

Abstract
Systemic exposure to lipopolysaccharides (LPS) produces a variety of effects, including movement-evoked hyperalgesia that can be measured using the grip force assay in mice. Because both lethality and enhanced sensitivity to cutaneous pain following exposure to endotoxins have each been attributed to inflammatory mediators, we explored the possibility that LPS-induced movement-evoked hyperalgesia is also sensitive to manipulations of glucocorticoids that regulate these other LPS responses. We found that the hyperalgesic effect of LPS (5 mg/kg s.c.) in mice that were adrenalectomized did not differ from that in control mice that were sham-operated, even though mortality after LPS was potentiated by adrenalectomy. The development of tolerance to the movement-evoked hyperalgesic effect of LPS also did not differ between adrenalectomized and sham-operated control mice. In addition, mifepristone (25 mg/kg s.c.), a glucocorticoid antagonist, did not attenuate the hyperalgesic effect of LPS (2 mg/kg s.c.), yet this dose of mifepristone was sufficient to enhance the incidence of lethality induced by LPS. Enhancement of glucocorticoid activity by two injections of dexamethasone (1 mg/kg s.c.) had no effect on the degree of hyperalgesia in mice injected with LPS (5 mg/kg s.c.), yet this dose of dexamethasone was sufficient to attenuate the incidence of mortality induced by LPS in adrenalectomized mice. Finally, morphine (10 mg/kg i.p.) reversed the decrease in grip force caused by LPS (5 mg/kg i.p.), supporting the interpretation that decreases in grip force produced by LPS reflect muscle hyperalgesia that is not sensitive to glucocorticoids.
Keywords: Mouse, grip force, corticosterone, endotoxemia, myalgia, glucocorticoid
1. Introduction
Lipopolysaccharides (LPS or endotoxins) are glycolipids that constitute the immunoreactive portion of gram-negative bacterial cell walls (Raetz, 1990). In addition to their ability to cause endotoxic shock, LPS in rats produces mechanical and thermal hyperalgesia measured with von Frey fibers (Walker et al., 1996; Cahill et al., 1998) and the tail flick assay (Maier et al., 1993; Watkins et al., 1994a; Wiertelak et al., 1994a, b). Administered centrally (Meller et al., 1994; Walker et al., 1996) or peripherally (Maier et al., 1993; Watkins et al., 1994a, b, c; Wiertelak et al., 1994a, b), a single dose is capable of decreasing mechanical nociceptive thresholds in rats and mice, consistent with the muscle aches and pains reported in humans during endotoxemia. Movement-evoked hyperalgesia induced by endotoxins can be modeled by decreases in grip force responses following parenteral injections of LPS in mice (Kehl et al., 2004). We and others have validated that the grip force assay detects hyperalgesia by the ability of narcotic analgesics to prevent decreases in grip force induced by algesic manipulations in rats (Kehl et al., 2000) and mice (Wacnik et al., 2003), including those induced by LPS in mice (Kehl et al., 2004).
Many acute effects of LPS have been studied extensively and are attributed to activation of genes encoding cytokines, chemokines, immune receptors and other pro-inflammatory compounds. LPS induces the release of interleukin-1 (IL-1), tumor necrosis factor alpha (TNF-α), and eicosanoids from macrophages (Nathan, 1987). TNF-α and IL-1 are responsible for LPS-induced shock leading to the lethal effects of endotoxemia (Beutler et al., 1985; Tracey et al., 1986). These mediators also participate in tactile hyperalgesia following peripheral injections of LPS (Ferreira et al., 1988; Maier et al., 1993; Watkins et al., 1994c; Rietschel et al., 1994, Safieh-Garabedian et al., 1997). Whether muscle pain and decreases in grip force responses that are produced by endotoxins also result from this same group of inflammatory mediators is not known.
Glucocorticoids are powerful immunosuppressors, especially to inflammatory reactions induced by endotoxins. They constitute an endogenous mechanism to avoid exaggerated immune responses. Adrenal hormones are released in response to LPS (Takemura et al., 1997) and adrenalectomized mice are more sensitive to lethal effects of LPS and its associated cytokines IL-1 and TNF-α (Bertini et al., 1988). In contrast, exogenous administration of glucocorticoids protects against lethality (Geller et al., 1954). Because of this inhibitory influence of glucocorticoids on inflammatory mediators, it is plausible that the adrenal might also protect against movement-evoked hyperalgesia associated with endotoxemia if it shares the same mediators.
The purpose of this study was to determine whether adrenal hormones influence LPS-induced muscle hyperalgesia similar to their well-characterized effects on lethality and cutaneous hyperalgesia. To accomplish this, we assessed the effects of mifepristone, a glucocorticoid antagonist, dexamethasone, a synthetic glucocorticoid agonist, and adrenalectomy on decreases in grip force responses induced by LPS in mice. If the hyperalgesic effect of LPS results from an inflammatory response, it is anticipated that glucocorticoids would attenuate and adrenalectomy exacerbate movement-evoked hyperalgesia.
