

Abstract
A clear understanding is developing concerning the importance of epigenetic-related molecular mechanisms in transcription-dependent long-term memory formation. Chromatin modification, in particular histone acetylation, is associated with transcriptional activation, and acetylation of histone 3 (H3) occurs in Area CA1 of the hippocampus following contextual fear conditioning training. Conversely, DNA methylation is associated with transcriptional repression, but is also dynamically regulated in Area CA1 following training. We recently reported that inhibition of the enzyme responsible for DNA methylation, DNA methyltransferase (DNMT), in the adult rat hippocampus blocks behavioral memory formation. Here we report that DNMT inhibition also blocks the concomitant memory-associated H3 acetylation, without affecting phosphorylation of its upstream regulator, extracellular signal-regulated kinase (ERK). Interestingly, the DNMT inhibitor-induced deficit in memory consolidation, along with deficits in long-term potentiation, can be rescued by pharmacologically increasing levels of histone acetylation prior to DNMT inhibition. These observations suggest that DNMT activity is not only necessary for memory and plasticity, but that DNA methylation may work in concert with histone modifications to regulate plasticity and memory formation in the adult rat hippocampus.
Both memory consolidation and an in vitro analog, long-term potentiation (LTP), require a cascade of signaling events that include activation of NMDA receptors, protein kinases and transcription factors; events which lead to changes in gene transcription. Recent evidence indicates that regulation of chromatin structure also serves as an additional level of control in this cascade. In particular, memory formation has been shown to be associated with histone acetylation (Alarcon et al., 2004; Guan et al., 2002; Korzus et al., 2004; Levenson et al., 2004; Vecsey et al., 2007; Wood et al., 2005). This process of histone acetylation relaxes chromatin structure, making it more accessible to transcriptional machinery (Lunyak et al., 2002; Turner et al., 2002; Varga-Weisz and Becker, 1998). Most recently, we have reported that an epigenetics-related mechanism, DNA (cytosine-5) methylation, is also important for synaptic plasticity and memory formation in the adult nervous system (Levenson et al., 2006; Miller and Sweatt, 2007). Methylation, a covalent chemical modification of DNA, is catalyzed by DNA (cytosine-5) methyltransferases (DNMTs) and, together with other modifications affecting chromatin structure, DNA methylation serves to regulate gene transcription. DNA methylation has been studied extensively in development, and has long been considered a static process following cell differentiation (Santos et al., 2005). However, high DNMT mRNA levels persist into adulthood in the brain and we have found that DNMT inhibition alters gene methylation in vitro and in vivo and prevents the induction of LTP and the consolidation of memory (Levenson et al., 2006; Miller and Sweatt, 2007). These observations suggest that DNA methylation is in fact rapidly and dynamically regulated in the adult nervous system. In the current study, we examined the possibility that DNA methylation influences memory consolidation, in part, by modulating chromatin structure.
To confirm our previous finding that DNMT activity is necessary for memory consolidation (Miller and Sweatt, 2007), we gave animals intra-CA1 infusions of the DNMT inhibitor, 5-aza-2-deoxycytidine (5-AZA) immediately after contextual fear conditioning, a hippocampus-dependent task. Infusions were administered post-training to avoid state-dependent effects of the drug. Infusion needle tips in the brains of animals examined were located well within Area CA1 in all cannulated animals (Supplementary Fig. S1). Confirming our earlier result, when memory was assessed twenty-four hours later, animals infused with 5-AZA displayed significantly less freezing than their vehicle-treated counterparts (F(1, 15) = 10.21, P < 0.01; Fig. 1A), indicating that hippocampal DNMT activity is indeed necessary for memory consolidation (Miller and Sweatt, 2007).
Figure 1.
