

Abstract
The reaction of Fe2(S2C2H4)(CO)6 with cis-Ph2PCH=CHPPh2 (dppv) yields Fe2(S2C2H4)(CO)4(dppv), 1(CO)4, wherein the dppv ligand is chelated to a single iron center. NMR analysis indicates that in 1(CO)4, the dppv ligand spans axial and basal coordination sites. In addition to the axial–basal isomer, the 1,3-propanedithiolate and azadithiolate derivatives exist as dibasal isomers. Density functional theory (DFT) calculations indicate that the axial–basal isomer is destabilized by nonbonding interactions between the dppv and the central NH or CH2 of the larger dithiolates. The Fe(CO)3 subunit in 1(CO)4 undergoes substitution with PMe3 and cyanide to afford 1(CO)3(PMe3) and (Et4N)[1(CN)(CO)3], respectively. Kinetic studies show that 1(CO)4 reacts faster with donor ligands than does its parent Fe2(S2C2H4)(CO)6. The rate of reaction of 1(CO)4 with PMe3 was first order in each reactant, k = 3.1 × 10− 4 M−1 s−1. The activation parameters for this substitution reaction, ΔH‡ = 5.8(5) kcal/mol and ΔS‡ = −48(2) cal/deg·mol, indicate an associative pathway. DFT calculations suggest that, relative to Fe2(S2C2H4)(CO)6, the enhanced electrophilicity of 1(CO)4 arises from the stabilization of a “rotated” transition state, which is favored by the unsymmetrically disposed donor ligands. Oxidation of MeCN solutions of 1(CO)3(PMe3) with Cp2FePF6 yielded [Fe2(S2C2H4)(μ-CO)(CO)2(dppv)(PMe3)(NCMe)](PF6)2. Reaction of this compound with PMe3 yielded [Fe2(S2C2H4)(μ-CO)(CO)(dppv)(PMe3)2(NCMe)](PF6)2.
Introduction
The design of first-row metal centers capable of catalyzing redox interconversions of hydrogen is a major goal of contemporary science.1 The structural analyses of the active sites of the two major families of hydrogenases has provided the information required for generating structural models. Replicating the reactivity of the enzymes has, however, proven far more challenging. More progress has been achieved in modeling the [Fe–Fe]-hydrogenases than modeling of the nickel–iron enzymes,2,3 and it is increasingly clear to us that further advances will follow from greater control of the ligands and the redox poise of the diiron center.
The active site of the [Fe–Fe]-hydrogenases feature a diiron tricarbonyl center that can be described as [Fe2(SR)2-(CO)3L3Hn]z (n = 0, 1; z = unknown) where two L’s are CN− and the third L is a μ-thiolate of a [4Fe–4S] cluster (Figure 1).3,4 Relevant to developing active site mimics are, therefore, trisubstituted derivatives of diiron(I) dithiolato complexes of the type Fe2(SR)2(CO)3L3.
Figure 1.
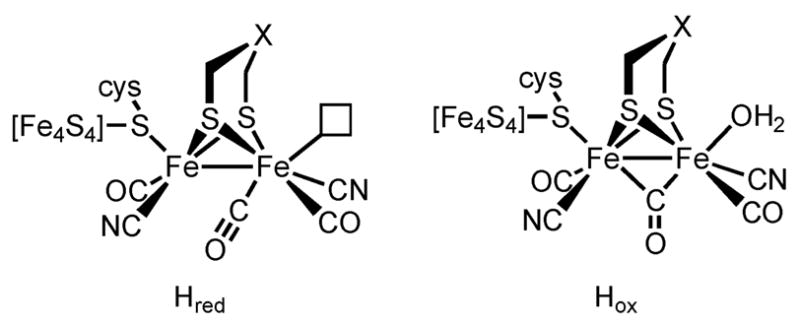
Active site of the [Fe–Fe]−hydrogenase in the reduced and oxidized forms.5
A few trisubstituted and even tetrasubstituted derivatives are known for ligands of small cone angles and modest donor ability, such as MeNC and P(OMe)3.6,7 We have, however, shown that cyanide will not substitute more than two CO ligands, despite its modest steric demands. This finding shows that both ligand size and basicity affect the degree of substitution.8 Cyanide also has a tendency to form linear M–CN–M′ ensembles,9 and metal cyanides are susceptible to N protonation. Phosphine complexes, on the other hand, exhibit more versatile solubility without the complicating tendencies of cyanide.10 Tertiary phosphine ligands are well-known to form mono- and disubstituted products, Fe2(SR)2-(CO)6–x(PR3)x (x = 1, 2),11 but strategies leading to trisubstituted [Fe(I)]2 species are rare.
One approach to controlling the degree of substitution entails the use of chelating ligands, which could stabilize complexes of the type Fe2(SR)2(CO)3(chel)L. Species such as Fe2(SR)2(CO)4(diphosphine) and [Fe2(SR)2(CO)5]2(diphosphine) have been previously investigated.12,13 Complexes where the diphosphine chelates to a single iron center have been only lightly studied.13,14
From the following studies of chelated diiron dithiolates, we have been able to extract detailed information on several points relevant to the behavior of the active site. Of particular interest was the influence of the dithiolate on the reactivity of the diiron center with respect to substitution and oxidation.
Results
Fe2(SR)2(CO)4(dppv)
Fe2(S2C2H4)(CO)4(dppv), 1(CO)4, was prepared in 80% yield by decarbonylation of Fe2(S2C2H4)(CO)6 in the presence of cis-1,2-bis(diphenylphosphino)ethylene (dppv). The dppv ligand was chosen because of its preference for chelating to a single iron center rather than bridging between the two iron centers.13 When the reaction was monitored by IR spectroscopy, we observed no intermediates. Similarly, we prepared the 1,3-propanedithiolate (pdt) Fe2(S2C3H6)(CO)4(dppv), 2(CO)4, and the azadithiolate (adt) Fe2[(SCH2)2NH](CO)4(dppv), 3(CO)4. All three compounds display similar IR spectroscopic features.
The IR spectrum of 1(CO)4 features three νCO bands, wherein the difference between the highest and lowest energy peaks, ΔνCO, is 101 cm−;1. DFT calculations (vide infra) suggest that the highest energy band (exptl, 2023 cm−1; theory, 2022 cm−1) can be approximated as the A mode of the Fe(CO)3 subunit and that the lowest energy band (exptl, 1915 cm−1; theory, 1930 cm−1) is associated with the Fe-(dppv)(CO) subunit. For comparison, ΔνCO is smaller (72cm−;1) for the symmetrcially substituted species Fe2(S2C3H6)(CO)4(PMePh2)2, whereas ΔνCO is 119 cm−;1for the monophosphine Fe2(S2C3H6)(CO)5(PMe2Ph).11 The large value of ΔνCO for 1(CO)4 is consistent with a structure wherein dppv is chelated to a single iron center, and the high ΔνCO value indicates that the second iron center should be susceptible to substitution.15
The geometry adopted by the dppv ligand (axial–basal versus dibasal) is influenced by the nature of the chelating dithiolate. For 1(CO)4, the 31P NMR spectrum displays a single resonance at δ 97.4 at 20 °C. Upon cooling of the sample to −60 °C, this peak splits into doublets at δ 97.2 and 99.5 (JP–P = 16 Hz), indicative of an axial–basal isomer.10 This species was further characterized crystallographically (Figure 2).
Figure 2.
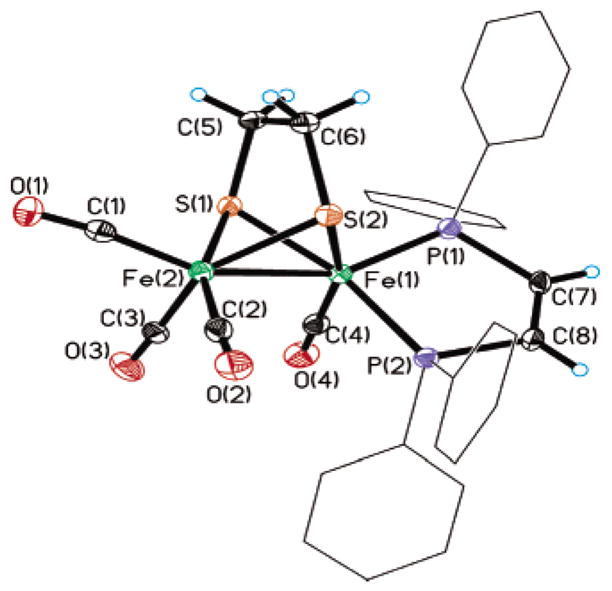
Structure of 1(CO)4, with thermal ellipsoids set at 35%. Phenyl ellipsoids and phenyl hydrogen atoms have been omitted. Selected bond lengths (Å) and angles (deg): Fe(1)–Fe(2), 2.5249(9); Fe(1)– S(1), 2.2451-(13); Fe(1)–S(2), 2.2575(13); Fe(1)–P(1), 2.1743(13); Fe(1)–P(2), 2.2070-(13); Fe(1)–C(4), 1.754(5); Fe(2)–C(1), 1.783(5); Fe(2)–C(2), 1.779(5); Fe(2)–C(3), 1.780(5); Fe(2)–Fe(1)–P(1), 154.05(5); Fe(2)–Fe(1)–P(2), 109.23(4); Fe(2)–Fe(1)–C(4), 105.11(15); P(1)–Fe(1)–P(2), 87.83(5); P(1)–Fe(1)–C(4), 94.32(15); P(2)–Fe(1)–C(4), 88.88(15).