Methods
Each animal was used only once. Animals received one treatment, as described in the figure legends, and repeated grip force measures were obtained to monitor the time-course of the drug effects. Observers were unaware of treatments throughout the experiments reported. All procedures were performed according to the guidelines of the International Association for the Study of Pain, the University of Minnesota Animal Care and Use Committee, and the Committee on Care and Use of Laboratory Animals of the Institute of Laboratory Animal Resources, National Research Council (DHEW Publication NIH 78–23, revised 1995).
2.1. Subjects
Adult female Swiss Webster mice weighing 20–25 g were housed four per cage and allowed to acclimate for at least one week prior to use. Mice were allowed free access to food and water, and housed in a room with a constant temperature of 23°C on a 12-hr light-dark cycle. To explore movement-evoked painful disorders in a gender in which these conditions are common in humans, such as fibromyalgia, we used female mice.
2.2. Drugs and Chemicals
LPS [Escherichia coli Serotype 0111:B4] was purchased from Sigma Chemical Company (St. Louis, MO). LPS was dissolved in LPS-free sterile saline (0.9% NaCl) immediately prior to each use and delivered at the doses indicated in volumes of 1 ml per 100 g body weight. Dexamethasone, a widely used synthetic glucocorticoid antagonist, was obtained from ICN Biochemicals, Inc. (Irvine, CA) and injected at a dose of 1 mg/kg s.c. immediately prior to and 8 hr after LPS. Mifepristone (RU38,486), a glucocorticoid and progesterone antagonist (Philibert and Teutsch, 1990), was purchased from Sigma Chemical Company (St. Louis, MO) and injected at 25 mg/kg s.c., a dose similar to that previously used to inhibit glucocorticoid activity in mice (Berki et al., 2002). Due to its low solubility, mifepristone was dissolved in a solution of 25% saline and 75% ethanol and injected s.c. 15 min after LPS.
2.3. Grip Force Assays
As a reflection of muscle hyperalgesia, forelimb grip force was measured using a grip force analyzer as described by Kehl and colleagues for use with rats (2000) and mice (Wacnik et al., 2003). This apparatus consists of a force transducer that is connected to a wire mesh grid (12 × 7 cm2 O.D. with an ~0.5 cm square wire grid) and positioned on top of an aluminum frame approximately 30 cm above the bench top. During testing, each mouse is held by its tail and gently passed in a horizontal direction over the wire grid until it grasps the grid with its forepaws. The peak force that is exerted by the forelimbs of each mouse when pulling on the grid, is recorded by the force transducer, to which the grid is attached. Three grip force measures are obtained at each time-point; the average of these measurements is used to represent each animal's forelimb grip force at that particular time. Animals were familiarized with the grip force apparatus for three days by grip force testing prior to initiation of the experiment. On the third day, grip force measurements were obtained prior to administration of LPS to establish baseline values for each animal. Then LPS was administered and grip force measured at the times indicated. Grip force data are represented as either raw data or as the reduction in grip force measured relative to pre-injection baseline values for that group.
2.4. Adrenalectomy
Mice were anesthetized using isoflurane and the skin shaved and sterilized using betadine. After a medial incision, the kidney and adrenal were approached by blunt dissection and externalized. The adrenal was removed prior to replacement of the kidney into the peritoneal cavity. The procedure was repeated on the opposite side. Muscle layers were sutured and skin closed using a wound clip after application of bupivacaine to the incision area. Butorphanol (2 mg/kg of Turbogesic s.c.) was administered twice at 8 hr intervals to provide post-surgical analgesia. Sham operated mice were treated identically except the adrenal was not removed prior to the replacement of the externalized kidney and adrenal into the peritoneal cavity.
2.5. Rota-rod assay
To test for the effect of LPS on motor coordination, one group of mice were first tested in the grip force assay and then evaluated for performance on the Rota-rod. Mice were trained for three days previously by undergoing three daily 5-min trials on an accelerating rotating rod (0–20 rpm). On the day of the experiment, each mouse was placed on the rotating rod for a 5-min interval. The time to the first fall was recorded and compared between groups.
2.6. Corticosterone assay
Whole trunk blood was collected in plastic tubes containing EDTA and stored on ice. Blood samples were centrifuged (3100 g) for 10 min at 4°C and the plasma stored at −20°C until assayed (Jasper and Engeland, 1991; Thrivikraman and Plotsky, 1993). Corticosterone was assayed in duplicate samples of mouse plasma by the ImmuChem™ Double Antibody Corticosterone 125I RIA kit from ICN Biochemicals Inc (Costa Mesa CA). Adrenalectomized mice were deemed incompletely adrenalectomized and excluded if their corticosterone value was not less than one standard deviation lower than the mean of the sham-operated control group.
