

Abstract
An enantioselective synthesis of the Hsp90 inhibitor geldanamycin was achieved in 20 linear steps and 2.0% overall yield from 2-methoxyhydroquinone. The synthesis is highlighted by a regio- and stereoselective hydroboration reaction; a Sc(OTf)3/Et3SiH-mediated pyran ring-opening reaction; an enantioselective crotylation to simultaneously install the C8–C9 (E)-trisubstituted olefin, the C10 and C11 stereocenters; a chelation-controlled asymmetric metallated acetylide addition; and an intramolecular copper(I)-mediated aryl amidation reaction to close the 19-membered macrolactam.
Heat shock protein 90 (Hsp90) is a molecular chaperone that regulates folding, transport, and degradation of client proteins. It also plays a key role in the conformational maturation of oncogenic signaling proteins.1 Hsp90 ATPase binding region’s role in cancer and protein maintenance as well as its broad range of functions make it a significant therapeutic target for anticancer drug development.1d,e
Geldanamycin, a natural product isolated from Streptomyces hygroscopicus var. geldanus var. nova in 1970,2 is the first reported Hsp90 inhibitor. It is currently under development as a therapeutic agent for cancers associated with abnormally elevated levels of receptor tyrosine kinase activity.3a Studies have shown that the Hsp90 client proteins can be destabilized when geldanamycin binds to the ATP-binding site of Hsp90 and inhibits the chaperone activity of the protein.3 Accordingly, several geldanamycin analogues are in various stages of development as novel antitumor agents and as chemotherapeutic agents in a number of diseases.4
Geldanamycin, together with herbimycin A,5 macbecin I,6 and reblastatin,7 are members of the ansamycin class of natural products (Figure 1). Geldanamycin differs at C6, C11, C15, and C17 from macbecin I and herbimycin A. Presently, one total synthesis of geldanamycin has been described.8 Herein, we report the total synthesis of geldanamycin as part of our ongoing studies of ansamycins.
Figure 1.
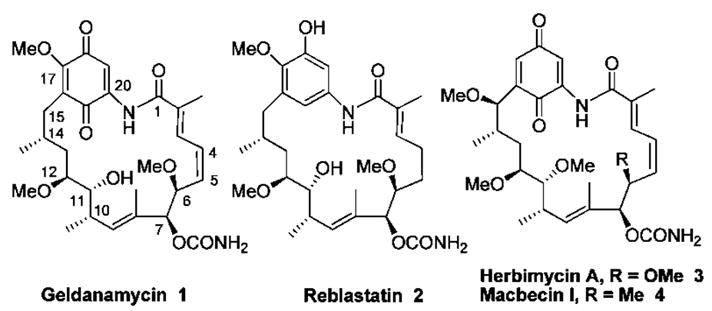
Geldanamycin and related ansamycin antibiotics.
Our strategy for the synthesis of geldanamycin (1) is shown in Scheme 1. We envisioned the 19-membered macrocycle would be formed from the acylic amide 5 through an intramolecular aryl amidation reaction similar to that described in our synthesis of reblastatin.7,9 The (E,Z)-diene could be installed by reduction of the enyne from alkynylation of precursors 6 and 7 which could be generated from easily accessible aldehyde 9 and chiral silane reagent 8. Organosilane 8 has unique features as it establishes the C10–C11 syn stereochemistry while simultaneously creating the C8–C9 (E)-trisubstituted olefin.
Scheme 1.