For the pdt and adt complexes, the 31P NMR spectrum again consists of a singlet at 20 °C, but upon cooling of the sample to −60 °C, new signals appeared, indicating the presence of four isomers. On the basis of coupling and shift patterns, we propose that two of these isomers are axial–basal and the other two isomers are dibasal. Singlets at δ 85.7 and 81.6 are attributed to a pair of dibasal isomers that differ with respect to the position of the nitrogen atom of the (SCH2)2NH ligand (Scheme 1 and Figure 3). A pair of partially resolved AB quartets (δ 100.3 and 95.0) is assigned to axial–basal isomers that again differ with respect to the orientation of the amine in (SCH2)2NH. A related pattern is observed for 2(CO)4, except that the dibasal isomers are more favored. At −80 °C, the dibasal isomer represents 18% abundance for the S2C3H6 derivative but only 8% abundance for the adt derivative. Thus, the central CH2 group of the pdt more strongly affects the orientation of the dppv ligand than does the NH of the adt. In general, we find that the 31P NMR shifts for the dppv ligand in [FeI]2 species correlate with stereochemistry: dppv ligands bound in the axial–basal fashion absorb at >δ90, whereas dppv ligands bound in the dibasal geometry absorb at <δ90.
Scheme 1.
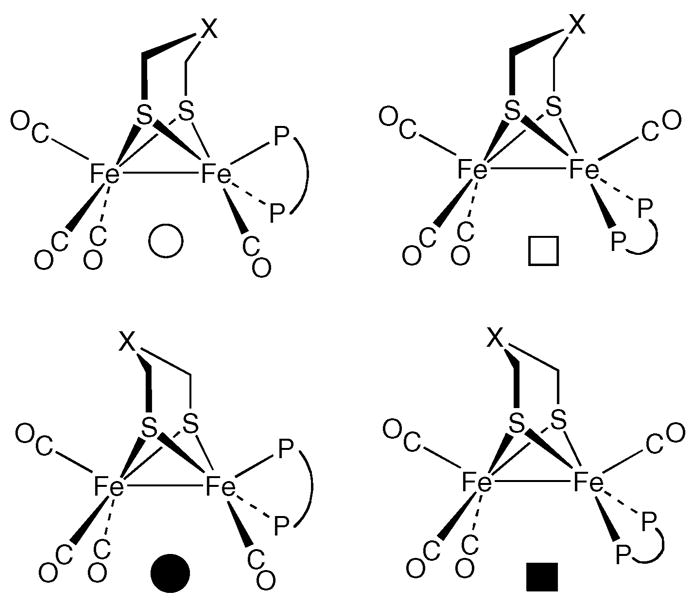
Figure 3.
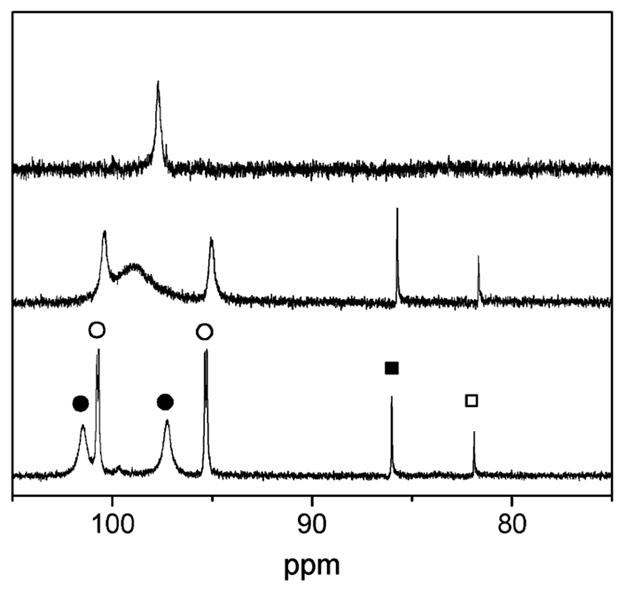
31P NMR spectra of Fe2[(SCH2)2NH](CO)4(dppv) in a CD2Cl2 solution at temperatures of +20 (top), −40, and −60 °C (bottom).
DFT Calculations on Fe2(SR)2(CO)4(dppv)
Using DFT calculations, we probed the stereochemistry of the dppv ligand for the three dithiolato complexes. For the 1,2-ethanedithiolate (edt) derivative 1(CO)4, computed structural parameters agree with experimental data. In particular, the discrepancy among computed and crystallographically determined bond distances is <0.05 Å. Similarly, only small deviations are observed for bond angles (not shown). Calculations are in good agreement with the experimental observation that the most stable isomer is axial–basal. In the axial–basal isomer, the dppv ligand does not clash with the edt or the carbonyl ligands on the second iron center. In contrast, the dibasal isomer would induce unfavorable steric crowding between the phenyl rings and the basal carbonyl ligands on the second iron center (Figure 4). The calculated values for νCO (2022.3, 1969.9, 1951.3, and 1930.3 cm−;1) compare favorably with the observed spectrum (2023, 1953 (br), and 1915 cm−1), where the middle band consists of two overlapping vibrational frequencies.
Figure 4.
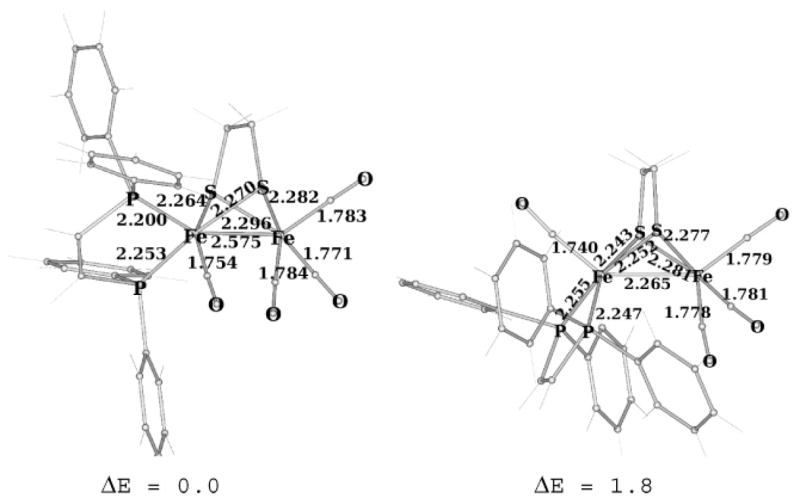
Axial–basal (left) and dibasal (right) isomers of Fe2(edt)(CO)4(dppv) as obtained by DFT calculations (distances in angstroms and computed energy differences in kilocalories per mole relative to the more stable isomer).
Relative to the edt derivative, adt destabilizes the axial–basal isomers (Figure 5). In one axial–basal isomer, the NH group is oriented toward the Fe(dppv)(CO) center and experiences steric crowding with one phenyl group. An attractive interaction between the polar N–Hδ+ and the π. system of the phenyl ring partially counterbalances steric problems. In the other axial–basal isomer, the NH group of adt points toward the axial CO on the Fe(CO)3 center. Even in this case, however, the CH2 groups of adt clash with the phenyl group of dppv. In all four conformers of Fe2[(SCH2)2-NH](CO)4(dppv), the N–H is axial because of the anomeric effect, in agreement with previous DFT studies.16 For the pdt derivative, 2(CO)4, the trimethylene group destabilizes the axial–basal isomer relative to the edt derivative because of steric clashes with one phenyl ring. In the most stable axial-basal isomer, the central CH2 of the pdt strap points away from the phenyl group (see the Supporting Information), whereas the axial–basal isomer in which the central CH2 points to the phenyl group (not shown) is less stable by 0.75 kcal/mol.
Figure 5.
