear egress (Mou et al., 2007).
The unique-short kinase has also been associated with blocking apoptosis in HSV-1 and PRV-infected cells (Aubert et al. 1999; Aubert et al., 2007; Deruelle et al., 2007; Nguyen and Blaho, 2007), and pUS3 is expressed from two overlapping transcripts, US3 and US3.5 (Demmin et al., 2001; Poon et al., 2006; Poon and Roizman, 2007; Van Minnebruggen et al., 2003). The encoded proteins mediate phosphorylation of cellular proteins but differ in their ability to block apoptosis and to aid in the translocation of virus particles from the nucleus to the cytoplasm (Poon et al., 2006). One other intriguing aspect of pUS3 is its ability to manipulate the cellular actin cytoskeleton by causing a destabilization of F-actin, a function demonstrated for PRV, HSV-2 and MDV (Murata et al., 2000; Schumacher et al., 2005; Van Minnebruggen et al., 2003).
In addition, PRV and HSV-2 pUS3 are also responsible for a corresponding change in cellular architecture and morphology, which, in the case of PRV, was reported to enhance viral spread by virions moving along actin-based projections that purportedly serve as causeways to move infectious virions from an infected to a neighboring uninfected cell (Favoreel et al., 2005). Possible interaction partners for pUS3 in this and other functions are key players in Rho GTPase signaling pathways involved in actin remodeling. In the case of HSV-2, pUS3 suppresses c-Jun N-terminal kinase (JNK) activation, thereby modulating signaling through Cdc42/Rac, which are both members of the Rho family of GTPases (Mori et al., 2003; Murata et al., 2000). These proteins function as molecular switches modulating organization of the actin cytoskeleton, their activity being regulated by phosphorylation (Bustelo et al., 2007). Cell rounding has been associated with the catalytic domain of pUS3 in the case of HSV-2 since kinase-dead mutants were unable to induce morphological changes (Murata et al., 2000).
Since pUS3 is involved in many diverse aspects of MDV infection, we aimed at dissecting what properties of the protein are linked to a functional catalytic domain of pUS3, and those that are not and hence possibly dependent on kinase-independent protein-protein interactions.
Our studies were based mainly on analyses of a recombinant virus in which a point mutation was introduced into the conserved kinase domain of MDV pUS3. This manipulation was shown to inhibit phosphorylation of an MDV-specific, lytic phosphoprotein, pp38, which has been shown to be critical for efficient lytic virus replication in vivo and exhibits anti-apoptotic properties (Gimeno et al., 2005; Reddy et al., 2002). The mutant virus exhibited virtually identical growth properties when compared to a US3 deletion mutant and primarily enveloped virions were found to accumulate at the nuclear rim. Remarkably, the disruption of the catalytic domain of pUS3, while abolishing its anti-apoptotic activity, did not have any effect on the ability of the protein to modulate the actin cytoskeleton. We concluded that the interactions of MDV pUS3 with cellular proteins, such as actin-regulating GTPases involved in actin remodeling, are independent of its ability to phosphorylate and are likely dependent on its structure.
Results
Construction and characterization of v20US3*K220A
To determine the functional role of the pUS3 kinase domain, we used two-step Red-mediated recombination to mutate the active site of pUS3 using the p20US3* infectious BAC clone (Schumacher et al., 2005). p20US3* is a recombinant clone that harbors sequences encoding a FLAG tag at the carboxyterminus of the US3 protein and was derived from parental BAC20 (Schumacher et al., 2000). Growth properties of the recombinant virus reconstituted from p20US3*, termed v20US3*, were shown to be virtually identical to those of parental v20 virus (Schumacher et al., 2005). Serine/threonine kinases share highly conserved subdomains with conserved individual functions (Hanks et al., 1988). We chose subdomain VIb, more specifically the catalytic loop, as a target to inhibit kinase activity. The lysine residue in this loop has been associated with the phosphotransfer, and an alignment of multiple amino acid sequences showed that the VIb subdomain is highly conserved among alphaherpesvirus US3 orthologues (Fig. 1A). An I-SceI-aphAI cassette was amplified by PCR with primers that allowed insertion of the desired K220A point mutation in the p20US3* genome. The genotype of the resulting p20US3*K220A BAC was confirmed by Southern blot analysis and nucleotide sequencing ensured that the point mutation in the kinase substituted the critical lysine residue required for kinase function with an alanine (Fig. 1B).
Figure 1. The unique-short kinase of MDV and mutagensis of the active site.

(A) Multiple amino acid sequence alignment of the VIB subdomain of MDV pUS3 and its orthologues in other alphaherpesviruses. The lysine that was altered in this study is shown in bold. Plus signs indicate similar amino acids and dots represent amino acids not shown in this alignment. (B) Nucleotide and deduced amino acid sequence before and after two-step Red en passant mutagenesis. The lysine residue at position 220 was substituted by an alanine by changing two adenines at position 657 and 658 into a guanine and a cytosine (see Materials and Methods).
Growth characteristics of v20US3*, v20ΔUS3 and v20US3*K220A
We first examined the growth properties in chicken embryo cells (CEC) of the generated virus in which the pUS3 kinase domain had been inactivated. In order to test functionality of the pUS3 kinase domain in direct cell-to-cell spread of MDV, plaque sizes of v20US3*, v20ΔUS3, or v20US3*K220A viruses were determined at 5 days p.i. A significant difference in plaque sizes between v20ΔUS3, v20US3*K220A on the one hand and the v20US3* parental virus was evident (Fig. 2A). Areas of plaques induced by either v20US3*K220A or v20ΔUS3 were comparable to each other, but reduced in size by about 65% when compared to v20US3* (Fig. 2B). The defects of v20US3*K220A were further corroborated by determining multi-step growth kinetics, which revealed a marked reduction in the ability of the v20US3*K220A mutant virus to replicate in cultured chicken cells (Fig. 2C). Consistent with the reduced plaque sizes, maximum titers of v20US3*K220A in CEC were comparable to those of v20ΔUS3. When compared to parental v20US3*, titers of the v20US3*K220A were reduced approximately 10-fold (Fig. 2C). The delayed viral growth and reduced plaque formation caused by 20US3*K220A indicated that the kinase activity of pUS3 is needed for efficient cell-to-cell spread, and we concluded that other putative functions and domains of pUS3 were not responsible for this growth defect.
Figure 2. Growth of US3 mutant viruses in cultured cells.