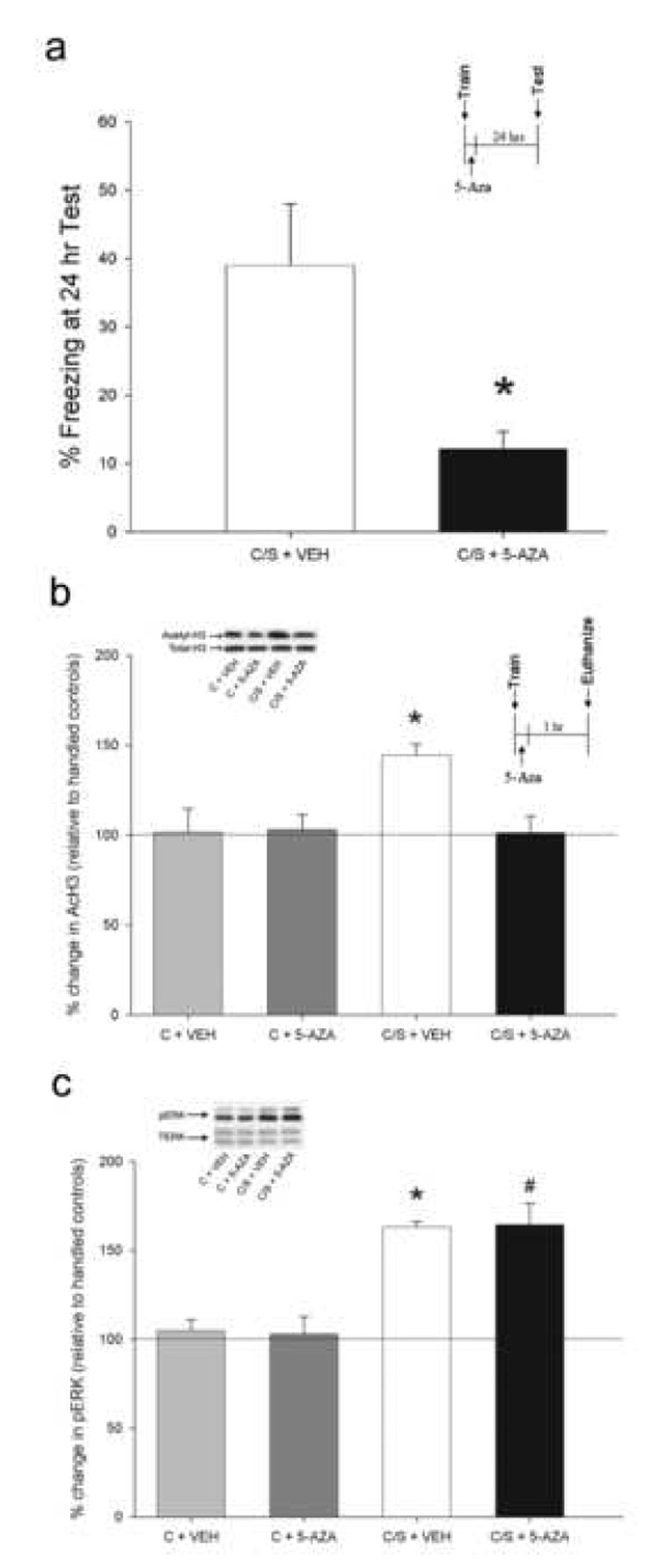
DNMT inhibition blocks memory consolidation and histone acetylation. (a) Intra-CA1 infusion of 5-AZA immediately following contextual fear conditioning blocked consolidation, as evidenced by a lack of freezing at the 24 h test (N = 7, C/S + VEH; N = 9, C/S + 5-AZA). (b) Intra-CA1 infusion of 5-AZA immediately following contextual fear conditioning (C/S + 5-AZA) blocked AcH3 one hr post-training, but had no effect on context only controls (C + 5-AZA; N = 7 per group). (c) Intra-CA1 infusion of 5-AZA had no effect on the ERK phosphorylation induced by contextual fear conditioning. (N = 7 per group.) * P < 0.05. Error bars represent s.e.m.
We and others have also previously reported that acetylation of histone 3 (H3) in Area CA1 is associated with consolidation of contextual fear memory and that memory can be enhanced by pre-treatment with histone deacetylase (HDAC) inhibitors, which pharmacologically raise levels of histone acetylation (Levenson et al., 2004; Wood et al., 2005; Vecsey et al., 2007; Fisher et al., 2007). In addition, changes in DNA methylation can affect levels of histone acetylation (Becker et al., 1987; Collins et al., 2004; Cross et al., 1997; Jones et al., 1998; Nan et al., 1997, 1998) and we have demonstrated in vitro that DNMT inhibition significantly attenuates PKC-induced H3 acetylation (Levenson et al., 2006). Therefore, we hypothesized that the blockade of memory consolidation by 5-AZA could be partially due to modulation of histone acetylation. To test this, we delivered intra-CA1 infusions of 5-AZA or vehicle immediately after contextual fear conditioning (C/S + 5-AZA or VEH) and assessed levels of acetylated H3 (AcH3) one hour later. Controls were infused (C + 5-AZA or VEH) after exposure to the novel context alone. There were no differences between vehicle and 5-AZA treatment for the context only controls (P > 0.05), indicating that 5-AZA alone does not have an effect on basal acetylation of H3. Context plus shock vehicle-treated animals showed the expected increase in AcH3 (P < 0.05 for all comparisons), but context plus shock 5-AZA-infused animals did not (FIG. 1B). This demonstrates that intra-CA1 5-AZA treatment immediately following fear conditioning blocks increased H3 acetylation, which may account in part for the 5-AZA induced memory deficit (Fig. 1A). No differences were observed in H4 acetylation for any of the groups (P > 0.05 for all comparisons), confirming and extending our previous findings (Levenson et al., 2004).
We examined contextual fear conditioning-associated ERK2 phosphorylation as a control for the non-specific effects of 5-AZA. There was no ERK activation in vehicle and 5-AZA treated animals with context only exposure, as expected (P > 0.05). However, context plus shock vehicle- and 5-AZA-treated animals both showed an identical elevation in pERK2 (P < 0.05; Fig. 1C). This observation suggests that 5-AZA infusion does not have non-specific effects on a wide variety of cellular and molecular processes upstream of ERK/MAPK activation in the hippocampus.
