

Abstract
Giant axonopathy (gaxp), an autosomal recessive mouse mutation, exhibits ataxia of the hind legs with a slight side-to-side wobble while walking. Within the genomic region of gaxp locus, a total of 94 transcripts were identified; the annotation of these genes using OMIM and PubMed yielded three potential candidate genes. By cDNA microarray analysis, 54 genes located on or near the gaxp locus were found to exhibit differential expression between gaxp and littermate controls. Based on microarray data and the known function of genes identified, slc12a6 was selected as the primary candidate gene and analyzed using the Reveal™ technology of SpectruMedix. A 17-base deletion was detected from within exon 4 of slc12a6. Reverse transcriptase polymerase chain reaction validated the difference in slc12a6 expression in different types of mice at the mRNA level, revealing a marked reduction in gaxp mice. Western blot analysis indicated that the protein product of slc12a6, the K+-Cl− cotransporter Kcc3, was not detectable in gaxp mice. The causative role of the exon 4 mutation within slc12a6 in the gaxp phenotype was further confirmed by screening multiple inbred strains and by excluding the mutation of nearby genes within the gaxp locus.
Keywords: genome, giant axonopathy, high-throughput screening, microarray, mouse, mutation
Introduction
Giant axonopathy (gaxp) is a new autosomal recessive mutation that arose spontaneously in a mutant strain on the C3H/HeDiSnJ background in the Mouse Mutant Resource at The Jackson Laboratory (TJL) (http://www.jax.org/mmr/MMR_Mutant_gaxp.html). Homozygous mutants exhibit ataxia of their hind legs, characteristically lifting their hind legs too high and wobbling side-to-side as they walk. When mutants are pulled backward by their tails, they curl their hind legs towards their bodies, whereas normal mice actively move their legs to resist being pulled backward. The swollen axons observed in gaxp/gaxp mice are a unique form of axonal dystrophy. Whereas the normal dystrophic axons are densely eosinophilic and are composed of densely packed organelles and filamentous material, the axons in these mutants contain only lightly packed organelles, suggesting that the swelling is due in part to an increased uptake of water. The inherited diseases of children characterized by dystrophic axons include infantile neuroaxonal dystrophy (INAD) and giant axonal neuropathy (GAN). These are characterized histopathologically by accumulation of the dense form of dystrophic axons. INAD (also known as Seitelberger’s disease) is a rare autosomal recessive hereditary neurodegenerative disease of humans [1–4]. GAN is an autosomal recessive neurologic disorder clinically characterized by a severe polyneuropathy, central nervous system abnormalities, and characteristic tightly curled hair; mutations in the gigaxonin gene have recently been identified as the underlying genetic defect [5–8]. However, the gigaxonin gene is not located within the murine gaxp locus; it is likely that the gaxp phenotype is caused by a new mutation in a gene that has not been functionally related to the dystrophic axons. Identifying the mutated gene in the gaxp locus is essential to understanding this mouse model and to investigating the molecular cause(s) of dystrophic axons.
According to TJL Webpage information, the gaxp mutation is on chromosome(Chr) 2. The most likely gene order places the mutation between D2Mit128 and D2Mit102 in 174 meioses tested. The recombination estimates with standard errors and best gene order are centromere-D2Mit386-3.04 +/−1.3-D2Mit249-1.18 +/− 0.83-D2Mit128-1.75 +/− 1.00-gaxp-D2Mit102-1.17 +/− 0.82-[D2Mit484, D2Mit17]-18.82 +/− 4.0 – D2Mit51.
The completion and annotation of the murine genome sequence has yielded a powerful tool for positional cloning [9–11]. First, all of the coding sequences within a given a chromosomal region of interest can be identified. Second, information on the introns and 5’ and 3’ regions of the sequences is generally available, making it possible for the gene to be analyzed thoroughly throughout the coding region and the regulatory region. Third, genomic sequence of a given region facilitates the analysis of nucleotide organization, gene ordering, gene expression patterns, and chromosomal structure. The availability of this information has coincided with the development of high throughput technologies for both mutation analysis and gene expression profiling. All of these developments will have considerable effect on the search for candidate genes in positional cloning.
Here we report the identification of the mutated gene in the gaxp model by using an integrated genomic strategy, encompassing high throughput screening of genomic elements [10–11], gene expression profiles, and gene function searching.
Results
Phenotype of gaxp mice
All gaxp/gaxp mice housed at the University of Tennessee Health Science Center exhibited ataxia of hind legs at the age of 7 days and a slight side-to-side wobble while walking. Two other tests distinguished gaxp/gaxp mice from unaffected littermates. First, as the mice walked, we exerted backward traction on their tails; the gaxp/gaxp mice were not able to use their hind legs to actively resist. Second, we found that the hind legs of gaxp/gaxp mice clamped together toward the body when held downward by the tail (Figure 1). No differences were noted between the gaxp/+ and +/+ mice.