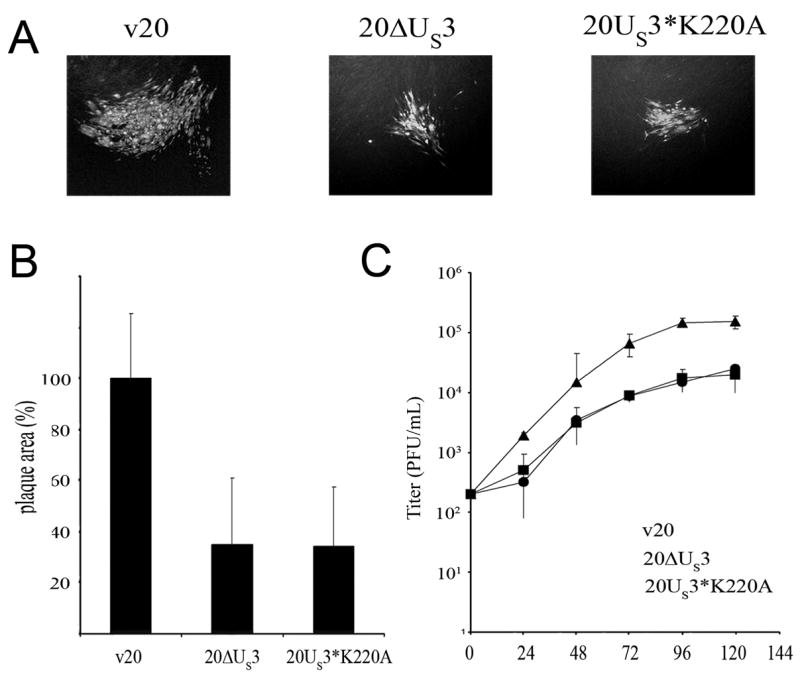
(A) CEC were infected with v20, v20US3 or v20US3*K220A. At 5 days p.i., cells were fixed with 90% aceton and analyzed by IIF using a convalescent serum from a chicken infected with MDV-1. Antibodies were detected using a secondary chicken IgG Alexa488® (Molecular Probes). (B) For each virus digital pictures of 100 plaques were taken and plaque sizes were measured. The mean plaque area of the v20 virus was set to 100%, and average relative plaque areas of the 20ΔUS3 and the 20US3*K220A virus were calculated. Plaque areas and standard deviations were determined in three independent experiments. (C) Multi-step growth kinetics of v20, v20ΔUS3 and v20US3*K220A virus recovered after transfection of BAC DNA. 1 × 106 CEC were infected with 200 plaque-forming units of the respective virus. At the given times p.i., cells were trypsinized, titrated and co-seeded with fresh CEC. Virus plaques were counted after IIF staining with a convalescent MDV chicken serum. Mean virus titers and standard deviations of the results of three independent experiments are shown.
We then examined growth of v20US3*K220A in CEC by electron microscopy and were able to detect accumulation of primarily enveloped virions at the nuclear rim, presumably trapped between the outer an inner leaflet of the nuclear membrane (Fig. 3). The phenotype presented itself in a fashion identical to that described for v20ΔUS3 that lacks the entire open reading frame (Schumacher et al., 2005). This finding suggested that loss of kinase activity causes a growth defect that is based on the inability to efficiently traverse through the endoplasmic reticulum, resulting in fewer virions in the cytoplasm, and, subsequently, a reduction in virus spread to neighboring cells. It has also been shown that HSV-1 pUS3 phosphorylates both pUL31 (Kato et al., 2005) and pUL34 (Purves et al., 2001 & 2002), although phosphorylation of at least pUL34 seems not to be required for proper localization of the protein at the nuclear rim (Ryckman and Roller, 2004).
Figure 3. Electron microscopical analyses of CEC infected with v20US3*K220A.
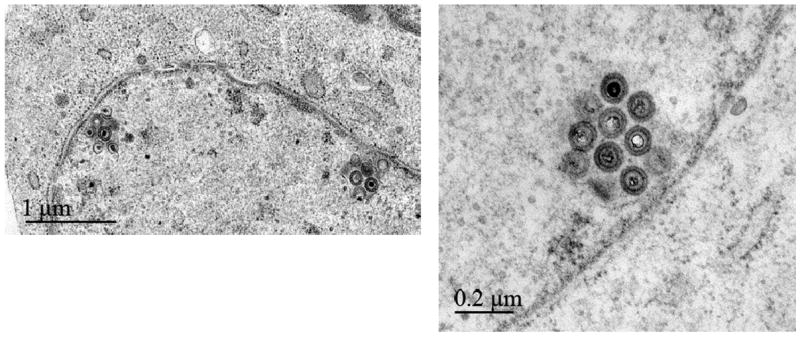
Cells were infected and fixed 3 days later. The v20US3*K220A kinase-negative mutant exhibited the same phenotype as the pUS3 null virus v20ΔUS3 (Schumacher et al., 2005). Primarily enveloped virions accumulated in the perinuclear space forming characteristic invaginations of the inner leaflet of the nuclear membrane. Bars represent 1 mm (left overview panel ) or 200 nm (right panel).
Identification of pp38 as a substrate of pUS3
To identify possible pUS3* substrates, immunoprecipitation with lysates from CEC infected with v20US3*, a recombinant virus in which pUS3 is tagged with a FLAG epitope, were performed. Co-precipitated proteins were analyzed by SDS-PAGE, silver staining, western blotting, and mass spectrometry. First, v20US3* cell lysates were incubated with ANTI-FLAG M2 agarose beads that bind to FLAG tagged pUS3*, and immunoprecipitates were eluted using 3X FLAG peptide (Sigma). Eluted proteins were separated by SDS-12%-PAGE and silver staining identified approximately 12 distinct protein bands that were co-precipitated with pUS3*. The co-precipitated proteins were found to be ~15 to >250 kDa in size and three of the most prominent protein bands were around 40 kDa (Fig. 4A). The size of approximately 40 kDa of co-precipitated proteins and the following mass spectrometry strongly suggested that the 40 kDa proteins represent the MDV-specific phosphoprotein pp38. Western blot analysis using the pp38-specific H19 antibody confirmed this assumption and the presence of pp38 of approximately 38, 41 and 43 kDa in pUS3* immunoprecipitates was detected (Fig. 4B). We were also able to detect the presence of pUS3* in immunoprecipitates obtained with the pp38-specific H19 antibody in cells infected with v20US3* and v20US3*K220A (Fig. 4C). It became apparent, however, that only the low molecular weight form of pp38 was precipitated in lysates of cells infected with the K220A mutant (Fig. 4C), indicating that pUS3 is responsible for or at least involved in pp38 phosphorylation.
Figure 4. Immunoprecipitation analyses using FLAG-tagged pUS3 expressed by recombinant v20US3* virus.
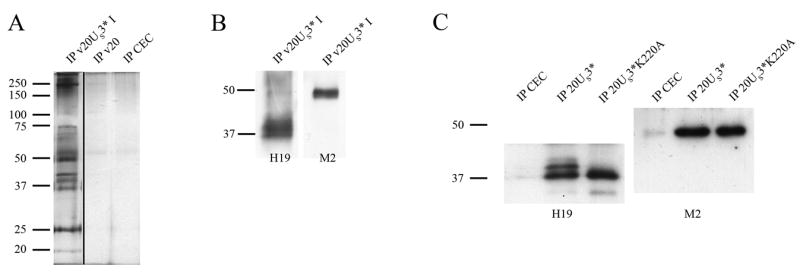
(A) Silver-stained SDS-polyacrylamide gel of immunoprecipitated proteins. Anti-FLAG® M2 affinity beads (Sigma-Aldrich) were used to precipitate pUS3* from CEC infected with v20US3*, v20 or mock infected cells. Cells were infected with the corresponding virus and harvested and lysed 4 days p.i. Lysates were separated by 12% SDS-PAGE and stained using SilverSNAP Stain Kit II (Pierce). (B) Western blot of proteins immunoprecipitated with anti-FLAG antibody beads and subsequently detected with antibodies H19 directed against pp38 or M2 to detect pUS3*. (C) Western blot detecting pp38 (left panel, H19 antibody) and pUS3* (right panel, M2 antibody) in immunoprecipitates obtained using antibody H19. Cell lysates infected with v20US3* or v20US3*K220A were used for the precipitations, which were then separated by SDS-12%-PAGE and transferred to nitrocellulose before western blot analyses were performed. The sizes of bands of a prestained molecular weight marker (Fermentas) are given in thousands.