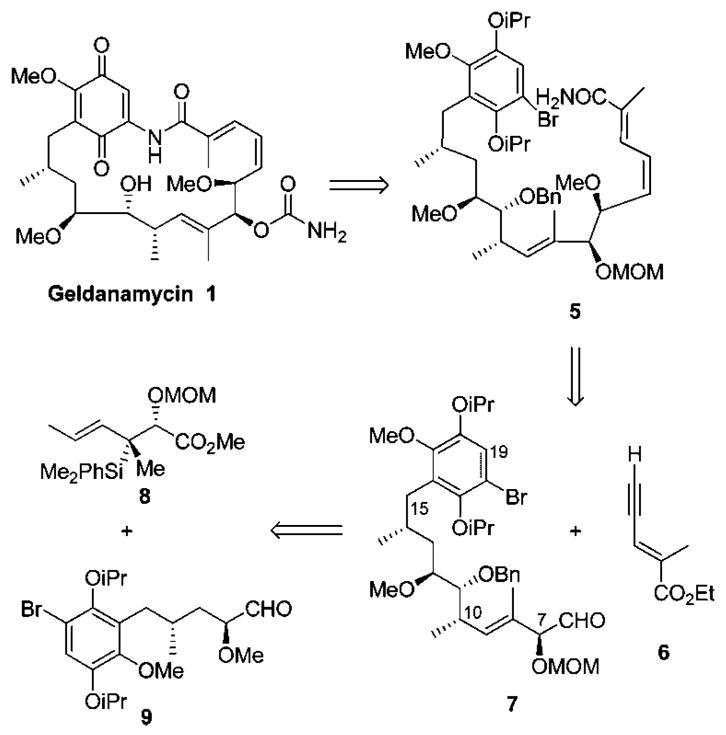
Retrosynthetic Analysis of Geldanamycin
Recently, we have reported the selective hydridic opening of aryl C-glycosides using a Sc(OTf)3/Et3SiH-mediated reduction of the benzylic C-O σ-bond resulting in the formation of a stereochemically well-defined acyclic system.10
The synthesis of the C6–C21 fragment 7 (Scheme 2) started from functionalized aromatic aldehyde 10, which was easily obtained in three steps from commercially available 2-methoxyhydroquinone in 55% overall yield. [4 + 2]-Annulation of this aldehyde with silane 11 provided a mixture of dihydropyrans 12 and 13 in 84% yield (trans/cis: 4:1).11 Regio- and stereoselective hydroboration of the pyran double bond provided the secondary alcohol, which was subsequently methylated with Meerwein’s reagent to provide tetrahydropyran 14 in good yield and excellent selectivity (dr > 20:1). Reductive opening of pyran 14 with Sc(OTf)3/Et3SiH gave a mixture of 15 (which was easily converted to 16 in 95% yield) and 16 in excellent yield, essentially deoxygenating C15 (cf. macbecin). Reduction of ester 16 with LiBH4 afforded the 1,2-diol, which was subjected to oxidative cleavage using sodium periodate to furnish the α-methoxyaldehyde 9.
Scheme 2.
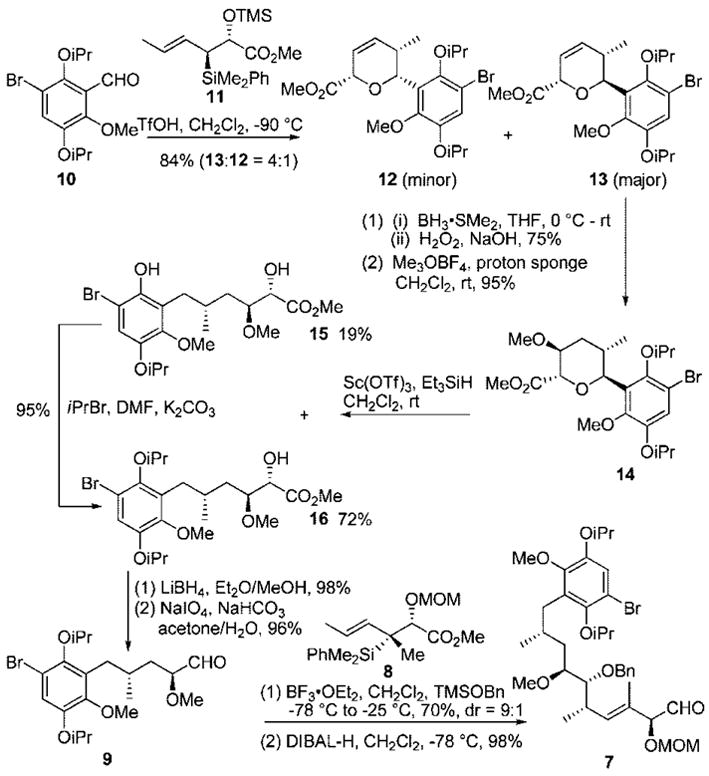
Synthesis of Aromatic C6–C21 Fragment 7
At this point, we turned toward the generation of the C7, C10, and C11 stereocenters and the installation of the trisubstituted alkene. Gratifyingly, after a variety of conditions were explored, a double-stereodifferentiating crotylation12 of silane 8 and aldehyde 9 promoted by BF3·OEt2 provided benzyloxy homoallylic ether bearing not only the three desired stereo-centers and trisubstituted alkene, but also a benzylic ether protecting group at C11. The reaction proceeded in 70% yield with high selectivity (dr = 9:1). Reduction of the product methyl ester with DIBAL-H yielded aldehyde 7.