Figure 1.

Phenotype of normal and gaxp littermates. A: The hind legs of a gaxp/gaxp mouse came together and its body curled when it was held upside down by the tail. B: A gaxp/gaxp mouse failed to positively resist when it was pulled backward by its tail. In both A and B, the normal mouse is the littermate of the gaxp mouse.
Target region of the mutation in the gaxp locus
To select candidate genes, we first identified all possible genes within the gaxp region. Previous genetic analysis showed that the gaxp mutation is located on mouse Chr 2, flanked by the molecular markers D2Mit128 and D2Mit102 (http://www.jax.org/mmr/gaxp.html). According to the Ensembl database, D2Mit128 is located between 106121793 and 106122031 bp, whereas D2Mit102 is located between 113984330 and 113984492 bp (Figure 2A). Genomic sequences within this region are complete in the Ensembl database. There are a total of 94 transcripts in this region, with 81 known genes and 13 that correspond to novel genes or pseudogenes.
Figure 2.

Schematic of the mutation identification in gaxp/gaxp mice. A: A genetic map of the gaxp locus showing the relative locations of microsatellite markers and the total number of candidate transcripts within the gaxp locus. B: PCR product analyses using the SpectruMedix system. The PCR products from different mice were compared to check the possible sequence difference between normal C3H/HeDiSnJ mice (+/+), homozygous gaxp/gaxp mice, and heterozygous +/gaxp mice. C: Expression level of Slc12a6 in gaxp and control mice. Amplification of Slc12a6 transcript from total RNA extracted from cerebella, cerebra, muscle, liver, and spleen of +/+ and gaxp/gaxp mice.
Informational search of gene functions within the targeted region
To prioritize the transcripts, we conducted a bioinformatics search of the function of every gene within the gaxp region to see whether any genes seemed functionally relevant to the gaxp phenotype. For each gene, we searched its function in OMIM (for key words anywhere in the gene description) and PubMed (words in the title or abstract) with the terms “ataxia”, “axons”, “wobble”, “eosinophilic”, and “neuropathy”. From those genes, we identified three candidates: brain-derived neurotrophic factor (BDNF), ryanodine receptor 3 (Ryr3) and solute carrier family 12, member 6 (Slc12a6) (Table 1) (ref 12–22).
Table 1.
Candidate genes based on bioinformatics literature search
| Search Term | Gene Symbol | Gene Name | OMIM | PubMed |
|---|---|---|---|---|
| ataxia | Bdnf | brain-derived neurotrophic factor | 0 | 7 (ref 12–15) |
| axons | Bdnf | brain-derived neurotrophic factor | 0 | 270 (ref 12–15) |
| Ryr3 | ryanodine receptor 3 | 0 | 1 (ref 16) | |
| eosinophilic | Bdnf | brain-derived neurotrophic factor | 0 | 1 (ref 16, 17) |
| neuropathy | Slc12a6 | solute carrier family 12, member 6 | 1 | 6 (ref 18–22) |
| Bdnf | brain-derived neurotrophic factor | 0 | 20 |
At the TJL Website, two genes, brain-derived neurotrophic factor precursor and voltage-gated potassium channel protein, shaker-related subfamily, member 4 (Kcna4), were suggested as candidate genes. However, our literature analysis did not identify Kcna4 as a likely candidate.
Gene expression levels of genes within the gaxp region
Abnormal gene expression has previously been used as a revealing fingerprint of disease genes [10, 11]. Using Affymetrix microarray chips, we analyzed the gene expression level of each of three mice from normal and gaxp/gaxp mice. Of 2642 probes on mouse Chr 2, 768 are located between 100 and 130 Mb; within this subset, the expression levels of 22 probes were down-regulated and 32 were up-regulated. The Slc12a6 gene is among those genes that are down-regulated in gaxp/gaxp mice, as detected by two probes on the chip; this result tagged Slc12a6 as a promising candidate for the gaxp gene. In contrast, the expression of Bdnf and Ryr3 appeared to be either unaffected or slightly up-regulated (Table 2), whereas there was no difference in Kcna4 expression between gaxp mice and controls.
Table 2.
Comparison of expression of candidate genes in gaxp and in normal mice
| Gene | Gaxp Signal | WT Signal | Gasp/WT Signal Log Ratio | Interpretation* |
|---|---|---|---|---|
| Bdnf | 415.6 | 566 | −0.0 | NC |
| 639.7 | 445.2 | 0.4 | I | |
| Ryr3 | 979.4 | 856.5 | 0.4 | NC |
| 339.6 | 183.2 | 0.7 | I | |
| Slc12a6 | 501.6 | 1200.4 | −1.1 | D |
| 51.8 | 234.4 | −1.9 | D | |
| Kcna4 | 274.3 | 226.1 | 0.6 | NC |
| 230.9 | 168.5 | 0.5 | NC |
NC = no change; I = increased expression level; D = decreased expression level.