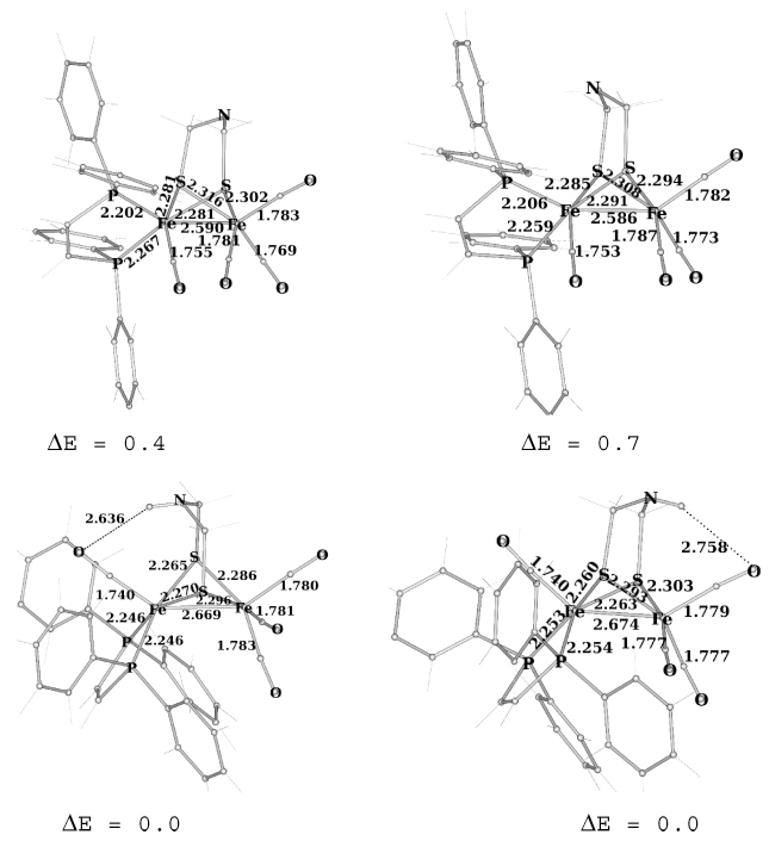
Axial–basal (top) and dibasal (bottom) isomers of Fe2(adt)(CO)4(dppv), as obtained by DFT calculations (distances in angstroms and computed energy differences in kilocalories per mole relative to the most stable isomer).
Substituted Derivatives of 1(CO)4
We found that 1(CO)4 reacted efficiently with PMe3 to give Fe2(S2C2H4)(CO)3(dppv)(PMe3), 1(CO)3(PMe3). Further substitution was not observed even with a 5-fold excess of PMe3. The compound 1(CO)4 also reacted efficiently with Et4NCN to give Et4N-[1(CN)(CO)3] (eq 1). The structures of 1(CO)3(PMe3) and Et4N[1(CN)(CO)3] were confirmed crystallographically (Figure 6).
Figure 6.
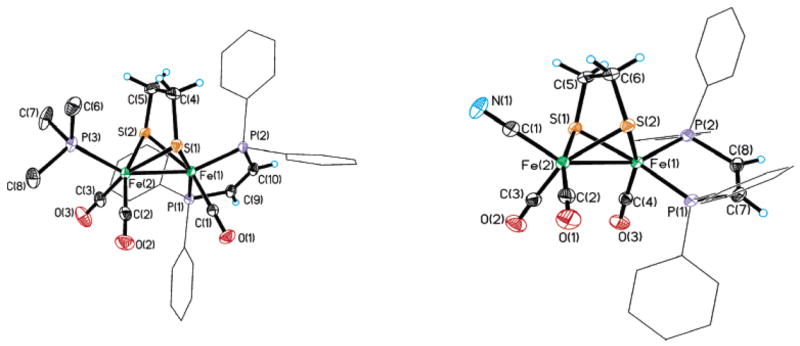
Left: structure of 1(CO)3(PMe3) with thermal ellipsoids set at 35%. Phenyl ellipsoids and phenyl hydrogen atoms were omitted for clarity. Selected bond lengths (Å) and angles (deg): Fe(1)–Fe(2), 2.5344(7); Fe(1)–S(1), 2.2633(11); Fe(1)–S(2), 2.2690(10); Fe(1)–P(1), 2.2006(11); Fe(1)–P(2), 2.1918(10); Fe(1)–C(1), 1.755(4); Fe(2)–P(3), 2.2312(11); Fe(2)–C(2), 1.767(4); Fe(2)–C(3), 1.758(4); Fe(2)–Fe(1)–P(1), 108.38(3); Fe(2)–Fe-(1)–P(2), 157.03(4); Fe(2)–Fe(1)–C(1), 101.08(12); P(2)–Fe(1)–P(1), 88.19(4); P(2)–Fe(1)–C(1), 93.97(12); C(1)–Fe(1)–P(1), 91.82(13). Right: structure of the anion in Et4N[1(CN)(CO)3], with thermal ellipsoids set at 35%. Phenyl ellipsoids, phenyl hydrogen atoms, and the Et4N+ groups are not shown. Selected bond lengths (Å) and angles (deg): Fe(1)–Fe(2), 2.5416(16); Fe(1)–S(1), 2.2453(12); Fe(1)–S(2), 2.2512(12); Fe(1)–P(1), 2.1828(13); Fe(1)–P(2), 2.1763(12); Fe(1)–C(4), 1.733(3); Fe(2)–C(1), 1.916(3); Fe(2)–C(2), 1.766(3); Fe(2)–C(3), 1.751(3); Fe(2)–Fe(1)–P(1), 112.81(5); Fe(2)–Fe(1)–P(2), 154.49(3); Fe(2)–Fe(1)–C(4), 101.07(10); P(2)–Fe(1)–P(1), 87.54(6); P(2)–Fe(1)–C(4), 94.50(10); P(1)–Fe(1)–C(4), 88.25(9).
The 31P NMR spectrum of 1(CO)3(PMe3) consists of singlets at δ 94.7 and 20.6 at room temperature, assigned to dppv and PMe3, respectively. Upon cooling of the sample to −90 °C, these peaks split to give two sets of three signals each, indicative of two isomers in an approximately 2:1 ratio. On the basis of peak positions, the dppv ligand is assigned to the axial–basal orientation in both isomers. Thus, the isomers differ with respect to the axial versus basal positions of the PMe3.
Kinetics of the Formation of Fe2(S2C2H4)(CO)3(dppv)(PMe3)
To our surprise, 1(CO)4 was found to react with PMe3 faster than the parent compound Fe2(S2C2H4)(CO)6. In a competition experiment, a solution containing a 1:1 mixture of Fe2(S2C2H4)(CO)6 and 1(CO)4 was treated with 3 equiv of PMe3. Interestingly, 1(CO)4 was consumed exclusively. The result is remarkable because not only is the hexacarbonyl less encumbered sterically but its effective concentration is twice that of 1(CO)4 because it has two equivalent Fe(CO)3 centers. We verified that the reaction of 1(CO)4 and PMe3 indeed proceeded via the expected bimolecular pathway.17,18 The rate of the 1(CO)4 + PMe3 reaction was examined under pseudo-first-order conditions over the temperature range 0 to 30 °C. Illustrative spectra for this conversion are presented in Figure 7. The temperature dependence of the rate indicated ΔH‡ = 5.8(5) kcal/mol and ΔS‡ = −48(2) cal/deg·mol. These values lead to a ΔG‡ that is smaller than that of previously reported reactions of Fe2(SR)2(CO)6 with tertiary phosphines.17,19 Upon a change of the dithiolate from edt to pdt, the rate of substitution decreased approximately 5-fold.
Figure 7.
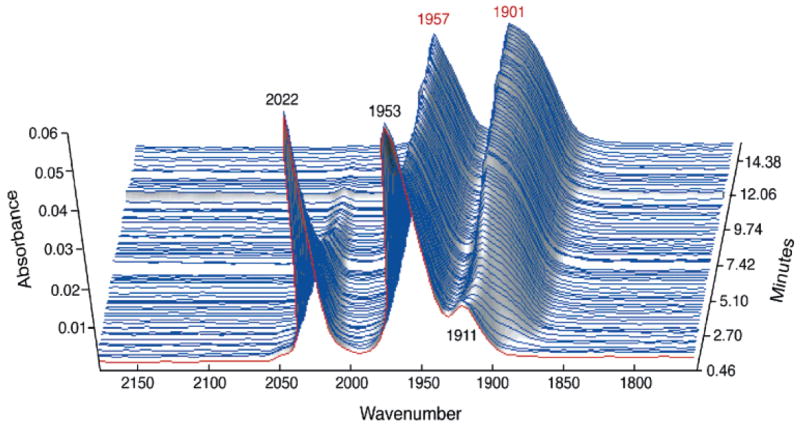
IR spectra for the reaction of Fe2(S2C2H4)(CO)4(dppv) (0.014 M) with 20 equiv of PMe3 in a CH2Cl2 solution at 20 °C.
DFT Analysis of the Substitution Process
As a complement to the experimental results, we employed DFT to examine the reactivity of 1(CO)4 and Fe2(S2C2H4)(CO)6. As a model nucleophile, we chose KCN to allow comparison with data previously obtained for related complexes.20 Energy barriers for the reactions KCN + 1(CO)4 and KCN + Fe2-(S2C2H4)(CO)6 were calculated to be 8.4 and 13.3 kcal/mol, respectively. A similar trend was observed with PMe3 as the nucleophile, with energy barriers = 10.3 and 14.8 kcal/mol for 1(CO)4 and Fe2(S2C2H4)(CO)6, respectively.21
The electron-rich character of the Fe(CO)(dppv) center induces rotation of the Fe(CO)3 group, favoring the formation of a μ-CO structure in the transition state. The (dppv)(OC)-Fe-μ-CO distance in the transition state is calculated as 2.020 Å. In contrast, the (OC)3Fe-μ-CO distance in the Fe2(S2C2H4)(CO)6 transition state is as long as 2.190 Å. The “rotated” structure is more susceptible to attack by nucleophiles than is the ground-state rotamer (Figure 8).20
Figure 8.