In light of the ability of DNMT inhibition to block memory-associated histone acetylation (Fig. 1B), we next investigated the possibility that the deficits in memory consolidation induced by intra-CA1 infusions of 5-AZA could be blocked by pharmacologically maintaining histone acetylation by inhibiting HDACs. To test this, we gave animals intra-CA1 infusions of the HDAC inhibitor sodium butyrate (NaB). Thirty minutes later, we trained animals for contextual fear conditioning and immediately followed this with an infusion of 5-AZA. Pre-training NaB infusions had no effect on freezing behavior observed during training, suggesting that NaB does not affect an animal’s ability to perceive and respond to footshock (P > 0.05; Fig. 2A). In addition, freezing levels at training were equal across all four groups of animals (vehicle = 46.5% ± 3.2, NaB = 51.8% ± 6.8, 5-AZA = 41.0% ± 4.0, NaB + 5-AZA = 42.6% ± 6.9; F(3,21)= 0.71, P > 0.05).
Figure 2.
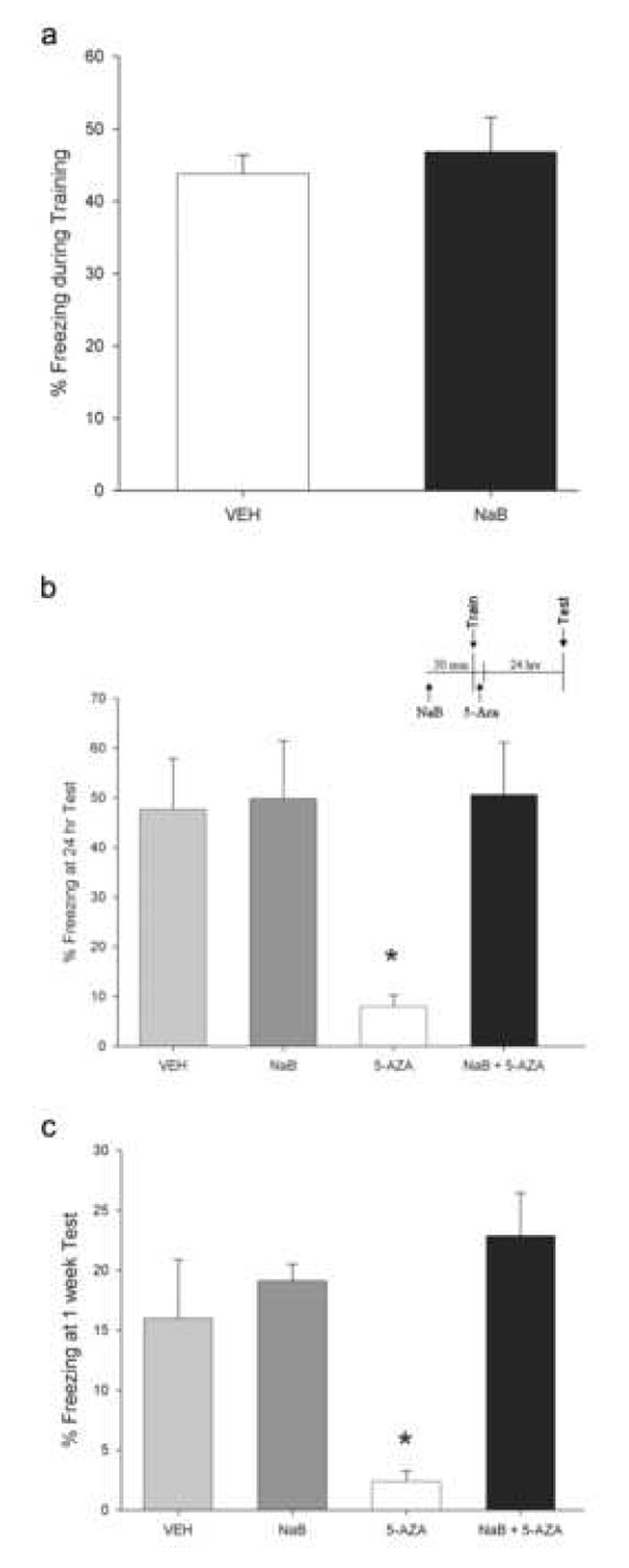
HDAC inhibition prevents the memory deficit induced by DNMT inhibition. (a) Pre-training intra-CA1 infusions of NaB do not affect an animal’s ability to perceive and respond to footshock during training. The “VEH” group refers to animals that received pre-training vehicle infusions, but contains animals that then received either vehicle or 5-AZA post-training, after the percent freezing during training measurements were taken. The “NaB” group refers to animals that received pre-training NaB infusions, but contains animals that received either vehicle or 5-AZA post-training. (b) Intra-CA1 infusion of NaB prior to training blocked the memory consolidation deficit produced by 5-AZA infusion. (N = 7–8 per group.) * P < 0.05. (c) The rescue effect of HDAC inhibition on memory for contextual fear conditioning is long-lasting; present at not only 24 hours (Fig. 2A), but also seven days later. * P < 0.05. Error bars represent s.e.m.