Initial identification of mutation in Slc12a6
Based on the results of our bioinformatics and microarray analyses, Slc12a6 was the first priority in our search for the mutated gene from this model, with Bdnf and Ryr3 as secondary candidates.
We first conducted polymerase chain reaction (PCR) amplification of each exon of Slc12a6, a large gene located in the genomic sequence between 112,067,658 and 112,163,995 on mouse Chr 2. Slc12a6 encompasses 25 exons, with a 2081-base untranslated 3’ end. Three kinds of PCR products were screened by Reveal™ (SpectruMedix): DNA fragments from gaxp, normal littermates, and a mixture of gaxp and normal mice. Among these amplificons, using primers flanking exon 4 from Slc12a6 we found that one fragment differed between gaxp and controls (Figure 2B).
Given this apparent difference in the Slc12a6 gene between gaxp and control mice, we further analyzed the expression level of Slc12a6 by reverse transcriptase polymerase chain reaction (RT-PCR). A pair of primers that flank exon 3–5 was used to amplify cDNA from several tissues. As shown in Figure 2C, the expression level of Slc12a6 was lower in gaxp mice than in controls.
To determine which nucleotide(s) was different between gaxp/gaxp mice and their littermate controls, we sequenced genomic DNA fragments from gaxp/gaxp and control mice. The data revealed a 17-base deletion within exon 4 in DNA from gaxp/gaxp mice (Figure 3A). To confirm this mutation at the cDNA level, we performed (T-PCR)on total RNAs from gaxp/gaxp, gaxp/+, and +/+ mice by using primers that covered the mRNA sequence from exon 3 to exon 5 of the Slc12a6 gene. The resultant RT-PCR products were sequenced using the SpectruMedix system, and the same deletion in gaxp/gaxp was found (Figure 3A, left panel).
Figure 3.

Sequence changes of Slc12a6 gene in gaxp mice. A: There are two sequencing panels in the figure. The right panel is genomic sequences of Slc12a6 from gaxp mice and the normal littermate as control; the left panel is cDNA sequences of Slc12a6 from gaxp mice and the normal littermate. In both cases, 17 base nucleotides are the deleted from gaxp mice. Notice that a T also was inserted when the 17 bases were deleted. B: Derived amino acid sequences of normal (black) and mutant Slc12a6 (brown). The mutation leads a frameshift change of 12 amino acids and a subsequent stop codon. C: The Slc12a6 mutation has dramatic effects at its translational levels. Upper panel, the expression of protein product, Kcc3, in kidney and cerebra in two gaxp mice and two wild type controls. Lower panel, the expression of beta-actin.
Analysis of KCC3, the protein product of Slc12a6, in gaxp mice
To detect the effect of the exon 4 deletion at the protein level, Western blot analysis was conducted using tissues from kidney and brain to detect the protein encoded by Slc12a6, the KCC3 K+-Cl− cotransporter. We used a well-characterized antibody specific for a peptide within exon 3 of Slc12a6 [24]; should a KCC3 protein product be generated in these mice, it should be detected by this antibody, given that the gaxp deletion is in exon 4 of Slc12a6. Whereas the KCC3 protein was detected in both tissues in normal littermates, it was not detected in gaxp mice (Figure 3C). The data suggested that the frameshift change of 12 amino acids may lead to nonsense-mediated decay (NMD) [11] of the gaxp KCC3 protein.
Confirmation of mutation
We conducted two experiments to further ensure the link between the mutation and the disease. First, we examined sequence polymorphism in exon 4 of Slc12a6 from nine additional inbred strains. As shown in Figure 4, while the mixture of DNA fragments from those 12 strains appeared as a single signal, the mixture with gaxp appeared as two signals, indicating the difference between gaxp/gaxp and the other strains. Second, we screened 121 other candidate genes within the gaxp region by using our SpectruMedix mutation detection system [10]; we detected no other potential mutations, indicating that Slc12a6 is the only gene with a coding sequence mutation in the gaxp locus.
Figure 4.