To access (E,Z)-unsaturated macrocycle 19 through the intermediacy of bromo ester 17, aldehyde 7 was homologated through diastereoselective addition13 of a metallated acetylide (Scheme 3). Accordingly, fragment 17 was obtained through chelation-controlled coupling of acetylene 6 and aldehyde 7 in 76% yield (dr = 10:1).14,15
Scheme 3.
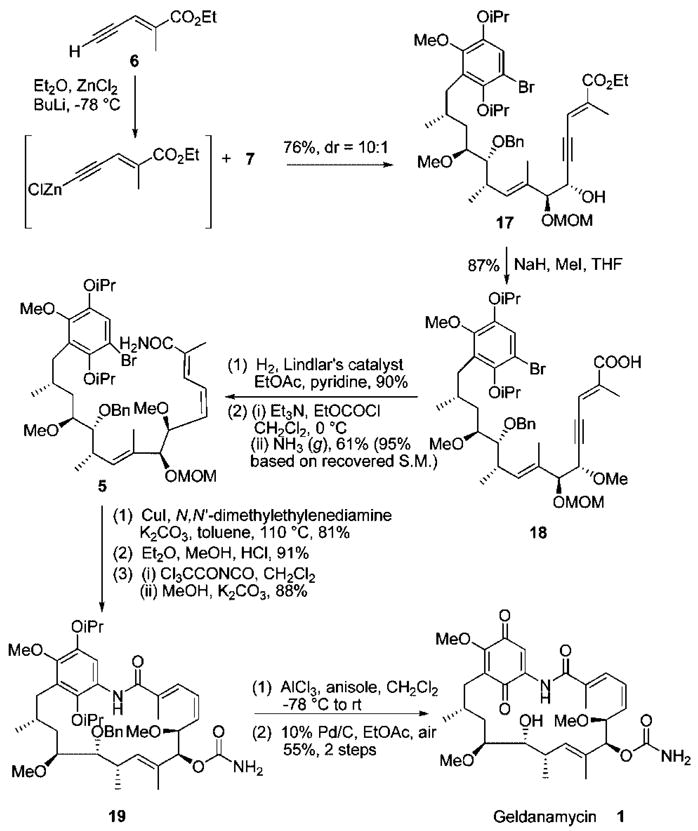
Completion of the Total Synthesis of Geldanamycin
To complete the synthesis, methylation of propargylic alcohol 17 using NaH and MeI occurred with simultaneous dealkylation of the ester to the corresponding acid16 and gave the enyne–acid 18 in 87% yield, which was subjected to Lindlar reduction.17 The resulting acid was subsequently converted to the (E,Z)-unsaturated amide 5. The amide 5 was then subjected to an intramolecular copper(I)-mediated aryl amidation reaction to provide the desired macrocyclic lactam in 81% yield. Deprotection of the MOM ether was achieved utilizing anhydrous HCl/Et2O/MeOH to obtain the secondary alcohol in nearly quantitative yield without affecting other protecting groups. The secondary alcohol was then converted to the desired carbamate 19 in 88% yield.7,18 Finally, deprotection of both the diisopropyl and benzyl ethers was accomplished with AlCl3 in the presence of anisole19 to generate dihydrogeldanamycin, which was immediately treated with catalytic palladium on carbon20 (10%) under air atmosphere to give geldanamycin in 55% yield over two steps. The spectroscopic data obtained for synthetic material were in agreement with those for authentic geldanamycin (optical rotation, 1H and 13C NMR, IR, and HRMS).21
In summary, we achieved the total synthesis of geldanamycin in 20 linear steps and 2.0% overall yield from commercially available 2-methoxyhydroquinone. Notable features of our synthetic route include the following: a concise synthesis of the C11–C21 fragment through reductive pyran-opening approach; an efficient and selective crotylation with silane 8 that simultaneously set two stereocenters (C10, C11), created an (E)-trisubstituted olefin, and put the stereocenter C7 in place; an asymmetric alkynylation to install stereocenter C6; and the first example of synthesis of the E,Z-diene of ansamycins through Lindlar reduction of the enyne precursor.22 An intramolecular copper(I)-mediated amidation reaction was used to close the 19-membered macrolactam.
Acknowledgments
Financial support for this research is obtained from NIH CA56304. J.S.P. is grateful to Amgen, AstraZeneca, Johnson & Johnson, Merck Co., Novartis, Pfizer, and GSK for financial support of our programs. We are grateful to Pfizer for the providing an authentic sample of geldanamycin. We are grateful to Mr. Jason Lowe (Boston University) and Ms. Iwona Wrona (Boston University) for helpful discussions.