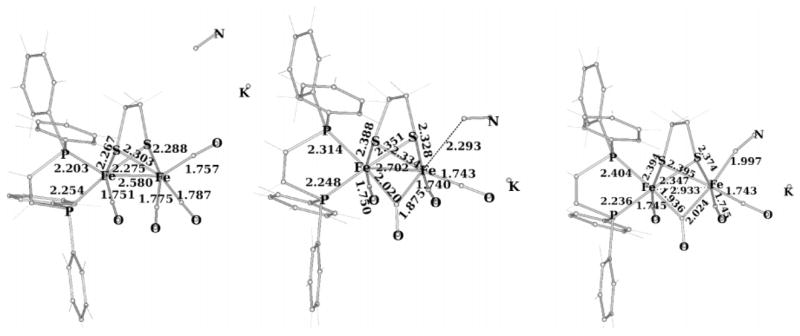
DFT-optimized reactant, transition state, and product for the reaction 1(CO)4 + KCN (distances in angstroms).
Diferrous Derivatives [Fe2(S2C2H4)(μ-CO)(CO)2(dppv)L-(NCMe)]2+
Oxidation of MeCN solutions of 1(CO)3(PMe3) with 2 equiv of Cp2FePF6 afforded the green diferrous derivative [Fe2(S2C2H4)(μ-CO)(CO)2(dppv)(PMe3)(NCMe)]-(PF6)2, [1(μ-CO)(CO)2(PMe3)(NCMe)](PF6)2. The IR spectrum exhibited a band at 1909 cm−1, assigned to νμ–CO. Other diferrous carbonyls exhibit νμ–CO bands of comparable high frequency.22,23 The 31P NMR data (δ 74.6 for the dppv and δ 36.4 for the PMe3) indicate a Cs-symmetric species, which requires that the PMe3 is axial and the dppv is dibasal (eq 2).
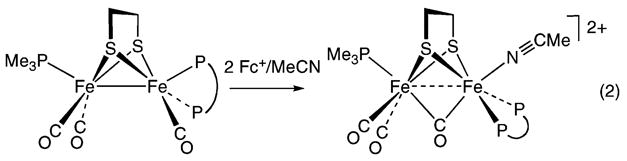
Compound 1(CO)3(PMe3) also smoothly oxidized with 2 equiv of Cp2FePF6 in an acetone solution at −40 °C. The initial product, assumed to be [Fe2(S2C2H4)(μ-CO)(CO)2-(dppv)(PMe3)(OCMe2)] 2+, was identified by FT-IR spectroscopy. The vCO portion of the spectrum (2059, 2015, and 1912 cm−1) is quite similar in pattern and energy to that seen for the MeCN adduct. The stabilities of the MeCN and acetone complexes, however differ sharply: the acetone complex decomposes upon warming of the reaction solution to room temperature, while the MeCN complex is stable both as a solid and in solution at room temperature for extended periods.
Complex [1(μ-CO)(CO)2(PMe3)(NCMe)]2+ is reactive toward further substitution, in contrast to the parent 1(CO)3-(PMe3). The addition of 1 equiv of PMe3 to a solution of [1(μ-CO)(CO)2(PMe3)(NCMe)](PF6)2 caused a color change from green to red. New vCO peaks appeared at 1983 and 1888 cm−1, with the former being the single terminal carbonyl and the latter the bridging carbonyl peak. On the basis of 1H and 31P NMR and IR spectroscopies and electrospray ionization mass spectrometry (ESI-MS), the new compound was identified as [1(μ-CO)(CO)(PMe3)2(NCMe)](PF6)2 (eq 3). The 31P NMR spectrum indicates that the dppv is dibasal and that the complex lacks symmetry. This species is similar to the recently described [Fe2(S2C2H4)(μ-CO)(CO)(PMe3)4-(NCMe)](PF6)2.10 Spectroscopic evidence suggests that oxidation of Et4N[1(CN)(CO)3] with 2 equiv of Cp2FePF6 resulted in the formation of [Fe2(S2C2H4)(μ-CO)(CN)(CO)2-(dppv)(NCMe)]PF6, [1(μ-CO)(CN)(CO)2(NCMe)]PF6. On the basis of its 31P NMR spectrum, which indicates equivalent phosphorus ligands, this species has Cs symmetry. Consistent with this conclusion, DFT calculations on these diferrous species indicate that donor ligands will preferentially occupy sites trans to μ-CO (Figure 9).
Figure 9.
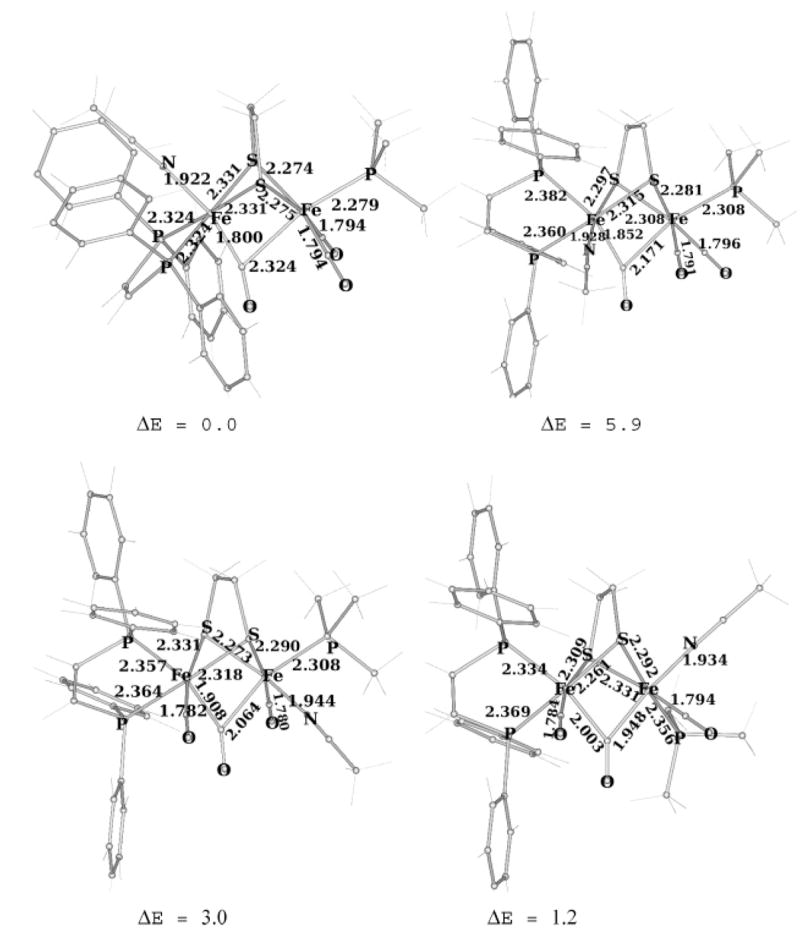
DFT-optimized structures of axial–basal and dibasal isomers of [Fe2(S2C2H4)(μ-CO)(CO)2(dppv)(PMe3)(NCMe)]2+ (distances in angstroms and computed energy differences in kilocalories per mole relative to the most stable isomer). For the sake of brevity, only the four most relevant (and most stable) isomers are shown.
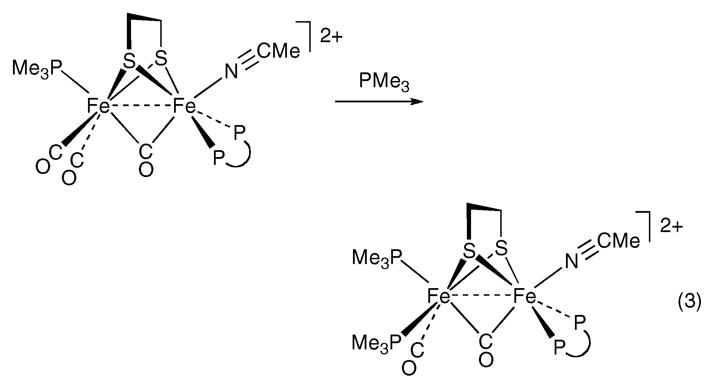
Oxidations of pdt Derivatives
To explore the 2e− oxidation of the pdt derivatives, we prepared Fe2(S2C3H6)-(CO)3(dppv)(PMe3), 2(CO)3(PMe3). The preparation proceeded analogously to that for the synthesis of 1(CO)3(PMe3). Low-temperature 31P NMR measurements show that this species exists as three isomers. Interestingly, the dibasal isomer is far less populated in this species than in its tetracarbonyl precursor, apparently as a result of the PMe3 ligand, probably axially coordinated, pushing the dibasal CO ligands toward the base and inducing nonbonded interactions with the phenyl rings on the dppv.