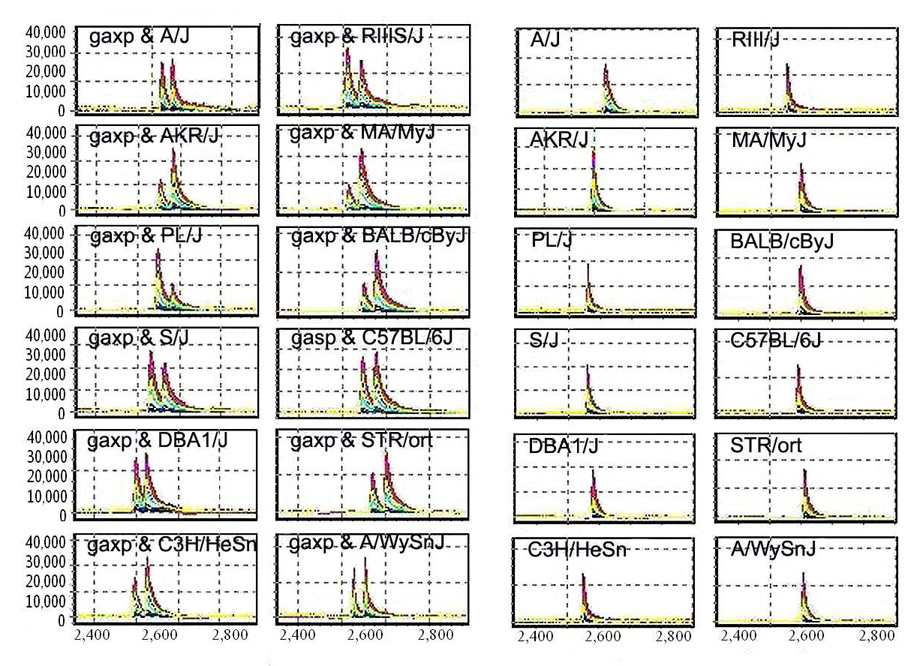
Size difference of a PCR product from gaxp mice and 12 mouse inbred strains. Left two panels, PCR products from 12 mouse inbred strains, C3H, DBA, B6, Balb/c, Str/ort, MA/HyJ, A/J, AKR/J, PL/J, S/J, RIIIS/J, and A/wySnJ. Right two panels, mixture of PCR products from gaxp/gaxp mice in 12 mouse inbred strains. The X-axis represents relative size of the PCR products. The Y-axis represents relative strength of signal or the amount of the PCR products.
Discussion
Using a combination of functional bioinformatics analysis and microarray screening of a large genome region (see Methods), we identified the mutant gene responsible for the gaxp phenotype. The initial mapping at TJL was conducted with only 21 mutant mice. Linkage of gaxp was first detected on Chr 2 with D2Mit128 and D2Mit102 by using the pooled sample. DNA samples were then typed for the individual 87 animals with these two and other markers and Chr 2 markers (http://www.jax.org/mmr/gaxp.html). The gaxp locus should be further mapped by using classical candidate positional cloning, because there is a multitude of candidate genes in this region of Chr 2. However, with the availability of mouse genome information and the rapidly evolving understanding of gene function [9–11], the likelihood of detecting candidate genes based on a rough genetic map is considerably increased. With our success at finding this mutation and others [10–11], we feel that in the near future we will no longer need fine mapping to identify mutated genes in many mouse strains with Mendelian phenotypes.
Several lines of evidence indicate that a 17-nucleotide deletion of Slc12a6 is causally associated with the gaxp phenotype. First, Slc12a6 is located within the genetic region of the gaxp locus. Second, the Slc12a6 mutation was the only defect detected in gaxp mice among candidate genes (Figure 2) within the gaxp locus. There were thus no other differences between gaxp/gaxp mice and their parental strains, so that the possibility of coding sequence mutations in other genes in this interval was ruled out. Third, cDNA sequence results agreed with the genomic DNA data. Fourth, gene network and real-time PCR indicated that Slc12a6 is the key that influences the differential expression of genes in gaxp mice. Finally, the 17-nucleotide deletion in Slc12a6 is predicted to delete some six amino acids from the KCC3 protein, leading to a stop codon and removing the entire 12-transmembrane hydrophobic core through which K+ and Cl− are transported. No KCC3 protein, truncated or full-length, was detected using an antibody directed against an epitope from within exon 3 of Slc12a6. These experiments strongly indicate that loss-of-function in Slc12a6 is the cause of the gaxp phenotype.