Footnotes
Supporting Information Available: Experimental details and selected spectral data for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Rutherford SL, Lindquist S. Nature. 1998;396:336. doi: 10.1038/24550. [DOI] [PubMed] [Google Scholar]; (b) Young JC, Moarefi I, Hartl FU. J Cell Biol. 2001;154:267. doi: 10.1083/jcb.200104079. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Picard D. Cell Mol Life Sci. 2002;59:1640. doi: 10.1007/PL00012491. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Le Brazidec JY, Kamal A, Busch D, Thao L, Zhang L, Timony G, Grecko R, Trent K, Lough R, Salazar T, Khan S, Burrows F, Boehm MF. J Med Chem. 2004;47 doi: 10.1021/jm0306125. [DOI] [PubMed] [Google Scholar]; (e) Cheng H, Cao X, Xian M, Fang L, Cai TB, Ji JJ, Tunac JB, Sun D, Wang PG. J Med Chem. 2005;48:645. doi: 10.1021/jm049693a. [DOI] [PubMed] [Google Scholar]
- 2.(a) De Boer C, Meulman PA, Wnuk RJ, Peterson DH. J Antibiot. 1970;23:442. doi: 10.7164/antibiotics.23.442. [DOI] [PubMed] [Google Scholar]; (b) Rinehart KL, Sasaki K, Slomp G, Grostic MF, Olson EC. J Am Chem Soc. 1970;92:7591. doi: 10.1021/ja00729a018. [DOI] [PubMed] [Google Scholar]
- 3.(a) Whitesell L, Mimnaugh EG, De Costa B, Myers CE, Neckers LM. Proc Natl Acad Sci USA. 1994;91:8324. doi: 10.1073/pnas.91.18.8324. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Schulte TW, An WG, Neckers LM. Biochem Biophys Res Commun. 1997;239:655. doi: 10.1006/bbrc.1997.7527. [DOI] [PubMed] [Google Scholar]; (c) Stebbins CE, Russo AA, Schneider C, Rosen N, Hartl FU, Pavletich NP. Cell. 1997;89:239. doi: 10.1016/s0092-8674(00)80203-2. [DOI] [PubMed] [Google Scholar]
- 4.(a) Schulte TW, Neckers LM. Cancer Chemother Pharmacol. 1998;42:273. doi: 10.1007/s002800050817. [DOI] [PubMed] [Google Scholar]; (b) Adams J, Elliott PJ. Oncogene. 2000;19:6687. doi: 10.1038/sj.onc.1204088. [DOI] [PubMed] [Google Scholar]; (c) Sausville EA, Tomaszewski JE, Ivy P. Curr Cancer Drug Targets. 2003;3:377. doi: 10.2174/1568009033481831. [DOI] [PubMed] [Google Scholar]
- 5.For syntheses of herbimycin, see: Nakata M, Osumi T, Ueno A, Kimura T, Tatsuta K. Tetrahedron Lett. 1991;32:6015.Nakata M, Osumi T, Ueno A, Kimura T, Tatsuta K. Bull Chem Soc Jpn. 1992;65:2974.Carter KD, Panek JS. Org Lett. 2004;6:55. doi: 10.1021/ol036092p.Canova S, Bellosta V, Bigot A, Mailliet P, Mignani S, Cossy J. Org Lett. 2007;9:145. doi: 10.1021/ol062642i.
- 6.For syntheses of macbecin, see: Baker R, Castro J. J Chem Soc, Chem Commun. 1989:378.Baker R, Castro J. J Chem Soc, Perkin Trans 1. 1990:47.Evans DA, Miller SJ, Ennis MD, Ornstein PL. J Org Chem. 1992;57:1067.Martin SF, Dodge JA, Burgess LE, Hartmann M. J Org Chem. 1992;57:1070.Evans DA, Miller SJ, Ennis MD. J Org Chem. 1993;58:471.Panek J, Xu F. J Am Chem Soc. 1995;117:10587.Martin SF, Dodge JA, Burgess LE, Limberakis C, Hartmann M. Tetrahedron. 1996;52:3229.Panek J, Xu F, Rondon AC. J Am Chem Soc. 1998;120:4113.
- 7.For the synthesis of reblastatin, see: Wrona IE, Gabarda AE, Evano G, Panek JS. J Am Chem Soc. 2005;127:15026. doi: 10.1021/ja055384d.