To elucidate the enzymatic relationship between pUS3 and pp38 and the presumed kinase-substrate interaction in more detail, we used the aforementioned recombinant viruses that lacked the gene or harbored a mutated version of pUS3*. In western blot analyses, we were able to detect the previously described 38, 41, and 43 kDa band in cells infected with parental v20 or v20US3*. In lysates from cells that were infected with the US3-negative v20ΔUS3, however, we only detected the 38 kDa band (Fig. 5A). To provide final proof that the catalysis of pp38 phosphorylation requires pUS3 kinase activity, we examined lysates of CEC infected with the v20US3*K220A virus. The immunoblot data showed that pp38 remains unmodified in cells infected with the kinase-dead v20US3*K220A mutant, similar to the results obtained with v20ΔUS3 (Fig 5B). We also monitored pp38 expression over several virus passages to examine whether the single point mutation at amino acid 220 abolishing kinase activity would be under evolutionary pressure to allow more effective growth, and, consequently, revert back to the original sequence. Over continuous passage, however, no such alterations were observed as evidenced by the lack of pp38 phoshporylation over three passages (Fig. 5B).
Figure 5. Western blot analyses to examine the enzymatic relationship between pUS3 and pp38.

(A) Western blot detecting pp38 in cell lysates from CEC cells infected with v20, v20ΔUS3 and v20US3*. (B) Detection of pp38 in cells infected with v20, v20ΔUS3 and multiple passages of v20US3*K220A. (C) Antibody H19 was used to detect pp38 in lysate of cells infected with v20US3*. Lysates were treated with λ phosphatase or mock treated for 30 or 60 minutes as detailed in Materials and Methods. The sizes of bands of a prestained molecular weight marker (Fermentas) are given in thousands.
To ensure that the 41 and 43 kDa bands detected with the H19 antibody in lysates of cells infected with pUS3-expressing viruses represented the phosphorylated forms of pp38, we performed a dephosphorylation assay on infected cell lysates. Aliquots of cell lysates were treated with 800 units of λ phosphatase for 30 min and 60 min. After 30 min we were unable to detect the 41 and 43 kDa bands and only the low molecular weight version of pp38 was present (Fig. 5C). This experiment clearly demonstrated that pp38 exists in three major forms in infected cells: a faster migrating, unphosphorylated moiety and two slower migrating forms that represent two presumably different stages of phosphorylation of pp38. In addition, we provide evidence that that pUS3 is responsible – directly or indirectly – for the phosphorylation of pp38 in MDV-infected cells.
Using purified HSV-1 pUS3 and synthetic polypeptides, Purves et al. identified a phosphorylation consensus sequence that is used by the unique-short protein kinase pUS3 (Leader et al., 1991; Purves et al., 1986). The consensus sequence was identified as RnX(S/T)YY, where n is greater than or equal to 2, X can be Arg, Ala, Val, Pro or Ser, and Y can be any except an acidic residue. The predicted amino acid sequence of MDV pp38 contains a motif resembling that established for HSV-1. We applied the same mutagenesis protocol that was used to construct the v20US3*K220A virus to engineer a recombinant virus in which the putative phosphorylation site within pp38 targeted by pUS3 was inactivated. A threonine residue at position 206 and a serine at position 209 were both substituted with an alanine. However, western blots performed with lysates obtained from cells infected with this recombinant virus showed a pp38 pattern that was indistinguishable from that of v20, indicating that the predicted phosphorylation site represented by T206 and S209 of pp38 is not the one targeted by pUS3 (data not shown). A recent report has shown that HSV-1 pUS3 is a promiscuous kinase that phosphorylates lamin A/C at multiple sites (Mou et al., 2007). This finding together with the results presented here suggests a broader spectrum of sites potentially targeted by the pUS3 kinase.
pUS3*K220A induces actin stress fiber breakdown similar to wild-type pUS3*
We had previously shown that MDV pUS3 mediates temporary and reversible actin stress fiber breakdown. Here, we investigated the role of the activity of the pUS3 kinase in actin rearrangements. Expression plasmids containing the US3*, US3*K220A, or glycoprotein B gene, which does not cause cytoskeleton disassembly and was used as a negative control, were transfected into CEC. Cells were fixed at 24 or 48 hours after transfection when pUS3* and its K220A variant were detected using a FLAG tag-specific mouse antibody, while gB was identified using monoclonal antibody 2K11 (kindly provided by Jean-Francois Vautherot, Institut National de la Recherche Agronomique, Tours, France). Phalloidin-Alexa 488 was used to visualize the actin cytoskeleton. Cells were inspected using confocal fluorescence microscopy and individual, transfected cells were scored for intact versus depolymerized actin stress fibers in a blinded fashion. Our results clearly show that pUS3*K220A induced cytoskeletal degeneration as efficiently as pUS3*. In both cases, only 10% of the cells contained an intact cytoskeleton after 24h (Fig. 4B and C) and the vast majority of pUS3*K220A- and pUS3*-expressing cells showed significant actin disassembly and absence of stress fibers. Consistent with earlier findings, actin stress fibers were regenerated after 48h (Fig. 4B and C), although expression of pUS3 and pUS3*K220A was readily detected at 48h, 72h, and 96h after transfection. We concluded that the ability of MDV pUS3 to mediate actin stress fiber breakdown is not dependent on its kinase activity. With regard to this property and the fact that actin depolymerization is transient in the case of MDV pUS3, this unique-short protein kinase differs from its orthologues in related viruses.
pUS3 but not pUS3*K220A inhibits induction of apoptosis
Finally, we investigated the anti-apoptotic activity of MDV pUS3, an activity that had previously been shown for HSV-1 and PRV pUS3. We also addressed the question whether kinase activity is required for the inhibition of apoptosis. Expression plasmids pcUS3*, pcUS3*K220A or pcORF9A, which encodes for a VZV small transmembrane protein and was used as a negative control, were transfected into CEC. After induction of apoptosis with staurosporine, apoptotic cells were detected with a rabbit antibody specific for cleaved caspase-3, a common marker for apoptosis. In order to eliminate systematic errors, the ratio of apoptotic cells expressing the respective transgene was related to the percentage of apoptotic cells not expressing a foreign protein within each sample. In four independent experiments we could clearly establish that MDV pUS3 has anti-apoptotic properties, which is not seen in cells transfected with the control plasmid (Fig. 7). Anti-poptotic activity of pUS3 is dependent on kinase activity, since the number and percentage of apoptotic cells transfected with the K220A variant was indistinguishable from that seen in the negative control (Fig 7). While K220A had no effect on apoptosis compared to the non-transfected cells (relative number 1.06), the apoptosis inhibition of pUS3 was statistically significant when compared with the K220A mutant (p=0.028). From these results we concluded that MDV pUS3, similar to its orthologues in HSV-1 and PRV, exhibits anti-apoptotic activity, which is strictly dependent on its ability to phosphorylate substrates.