Prior published reports have identified a spectrum of disease phenotypes associated with loss of KCC3 function. The first study indicated that single-nucleotide mutations in human SLC12A6 cause a peripheral neuropathy associated with agenesis of the corpus callosum (ACCPN), a severe sensorimotor neuropathy associated with mental retardation, dysmorphic features, and complete or partial agenesis of the corpus callosum [20]. In the same report, mice with a targeted deletion in exon 3 of Slc12a6 had a locomotor deficit, peripheral neuropathy, and a sensorimotor gating deficit. A second knockout was independenty generated by fusing the first conserved exon in-frame to β-galactosidase [23]; these mice suffered from deafness, neurodegeneration, and reduced seizure threshold. Notably, central neurodegeneration was not identified in the mice with a targeted deletion of exon 3, despite careful histopathology [20]. Most recently, Uyanik et al. [18] showed that not only truncating but also missense mutations of the KCC3 gene are associated with ACCPN,with a spectrum of mental retardation and progressive sensorimotor neuropathy, depending on the specific disease-associated mutation in SLC12A6. As Slc12a6 contains 25 exons and encodes severaldifferent KCC3 isoforms, varying in the composition of the cytoplasmic N-terminal domain [21, 24–27], it is possible that it has multiple functions in a variety of pathways. Notably, however, swelling-activated K+-Cl− cotransport mediated by KCC3 appears to play a critical role in neuronal volume regulation [23], and a recent report has implicated axonal and periaxonal swelling in the peripheral neuropathy associated with KCC3 deficiency [28]. The differences in the phenotypes of gaxp mice, the two published knockout mice, and humans with truncating versus nonsense mutations in SLC12A6 are provocative, and suggest that several other factors modulate the effect of KCC3 deficiency in both the central and peripheral nervous systems.
The protein product, KCC3, was undetectable in gaxp mice, suggestive of nonsense-mediated decay (NMD). NMD, first documented by Losson and Lacroute over 20 years ago [29], is a proofreading mechanism that enables eukaryotic cells to detect and degrade mRNAs that contain premature termination codons (PTCs). About one-third of inherited genetic disorders and many forms of cancer are caused by frame-shift or nonsense mutations, which result in the generation of PTCs [30, 31]. Although mRNA containing a PTC may initially be translated into a truncated protein, cells can initiate the NMD mechanism to recognize and degrade the mutant transcripts if the truncated protein is deleterious, as we previously reported in a waddles mouse model [11].
Slc12a6 was not in the list of candidate genes for allelism tests at TJL. According to the information on TJL Webpage (http://www.jax.org/mmr/lbab.html), allelisms tested were anorexia, with a ratio of disease/total at 0/50 progeny born, and hotfoot, with a ratio of 0/17 progeny born. In addition, Kcna4 and Bdnf, but not Slc12a6, were listed as candidate genes. In aggregate, we feel our study suggests that it is extremely important to conduct a comprehensive expression and bioinformatics analysis for any known disease locus for which the mutated gene has not yet been discovered.
Methods
Strategy of mutation analysis
Figure 5 shows the strategy we used to identify mutated genes from mouse disease models. The high-throughput technology for screening a large number of genes by using the SpectruMedix system identified three other mutations. In the current study, bioinformatics searching and gene expression profiles made the screening simpler and more efficient.
Figure 5.

Schematic illustration of strategy of mutation identification in our laboratory. In Step 1, transcripts are identified based on the Ensembl database, http://www.ensembl.org; In Step 2, functional information of candidate genes are obtained from the Online Mendelian Inheritance in Man (OMIM) Website (http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=OMIM) and PubMed (http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=PubMed) while the gene expression profile comes from the whole genome microarray analysis. In Steps 3, 4, and 5, screening of possible mutations are done with SpectruMedix high-throughput system. In case a mutation is identified, any of the steps goes directly to Step 6. In Step 7, multiple sequence comparison can be done through multiple mouse strains or large population from a segregated F2 or recombinant inbred lines. In Step 8, the effect of a mutation can be predicted through multiple analyses such as sequence analysis, Western blot, and histobiochemistry analysis.
Mice
A heterozygous (gaxp/+) breeding pair of mice was purchased from TJL, and a breeding colony was established at the research animal facility of the University of Tennessee Health Science Center. Homozygous mutation gaxp/gaxp, heterozygous gaxp/+, and homozygous normal +/+ mice produced from the heterozygous parents were used in this study for mutation identification and protein analysis. Experimental animal procedures and mouse husbandry were performed in accordance with the National Institutes of Health’s Guide for the Care and Use of Laboratory Animals and approved by the UTHSC Institutional Animal Care and Use Committee.
Mutation screening
Based on the Ensembl and National Center for Biotechnology Information (NCBI) databases as of March 07, 2007, primer pairs flanking the exons of known and predicted genes (including expressed sequence tags) within the gaxp locus were designed using Primer3 software (http://www-genome.wi.mit.edu/cgi-bin/primer/primer3_www.cgi). Primers were located approximately 100 bp 5’ or 3’ to each exon and, in general, produced 300–400 bp amplicons. For large exons that required multiple pairs of primers, primers were designed to allow neighbouring DNA fragments to overlap each other by at least 50 bp. Primer pairs were synthesized by Illumina (www.illumina.com). PCR amplification of gDNA was performed in a 96-well plate format and consisted of 30–35 cycles at three temperatures: strand denaturation at 96°C for 30 sec, primer annealing at 54–60°C for 60 sec, and primer extension at 72°C for 120 sec.