Treatment of 2(CO)3(PMe3) with 2 equiv of Cp2FePF6 at −40 °C resulted in an immediate change in the νCO region of the IR spectrum to a pattern resembling that for [1(μ- CO)(CO)2(PMe3)(NCMe)]2+, i.e., one bridging (νμ–CO = 1922 cm−;1) and two terminal (νCO–t = 2055 and 2009 cm−1) bands. The shift of νCO–t to higher energies is typical for the oxidation to the diferrous state, and the broad, low-intensity band is typical for νμ–CO. The energies of these bands were, however, quite different for the pdt versus edt derivatives: the μ-CO band occurs at 1921 cm−1 for the pdt derivative, 12 cm−1 higher than that for the edt derivative. This shift is compensated for by opposite shifts in νCO–t, which are each ca. 7 cm−1 lower than those for the edt derivative. Thus, the data indicate that the terminal CO ligands are relatively better π acceptors for the pdt derivative, whereas the μ-CO ligand is relatively more of a π acceptor in the edt derivative. The effect of the dithiolate on the stability of the oxidized derivatives is profound: whereas [1(μ-CO)(CO)2(PMe3)(NCMe)]2+ is robust at room temperature, [2(μ-CO)(CO)2(PMe3)(NCMe)]2+ decomposed within ca. 30 min at −40 °C. The addition of PMe3 to a solution of [2(μ-CO)(CO)2(PMe3)(NCMe)]2+ at −40 °C did not yield an isolable derivative. Oxidation of the adt derivative Fe2[(SCH2)2NH](CO)3(PMe3)(dppv) did not yield stable or observable diferrous derivatives.
Discussion
Reactivity of Fe2(SR)2(CO)4(dppv)
Ligands substitute 1(CO)4 exclusively at the Fe(CO)3 subunit under mild conditions. This reactivity enabled the preparation of otherwise rare trisubstituted diiron(I) dithiolato complexes. Kinetic studies indicate that 1(CO)4 is more electrophilic than the parent hexacarbonyl. According to the prevailing mechanistic paradigm,20 substitution at Fe2(SR)2(CO)6–nLn proceeds via attack of the nucleophile on a rotamer that features an exposed coordination site on one Fe(CO)3 subunit, trans to the Fe–Fe bond (Scheme 2). The binding of a ligand L to this “rotated isomer” ruptures the Fe–Fe bond, forming the 36e− adduct Fe2(SR)2(μ-CO)(CO)3L3. Such 36e− adducts readily decarbonylate concomitant with reformation of the Fe–Fe bond and subsequent rotation of the Fe(CO)2L to its ground-state orientation.
Scheme 2.
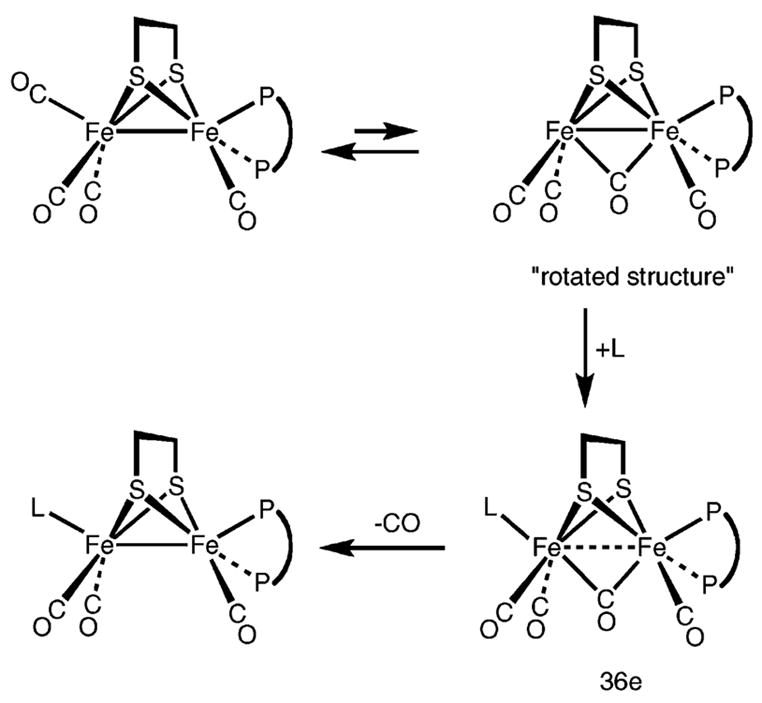
Pickett et al. have spectroscopically detected an adduct of the type Fe2(SR)2(μ-CO)(CO)6–xLx in the cyanation of Fe2[(SCH2)2C(Me)CH2SMe](CO)6.19 In their case, a pendent SMe ligand stabilizes the 36e−intermediate. Recent DFT calculations suggest that the “rotated” geometry is stabilized by the presence of donor ligands in unsymmetrically substituted complexes.24,25 Darensbourg and Hall calculate that unsymmetrical substitution of the diiron center stabilizes the rotated structure, thereby accelerating substitution.25 Our results support the role of the rotated structure. Our calculations agree with these previous predictions: the dppv ligand in 1(CO)4 stabilizes the rotated transition state by about 5 kcal/mol. For the reaction Fe2(S2C2H4)(CO)6 + PBu3, ΔH‡ = 13.6 kcal/mol and ΔS‡ = −31 cal/deg·mol, whereas the presently discussed 1(CO)4 + PMe3 reaction proceeds with ΔH‡ = 5.8(5) kcal/mol and ΔS‡ = −48(2) cal/deg·mol. The enthalpic advantage for the dppv complexes is attributed to the stabilization of the rotated structure, which precedes attack by the nucleophile. We do not presently understand the characteristics of the ligands that will lead to such rate accelerations.
Effect of the Dithiolate (edt vs pdt vs adt) on Subferrous Species
A general question arising from studies on diiron dithiolato carbonyls concerns the influence of the dithiolate on the reactivity and structure of the underlying Fe2(CO)6–xLx subunit.6,18,22 An understanding of how the dithiolate influences the reactivity of the underlying diiron centers is relevant to the design of new dimetallic reaction centers based on dithiolato bridges.
The rate of substitution was found to depend upon the dithiolate ligand. Differences in rates must reflect differences in the steric accessibility of the axial position because the electronic properties of the Fe2(SR)2(CO)4(dppv) cores are virtually identical, as indicated by νCO values. The reaction of 1(CO)4 with PMe3 was ca. 5 times faster than the corresponding reaction for the pdt derivative. This difference is attributed to increased steric protection provided by the pdt ligand, which positions its central CH2 group over the axial sites on the diiron framework.
The relative steric bulk of the dithiolates is indicated by the ratio of the dibasal versus axial–basal isomers for Fe2-(xdt)(CO)4(dppv) (xdt = adt, edt, pdt). At comparable temperatures, the ratio of axial–basal/dibasal was >20:1 (edt), 12:1 (adt), and 7:1 (pdt). Thus, pdt and adt are sterically more imposing than edt. The steric bulk of pdt versus adt does not appear to influence the barrier for the interconversion of the dibasal and axial–basal isomers. The turnstile rotation of related compounds has been well studied.18,26
Impact of Dithiolate on Diferrous Species
Previous work had shown that oxidative decarbonylation of Fe2(SR)2-(CO)6–xLx (L = PR3, RNC) gives diferrous derivatives.22,23 In general, larger ligands (PMe3) are not tolerated for pdt derivatives; thus, nature’s use of small ligands (CO and CN−) on the “distal” iron (location of the variable coordination site) is understandable. Consistent with this view, the nature of the dithiolate strap was found to strongly affect the stability of the derivatives in the series [Fe2(xdt)(μ-CO)(CO)2-(dppv)(PMe3)(NCMe)]2+ [xdt = edt, adt, pdt]. The edt species was robust at room temperature, the pdt derivative was observable at low temperature, and the adt derivative could not be generated. Because adt is less bulky than pdt, the instability of this oxidized compound indicates that a combination of steric and electronic effects are relevant.
We had previously shown that Fe2(S2C2H4)(CO)4(PMe3)2 could be oxidized to give derivatives [FeII2(S2C2H4)(μ-CO)-(CO)2(PMe3)3(NCMe)]2+ and [FeII2(S2C2H4)(μ-CO)-(PMe3)4(NCMe)]2+. These reactions require the addition of trapping donor ligands such as PMe3, which displace one or two terminal CO ligands, stabilizing the higher oxidation state. Being already trisubstituted, Fe2(SR)2(CO)3(dppv)L underwent efficient oxidation without the need for exogenous donor ligands (aside from the MeCN provided by the solvent). Oxidation proceeded in acetone, but the adduct was thermally unstable. In the diferrous state of the enzyme, the so-called Hoxair, an axial site is proposed to be occupied by water, which we propose is simulated by MeCN in our case. In this and related work, we have not observed coordination of water.