Figure 7. pUS3 but not pUS3*K220A inhibits induction of apoptosis.
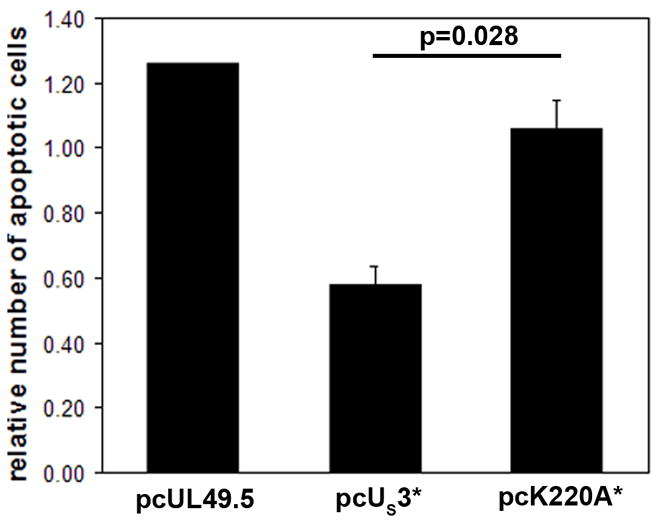
CEC were transfected with pcUS3*, pcUS3*K220A or a control plasmid (pcORF9A) and apoptosis was induced with staurosporine. Apoptotic cells were detected with cleaved caspase-3 specific rabbit antibody, while transfected cells were identified using anti-FLAG or anti 6xHIS specific antibodies. The bar graph shows the ratio of apoptotic to non-apoptotic cells expressing the respective protein, normalized to the same ratio in non-transfected cells in the same experiment. Shown are means and standard deviations (error bars) from three independent experiments. The p value was computed using an unpaired Student’s t test.
Discussion
Previous findings regarding the alphaherpesviral US3-encoded serine/threonine protein kinase revealed that it is involved in several aspects of virus replication, including modification of viral proteins, direct cell-to-cell spread of infectious virus, and actin disassembly. While MDV pUS3 was shown not to be essential for growth, virus mutants lacking US3 exhibited greatly reduced growth properties and induced smaller plaques when compared to parental virus. Additionally, a clear role of pUS3 in actin stress fiber breakdown was established for several alphaherpesviruses, although the mechanisms and timely regulation seemed variable between individual viruses of the subfamily (Favoreel et al., 2005; Schumacher et al., 2005; Van Minnebruggen et al., 2003). Inhibiting the kinase activity of MDV pUS3 allowed us to further specify the role of the functionality of pUS3 kinase activity in virus replication and egress, separately from the structural role the protein might play. We expected most of the effects of pUS3 to stem from its function as a protein kinase through phosphorylation of viral and cellular proteins. Indeed, the 20US3*K220A mutant exhibited most of the characteristics of the MDV mutant lacking the US3 gene. The growth properties of both mutants were identical as evidenced by defects in multi-step growth kinetics and reduced plaque sizes (Schumacher et al., 2005). To our surprise, transfecion of a plasmid encoding US3*K220A was still able to cause temporary actin stress fiber breakdown in chicken embryo cells and did so as efficiently as wild-type pUS3. The results are an indication that kinase activity may not be required for pUS3-mediated breakdown of stress fibers that was observed in infected cells. The effects of the mutation were unexpected, but a similar behavior has been described for the vaccinia virus protein encoded by the F11L open reading frame. A GFP tagged version of this protein induces a loss of actin stress fibers in transfected BSC-1 cells, and the F11L protein interacts directly with RhoA via a domain exhibiting some homology to one identified in the Rho-associated kinase (ROCK).
F11L binding inhibits downstream signaling by blocking interaction of RhoA with ROCK but does not involve phosphorylation of RhoA (Valderrama et al., 2006). It is conceivable that MDV pUS3 results in actin depolymerization through the RhoA-ROCK pathway that is independent of the kinase activity of the viral protein. We are currently investigating the functional role of pUS3 in actin stress fiber breakdown as well as the transient nature of depolymerization of actin filaments. Special emphasis here is put on the potential inhibition of proteins involved with stress fiber maintenance and dynamics, including the Ras/Raf/MEK/ERK signaling pathway.
We demonstrated, that cells transfected with pcUS3*K220A had restored actin stress fibers at 48 h after transfection. This phenomenon could be explained by a cellular stress response induced by the expression of the viral protein. Cellular stress responses can result in increased actin polymerization, which might explain the reestablishment of an intact cytoskeleton at later time points (Bustelo et al., 2007; Valderrama et al., 2006). The pUS3-mediated effects did not influence cell viability, as we did not notice a significant difference in the number of transfected cells at the 24 h and 48 h time points. Our investigation of potential actin filament maintaining signaling pathways that are abrogated by pUS3 binding will, therefore, focus on those not associated with apoptosis and direct or indirect interaction with RhoA.
Our results also showed that MDV pUS3 fulfills functions that are solely based on its kinase activity, namely its involvement in nuclear egress, which likely is mediated by its role in proper co-localization of pUL31 and pUL34 to the nuclear membrane through phophorylation. This co-localization in turn has been shown to be critical for proper envelopment of nuclear virions and exit from the perinuclear space. The catalytic relationship between pUS3 and pUL34 has been analyzed in great detail in HSV-1 (Purves et al., 2001 and 2002; Ryckman and Roller, 2004). We report here that MDV with a mutated pUS3 kinase domain exhibit an ultrastructural phenotype that is characterized by an accumulation of virions at the nuclear rim, which is indistinguishable to that seen with a virus devoid of the entire open reading frame. Since formation of invaginations of the inner leaflet of the nuclear membrane and entrapment of primarily enveloped virions is closely associated with mislocalization of pUL34 we surmise such mislocalization in the absence of the pUS3 kinase domain in the case of MDV as well. Interestingly, however, the defect in de-envelopment and cell-to-cell spread seems to be independent of pUS3-mediated destabilization of the actin cytoskeleton, indicating a different, pUS3-independent mechanism to be involved in actin-mediated enhancement of virus cell-to-cell spread that we reported earlier (Schumacher et al., 2005).