2.7. Statistics
The grip force data were analyzed statistically using paired, two-tailed Student’s t-tests or ANOVA followed by a post hoc analysis using the Fisher Protected Least Squares Difference (PLSD) test to determine differences at individual time-points. A difference was considered significant if the probability that it occurred because of chance alone was less than 5% (P < 0.05).
Results
3.1. Mifepristone and grip force responses to LPS
To assess the influence of endogenous glucocorticoid activity on the hyperalgesic effect of LPS, we pretreated mice with mifepristone at a dose of 25 mg/kg s.c. immediately prior to the s.c. injection of 2 mg/kg of LPS. To maintain the antagonism of the glucocorticoid receptor, a second dose of mifepristone or vehicle was also delivered 24 hr later, immediately after grip force responses were recorded. The dose of LPS used was previously found to decrease grip force responses in male and female mice (Kehl et al., 2004). Injection of mifepristone alone had no effect on grip force responses taken 24 hr later compared to mice injected with vehicle only (Fig. 1A). In contrast, the grip force of mice injected with mifepristone plus LPS was significantly less than mice not injected with LPS and did not differ from mice injected with LPS only.
Fig. 1. The influence of mifepristone on the ability of LPS to decrease forelimb grip force measurements and to induce lethality in mice.
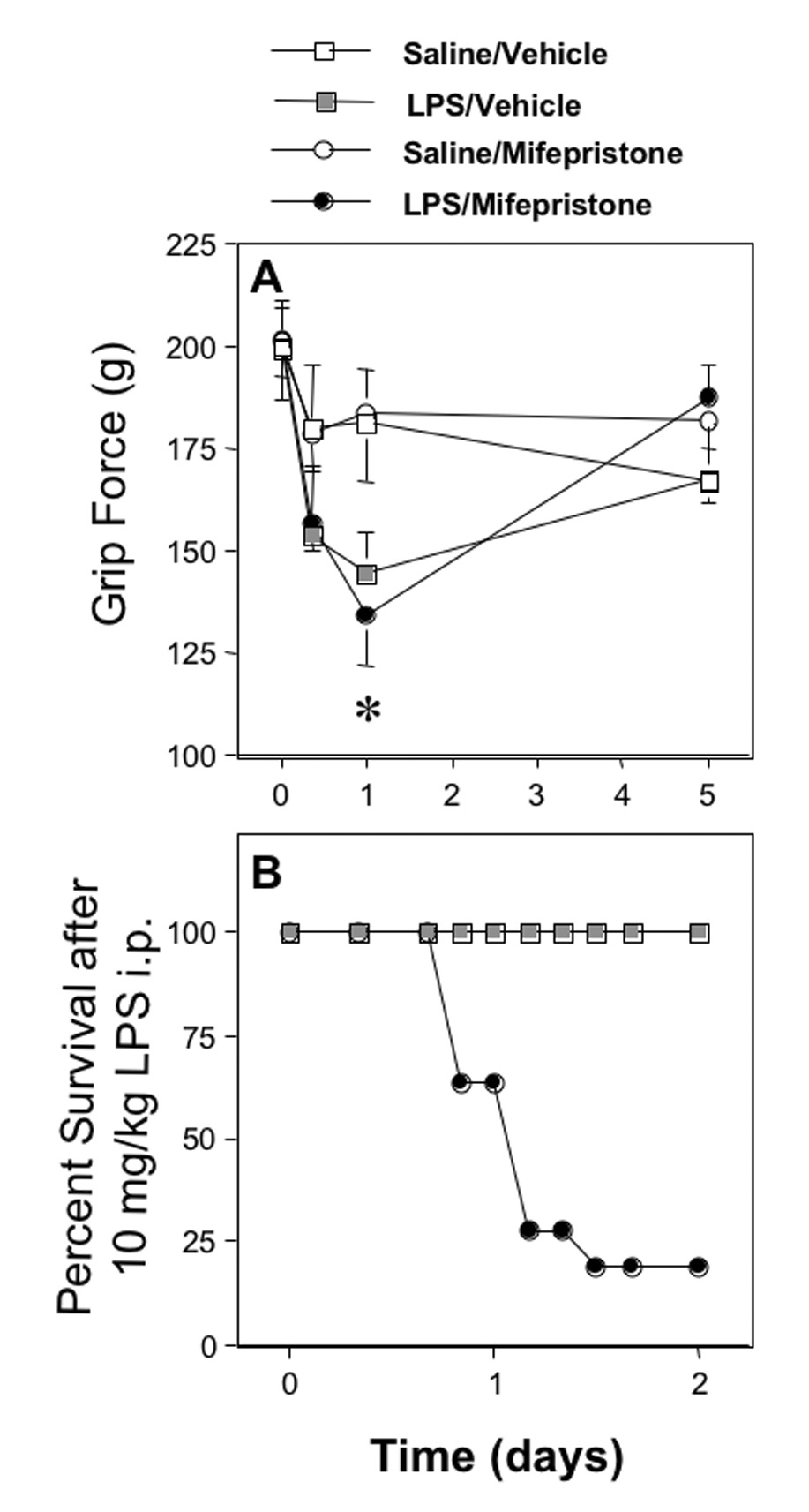
A. Values in the upper panel represent the effect of mifepristone (25 mg/kg s.c.) or vehicle on grip force measurements (mean±SEM) in mice measured before (0) and at the times indicated after a s.c. injection of 2 mg/kg of LPS or saline. Twenty-four hr after LPS, the mean grip force response of the group injected with LPS plus mifepristone was significantly less than that of either group not injected with LPS, as indicated by the asterisk, but did not differ from the group injected with saline plus LPS. Significant differences (P < 0.05) between the groups (n=7) were determined at each time interval using analysis of variance (ANOVA) followed by Fisher Protected Least Squares Difference (PLSD) test to determine significance at P < 0.05, which is indicated by the asterisk.