Conclusions
Studies on diiron dithiolato complexes with unsymmetrical coordination environments have led to a better understanding of the influence of the dithiolate on the stereochemistry of these complexes. The unsymmetrical coordination environment enhances the overall reactivity of the subferrous complexes, allowing a third ligand to be easily installed. Trisubstituted complexes undergo oxidation to the diferrous derivatives under highly controlled conditions, providing the foundation for future detailed studies on the redox properties of diiron dithiolates.
Experimental Section
Unless otherwise indicated, reactions were conducted using Schlenk techniques at room temperature. Chemicals were purchased from Aldrich, and solvents were either HPLC-grade from an alumina filtration system or distilled under nitrogen over an appropriate drying agent. NMR spectra were recorded at room temperature on a Varian Mercury 500 MHz spectrometer. NMR chemical shifts are quoted in ppm; spectra are referenced to tetramethylsilane for 1H and 13C{1H} NMR and 85% H3PO4 for 31P{1H} NMR spectra. FTIR spectra were recorded on a Mattson Infinity Gold FTIR spectrometer. Fe2(S2C2H4)(CO)6, Fe2(S2C3H6)-(CO)6, and Fe2[(SCH2)2NH](CO)627 were prepared according to literature procedures. PMe3 was distilled prior to use on a high-vacuum line. Et4NCN and dppv were obtained from Aldrich. ONMe3 was obtained from Aldrich and dried by repeated vacuum sublimation.
Fe2(S2C2H4)(CO)4(dppv), 1(CO)4
A solution of 2.70 g (7.24 mmol) of Fe2(S2C2H4)(CO)6 in 35 mL of toluene was treated with a solution of 0.54 g (7.24 mmol) of Me3NO in 15 mL of MeCN. After stirring for 10 min, the reaction mixture was treated with a solution of 2.87 g (7.24 mmol) of dppv in 20 mL of toluene. After 5 h, the solvent was removed in vacuum. The residue, a brown solid, was extracted into 10 mL of CH2Cl2, and the product precipitated as a light-brown powder upon the addition of 100 mL of hexanes. The product was rinsed with ~60 mL of hexanes. Crystals were grown via slow evaporation of a MeCN–toluene solution. Yield: 4.57 g (82%). 1H NMR (CD2Cl2): δ 7.7–7.1 (m, 20H, C6H5), 4.3 (s, 2H, PCH), 1.6 (m, 2H, SCH2), 1.1 (m, 2H, SCH2). 31P NMR (CD2Cl2): δ 97.4 at 20 °C; 99.5 (d, JP–P = 16 Hz), 97.2 (d, JP–P = 16 Hz) at −60 °C. IR (CH2Cl2): νCO 2023, 1953, 1915 cm−1. FD-MS: m/z 712.02 (1(CO)4). Anal. Calcd for C32H26Fe2O4P2S2 (found): C, 53.94 (53.73); H, 3.68 (3.63).
Fe2(S2C3H6)(CO)4(dppv), 2(CO)4, was prepared similarly. Yield: 57%. 1H NMR (CD2Cl2): δ 7.7 – 7.1 (m, 20H, C6H5), 4.3 (s, 2H, PCH), 1.59 (m, 4H, SCH2), 1.29 (m, 2H, SCH2), 0.97 (m, 2H, CH2CH2CH2). 31P NMR (CD2Cl2): δ 96.9, 82.9 at 20 °C; 95.9 at 90 °C; 103.2, 100.0, 96.0, 95.5, 85.7, 82.3 at −90 °C. IR (CH2-Cl2): νCO 2021, 1950, 1912 cm−1. FD-MS: m/z 726.5 (2(CO)4). Anal. Calcd for C33H28Fe2O4P2S2 (found): C, 54.55 (54.60); H, 3.89 (3.87).
Fe2(S2(CH2)2NH)(CO)4(dppv), 3(CO)4, was prepared similarly. Yield: 26%. 1H NMR (toluene-d8): δ 7.7–7.1 (m, 20H, C6H5), 4.3 (s, 2H, PCH), 3.04 (m, 4H, SCH2N). 31P NMR (CD2Cl2, 20 °C): δ 97.6. 31P NMR (CD2Cl2, −60 °C): δ 101.4, 100.7 (JP–P = 20 Hz), 97.2, 95.3 (JP–P = 20 Hz), 86.0, 82.9. IR (CH2Cl2): νCO 2021, 1953, 1915 cm−1. FD-MS: m/z 727.0 (3(CO)4).
Fe2(S2C2H4)(CO)3(dppv)(PMe3), 1(CO)3(PMe3)
A solution of 0.30 g (0.421 mmol) of 1(CO)4 in 20 mL of toluene was treated with a solution of 0.031 g (0.421 mmol) of Me3NO in 5 mL of MeCN. To this solution was added 0.90 mL (0.450 mmol) of a 0.50 M solution of PMe3 in toluene. After 12 h, the solvent was removed in vacuum. The dark-red residue was extracted into 5 mL of CH2Cl2, and the product was precipitated as a dark-red powder upon the addition of 50 mL of hexanes. Crystals were grown via slow diffusion of hexanes into a CH2Cl2 solution. Yield: 0.28 g (88%). 1H NMR (CD2Cl2): δ 8.0–7.2 (m, 20H, C6H5), 4.3 (s, 2H, PCH), 1.6 (m, 2H, SCH2), 1.2 (d, JP–H = 8 Hz, 9H, PCH3), 0.8 (m, 2H, SCH2). 31P NMR (CD2Cl2, 20 °C): δ 94.7, 20.6. 31P NMR (CD2Cl2, −90 °C): δ 101.1, 96.3, 94.0, 92.7, 28.4, 11.5. IR (CH2-Cl2): νCO 1943, 1892 cm−1. FD-MS: m/z 760.00 ([Fe2(S2C2H4)-(CO)3(dppv)(PMe3)]). Anal. Calcd for C34H35Fe2O3P3S2 (found): C, 53.68 (53.82); H, 4.64 (4.51).
Reaction of Fe2(S2C2H4)(CO)6 and 1(CO)4 with PMe3
A solution of 0.050 g (0.134 mmol) of Fe2(S2C2H4)(CO)6 and 0.096 g (0.134 mmol) of 1(CO)4 in 10 mL of toluene was treated with 0.41 mL (0.402 mmol) of a 0.98 M solution of PMe3 in toluene. After 1 h, the following IR spectrum was obtained: νCO 2075, 2035, 2002, and 1991 cm−1 for Fe2(S2C2H4)(CO)6 and νCO 1960, 1949, 1911, and 1895 cm−1 for 1(CO)3(PMe3).
Et4N[Fe2(S2C2H4)(CN)(CO)3(dppv)], Et4N[1(CN)(CO)3]
A solution of 0.750 g (1.03 mmol) of 1(CO)4 in 15 mL of MeCN was treated with a solution of 0.161 g (1.03 mmol) of Et4NCN in 10 mL of MeCN. The solution was stirred for 3 h and the solvent removed in vacuum. The residue, a dark-red solid, was recrystallized by extraction into 5 mL of CH2Cl2, followed by the addition of 50 mL of hexanes. Crystals were grown via slow diffusion of hexanes into a CH2Cl2 solution. Yield: 0.65 g (75%). 1H NMR (MeCN-d3): δ 8.1–7.2 (m, 20H, C6H5), 5.4 (s, 2H, PCH), 3.5 (q, 16H, NCH2CH3), 1.6 (m, 2H, SCH2), 1.4 (t, 24H, NCH2CH3), 1.2 (m, 2H, SCH2). 31P NMR (MeCN-d3, 20 °C): δ 94.7. IR (MeCN): νCN 2072 cm−1; νCO 1943, 1890 cm−1. ESI-MS: m/z 710.30 ([Fe2(S2C2H4)(CN)(CO)3(dppv)]−). Anal. Calcd for C41H48Fe2N2O3P2S2Cl2 (found): C, 53.20 (52.97); H, 5.06 (5.22); N, 3.06 (3.02).
Kinetics
Pseudo-first-order reaction conditions were employed for all kinetics studies, using 10 equiv or more of PMe3. Reactions were performed in a Schlenk vessel designed to contain the in situ IR probe (React IR). Typically, the reactor, a 50 mL flask, was charged with 0.050 g (0.070 mmol) of 1 and flushed with N2 before the addition of 5.0 mL of CH2Cl2. The IR probe was then inserted into the 0.014 M reaction mixture, and the reaction mixture was placed into a thermostatted bath. Following thermal equilibration, a 0.78 M solution PMe3 in CH2Cl2 was injected rapidly. While being magnetically stirred, the solution was analyzed by IR spectroscopy typically at intervals of 15 s until the reaction was judged complete and then at 30 s intervals for an additional 30 min. The rates of reaction were analyzed using the program Concert (Mettler-Toledo). Pseudo-first-order rate constants were the slopes of plots of ln(1(CO)4) vs time (seconds). Rates were determined for the following concentrations of PMe3: 10, 20, 30, and 40 equiv at 20 °C. The slope of the graph of kobs vs [PMe3] gave the second-order rate constant. Activation parameters were deduced from a graph of ln(kT−1) vs T−1 for T = 0, 10, 20, and 30 °C for reactions employing a 20 times excess of PMe3. The slope was taken as ΔH‡/R and the y intercept as ΔS‡/R + 23.8 [=ln(kb/h)]. We estimate 5% uncertainty in the rate constants. With this assumption, using only the rate constants for the 273 and 303 K reactions, we determined uncertainties in ΔH‡ and ΔS‡.