Twenty-four hours after training, we tested animals for their freezing behavior. Post hoc analyses revealed a confirmation of our earlier results (Fig. 1A), again demonstrating a memory consolidation deficit induced by 5-AZA compared to vehicle-treated context plus shock controls (Fig. 2B, P < 0.05). Animals treated with NaB alone showed levels of freezing equivalent to vehicle-treated controls (P > 0.05). This indicates that with this training procedure and dose of NaB, NaB does not affect baseline memory formation. It is worth commenting on the lack of HDAC inhibitor-induced memory enhancement in the current experiment (Fig. 2B), versus what has been previously reported (Levenson et al., 2004; Vecsey et al., 2007; Fisher et al., 2007). The differing result is likely due to two factors: our use of a low concentration and dose of NaB delivered in a site-specific (intra-CA1), rather than systemic fashion, and our use of a 3-shock fear conditioning protocol, rather than the single shock protocol utilized by Levenson et al. (2004), Fischer et al. (2007), and Vecsey et al. (2007). The current experiments were designed to specifically probe for an interaction between DNMTs and histone acetylation at normal levels of learning.
Interestingly, animals that were treated with NaB plus 5-AZA displayed freezing equivalent to the vehicle and NaB-treated animals (P < 0.05 for all comparisons; Fig. 2B). These same effects on memory were present when we tested the animals again 1 week later (P < 0.05; Fig. 2C). This demonstrates that preserved and/or enhanced histone acetylation in Area CA1 during the memory consolidation period immediately after training is sufficient to overcome the memory deficit produced by DNMT inhibition, and is consistent with the hypothesis that regulation of histone acetylation is one mechanism of action of DNA methylation in the adult nervous system.
We next investigated the possibility that, as with memory consolidation, θ-burst stimulus-induced LTP could be rescued by treating slices with an HDAC inhibitor prior to application of the DNMT inhibitor. For this experiment, we used Trichostatin A (TSA), an HDAC inhibitor that can be used at a lower concentration than NaB in vitro in a dilute aqueous solution. We have previously demonstrated that both TSA and NaB enhance LTP and histone acetylation in acute hippocampal slices (Levenson et al., 2004). θ-burst stimulation resulted in the induction of robust LTP (Fig. 3A). Confirming our earlier findings, exposure of slices to 5-AZA resulted in an immediate and significant reduction in LTP (P < 0.01), while application of TSA resulted in an enhancement of LTP (P < 0.01) (Levenson et al., 2004, 2006). Slices that were pre-treated with TSA, followed by 5-AZA 20 minutes later, displayed LTP that was equivalent to slices treated with vehicle (P > 0.05; Fig. 3A). We also examined basal synaptic transmission and found no significant difference between slices treated with 5-Aza, TSA or co-application of 5-Aza and TSA compared to their respective vehicles for the input-output function (P > 0.05; Fig 3B). Together these data demonstrate that the deficit in synaptic plasticity produced by DNMT inhibition can be overcome to a significant extent by enhancing levels of histone acetylation, consistent with what we observed in our behavioral memory studies.