B. Values in the lower panel represent the percent of mice that survived when mice were tested with an injection of 10 mg/kg of LPS delivered i.p. While the 10 mice pretreated with saline were completely resistant to the lethal effects of LPS, 9 of the 11 mice injected with mifepristone (25 mg/kg s.c.) plus LPS died by the second day.
3.2. Effect of mifepristone on the lethal effect of LPS
To ensure that the dose of mifepristone used was sufficient to inhibit the actions of endogenously derived glucocorticoids, we examined the effect of this same dose of mifepristone on the incidence of lethality induced by LPS. Mifepristone (25 mg/kg s.c.) delivered immediately prior to LPS (10 mg/kg i.p.) resulted in lethality in 80% of mice whereas none of the mice died when pretreated with vehicle prior to LPS (Fig. 1B).
3.3. Adrenalectomy and grip force responses to LPS
To determine whether endogenous adrenal hormones influence the hyperalgesic effect of LPS, we adrenalectomized mice 2 weeks prior to testing and compared the effect of LPS in those mice to sham-operated controls. We found that LPS (5 mg/kg s.c.) produced the same magnitude of decrease in grip force responses in adrenalectomized mice as in sham-operated controls (Fig. 2A).
Fig. 2. Effect of adrenalectomy on the ability of LPS to decrease forelimb grip force measurements and to induce lethality in mice.
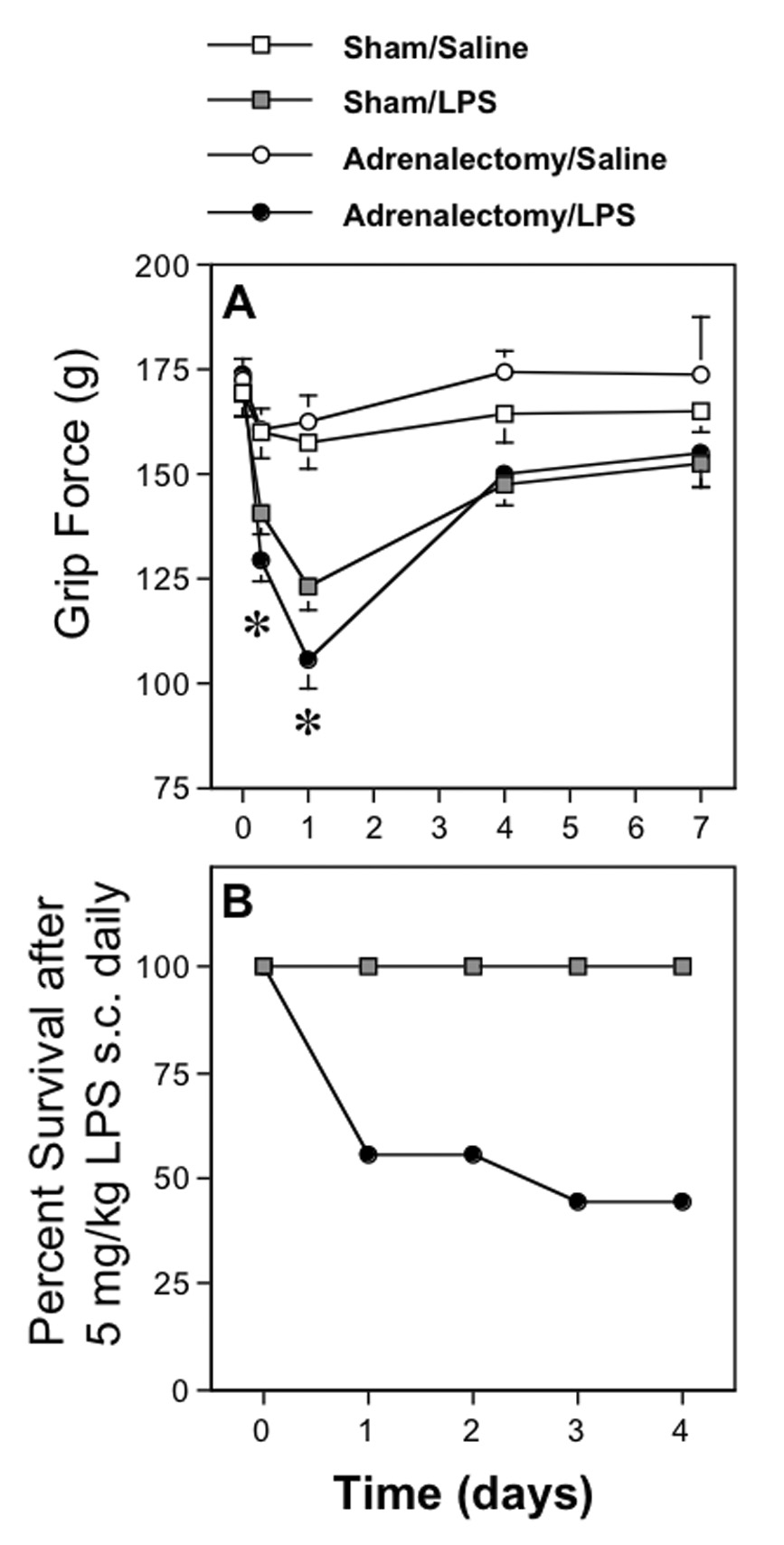
A. Values in the upper panel represent grip force responses (mean±SEM) taken at the times indicated after s.c. injections of 5 mg/kg of LPS or saline in mice that were subjected to either adrenalectomy (ADX) or sham-surgery two weeks before injection. The asterisks indicate the time-intervals at which values from LPS-injected ADX mice (8 hr) and the values from the two groups injected with LPS (24 hr) were significantly less than those in the remaining groups, but not significantly different from each other. Significant differences (P < 0.05) between the groups (n=7) were determined using ANOVA followed by PLSD. Throughout the grip force values shown, standard errors were calculated but are in some cases too small to be depicted.