A TGCE device made by SpectruMedix was used to analyze amplicons from +/+, +/gaxp, and gaxp/gaxp mice. The SpectruMedix system includes a high-throughput capillary electrophoresis instrument capable of analyzing 96 samples every 140 min. Heteroduplex analysis was subsequently performed using SpectruMedix software. Amplicons from gaxp mice were sequenced if they differed from normal ones. Heteroduplex analysis was done with our previously reported protocol [10–11].
Genotyping
A pair of primers flanking the position of a single nucleotide mutation of Slc12a6 gene (forward primer: CCCTCAGCTCCACACAGTCT; reverse primer: TTCTTCACATGGATGGATGC) was used to genotype +/+, gaxp/+, and gaxp/gaxp mice. The PCR products from heterozygous and homozygous mice were mixed with those from normal mice. The individual and mixed PCR products were then run on TGCE to detect possible sequence variations. PCR amplification was performed in a total volume of 25 µl at a final concentration of 1.5 mM MgCl2 concentration and 0.2 mmol/L each dNTP, 0.2 µmol/L oligonucleotide primer, 100 ng template DNA, and 0.7 units Taq polymerase (Fisher Scientific, Pittsburgh, PA) for 35 cycles of 94°C for 1 min, 50–55°C for 1 min, and 72°C for 1 min.
RT-PCR amplification of Slc12a6
One-step RT-PCR kit (Invitrogen, Carlsbad, CA) was used to detect the expression of mutated Slc12a6 at the mRNA level. Reactions were performed in a total volume of 50 µl with 8 ng/µl of total RNA, and 0.2 µM forward (ATTCAGGATCCCCAAGAACC) and reverse (AAGACGCAGGAAGAGGATCA) primers were used to amplify exons 2 to 3 of Nppc. First, cDNA synthesis and predenaturation were performed in single cycles at 50°C for 40 min and 94°C for 2 min. Next, PCR amplification was performed for 35 cycles: 94°C for 30 sec, 54–58°C for 36 sec, and 72°C for 2 min. Gapdh was used as control with the same amplification protocol and cycle number.
DNA sequencing
DNA sequencing was conducted to verify the mutation in the gDNA and cDNA of Slc12a6 from different species of mice. PCR products from both gDNA and cDNA were purified using an AMPure PCR Purification Kit (Agencourt, Beverly, MA ), and the resultant products were sequenced using the BigDye® Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, CA). A total volume of 5 µl sequencing reactions including 2 µl Big Dye (plus Half-BD), 10 to 23 ng of purified DNA template, and 1 to 3 ρmoles of either forward or reverse universal sequencing primers was incubated for 37 cycles at 96°C for 180 sec, 50°C for 30 sec, and 60°C for 180 sec. Unreacted primers were removed by ethanol-acetate precipitation (3.75% 3M NaOAc, 87.5% nondenatured 100% EtOH, and 8.75% dH2O, pH 4.6). The labeled products were dissolved in 0.02 mM EDTA in HiDi formamide prior to electrophoretically loading onto the SpectruMedix 96 capillary sequencing system. The same primers in the amplification of DNA fragments from either genomic DNA or mRNA were also used in the sequencing. Sequencing was conducted two times to verify the results for either gDNA or cDNA.
Western blot analysis
Cerebra and kidney from two pairs of wild type C3H/HensJ and gaxp/gaxp mice were dissected and homogenized in sucrose buffer (0.32 M sucrose, 5 mM Tris±HCl, pH 7.5, 2 mM EDTA) and ground with a Dellco pestle. Protein was obtained by centrifugation at 25,000g, 60 min, a total of 40 µg/well were loaded on a 6–10% gradient SDS-PAGE gel. Proteins were then electrophoretically transferred to PVDF membranes (Bio-Rad, Hercules, CA). The antibody to KCC3 was specific for a peptide epitope from within exon 3 of Slc12a6; membranes were probed with affinity-purifed KCC3 antibody at a dilution of 1:1000. Proteins were detected using ECL Plus detection system (Amersham, Arlington Heights, IL) [27].
Microarray analysis
Individual total RNA samples from wild type and gaxp mice (three each) were pooled separately into two groups for a pilot microarray assay. Five micrograms of total RNA were used for each chip. Microarray hybridization and data generation were performed according to Affymetrix guidelines using GeneChip® Mouse Genome 430 2.0 arrays, cDNA and cRNA synthesis kits, and GCOS 1.4 software (www.affymetrix.com). Raw data were normalized using MAS5 algorithm, and comparative analysis was carried out between wild type and mutant datasets using GCOS 1.4. Functional clustering was performed with DAVID tools (http://david.abcc.ncifcrf.gov).