Figure 3.
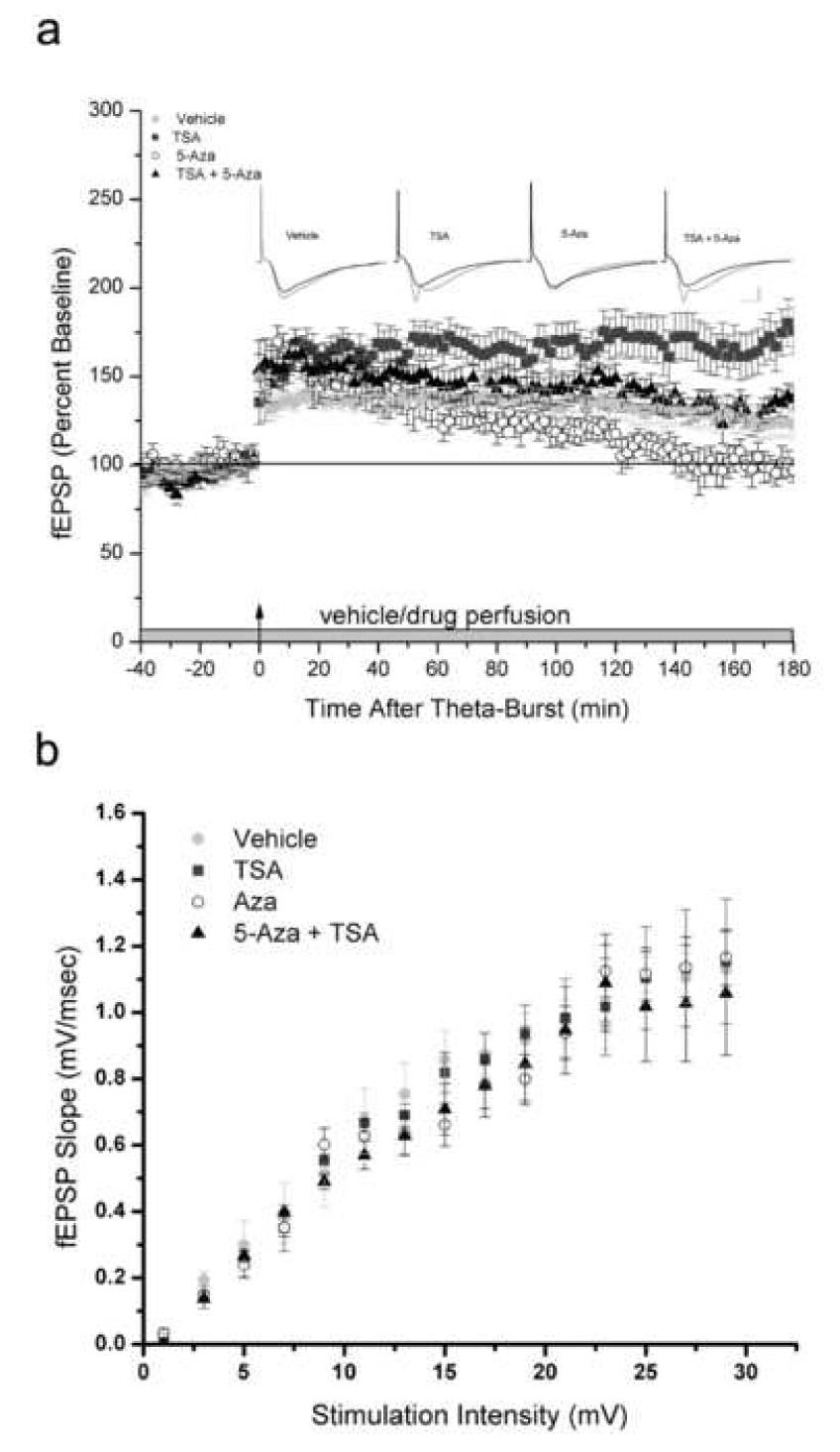
HDAC inhibition prevents the LTP deficit induced by DNMT inhibition. (a) LTP was rescued by application of the HDAC inhibitor, TSA, prior to 5-AZA. (N = 10–17 per group; calibration is 1 mV and 5 ms.) (b) Input-output synaptic relation is normal in slices pretreated with 5-Aza, TSA or co-application of 5-Aza and TSA relative to their respective vehicles. Error bars represent s.e.m.
DNA methylation is associated with transcriptional repression and, indeed, we have recently reported that inhibition of the DNA methyltransferase enzyme not only blocks memory consolidation, but also results in the aberrant transcription of a memory suppressor gene, protein phosphatase 1 (PP1). In the absence of normal DNMT activity, memory-associated PP1 gene methylation no longer occurs, resulting in its aberrant transcription and interference with memory formation. In the present study, we were able to rescue the DNMT inhibitor-induced memory deficit by elevating histone acetylation levels with HDAC inhibition. Interestingly, Vecsey and colleagues (2007) have recently reported that HDAC inhibition does not result in global increases in acetylation. Rather, HDAC inhibition increases the acetylation associated with just a subset of genes. Therefore, it seems likely that NaB overcomes the memory deficit produced by 5-AZA by increasing the transcriptional activity of memory promoter genes, such as Nr4a1, whose transcription is upregulated by HDAC inhibition (Vecsey et al., 2007). These genes may then overcome the memory suppressing effects of genes like PP1 that were made aberrantly active by the DNMT inhibition, ultimately resulting in normal memory formation.
This is the first study to present evidence suggesting that DNA methylation, once thought to be a static process after cellular differentiation, might dynamically regulate memory formation in the adult nervous system through modulation of chromatin structure. We have confirmed that DNA methyltransferase activity is necessary for memory consolidation in the hippocampus and demonstrated that DNMT inhibition blocks memory-associated histone acetylation. In addition, modifying chromatin structure with an HDAC inhibitor is sufficient to rescue the memory and synaptic plasticity deficits induced by DNA methyltransferase interference. The current findings indicate that DNA methylation and histone acetylation play an important and interacting role in memory consolidation and synaptic plasticity. We speculate that future studies of the adult CNS will likely reveal that these two epigenetics-related modifications effect memory-associated processes through a complex set of bidirectional interactions.
MATERIALS AND METHODS
Animals
Adult male Sprague-Dawley rats weighing 250–300g were used for all experiments. Rats were housed under 12:12 light/dark cycles, with food and water available ad libidum. All procedures were performed in accordance with the University of Alabama at Birmingham Institutional Animal Care and Use Committee and with national regulations and policies. All animals used for behavioral experiments were handled for 3–5 days prior to the start of behavioral conditioning.