B. The lower panel illustrates the effect of LPS on lethality in mice 2 weeks after ADX or sham surgery. Values represent the percent of 9 ADX or sham-operated mice that survived each injection of 5 mg/kg of LPS delivered s.c. daily. While sham-operated mice are remarkably resistant to the lethal effects of LPS, several adrenalectomized mice died after the first two injections, illustrating the high sensitivity of ADX mice to endotoxemia.
3.4. Effect of adrenalectomy on the lethal effect of LPS
In spite of the inability of adrenalectomies to influence the hyperalgesic effect of LPS, when additional groups of mice were examined, we found that daily injections of LPS (5 m/kg s.c.) were sufficient to induce lethality in some adrenalectomized mice, but had no effect in sham-operated mice (Fig. 2B).
3.5. Adrenalectomy and the development of tolerance to LPS
We have previously found that repeated injections of LPS to mice result in complete tolerance to the hyperalgesic effect measured using the grip force assay. Because adrenalectomized mice do not become tolerant to the ability of LPS to induce TNF-α at 1 hr after injection (Evans and Zuckerman, 1991), we continued to inject and test the adrenalectomized and sham-operated mice from the experiments described in figure 2A at weekly intervals to evaluate tolerance in our paradigm. The mean grip force values were taken 24 hr after each s.c. injection of 5 mg/kg LPS or saline to monitor the previously characterized development of tolerance to decreases in grip force measures (Kehl et al., 2004). We found that tolerance developed rapidly in both adrenalectomized and sham-operated mice, as it was clearly present by week 2 (Fig. 3).
Fig. 3. Tolerance to the hyperalgesic effect of LPS in adrenalectomized and sham-operated mice.