- 8.(a) Andrus MB, Meredith EL, Simmons BL, Sekhar BBVS, Hicken EJ. Org Lett. 2002;4:3549. doi: 10.1021/ol0267432. [DOI] [PubMed] [Google Scholar]; (b) Andrus MB, Meredith EL, Hicken EJ, Simmons BL, Glancey RR. J Org Chem. 2003;68:8162. doi: 10.1021/jo034870l. [DOI] [PubMed] [Google Scholar]
- 9.Klapars A, Huang X, Buchwald SL. J Am Chem Soc. 2002;124:7421. doi: 10.1021/ja0260465. [DOI] [PubMed] [Google Scholar]
- 10.Qin HL, Lowe JT, Panek JS. J Am Chem Soc. 2007;129:38. doi: 10.1021/ja067234o. [DOI] [PubMed] [Google Scholar]
- 11.(a) Lowe JT, Panek JS. Org Lett. 2005;7:1529. doi: 10.1021/ol0501875. [DOI] [PubMed] [Google Scholar]; (b) Huang H, Panek JS. Org Lett. 2003;5:1991. doi: 10.1021/ol034582b. [DOI] [PubMed] [Google Scholar]; (c) Huang H, Panek JS. J Am Chem Soc. 2000;122:9836. [Google Scholar]
- 12.(a) Jain NF, Takenaka N, Panek JS. J Am Chem Soc. 1996;118:12475. [Google Scholar]; (b) Masse CE, Panek JS. Chem Rev. 1995;95:1293. [Google Scholar]
- 13.The use of recently developed methods for the preparation of propargylic alcohols were unsuccessful with the illustrated reaction partners: Frantz DE, Fässler R, Carreira EM. J Am Chem Soc. 2000;122:1806.Anand NK, Carreira EM. J Am Chem Soc. 2001;123:9687. doi: 10.1021/ja016378u.Takita R, Yakura K, Ohshima T, Shibasaki M. J Am Chem Soc. 2005;127:13760. doi: 10.1021/ja053946n.Shahi SP, Koide K. Angew Chem, Int Ed. 2004;43:2525. doi: 10.1002/anie.200353400.Gao G, Wang Q, Yu XQ, Xie MG, Pu L. Angew Chem, Int Ed. 2006;45:122. doi: 10.1002/anie.200500469.
- 14.(a) Mead KT. Tetrahedron Lett. 1987;28:1019. [Google Scholar]; (b) Guillarme S, Haudrechy A. Tetrahedron Lett. 2005;46:3175. [Google Scholar]
- 15.Midland MM, Tramontano A, Cable JR. J Org Chem. 1980;45:28. [Google Scholar]
- 16.(a) Xu H, Sun X. Process for Preparation of N-(fluorenyl-methoxy-carbonyl)-N-methyl-O-tert-butyl-serine. Chinese Pat. CN1837189, 2006. Chem Abstr. 2006;145:397780. [Google Scholar]; (b) Srikrishna A, Kumar PR. Tetrahedron Lett. 2004;45:6867. [Google Scholar]
- 17.(a) Lipshutz BH, Clososki GC, Chrisman W, Chung DW, Ball DB, Howell J. Org Lett. 2005;7:4561. doi: 10.1021/ol051406p. [DOI] [PubMed] [Google Scholar]; (b) Siegel S. In: ComprehensiVe Organic Synthesis. Trost BM, Fleming I, editors. Vol. 8. Pergamon Press; New York: 1991. pp. 417–441. [Google Scholar]
- 18.Kocovsky P. Tetrahedron Lett. 1986;27:5521. [Google Scholar]
- 19.Akiyama T, Hirofuji H, Ozaki S. Tetrahedron Lett. 1991;32:1321. Other conditions such as BCl3, AlCl3 alone, BCl3·SMe2, BBr3, and other Lewis acids or hydrolysis were screened which resulted in either incomplete reaction or decomposition. [Google Scholar]
- 20.(a) Luly JR, Rapoport H. J Org Chem. 1984;49:1671. [Google Scholar]; (b) Andrus MB, Hicken EJ, Meredith EL, Simmons BL, Cannon JF. Org Lett. 2003;5:3859. doi: 10.1021/ol035400g. [DOI] [PubMed] [Google Scholar]
- 21.Authentic geldanamycin was provided by Pfizer, and experiments to obtain spectroscopic data were carried out at Boston University Chemical Instrumentation Center.
- 22.During the review of this work, Belardi and Micalizio reported a related Lindlar reduction to access the E,Z-dienoate of macbecin: Belardi JK, Micalizio GC. Angew Chem Int Ed. 2008;47 doi: 10.1002/anie.200800400. Early View.