[Fe2(S2C2H4)(μ-CO)(CO)2(dppv)(PMe3)(NCMe)](PF6)2, [1(CO)3-(PMe3)(NCMe)](PF6)2
A solution of 0.200 g (0.263 mmol) of 1(CO)3(PMe3) in 10 mL of MeCN was treated with a solution 0.174 g (0.526 mmol) of Cp2FePF6 in 5 mL of MeCN. The reaction solution changed from dark red to green immediately. Solvent was removed in vacuum, and the green-colored residue was rinsed with ~30 mL of hexane. The reaction mixture was extracted into 3 mL of MeCN, and the product precipitated as a green powder upon the addition of 35 mL of Et2O. Yield: 0.214 g (75%). 1H NMR (MeCN-d3): δ 8.8–7.5 (m, 20H, C6H5), 4.40 (s, 2H, PCH), 3.40 (m, 2H, SCH2), 3.03 (m, 2H, SCH2) 1.85 (d, JP–H = 12 Hz, 9H, PCH3). 31P NMR (MeCN-d3): δ 74.6 (d, JP–P = 5 Hz), 36.4 (t, JP–P = 5 Hz). IR (MeCN): νCO 2061, 2018, 1909 cm−1. ESI-MS: m/z 946.2 ([Fe2(S2C2H4)( μ-CO)(CO)2(dppv)(PMe3)(NCMe)]PF6+). Anal. Calcd for C36H38Fe2NF12O3P5S2 (found): C, 39.60 (39.31); H, 3.51 (3.45); N, 1.28 (1.24).
When a solution of 0.020 g (0.0263 mmol) of 1(CO)3(PMe3) in 5 mL of acetone was treated with 0.017 g (0.0526 mmol) of Cp2-FePF6 in 5 mL of acetone at −40 °C, the solution color changed from red to green. The IR spectrum indicated [Fe2(S2C2H4)(μ-CO)-(CO)2(dppv)(PMe3)(OCMe2)]2+. IR (OCMe2): νCO 2059, 2015, 1912 cm−1. Upon warming of the solution to 20 °C, the IR spectrum revealed that the complex had decomposed.
[Fe2(S2C2H4)(μ-CO)(CO)(dppv)(PMe3)2(NCMe)](PF6)2, [1(CO)2-(PMe3)2(NCMe)](PF6)2
A solution of 0.280 g (0.368 mmol) of 1(CO)3(PMe3) in 10 mL of MeCN was treated with a solution of 0.243 g (0.736 mmol) of Cp2FePF6 in 5 mL of MeCN. The reaction mixture became green and was immediately treated with 1.2 mL (0.736 mmol) of a 0.59 M solution of PMe3 in MeCN. The solvent was removed in vacuum, and the dark-red residue was rinsed with ~30 mL of hexane. The solid was extracted into 3 mL of MeCN; the product precipitated as a brown powder upon the addition of 35 mL of Et2O. Yield: 0.285 g (68%). 1H NMR (MeCN-d3): δ 8.7–7.3 (m, 20H, C6H5), 4.40 (s, 2H, PCH), 3.40 (m, 2H, SCH2), 2.91 (m, 2H, SCH2), 2.53 (m, 3H, Fe-NCCH3), 1.43 (d, JP–H = 10 Hz, 9H, PCH3), 1.20 (d, JP–H = 11 Hz, 9H, PCH3). 31P NMR (MeCN-d3): δ 74.5, 68.7, 24.3 (t, JP–P = 27 Hz), 17.2 (t, JP–P = 27 Hz). IR (MeCN): νCO 1983, 1888 cm−1. ESI-MS: m/z 994.4 ([Fe2(S2C2H4)(μ-CO)(CO)(dppv)(PMe3)(NCMe)](PF6)+).
Oxidation of Et4N[1(CN)(CO)3] with Cp2FePF6
A solution of 0.030 g (0.0357 mmol) of Et4N[1(CN)(CO)3] in 10 mL of MeCN was treated with a solution of 0.023 g (0.0714 mmol) of Cp2FePF6 in 5 mL of MeCN at −40 °C under a CO atmosphere. The solution was warmed to room temperature and the product spectroscopically identified as [Fe2(S2C2H4)(μ-CO)(CN)(CO)2(dppv)(NCMe)]+. 1H NMR (MeCN-d3): δ 8.8–7.3 (m, 20H, PC6H5), 4.17 (s, 2H, PCH), 3.4 (m, 2H, SCH2), 3.18 (m, 2H, SCH2). 31P NMR (MeCN-d3): δ 75.3. IR (MeCN): νCN 2139, 2121 cm−1. νCO 2068, 2028, 1895 cm−1. ESI-MS: m/z 751 ([Fe2(S2C2H4)(μ-CO)(CN)(CO)2(dppv)(NCMe)]+).
Fe(S2C3H6)(CO)3(dppv)(PMe3), 2(CO)3(PMe3), and Its Oxidation
The preparation followed that described above for 1(CO)3-(PMe3). Yield: 0.29 g (90%). 1H NMR (CDCl2): δ 8.0–7.3 (m, 20H, PC6H5), 4.2 (s, 2H, PCH), 1.8 (m, 2H, SCH2), 1.5 (m, 2H, SCH2), 1.36 (d, JP–H = 8 Hz, 9H, PCH3), 1.2 (m, 2H, CH2CH2-CH2). 31P NMR (CD2Cl2, 20 °C): δ 92.9, 26.9. 31P NMR (CD2-Cl2, −90 °C): δ 101.6, 96.4, 91.7, 90.2, 84.8, 34.2, 28.3, 26.7. IR (CH2Cl2): νCO 1943, 1893 cm−1. FD-MS: m/z 774.6 ([Fe(S2C3H6)-(CO)3(dppv)(PMe3)]+).
When a solution of 0.075 g (0.097 mmol) of 2(CO)3(PMe3) in 10 mL of MeCN was treated with 0.064 g (0.194 mmol) of Cp2-FePF6 in 5 mL of MeCN at −40 °C under a CO atmosphere, the solution color changed from red to green. The IR spectrum indicated [Fe2(S2C3H6)(μ-CO)(CO)2(PMe3)(dppv)(NCMe)]2+. IR (MeCN): νCO 2055, 2009, 1922 cm−1. Upon warming of the solution to 20 °C, the IR spectrum revealed that the complex had decomposed.
X-ray Crystallography
Crystals were mounted to a thin glass fiber using Paratone-N oil (Exxon). Data, collected at 198 K on a Siemens CCD diffractometer, were filtered to remove statistical outliers. The integration software (SAINT) was used to test for crystal decay as a bilinear function of X-ray exposure time and sin(Θ). The data were solved using SHELXTL by direct methods (Table 1); atomic positions were deduced from an E map or by an unweighted difference Fourier synthesis. Hydrogen atom U’s were assigned as 1.2Ueq for adjacent carbon atoms. Non-hydrogen atoms were refined anisotropically. Successful convergence of the full-matrix least-squares refinement of F 2 was indicated by the maximum shift/error for the final cycle.
Table 1.