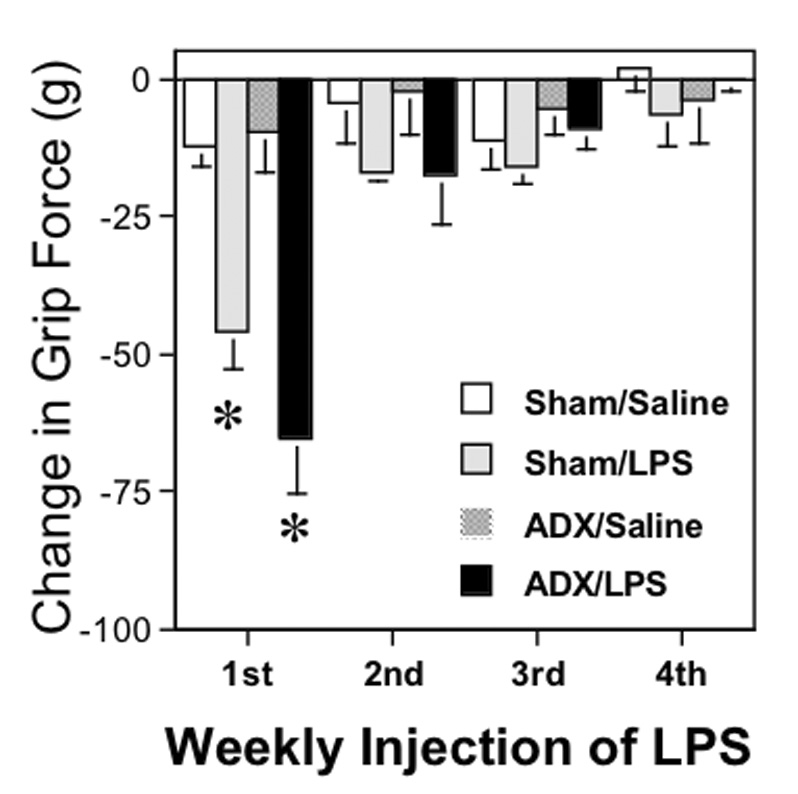
Data represent the mean change in grip force responses from their original control values prior to each injection of LPS (mean ± SEM) 24 hr after each of 4 weekly s.c. injections of either 5 mg/kg of LPS or saline in the same adrenalectomized (ADX) and sham-operated mice as in figure 2A. Asterisks represent significant decreases (P < 0.05) in the responses following the injection of LPS in each of the two groups injected with LPS compared to those injected with saline, as determined using ANOVA followed by PLSD. By the second week of injections, tolerance to the effect of LPS was evident by the lack of significant decreases in grip force following LPS.
3.6. Effect of adrenalectomy on circulating concentrations of corticosterone
To verify the completeness of the adrenalectomies performed, we examined the concentration of circulating corticosterone in additional groups of mice injected s.c. with either LPS (5 mg/kg) or saline. Grip force values 24 hr after LPS were significantly less than in mice injected with saline (Fig. 4A). Mice were then subjected to a 5-min forced swim stress (31°C) to activate the adrenal and better differentiate between adrenalectomized and sham-operated groups, and killed immediately. The mean circulating corticosterone value in the 15 adrenalectomized mice (161.9±16.9 ng/ml) was significantly less than in the 17 sham-operated control mice (407.1±31.1), as detailed in Fig. 4B. The corticosterone remaining after adrenalectomy was sufficiently low that it is likely due to synthesis from extra-adrenal sources, described previously (Davies and MacKenzie, 2003). In summary, the grip force values of adrenalectomized mice injected with LPS did not differ from those in sham-operated control mice even though the concentration of corticosterone after adrenalectomy was less than in sham-operated mice.
Fig. 4. Effect of adrenalectomy on grip force and circulating corticosterone.
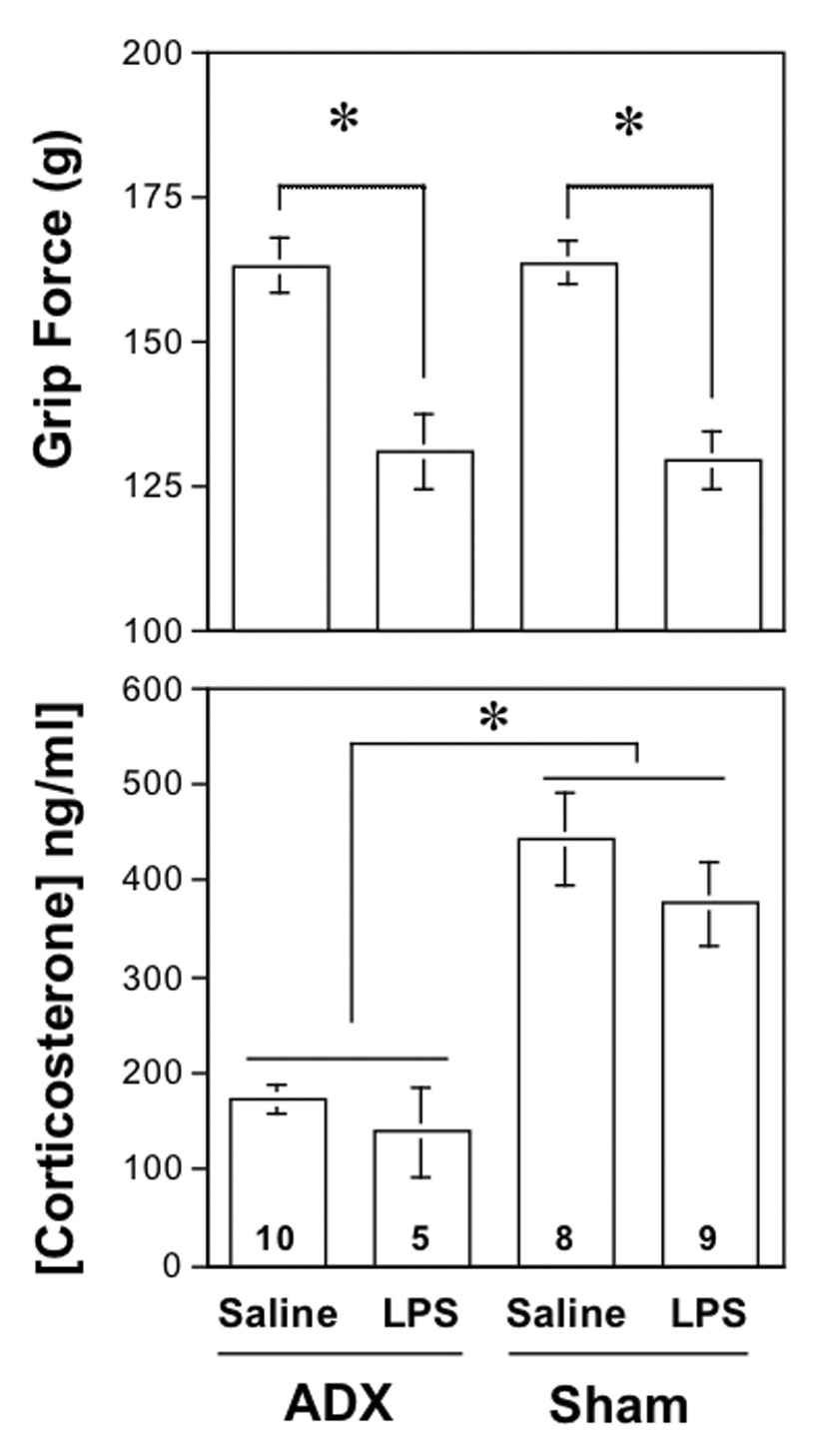