Lastly, our analyses focused on identifying viral proteins targeted by the protein kinase. Based on pull-down assays and mass spectrometric analyses, one MDV-specific phophoprotein, pp38, seemed to be predominantly associated with pUS3. In a series of experiments we were able to show that pp38 phosphorylation is dependent on pUS3 kinase activity, because cell lysates obtained after infection with the kinase-inactive form of pUS3 did not contain phosphorylated pp38 species. This result not only suggests disruption of the kinase activity of pUS3 in the K220A mutant, but also sheds new light on the role of pp38, which is a multifunctional protein detected early during MDV lytic replication. The MDV-specific phosphoprotein apparently is, however, also expressed during the latent and tumor phase of infection (Gimeno et al., 2005; Reddy et al., 2002; Shek et al., 1983). Since according to our current knowledge expression of pUS3 is restricted to the lytic replication cycle and not detected during latency or in lymphoblastoid cell lines derived from MDV-induced tumors, it seems logical that pp38 activity in different phases of infection is regulated through phosphorylation by pUS3. Of particular interest in this context is the fact that pp38 exhibits anti-apoptotic properties, a function also mediated by MDV pUS3 itself and its orthologues in HSV-1 and PRV (Aubert et al., 1999; Aubert et al., 2007; Calton et al., 2004; Deruelle et al., 2007; Nguyen and Blaho, 2007). Interestingly, the inhibition of apoptosis is dependent on the kinase activity of pUS3 which plays a critical role in pp38 phosphorylation, possibly suggesting that the anti-apoptotic function of pUS3 is mediated by phosphorylation of as of yet unknow cellular substrates and viral proteins such as pp38. Unfortunately, we were unable to specifically disrupt phosphorylation of pp38 because pUS3 is apparently capable of phosphorylating at motifs that differ from that identified for the HSV-1 orthologue, but the relationship of pp38’s anti-apoptotic functions with its phosphorylation status is of particular interest. The phosphorylation of pp38 will be addressed by the generation of further virus mutants that will be designed to reveal the consensus motif targeted by MDV pUS3.
Materials and Methods
Virus and cells
Primary chicken embryo cells (CEC) were prepared from 11-day old chicken embryos using standard protocols (Osterrieder, 1999) and maintained at 37°C under a 5% CO2 atmosphere in minimal essential medium with Earle’s salts (EMEM), supplemented with 1 to 10 % fetal bovine serum (FBS). MDV was reconstituted from parental or mutated infectious BAC20 clones (Morgan et al., 1990). Virus was harvested five days after transfection of 1 μg of BAC DNA into CEC by the calcium phosphate precipitation method, unless otherwise stated (Schumacher et al., 2000). Transfections of expression plasmids pcUS3* (Schumacher et al., 2005) or pcUS3*K220A were performed accordingly using ~100 ng of plasmid DNA.
Apoptosis assay
CEC were transfected with 2 μg of pcUS3*, pcUS3*K220A or pcORF9A, a pcDNA3.1/V5-His TOPO® control plasmid expressing a VZV small transmembrane protein encoded by ORF9A, using Lipofectamin2000 (Invitrogen) according to manufacturer’s instructions. 24 hours after transfection, apoptosis was induced by the addition of 1 μM staurosporine (Sigma) to the media and incubation for 4 h. Subsequently, supernatant was collected and attached cells were harvested by trypsinization. Cells were washed with 1x PBS, fixed with 2% paraformaldehyde and permeabilized with 0.1% saponin. Primary antibodies, rabbit polyclonal anti-cleaved caspase-3 (Cell Signaling Technology) and anti-FLAG mouse MAb M2, for the detection of pcUS3* and pcUS3*, or anti- 6xHIS mouse MAb (Qiagen), for pcORF9A, were diluted 1:50 or 1:200 respectively in FACS buffer (1x PBS with 1% NCS; 0.1% saponin and 0.01% sodium azide). Cy5-conjugated goat anti-mouse and AlexaFluor488-conjugated goat anti-rabbit (Molecular Probes) were diluted 1:200 in FACS buffer. After the staining procedure, cells were analyzed using the FACSCalibur flow cytometer and the CellQuest software (BD Biosciences).
Mutagenesis of p20US3*, generation of rescuant virus, and plasmid construction
A fragment containing an I-SceI-kan cassette was obtained by using plasmid pEPkan-S (Tischer et al., 2006) as a template and used for two-step Red recombination. Primers US3-K220A-fw (5′-ATATATCCACGAAAAGGGTATAATACATCGTGATGTAGCAACTGAAAATATATTTTTAGG GATAACAGGGTAATCGATTT- 3′) and US3-K220A-rv (5′–TACATTTTCAGGTTTGTCCAAAAATATATTTTCAGTTGCTACATCACGATGTATTAGCCAGTGTTACAACCAATTAACC-3′) contain 23 bp that are homologous to the I-SceI-kan plasmid. The primers also contained 56 bp of sequences with homology to the targeted locus (underlined). Finally, each of the primers contained 37 bp of modified target sequence (italics, changed nucleotides in bold face), which were reverse complementary to each other and used for the second step of markerless mutagenesis that results in the excision of the antibiotic resistance gene (see below). Recombination and electrocompetent Escherichia coli EL250 cells (Copeland et al., 2001) harboring recombinant BAC p20US3*, a recombinant harboring a FLAG tag at the C terminus of pUS3 (Schumacher et al., 2005), were used for manipulations. 300 ng of a purified PCR fragment, designed to substitute the lysine at position 220 of pUS3 with an alanine, and 100 ng of pBAD-I-SceI were electroporated into 40 μl of heat-induced, electrocompetent cells using standard electroporation conditions (1.25 kV/cm, 200 Ω, 25μF). Cells were then grown in 1 mL of LB for 60 min at 32°C and plated onto LB agar plates containing 30 μg/mL chloramphenicol, 100 μg/mL ampicillin and 30 μg/mL kanamycin. The correct insertion of the kanamycin resistance gene into the BAC after the first recombination was analyzed by restriction enzyme digestion with HindIII (NEB) and inspection of the fragments on a 0.8 % agarose gel. DNA fragments were transferred to positively charged nylon membranes (Pharmacia-Amersham), and Southern blot hybridization was performed using a digoxigenin-labeled kanR probe (Osterrieder, 1999). Chemoluminescent detection of DNA-DNA hybrids using CSPD® was done according to the supplier’s instructions (Roche Biochemicals). The second Red recombination resulted in the excision of the kanamycin resistance gene via cleavage at the I-SceI site and recombination of the duplicated sequences (italics) (Tischer et al., 2006). I-SceI expression was induced by adding 1% arabinose to the media before heat induction of the Red recombination system. After recombination was allowed to occur, cells were spread on plates containing 30 μg/mL chloramphenicol and 1% arabinose. Kanamycin-sensitive clones were picked, extrachromosomal BAC DNA was extracted, digested with HindIII, and analyzed by 0.8% agarose gel electrophoresis as described above. The mutated region was amplified by standard PCR and sequenced to verify the correct point mutations.
The modified open reading frame (ORF) US3*K220A was amplified from the recombinant p20US3*K220A BAC, using primers US3*amp1 (5′-TAATAGACTGGATGTCTTCG-3′), US3*amp2 (5′-TTACTTATCGTCGTCATCCTTG-3′) and cloned into expression vector pcDNA3.1, using the pcDNA3.1/V5-His TOPO® vector according to the manufacturer’s recommendations (Invitrogen). The generated plasmid was labeled pcUS3*K220A.