Details of Data Collection and Structure Refinement for Crystallography
| Complex | 1(CO)4 | 1(CO)3(PMe3) | Et4N[1(CN)(CO)3] |
| chemical formula | C32H26O4P2S2Fe2 | C34H35O3P3S2Fe2 | C40H46Fe2N2O3P2S2 |
| T (K) | 193 (2) | 193 (2) | 193 (2) |
| cryst size (mm3) | 0.46 × 0.08 × 0.06 | 0.16 × 0.12 × 0.01 | 0.48 × 0.36 × 0.14 |
| cryst syst | triclinic | triclinic | Triclinic |
| space group | P1 ¯ | P1 ¯ | P1 ¯ |
| a (Å) | 9.5119(15) | 10.4617(10) | 11.219(7) |
| b (Å) | 12.1329(19) | 12.3125(10) | 11.505(7) |
| c (Å) | 28.710(5) | 15.2256(13) | 18.994(12) |
| α (deg) | 79.242(3) | 76.276(5) | 88.297(10) |
| β (deg) | 89.738(3) | 88.122(5) | 74.230(10) |
| γ (deg) | 83.877(3) | 89.143(4) | 66.601(9) |
| V (Å3) | 3236.1(9) | 1904.1(3) | 2157(2) |
| Z | 1 | 2 | 2 |
| Dcalcd (Mg m−3) | 1.509 | 1.474 | 1.425 |
| μ (Mo Kα) (mm−1) | 0.71073 | 0.71073 | 0.71073 |
| max/min transmn | 0.9386/0.4782 | 0.9767/0.8247 | 0.8773/0.5901 |
| reflns measd/indep | 34970/12272 | 58398/9444 | 18473/7884 |
| data/restraints/parameters | 12272/0/770 | 9444/0/427 | 7884/66/519 |
| GOF on F2 | 0.916 | 1.048 | 1.026 |
| Rint | 0.0931 | 0.0513 | 0.0335 |
| R1 [I > 2σ] (all data)a | 0.0513 (0.1268) | 0.0518 (0.0824) | 0.0374 (0.0603) |
| wR2 [I > 2σ] (all data)b | 0.0901 (0.1070) | 0.1357 (0.1532) | 0.0862 (0.0944) |
| max peak/hole (e−/Å3) | 0.565/−0.411 | 0.834/−1.189 | 0.569/−0.335 |
DFT Calculations
DFT calculations used both the BP8628,29 functional and a valence triple-ζbasis set with polarization on all atoms (TZVP).30 Stationary points of the energy hypersurface were located using energy gradient techniques, and full vibrational analysis has been carried out to further characterize each stationary point. Free energy values have been obtained from the electronic self-consistent-field energy considering three contributions to the total partition function (Q), namely, qtranslational, qrotational, qvibrational, under the assumption that Q may be written as the product of such terms. Solvation effects (CH3CN;ε = 37.5) have been evaluated according to the COSMO approach.
Acknowledgments
This work was supported by the National Institutes of Health and the Petroleum Research Fund. We thank Terésa Prussak-Wieckowska for assistance with the crystallographic analyses.
Footnotes
Supporting Information Available: X-ray crystallographic data for 1(CO)4, (Et4N)[1(CN)(CO)3], and 1(CO)3(PMe3), details for DFT calculations, graphs for kinetics, and determination of the activation parameters. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Basic Research Needs for the Hydrogen Economy. Report of the Basic Energy Sciences Workshop on Hydrogen Production, Storage, and Use.
- 2.Tye JW, Hall MB, Darensbourg MY. Proc Natl Acad Sci. 2005;102:16911–16912. doi: 10.1073/pnas.0508740102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Linck RC, Rauchfuss TB. In: Bioorganometallics: Biomolecules, Labeling, Medicine. Jaouen G, editor. Wiley-VCH; Weinheim, Germany: 2005. [Google Scholar]
- 4.Cammack R, Frey M, Robson R. Hydrogen as a Fuel: Learning from Nature. Taylor & Francis; London: 2001. [Google Scholar]
- 5.Frey M. Chem Bio Chem. 2002;3:153–160. [Google Scholar]
- 6.Lawrence JD, Rauchfuss TB, Wilson SR. Inorg Chem. 2002;41:6193–6195. doi: 10.1021/ic025919t. [DOI] [PubMed] [Google Scholar]
- 7.de Beer JA, Haines RJ. J Organomet Chem. 1972;36:297–312. [Google Scholar]
- 8.Lyon EJ, Georgakaki IP, Reibenspies JH, Darensbourg MY. Angew Chem, Int Ed. 1999;38:3178–3180. [PubMed] [Google Scholar]; Schmidt M, Contakes SM, Rauchfuss TB. J Am Chem Soc. 1999;121:9736–9737. [Google Scholar]
- 9.Vahrenkamp H, Geiβ A, Richardson GN. J Chem Soc Dalton Trans. 1997:3643–3651. [Google Scholar]
- 10.van der Vlugt JI, Rauchfuss TB, Wilson SR. Chem –Eur J. 2005;12:90–98. doi: 10.1002/chem.200500752. [DOI] [PubMed] [Google Scholar]
- 11.Li P, Wang M, He C, Li G, Liu X, Chen C, Åkermark B, Sun L. Eur J Inorg Chem. 2005:2506–2513. [Google Scholar]
- 12.Song LC, Yang ZY, Bian HZ, Hu QM. Organometallics. 2004;23:3082–3084. [Google Scholar]; Song LC, Yang ZY, Bian HZ, Liu Y, Wang HT, Liu XF, Hu QM. Organometallics. 2005;24:6126–6135. [Google Scholar]; Hogarth G, O’Brien M, Tocher DA. J Organomet Chem. 2003;672:29–33. [Google Scholar]; de Beer JA, Haines RJ, Greatrex R, Greenwood NN. J Chem Soc. 1971:3271–3282. [Google Scholar]; Hossain GMG, Hyder MI, Kabir SE, Abdul Malik KM, Miah MA, Siddiquee TA. Polyhedron. 2003;22:633–640. [Google Scholar]
- 13.de Beer JA, Haines RJ, Greatex R, Greenwood NN. J Organomet Chem. 1971;27:C33–C35. [Google Scholar]
- 14.Song LC, Yan CG, Mao XA. Gaodeng Xuexiao Huaxue Xuebao. 1997;18:1093–1097. [Google Scholar]
- 15.Tye JW, Lee J, Wang HW, Mejia-Rodriguez R, Reibenspies JH, Hall MB, Darensbourg MY. Inorg Chem. 2005;44:5550–5552. doi: 10.1021/ic050402d. [DOI] [PubMed] [Google Scholar]; Ekström J, Ott S, Sun L, Åkermark B, Eriksson L. Acta Crystallogr, Sect E. 2005;E61:m852–m853. [Google Scholar]
- 16.Lawrence JD, Li H, Rauchfuss TB, Bénard M, Rohmer M-M. Angew Chem Int Ed. 2001;40:1768–1771. doi: 10.1002/1521-3773(20010504)40:9<1768::aid-anie17680>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 17.Ellgen PC, Gerlach JN. Inorg Chem. 1973;12:2526–2532. [Google Scholar]
- 18.Lyon EJ, Georgakaki IP, Reibenspies JH, Darensbourg MY. J Am Chem Soc. 2001;123:3268–3278. doi: 10.1021/ja003147z. [DOI] [PubMed] [Google Scholar]
- 19.Zampella G, Bruschi M, Fantucci P, Razavet M, Pickett CJ, De Gioia L. Chem– Eur J. 2005;11:509–520. doi: 10.1002/chem.200400442. [DOI] [PubMed] [Google Scholar]
- 20.Georgakaki IP, Thomson LM, Lyon EJ, Hall MB, Darensbourg MY. Coord Chem Rev. 2003;238–239:255–266. [Google Scholar]
- 21.Corrections to energy barriers led, for the reaction between 1(CO)4 and PMe3, to a computed ΔG‡ at 298 K of 24.4 kcal/mol, which is in reasonable agreement with experimental data, considering that the BP86 functional is known to underestimate reaction barriers.
- 22.Boyke CA, Rauchfuss TB, Wilson SR, Rohmer MM, Bénard M. J Am Chem Soc. 2004;126:15151–15160. doi: 10.1021/ja049050k. [DOI] [PubMed] [Google Scholar]
- 23.Boyke CA, van der Vlugt JI, Rauchfuss TB, Wilson SR, Zampella G, De Gioia L. J Am Chem Soc. 2005;127:11010–11018. doi: 10.1021/ja051584d. [DOI] [PubMed] [Google Scholar]
- 24.Bruschi M, Fantucci P, De Gioia L. Inorg Chem. 2004;43:3733–3741. doi: 10.1021/ic035326y. [DOI] [PubMed] [Google Scholar]
- 25.Tye JW, Darensbourg MY, Hall MB. Inorg Chem. 2006;45:1552–1559. doi: 10.1021/ic051231f. [DOI] [PubMed] [Google Scholar]
- 26.Adams RD, Cotton FA, Cullen WR, Hunter DL, Mihichuk L. Inorg Chem. 1975;14:1395–1399. [Google Scholar]; Flood TC, DiSanti FJ, Campbell KD. Inorg Chem. 1978;17:1643–1646. [Google Scholar]
- 27.Li H, Rauchfuss TB. J Am Chem Soc. 2002;124:726–727. doi: 10.1021/ja016964n. [DOI] [PubMed] [Google Scholar]
- 28.Becke AD. Phys Rev A: Gen Phys. 1988;38:3098–3100. doi: 10.1103/physreva.38.3098. [DOI] [PubMed] [Google Scholar]; Perdew JP. Phys Rev. 1986;B33:8822–8824. doi: 10.1103/physrevb.33.8822. [DOI] [PubMed] [Google Scholar]
- 29.Schaefer A, Huber C, Ahlrichs R. J Chem Phys. 1994;100:5829–5835. [Google Scholar]
- 30.Klamt A. J Phys Chem. 1996;100:3349. [Google Scholar]; Klamt A. J Phys Chem. 1995;99:2224. [Google Scholar]