A. Values in the upper panel represent the grip force responses (mean±SEM) measured at 24 hr after LPS (5 mg/kg s.c.) or saline when administered to mice that were previously adrenalectomized (ADX) or sham-operated (Sham) two weeks previously. The asterisk represents a significant difference (P < 0.05) between the groups indicated, as determined using ANOVA followed by PLSD.
B. Values in the lower panel represent the concentration of circulating corticosterone (mean±SEM) in the same adrenalectomized and sham-operated mice as in the panel above, measured 24 hr after the injection of LPS (5 mg/kg s.c.) or saline. Mice were subjected to a 5-min forced swim at room temperature immediately prior to death to activate adrenal tissue, where present. The asterisk represents a significant difference (P < 0.05) between the groups indicated, as determined using ANOVA followed by PLSD.
3.7. Dexamethasone and grip force responses to LPS
To evaluate whether an increase in glucocorticoid activity protects against LPS-induced decreases in grip force responses, we pretreated mice with 1 mg/kg of dexamethasone injected s.c. at 0 and 8 hr after injection of 5 mg/kg of LPS s.c. (Fig. 5A). This dose was selected because it is even higher (ten times) than that previously found to inhibit the hyperalgesic effect of LPS injected directly into the paw of mice (Raghavendra et al., 2000). This dose should, therefore, be sufficient to inhibit hyperalgesia induced by a similar mechanism of action. However, dexamethasone had no effect on grip force responses compared to vehicle-injected mice and the decrease in grip force responses produced by LPS alone did not differ from that after LPS plus dexamethasone.
Fig. 5. Effect of dexamethasone on the ability of LPS to decrease grip force measurements and to produce lethality.
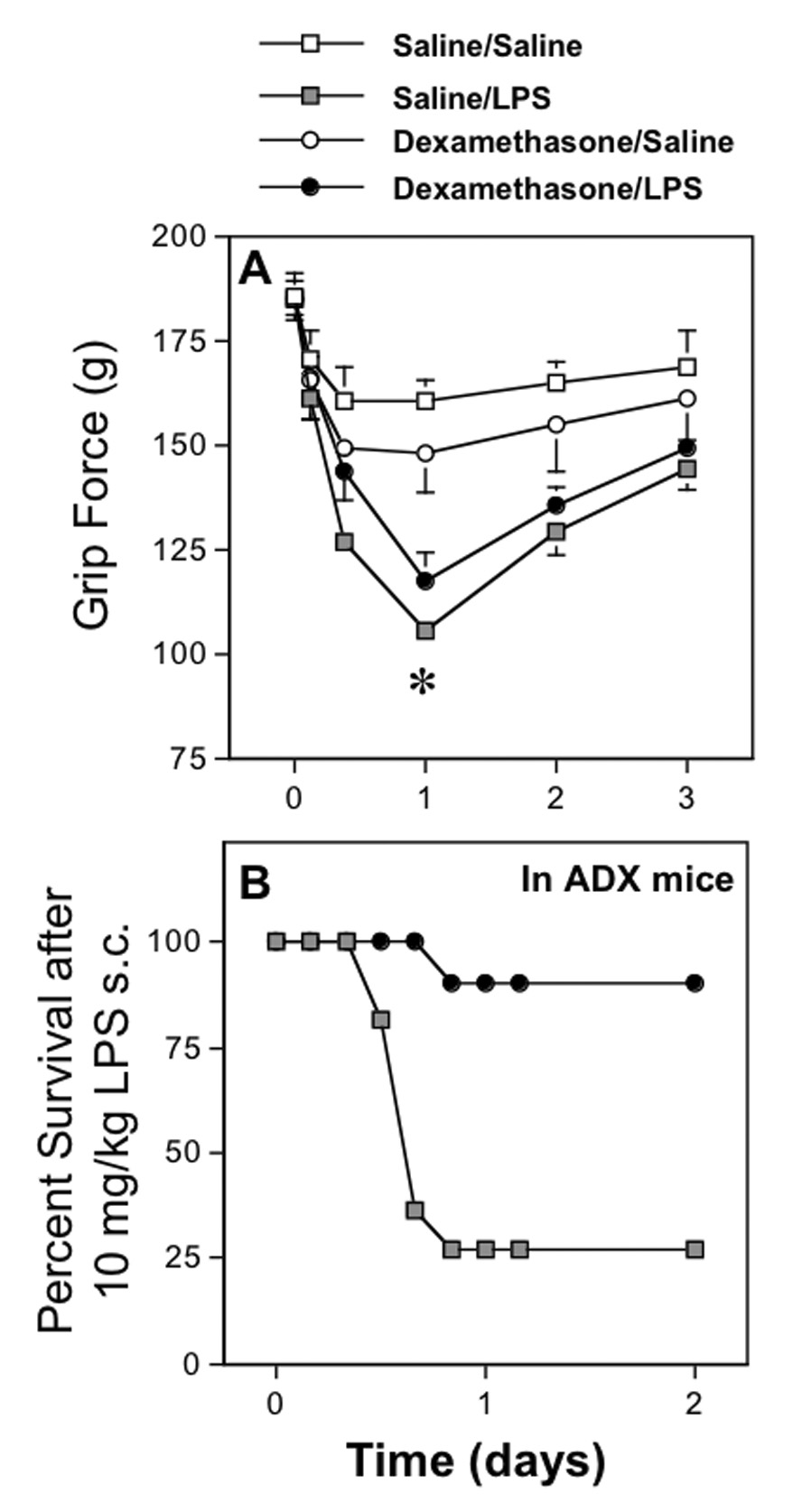
A. Values in the upper panel represent the mean decrease in grip force responses (mean±SEM) measured at various times before (0) and after LPS (5 mg/kg s.c.) or saline when pretreated with either dexamethasone (1 mg/kg s.c.) or saline. Mice were re-injected with either saline or dexamethasone 8 hr later to maintain a high circulating concentration of this drug. Values reflect groups of at least 6 mice per group. The asterisk represents a significant difference (P < 0.05) between the two groups injected with LPS and their saline- or dexamethasone-pretreated controls, as determined using ANOVA followed by PLSD.