Western blotting, dephosphorylation reactions and immunoprecipitation of FLAG-tagged pUS3*
CEC were transfected with p20US3*, p20ΔUS3, or p20US3*K220A DNA as described above. After 5 days, transfected cells were trypsinized and co-seeded with fresh CEC, from which lysates were prepared at 7 days post infection (p.i.). Samples were separated by sodium dodecyl sulfate (SDS)–12% polyacrylamide gel electrophoresis (PAGE) and transferred to nitrocellulose membranes (Biorad) by the semidry method (Kyhse-Andersen, 1984). Membranes were incubated in 5% skim milk in phosphate-buffered saline (PBS) containing 0.03% Tween (PBST), followed by incubation with anti-FLAG monoclonal antibody (MAb) M2 (1:5,000 dilution in PBST) or anti-pp38 MAb H19 (1:10,000 dilution in PBST). Bound antibodies were detected with anti-mouse immunoglobulin G (IgG) peroxidase conjugates (Sigma-Aldrich), visualized by enhanced chemoluminescence (ECL™, Pharmacia-Amersham) and recorded on X-ray films (Amersham Biosciences) as described (Sambrook and Maniatis, 1989). For dephosphorylation reactions, aliquots of cell lysates were exposed to a reaction mixture consisting of 800 units of λ phosphatase (New England Biolabs), 2 mM MnCl2, and λ phosphatase buffer (New England Biolabs) for 30 or 60 min at 60°C. Control samples were treated in the exact same manner, but reaction mixtures did not contain λ phosphatase. After the reaction loading buffer was added and the treated lysates were heated for 2 minutes at 95°C before being loaded on the gel.
Immunoprecipitation was performed with lysates from cells infected with v20US3* using anti-FLAG® M2 affinity beads (Sigma-Aldrich) according to the manufacturer’s protocol. Bound proteins were eluted from the beads using 3xFLAG peptide (Sigma-Aldrich) at a final concentration of 150ng/μL.
Indirect immunofluorescence, single-step growth kinetics and plaque area determinations
For indirect immunofluorescence (IIF), cells grown on glass coverslips were transfected with recombinant plasmids and fixed with 90% acetone after 24 or 48 h. Free binding sites were blocked with PBS-10% FBS, and a convalescent anti-MDV chicken antiserum, anti-gB MAb 2K11 (1:250) or a FLAG-tag specific mouse serum (Stratagene) was added at a 1:500 dilution in PBS-10% FBS for 30 min. After two washing steps in PBST, Alexa 488-conjugated anti-mouse or chicken IgG antibodies were added for 30 min. Polymerized actin was detected by staining with 1 U per reaction of AlexaFluor 488 phalloidin (Molecular Probes). After two final washing steps using PBST, coverslips were mounted onto glass slides using Fluoromount-G (Southern Biotechnology Associates, Inc.) and viewed by confocal laser scan microscopy (CLSM) using a laser confocal microscope (Olympus Fluoview 500). Green and red fluorescence signals were recorded separately using appropriate filters for excitation and detection. Differential cell counts were done, distinguishing transfected cells with degenerated actin cytoskeletons from those with intact fibers in a blinded fashion, i.e., the person recording was unaware of the plasmid that was used for transfection. Confocal images were processed using Adobe Photoshop 7.0 (Adobe Systems Incorporated).
Multi-step virus growth kinetics were determined after infection of CEC with 200 PFU of v20US3*, v20ΔUS3, or v20US3*K220A virus. At 24, 48, 72, 96, and 120 h p.i., cells were trypsinized and titers were determined by plating infected onto fresh CEC in serial 10-fold dilutions. Plaque areas were measured after plating of viruses and subsequently incubated at 37°C and 5% CO. Cells were fixed after 5 days and analyzed by IIF with an MDV-specific polyclonal chicken serum (Schumacher et al., 2000). For each virus, 100 plaques were measured by taking digital pictures of individual plaques. Plaque areas were computed using the documentation software ImageJ (http://rsb.info.nih.gov/ij/index.html). Average percentages of plaque areas and standard deviations were determined exactly as described earlier (Schumacher et al., 2000).
Electron microscopy
Uninfected or infected CEC were fixed after 3 days of incubation at 37°C for 30 min with 2.5% glutaraldehyde buffered in 0.1 M Na-cacodylate (300 mOsmol, pH 7.4). After washing with 0.1 M Na-cacodylate, cells were scraped off of the plate, pelleted by low-speed centrifugation (1250 rpm), post-fixed in 1% aqueous OsO4 (Electron Microscopy Science), and finally stained with uranyl acetate. After stepwise dehydration in ethanol, cells were embedded in epon araldite (Electron Microscopy Science) and polymerized at 60°C overnight. Ultrathin sections of embedded material were counterstained with uranyl acetate and lead salts, and examined in an electron microscope (Philips EM 400 Tecnai).
Figure 6. Kinase activity of pUS3 is not required for depolymerization of actin stress fibers.
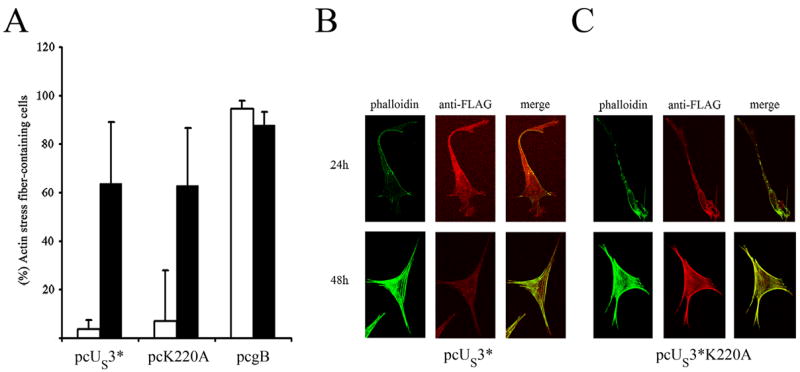
(A) Percent-age of cells containing an intact actin cytoskeleton at 24h and 48h after the transfection of plasmids expressing FLAG-tagged pUS3*, FLAG-tagged US3*K220A or gB. CEC were transfected and fixed at 24 or 48 h after transfection. Expressing cells were detected using antibody M2 to detect FLAG-tagged versions of pUS3* or 2K11 to detect gB (red). Actin (green) was stained with phalloidin-Alexa488® (Molecular Probes). Data were obtained by analyzing 300 transfected cells for each plasmid and time point in three individual experiments in a blinded manner. (B) Disassembly of the actin cytoskeleton in pcUS3* and pUS3*K220A transfected cells after 24h. Cells were fixed at the given times after transfection. pUS3* and actin were stained as stated above. Red and green fluorescence signals were recorded separately by using appropriate filters with a confocal microscope (Olympus) before assembly using Adobe Photoshop. Overlay of the pUS3 (red) and actin (green) fluorescent signals are shown in the righost panels (merge).