Acknowledgements
We thank David Armbruster, author’s editor (University of Tennessee Health Science Center), for kindly editing this manuscript. This work was supported by the Center of Genomics and Bioinformatics (WG) and Center in Connective Tissue Research (WG) at the University of Tennessee Health Science Center; Veterans Administration Medical Centers in Memphis (WG) and Boston (DBM); National Institute of Arthritis and Musculoskeletal and Skin Diseases, National Institutes of Health (R01 AR51190 to WG); National Institute of Diabetes and Digestive and Kidney Diseases (RO1 DK57708 to DBM).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hortnagel K, et al. Infantile neuroaxonal dystrophy and pantothenate-kinase-associated neurodegeneration: locus heterogeneity. Neurology. 2004;14:922–924. doi: 10.1212/01.wnl.0000137046.28085.b2. [DOI] [PubMed] [Google Scholar]
- 2.Umemura A, Jaggi JL, Dolinskas CA, Stern MB, Baltuch GH. Pallidal deep brain stimulation for longstanding severe generalized dystonia in Hallervorden-Spatz syndrome (Case report) J Neurosurg. 2004;100:706–709. doi: 10.3171/jns.2004.100.4.0706. [DOI] [PubMed] [Google Scholar]
- 3.Marotti JD, Tobias S, Fratkin JD, Powers JM, Rhodes CH. Adult onset leukodystrophy with neuroaxonal spheroids and pigmented glia: report of a family, historical perspective, and review of the literature. Acta Neuropathol (Berl.) 2004;107:481–488. doi: 10.1007/s00401-004-0847-x. Epub 2004 Apr 6. [DOI] [PubMed] [Google Scholar]
- 4.Thomas M, Hayflick SJ, Jankovic J. Clinical heterogeneity of neurodegeneration with brain iron accumulation (Hallervorden-Spatz syndrome) and pantothenate kinase-associated neurodegeneration Mov Disord. 2004;19:36–42. doi: 10.1002/mds.10650. [DOI] [PubMed] [Google Scholar]
- 5.Allen E, et al. Gigaxonin-controlled degradation of MAP1B light chain is critical to neuronal survival. Nature. 2005;438:224–228. doi: 10.1038/nature04256. Epub 2005 Oct 16. [DOI] [PubMed] [Google Scholar]
- 6.Cullen VC, et al. Gigaxonin is associated with the Golgi and dimerises via its BTB domain. Neuroreport. 2004;15:873–876. doi: 10.1097/00001756-200404090-00028. [DOI] [PubMed] [Google Scholar]
- 7.Bomont P, et al. Identification of seven novel mutations in the GAN gene. Hum Mutat. 2003;21:446. doi: 10.1002/humu.9122. [DOI] [PubMed] [Google Scholar]
- 8.Bomont P, et al. The gene encoding gigaxonin, a new member of the cytoskeletal BTB/kelch repeat family, is mutated in giant axonal neuropathy. Nat Genet. 2000;26:370–374. doi: 10.1038/81701. [DOI] [PubMed] [Google Scholar]
- 9.Mouse Genome Sequencing Consortium. Initial sequencing and comparative analysis of the mouse genome. Nature. 2002;420:520–562. doi: 10.1038/nature01262. [DOI] [PubMed] [Google Scholar]
- 10.Jiao Y, et al. Identification of a deletion causing spontaneous fracture by screening a candidate region of mouse chromosome 14. Mammalian Genom. 2005;16:20–31. doi: 10.1007/s00335-004-2414-0. [DOI] [PubMed] [Google Scholar]
- 11.Jiao Y, et al. Carbonic anhydrase-related protein VIII deficiency causes a distinctive lifelong gait disorder in waddles mice. Genetics. 2005;171:1239–1246. doi: 10.1534/genetics.105.044487. Epub 2005 Aug 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Meng H, Walker N, Su Y, Qiao X. Stargazin mutation impairs cerebellar synaptogenesis, synaptic maturation and synaptic protein distribution. Brain Res. 2006;1124:197–207. doi: 10.1016/j.brainres.2006.09.086. Epub 2006 Oct 27. [DOI] [PubMed] [Google Scholar]
- 13.Richardson CA, Leitch B. Cerebellar Golgi, Purkinje, and basket cells have reduced gamma-aminobutyric acid immunoreactivity in stargazer mutant mice. J Comp Neurol. 2002;453:85–99. doi: 10.1002/cne.10406. [DOI] [PubMed] [Google Scholar]
- 14.Olson JM, et al. NeuroD2 is necessary for development and survival of central nervous system neurons. Dev Biol. 2001;234:174–187. doi: 10.1006/dbio.2001.0245. [DOI] [PubMed] [Google Scholar]