Behavioral Procedures
For contextual fear conditioning, animals were placed into the training chamber and allowed to explore for 2 min, after which they received an electric shock (1 s, 0.5 mA). The 2 min/1 s shock paradigm was repeated for a total of three shocks. After the final shock, animals remained in the training chamber for an additional 1 minute. Control animals were exposed to the fear conditioning context during the training period, but received no shock. When appropriate, intra-CA1 infusions of sodium butyrate were administered 30 min prior to fear conditioning training and infusions of the DNMT inhibitor, 5-AZA, were performed immediately post-training. For experiments in which CA1 tissue was to be used for biochemistry, a subgroup of animals was allowed to survive and was tested for retention of the fear memory 24 hours later to confirm the memory effects.
Cannula Implantation
For stereotaxic surgery, rats were anesthetized with ketamine and xylazine and secured in a Kopf stereotaxic apparatus. Bilateral stainless steel guide cannulae (26G; Plastics One, Roankoke, VA) were aimed at area CA1 of the hippocampus (AP: −3.6 mm relative to bregma; ML: ± 1.7 mm; DV: −2.6 mm from skull; Paxinos and Watson, 1998). Clearance through the guide cannulae was maintained with 33G obdurators (Plastics One) cut to project 1 mm beyond the tip of the guide. Animals were habituated to dummy cannula removal and given 5 days of recovery and handling before the start of behavioral conditioning. To ensure accurate cannula placement, brains were collected from those animals given both fear conditioning training and a retention test. 40 µm sections were collected through area CA1 and stained with cresyl violet to verify the location of the infusion needle tips. Infusion needle tips were found to be located well within area CA1 in all cannulated animals.
Drugs
5-aza-deoxyctidyne (VWR) was dissolved in 0.8% acetate and diluted to a concentration of 2 ng/µl in sterile saline. Sodium butyrate (Sigma) was dissolved and diluted to a concentration of 2 µg/µl in sterile saline. Both drugs were infused at a rate of 0.25 µl/min for 2 min and the infuser was left in place for an additional minute to allow for diffusion of the drug. Trichostatin A (Sigma) was dissolved in 100% ethanol and diluted to a concentration of 1.65 µM in artificial cerebro-spinal fluid (ACSF).
Isolation of Area CA1
For isolation of area CA1 from whole brain, brains were immersed in oxygenated (95%/5% O2/CO2) ice-cold cutting saline (CS; in mM: 110 sucrose, 60 NaCl, 3 KCl, 1.25 NaH2PO4, 28 NaHCO3, 0.5 CaCl2, 7 MgCl2, 5 glucose, 0.6 ascorbate) immediately after rapid decapitation and removal of the brain. Area CA1 was dissected away from other hippocampal subfields under a dissecting scope and subsequently frozen on dry ice and stored at −80°C overnight.
Histone Extraction
Histone extractions were performed on area CA1 tissue from animals that were euthanized 1 hour after fear conditioning training. All procedures were performed on ice and all solutions were chilled to 4°C prior to use unless otherwise indicated. All centrifugation steps were performed at 4°C. Tissue homogenates were centrifuged at 7,700 × g for 1 min. The supernatant (cytoplasmic fraction) was aspirated and stored at −80°C. The pellet (nuclear fraction) was resuspended in 1 ml of 0.4 N H2SO4. Histones were acid-extracted from the nuclear fraction for 30 min. Acid extracts were centrifuged at 14,000 × g for 10 min. The supernatant was transferred to a fresh tube, and proteins were precipitated with the addition of 100% trichloroacetic acid containing deoxycholic acid (Na+ salt, Sigma) for 30 min. Precipitated proteins were collected by centrifugation at 14,000 × g for 30 min. The supernatant was discarded and the protein pellet was washed with 1 ml of acidified acetone (0.1% HCl) followed by 1 ml acetone for 5 min each. Protein precipitates were collected between washes by centrifugation (14,000 × g, 5 min). The resulting purified proteins were resuspended in 50 mM Tris (pH 8.0) and stored at −80°C.
Western Blotting
Acetylation levels of histone extracts were assayed by Western blotting. Loading buffer was added (final concentration: 6.25 mM Tris, pH 6.8, 2% SDS, 10% glycerol, 1.25% 2-mercaptoethanol, 0.1% bromophenol blue) and protein extracts (1 µg) were separated by SDS-PAGE on a 15% resolving gel with a 4% stacking gel and transferred to polyvinylidene diflouride membranes for immunoblotting. Membranes were briefly rinsed with 100% methanol, air-dried for 15 min, and washed with TBST (in mM: 150 NaCl, 20 Tris, pH 7.5, 0.05% Tween 20) for 5 min at room temperature. The membranes were then blocked in 3% bovine serum albumin (BSA) in TBST for 45 min at room temperature and then incubated in primary antibody (1:1000 for all primary antibodies used) containing TBST and 5% BSA for 1 hour at room temperature. This was followed by three washes in TBST and incubation in horseradish peroxidase-conjugated anti-rabbit secondary antibody (1:10,000) for 45 min at room temperature. The membranes were again rinsed in TBST and immunolabeling of membranes was detected via chemiluminescence (ECL, Pierce). The anti-rabbit primary antibodies used were as follows: anti-histone H3 (Upstate), anti-acetyl histone H3 (Lys-14; Upstate), anti-histone H4 (Upstate), anti-acetyl-H4 (Lys-5/Lys-8/Lys-12/Lys-16; Upstate), anti-p44/42 MAPK (Upstate), anti-phospho-p44/42 MAPK (Cell Signaling).