Acknowledgments
We thank Drs. Lucy Lee (ARS-ADOL, East Lansing, MI) and Jean-François Vautherot (INRA, Nouzilly, France) for generously providing monoclonal antibodies. This work was supported in part by the National Research Initiative of the USDA Cooperative State Research, Education and Extension Service, grant number 2003-02234, and by PHS grant AI063048A to NO.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aubert M, O’Toole J, Blaho JA. Induction and prevention of apoptosis in human HEp-2 cells by herpes simplex virus type 1. J Virol. 1999;73:10359–70. doi: 10.1128/jvi.73.12.10359-10370.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aubert M, Pomeranz LE, Blaho JA. Herpes simplex virus blocks apoptosis by precluding mitochondrial cytochrome c release independent of caspase activation in infected human epithelial cells. Apoptosis. 2007;12(1):19–35. doi: 10.1007/s10495-006-0330-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baigent SJ, Ross LJ, Davison TF. Differential susceptibility to Marek’s disease is associated with differences in number, but not phenotype or location, of pp38+ lymphocytes. J Gen Virol. 1998;79:2795–2802. doi: 10.1099/0022-1317-79-11-2795. [DOI] [PubMed] [Google Scholar]
- Bustelo XR, Sauzeau V, Berenjeno IM. GTP-binding proteins of the Rho/Rac family: regulation, effectors and functions in vivo. Bioessays. 2007;29(4):356–370. doi: 10.1002/bies.20558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calnek BW. Marek’s disease--a model for herpesvirus oncology. Crit Rev Microbiol. 1986;12(4):293–320. doi: 10.3109/10408418509104432. [DOI] [PubMed] [Google Scholar]
- Calnek BW, Adldinger HK, Kahn DE. Feather follicle epithelium: a source of enveloped and infectious cell-free herpesvirus from Marek’s disease. Avian Dis. 1970;14(2):219–233. [PubMed] [Google Scholar]
- Calnek BW, Hirai K. Pathogenesis of Marek’s disease virus infection. Vol. 255. Springer; Berlin: 2001. Marek’s disease; pp. 25–55. [DOI] [PubMed] [Google Scholar]
- Calton CM, Randall JA, Adkins MW, Banfield BW. The pseudorabies virus serine/threonine kinase Us3 contains mitochondrial, nuclear and membrane localization signals. Virus Genes. 2004;29(1):131–45. doi: 10.1023/B:VIRU.0000032796.27878.7f. [DOI] [PubMed] [Google Scholar]
- Churchill AE, Biggs PM. Agent of Marek’s disease in tissue culture. Nature. 1967;215:528–530. doi: 10.1038/215528a0. [DOI] [PubMed] [Google Scholar]
- Copeland NG, Jenkins NA, Court DL. Recombineering: a powerful new tool for mouse functional genomics. Nat Rev Genet. 2001;2(10):769–779. doi: 10.1038/35093556. [DOI] [PubMed] [Google Scholar]
- de Wind N, Domen J, Berns A. Herpesviruses encode an unusual protein-serine/threonine kinase which is nonessential for growth in cultured cells. J Virol. 1992;66(9):5200–5209. doi: 10.1128/jvi.66.9.5200-5209.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demmin GL, Clase AC, Randall JA, Enquist LW, Banfield BW. Insertions in the gG gene of pseudorabies virus reduce expression of the upstream Us3 protein and inhibit cell-to-cell spread of virus infection. J Virol. 2001;75:10856–69. doi: 10.1128/JVI.75.22.10856-10869.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deruelle M, Geenen K, Nauwynck HJ, Favoreel HW. A point mutation in the putative ATP binding site of the pseudorabies virus US3 protein kinase prevents Bad phosphorylation and cell survival following apoptosis induction. Virus Res. 2007 doi: 10.1016/j.virusres.2007.04.006. [DOI] [PubMed] [Google Scholar]
- Favoreel HW, Van Minnebruggen G, Adriaensen D, Nauwynck HJ. Cytoskeletal rearrangements and cell extensions induced by the US3 kinase of an alphaherpesvirus are associated with enhanced spread. Proc Natl Acad Sci USA. 2005;102(25):8990–8995. doi: 10.1073/pnas.0409099102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuchi K, Sudo M, Lee YS, Tanaka A, Nonoyama M. Structure of Marek’s disease virus DNA: detailed restriction enzyme map. J Virol. 1984;51(1):102–109. doi: 10.1128/jvi.51.1.102-109.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gimeno IM, Witter RL, Hunt HD, Reddy SM, Lee LF, Silva RF. The pp38 gene of Marek’s disease virus (MDV) is necessary for cytolytic infection of B cells and maintenance of the transformed state but not for cytolytic infection of the feather follicle epithelium and horizontal spread of MDV. J Virol. 2005;79(7):4545–4549. doi: 10.1128/JVI.79.7.4545-4549.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanks SK, Quinn AM, Hunter T. The protein kinase family: conserved features and deduced phylogeny of the catalytic domains. Science. 1988;241(4861):42–52. doi: 10.1126/science.3291115. [DOI] [PubMed] [Google Scholar]
- Ichijo K, Isogai H, Okada K, Fujimoto Y. Initial proliferation site of Marek’s disease tumor cells in the spleen. Zentralbl Veterinarmed B. 1981;28(3):177–189. doi: 10.1111/j.1439-0450.1981.tb01752.x. [DOI] [PubMed] [Google Scholar]
- Kato A, Yamamoto M, Ohno T, Kodaira H, Nishiyama Y, Kawaguchi Y. Identification of proteins phosphorylated directly by the Us3 protein kinase encoded by herpes simplex virus 1. J Virol. 2005;79:9325–9331. doi: 10.1128/JVI.79.14.9325-9331.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingham BF, Zelnik V, Kopacek J, Majerciak V, Ney E, Schmidt CJ. The genome of herpesvirus of turkeys: comparative analysis with Marek’s disease viruses. J Gen Virol. 2001;82:1123–1135. doi: 10.1099/0022-1317-82-5-1123. [DOI] [PubMed] [Google Scholar]
- Kyhse-Andersen J. Electroblotting of multiple gels: a simple apparatus without buffer tank for rapid transfer of proteins from polyacrylamide to nitrocellulose. J Biochem Biophys Methods. 1984;10(3–4):203–209. doi: 10.1016/0165-022x(84)90040-x. [DOI] [PubMed] [Google Scholar]
- Leader DP, Deana AD, Marchiori F, Purves FC, Pinna LA. Further definition of the substrate specificity of the alpha-herpesvirus protein kinase and comparison with protein kinases A and C. Biochim Biophys Acta. 1991;1091(3):426–431. doi: 10.1016/0167-4889(91)90210-o. [DOI] [PubMed] [Google Scholar]
- Lee LF, Wu P, Sui DX, Ren DL, Kamil J, Kung HJ, Witter RL. The complete unique long sequence and the overall genomic organization of the GA strain of Marek’s disease virus. Proc Natl Acad Sci USA. 2000;97:6091–6096. doi: 10.1073/pnas.97.11.6091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marek J. Multiple NervenentzÅndung (Polyneuritis) bei HÅhnern. Dtsch Tierärztl Wschrf. 1907;15:417–421. [Google Scholar]
- Morgan RW, Cantello JL, McDermott CH. Transfection of chicken embryo fibroblasts with Marek’s disease virus DNA. Avian Dis. 1990;34(2):345–351. [PubMed] [Google Scholar]