- 15.Jones KR, Farinas I, Backus C, Reichardt LF. Targeted disruption of the BDNF gene perturbs brain and sensory neuron development but not motor neuron development. Cell. 1994;76:989–999. doi: 10.1016/0092-8674(94)90377-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kubota M, et al. Type-3 ryanodine receptor involved in Ca(2+)-induced Ca2+ release and transmitter exocytosis at frog motor nerve terminals. Cell Calcium. 2005;38:557–567. doi: 10.1016/j.ceca.2005.07.008. Epub 2005 Sep 12. [DOI] [PubMed] [Google Scholar]
- 17.Nassenstein C, et al. The neurotrophins nerve growth factor, brain-derived neurotrophic factor, neurotrophin-3, and neurotrophin-4 are survival and activation factors for eosinophils in patients with allergic bronchial asthma. J Exp Med. 2003;198:455–467. doi: 10.1084/jem.20010897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Uyanik G, et al. Novel truncating and missense mutations of the KCC3 gene associated with Andermann syndrome. Neurology. 2006;66:1044–1048. doi: 10.1212/01.wnl.0000204181.31175.8b. [DOI] [PubMed] [Google Scholar]
- 19.Meyer J, et al. Rare variants of the gene encoding the potassium chloride co-transporter 3 are associated with bipolar disorder. Int J Neuropsychopharmacol. 2005;8:495–504. doi: 10.1017/S1461145705005821. Epub 2005 Aug 5. [DOI] [PubMed] [Google Scholar]
- 20.Howard HC, et al. The K-Cl cotransporter KCC3 is mutant in a severe peripheral neuropathy associated with agenesis of the corpus callosum. Nat Genet. 2002;32:384–392. doi: 10.1038/ng1002. Epub 2002 Oct 7, Erratum in: Nat Genet 32 (2002) 681. [DOI] [PubMed] [Google Scholar]
- 21.Hebert SC, Mount DB, Gamba G. Molecular physiology of cation-coupled Cl- cotransport: the SLC12 family Pflugers Arch. 2004;447:580–593. doi: 10.1007/s00424-003-1066-3. Epub 2003 May 9. [DOI] [PubMed] [Google Scholar]
- 22.Dupre N, et al. Hereditary motor and sensory neuropathy with agenesis of the corpus callosum. Ann Neurol. 2003;54:9–18. doi: 10.1002/ana.77777. [DOI] [PubMed] [Google Scholar]
- 23.Boettger T, et al. Loss of K-Cl co-transporter KCC3 causes deafness, neurodegeneration and reduced seizure threshold. EMBO J. 2003;22:5422–5434. doi: 10.1093/emboj/cdg519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mercado A, et al. NH2-terminal heterogeneity in the KCC3 K+-Cl− cotransporter. Am J Physiol Renal Physiol. 2005;289:F1246–F1261. doi: 10.1152/ajprenal.00464.2004. Epub 2005 Jul 26. [DOI] [PubMed] [Google Scholar]
- 25.Shen MR, et al. The KCl cotransporter isoform KCC3 can play an important role in cell growth regulation. Proc Natl Acad Sci U S A. 2001;98:14714–14719. doi: 10.1073/pnas.251388798. Epub 2001 Nov 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Klein T, Cooper TG, Yeung CH. The role of potassium chloride cotransporters in murine and human sperm volume regulation. Biol Reprod. 2006;75:853–858. doi: 10.1095/biolreprod.106.054064. Epub 2006 Aug 30. [DOI] [PubMed] [Google Scholar]
- 27.Pearson MM, Lu J, Mount DB, Delpire E. Localization of the K(+)-Cl(−) cotransporter, KCC3, in the central and peripheral nervous systems: expression in the choroid plexus, large neurons and white matter tracts. Neuroscience. 2001;103:481–491. doi: 10.1016/s0306-4522(00)00567-4. [DOI] [PubMed] [Google Scholar]
- 28.Byun N, Delpire E. Axonal and periaxonal swelling precede peripheral neurodegeneration in KCC3 knockout mice. Neurobiol Dis. 2007;28:39–51. doi: 10.1016/j.nbd.2007.06.014. Epub 2007 Jun 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Losson R, Lacroute F. Interference of nonsense mutations with eukaryotic messenger RNA stability. Proc Natl Acad Sci U S A. 1979;76:5134–5137. doi: 10.1073/pnas.76.10.5134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Frischmeyer PA, Dietz HC. Nonsense-mediated mRNA decay in health and disease. Hum Mol Genet. 1999;8:1893–1900. doi: 10.1093/hmg/8.10.1893. Review. [DOI] [PubMed] [Google Scholar]
- 31.Holbrook JA, Neu-Yilik G, Hentze MW, Kulozik AE. Nonsense-mediated decay approaches the clinic. Nat Genet. 2004;36:801–808. doi: 10.1038/ng1403. [DOI] [PubMed] [Google Scholar]