Hippocampal Slice Preparation
Animals were euthanized by rapid decapitation. Brains were immersed in oxygenated ice-cold CS prior to isolation of the hippocampus. Transverse slices (400 µm) were prepared with a Vibratome (Vibratome 1000; The Vibratome Co., St. Louis, MO). During isolation, hippocampal slices were stored in oxygenated ice-cold CS. After isolation, hippocampal slices were equilibrated in an oxygenated mixture of 50% CS and 50% artificial cerebrospinal fluid (ACSF; in mM: 125 NaCl, 2.5 KCl, 1.25 NaH2PO4, 25 NaHCO3, 2 CaCl2, 1 MgCl2, 25 glucose) at room temperature for 45 min prior to use.
Slice Electrophysiology
Electrophysiology was performed in an interface chamber (Fine Science Tools, Foster City, CA). Oxygenated ACSF (95% O2, 5% CO2, 30°C) was perfused into the recording chamber at a rate of 1 ml/min. Signals were acquired using an A–M Systems amplifier (model 1800; Sequim, WA) controlled by Axon Instruments Clampex 8.0 software via Digidata 1321A and 1200B interfaces (Union City, CA), and stored on a PC. Extracellular stimuli were delivered (model 2200 stimulus isolator; A–M systems) by placing isonel enamel-coated bipolar platinum-tungsten (92%:8%, 0.0011-inch diameter) electrodes on the border of Areas CA3 and CA1 along the Schaffer collaterals. fEPSPs were recorded in stratum radiatum with an ACSF-filled glass recording electrode (1–3 megaohms). All analyses of electrophysiologic traces were performed using Clampfit (Axon Instruments). Experimental stimuli were set to an intensity that evoked a fEPSP that had a left slope of 50% of the maximum fEPSP left slope. LTP was induced by administering three trains of theta-burst stimulation. Each train consisted of 10 sets of bursts (4 stimuli, 100 Hz) with an interburst interval of 200 ms. There were 20 s between each stimulus train. Synaptic efficiency was monitored 30 min prior to and 180 min following induction by recording fEPSPs every 20 s (traces were averaged for every 2 min interval). Drug or vehicle was administered to slices for the duration of the experiment beginning 20 min prior to LTP induction. Slices that did not exhibit stable fEPSP slopes during the first 10 min of recording were excluded from the study. Normalized fEPSP slope values were averaged and plotted as the percent change from baseline.
Statistical Analysis
One-way analysis of variance was used to analyze all data. The Tukey-Kramer post hoc test was used when necessary. Significance was set at P ≤ 0.05 for all tests.
Supplementary Material
Supplementary Figure 1. Location of needle tips for intra-CA1 infusions. Diagram represents histology from animals whose behavioral data are depicted in Figure 1A and 2. Because of the extensive overlap between the infusion needle tips of these animals, not all tip locations are resolvable on this diagram.
ACKNOWLEDGEMENTS
The authors would like to thank an anonymous reviewer for his or her helpful comments on the manuscript. This work was supported by the NIMH, NINDS, NARSAD, NIH Blueprint for Neuroscience Research, American Health Assistance Foundation and the Evelyn F. McKnight Research Foundation. C.A.M. is a Civitan Emerging Scholar.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Alarcon JM, Malleret G, Touzani K, Vronskaya S, Ishii S, Kandel ER, Barco A. Chromatin acetylation, memory, and LTP are impaired in CBP+/− mice: a model for the cognitive deficit in Rubinstein-Taybi syndrome and its amelioration. Neuron. 2004;42:947–959. doi: 10.1016/j.neuron.2004.05.021. [DOI] [PubMed] [Google Scholar]
- Becker PB, Ruppert S, Schutz G. Genomic footprinting reveals cell type-specific DNA binding of ubiquitous factors. Cell. 1987;51:435–443. doi: 10.1016/0092-8674(87)90639-8. [DOI] [PubMed] [Google Scholar]
- Collins AL, Levenson JM, Vilaythong AP, Richman R, Armstrong DL, Noebels JL, Sweatt JD, Zoghbi HY. Mild overexpression of MeCP2 causes a progressive neurological disorder in mice. Human Molecular Genetics. 2004;13:2679–2689. doi: 10.1093/hmg/ddh282. [DOI] [PubMed] [Google Scholar]