- Mori I, Goshima F, Koshizuka T, Koide N, Sugiyama T, Yoshida T, Yokochi T, Kimura Y, Nishiyama Y. The US3 protein kinase of herpes simplex virus attenuates the activation of the c-Jun N-terminal protein kinase signal transduction pathway in infected piriform cortex neurons of C57BL/6 mice. Neurosci Lett. 2003;351(3):201–205. doi: 10.1016/j.neulet.2003.08.033. [DOI] [PubMed] [Google Scholar]
- Mou F, Forest T, Baines JD. US3 of herpes simplex virus type 1 encodes a promiscuous protein kinase that phosphorylates and alters localization of lamin A/C in infected cells. J Virol. 2007;81:6459–6470. doi: 10.1128/JVI.00380-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murata T, Goshima F, Daikoku T, Takakuwa H, Nishiyama Y. Expression of herpes simplex virus type 2 US3 affects the Cdc42/Rac pathway and attenuates c-Jun N-terminal kinase activation. Genes Cells. 2000;5:1017–1027. doi: 10.1046/j.1365-2443.2000.00383.x. [DOI] [PubMed] [Google Scholar]
- Nazerian K, Solomon JJ, Witter RL, Burmester BR. Studies on the etiology of Marek’s disease. II. Finding of a herpesvirus in cell culture. Proc Soc Exp Biol Med. 1968;127(1):177–182. doi: 10.3181/00379727-127-32650. [DOI] [PubMed] [Google Scholar]
- Nguyen ML, Blaho JA. Apoptosis during herpes simplex virus infection. Adv Virus Res. 2007;69:67–97. doi: 10.1016/S0065-3527(06)69002-7. [DOI] [PubMed] [Google Scholar]
- Osterrieder N. Sequence and initial characterization of the U(L)10 (glycoprotein M) and U(L)11 homologous genes of serotype 1 Marek’s Disease Virus. Archives of Virology. 1999;144:1853–1863. doi: 10.1007/s007050050710. [DOI] [PubMed] [Google Scholar]
- Osterrieder N, Kamil JP, Schumacher D, Tischer BK, Trapp S. Marek’s disease virus: from miasma to model. Nat Rev Microbiol. 2006;4:283–294. doi: 10.1038/nrmicro1382. [DOI] [PubMed] [Google Scholar]
- Osterrieder N, Vautherot JF. The genome content of Marek’s disease-like viruses. In: Davison TF, Nair V, Pastoret pp, editors. Marek’s disease. Elsevier; London: 2004. [Google Scholar]
- Poon AP, Benetti L, Roizman B. U(S)3 and U(S)3.5 protein kinases of herpes simplex virus 1 differ with respect to their functions in blocking apoptosis and in virion maturation and egress. J Virol. 2006;80:3752–3764. doi: 10.1128/JVI.80.8.3752-3764.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poon AP, Gu H, Roizman B. ICP0 and the US3 protein kinase of herpes simplex virus 1 independently block histone deacetylation to enable gene expression. Proc Natl Acad Sci USA. 2006;103:9993–9998. doi: 10.1073/pnas.0604142103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poon AP, Roizman B. Mapping of key functions of the herpes simplex virus 1 U(S)3 protein kinase: the U(S)3 protein can form functional heteromultimeric structures derived from overlapping truncated polypeptides. J Virol. 2007;81:1980–1989. doi: 10.1128/JVI.02265-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purves FC, Deana AD, Marchiori F, Leader DP, Pinna LA. The substrate specificity of the protein kinase induced in cells infected with herpesviruses: studies with synthetic substrates [corrected] indicate structural requirements distinct from other protein kinases. Biochim Biophys Acta. 1986;889:208–215. doi: 10.1016/0167-4889(86)90106-0. [DOI] [PubMed] [Google Scholar]
- Purves FC, Longnecker RM, Leader DP, Roizman B. Herpes simplex virus 1 protein kinase is encoded by open reading frame US3 which is not essential for virus growth in cell culture. J Virol. 1987;61:2896–2901. doi: 10.1128/jvi.61.9.2896-2901.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy SM, Lupiani B, Gimeno IM, Silva RF, Lee LF, Witter RL. Rescue of a pathogenic Marek’s disease virus with overlapping cosmid DNAs: use of a pp38 mutant to validate the technology for the study of gene function. Proc Natl Acad Sci USA. 2002;99(10):7054–7059. doi: 10.1073/pnas.092152699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds AE, Ryckman BJ, Baines JD, Zhou Y, Liang L, Roller RJ. U(L)31 and U(L)34 proteins of herpes simplex virus type 1 form a complex that accumulates at the nuclear rim and is required for envelopment of nucleocapsids. J Virol. 2001;75:8803–8817. doi: 10.1128/JVI.75.18.8803-8817.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds AE, Wills EG, Roller RJ, Ryckman BJ, Baines JD. Ultrastructural localization of the herpes simplex virus type 1 UL31, UL34, and US3 proteins suggests specific roles in primary envelopment and egress of nucleocapsids. J Virol. 2002;76:8939–8952. doi: 10.1128/JVI.76.17.8939-8952.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryckman BJ, Roller RJ. Herpes simplex virus type 1 primary envelopment: UL34 protein modification and the US3-UL34 catalytic relationship. J Virol. 2004;78:399–412. doi: 10.1128/JVI.78.1.399-412.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J, Maniatis T. Molecular cloning: a laboratory manual. Cold Spring Harbor Press; Cold Spring Harbor, N.Y: 1989. [Google Scholar]
- Schumacher D, Tischer BK, Fuchs W, Osterrieder N. Reconstitution of Marek’s disease virus serotype 1 (MDV-1) from DNA cloned as a bacterial artificial chromosome and characterization of a glycoprotein B-negative MDV-1 mutant. J Virol. 2000;74:11088–11098. doi: 10.1128/jvi.74.23.11088-11098.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumacher D, Tischer BK, Trapp S, Osterrieder N. The protein encoded by the US3 orthologue of Marek’s disease virus is required for efficient de-envelopment of perinuclear virions and involved in actin stress fiber breakdown. J Virol. 2005;79:3987–3997. doi: 10.1128/JVI.79.7.3987-3997.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shek WR, Calnek BW, Schat KA, Chen CH. Characterization of Marek’s disease virus-infected lymphocytes: discrimination between cytolytically and latently infected cells. J Natl Cancer Inst. 1983;70:485–491. [PubMed] [Google Scholar]
- Tischer BK, von Einem J, Kaufer B, Osterrieder N. Two-step red-mediated recombination for versatile high-efficiency markerless DNA manipulation in Escherichia coli. Biotechniques. 2006;40:191–197. doi: 10.2144/000112096. [DOI] [PubMed] [Google Scholar]
- Tulman ER, Afonso CL, Lu Z, Zsak L, Rock DL, Kutish GF. The genome of a very virulent Marek’s disease virus. J Virol. 2000;74:7980–7988. doi: 10.1128/jvi.74.17.7980-7988.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valderrama F, Cordeiro JV, Schleich S, Frischknecht F, Way M. Vaccinia virus-induced cell motility requires F11L-mediated inhibition of RhoA signaling. Science. 2006;311:377–381. doi: 10.1126/science.1122411. [DOI] [PubMed] [Google Scholar]
- Van Minnebruggen G, Favoreel HW, Jacobs L, Nauwynck HJ. Pseudorabies virus US3 protein kinase mediates actin stress fiber breakdown. J Virol. 2003;77:9074–9080. doi: 10.1128/JVI.77.16.9074-9080.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]