- Cross SH, Meehan RR, Nan X, Bird A. A component of the transcriptional repressor MeCP1 shares a motif with DNA methyltransferase and HRX proteins. Nature Genetics. 1997;16:256–259. doi: 10.1038/ng0797-256. [DOI] [PubMed] [Google Scholar]
- Fischer A, Sananabenesi F, Wang X, Dobbin M, Tsai LH. Recovery of learning and memory is associated with chromatin remodelling. Nature. 2007;447:178–182. doi: 10.1038/nature05772. [DOI] [PubMed] [Google Scholar]
- Guan Z, Giustetto M, Lomvardas S, Kim JH, Miniaci MC, Schwartz JH, Thanos D, Kandel ER. Integration of long-term-memory-related synaptic plasticity involves bidirectional regulation of gene expression and chromatin structure. Cell. 2002;111:483–493. doi: 10.1016/s0092-8674(02)01074-7. [DOI] [PubMed] [Google Scholar]
- Jones PL, Veenstra GJ, Wade PA, Vermaak D, Kass SU, Landsberger N, Strouboulis J, Wolffe AP. Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nature Genetics. 1998;19:187–191. doi: 10.1038/561. [DOI] [PubMed] [Google Scholar]
- Korzus E, Rosenfeld MG, Mayford M. CBP histone acetyltransferase activity is a critical component of memory consolidation. Neuron. 2004;42:961–972. doi: 10.1016/j.neuron.2004.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levenson JM, O’Riordan KJ, Brown KD, Trinh MA, Molfese DL, Sweatt JD. Regulation of histone acetylation during memory formation in the hippocampus. Journal of Biological Chemistry. 2004;279:40545–40559. doi: 10.1074/jbc.M402229200. [DOI] [PubMed] [Google Scholar]
- Levenson JM, Roth TL, Lubin FD, Miller CA, Huang I, Desai P, Malone LM, Sweatt JD. Evidence that DNA (cytosine-5) methyltransferase regulates synaptic plasticity in the hippocampus. Journal of Biological Chemistry. 2006;281:15763–15773. doi: 10.1074/jbc.M511767200. [DOI] [PubMed] [Google Scholar]
- Lunyak VV, Burgess R, Prefontaine GG, Nelson C, Sze SH, Chenoweth J, Schwartz P, Pevzner PA, Glass C, Mandel G, Rosenfeld MG. Corepressor-dependent silencing of chromosomal regions encoding neuronal genes. Science. 2002;298:1747–1752. doi: 10.1126/science.1076469. [DOI] [PubMed] [Google Scholar]
- Miller CA, Sweatt JD. Covalent modification of DNA regulates memory formation. Neuron. 2007;53:857–869. doi: 10.1016/j.neuron.2007.02.022. [DOI] [PubMed] [Google Scholar]
- Nan X, Campoy FJ, Bird A. MeCP2 is a transcriptional repressor with abundant binding sites in genomic chromatin. Cell. 1997;88:471–481. doi: 10.1016/s0092-8674(00)81887-5. [DOI] [PubMed] [Google Scholar]
- Nan X, Ng HH, Johnson CA, Laherty CD, Turner BM, Eisenman RN, Bird A. Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature. 1998;393:386–389. doi: 10.1038/30764. [DOI] [PubMed] [Google Scholar]
- Santos KF, Mazzola TN, Carvalho HF. The prima donna of epigenetics: the regulation of gene expression by DNA methylation. Brazilian Journal of Medical and Biological Research. 2005;38:1531–1541. doi: 10.1590/s0100-879x2005001000010. [DOI] [PubMed] [Google Scholar]
- Turner BM. Cellular memory and the histone code. Cell. 2002;111:285–291. doi: 10.1016/s0092-8674(02)01080-2. [DOI] [PubMed] [Google Scholar]
- Varga-Weisz PD, Becker PB. Chromatin-remodeling factors: machines that regulate? Current Opinion in Cell Biology. 1998;10:346–353. doi: 10.1016/s0955-0674(98)80010-0. [DOI] [PubMed] [Google Scholar]
- Vecsey CG, Hawk JD, Lattal KM, Stein JM, Fabian SA, Attner MA, Cabrera SM, McDonough CB, Brindle PK, Abel T, Wood MA. Histone deacetylase inhibitors enhance memory and synaptic plasticity via CREB: CBP-dependent transcriptional activation. Journal of Neurosience. 2007;27:6128–6140. doi: 10.1523/JNEUROSCI.0296-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood MA, Kaplan MP, Park A, Blanchard EJ, Oliveira AM, Lombardi TL, Abel T. Transgenic mice expressing a truncated form of CREB-binding protein (CBP) exhibit deficits in hippocampal synaptic plasticity and memory storage. Learning & Memory. 2005;12:111–119. doi: 10.1101/lm.86605. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Location of needle tips for intra-CA1 infusions. Diagram represents histology from animals whose behavioral data are depicted in Figure 1A and 2. Because of the extensive overlap between the infusion needle tips of these animals, not all tip locations are resolvable on this diagram.