B. Values in the lower panel represent the percent of adrenalectomized (ADX) mice that survived an injection of LPS (10 mg/kg s.c.) delivered immediately after an injection of either dexamethasone (1 mg/kg s.c.) or its vehicle. A second injection of either vehicle or dexamethasone was delivered 8 hr later to maintain the blood concentration of this drug. We assessed the adequacy of dexamethasone, at the dose used in the grip force studies, to protect against the lethal effects of LPS. By two days after the injection of LPS, 90% of the ADX mice injected with dexamethasone survived, whereas only 27% of the ADX mice injected with vehicle survived the effect of LPS.
3.8. Effect of dexamethasone on the lethal effect of LPS in adrenalectomized mice
To assess the efficacy of this dose of dexamethasone, we evaluated its ability to replace naturally occurring glucocorticoids in adrenalectomized mice and protect them from the lethal effects of LPS (10 mg/kg s.c.). Using the same injection schedule for dexamethasone as that in the experiments on grip force, dexamethasone (1 mg/kg of dexamethasone injected s.c. at 0 and 8 hr after injection of LPS) provided a substantial degree of protection from lethality following LPS (10 mg/kg s.c.) in adrenalectomized mice compared to that in adrenalectomized mice pretreated with vehicle (Fig. 5B). The decrease in grip force responses produced by this higher dose of LPS (10 mg/kg i.p.) in un-operated mice was no different in mice pretreated with 1 mg/kg of dexamethasone (−75.6±10.5 g, n=11) than in mice pretreated with saline (−71.1±8.2 g, n=18) when measured 24 hr after injection of LPS.
3.9. Effect of LPS on Rota-rod performance
Using the Rota-rod, we monitored the possible influence of LPS on motor coordination, fatigue and weakness that may influence the animals’ ability to function in the grip force assay. Mice were injected with LPS (5 mg/kg s.c.) or saline. Twenty-four hours later, mice were tested for the time that they were able to remain on the rotating cylinder during a 5-min trial. No significant difference was found between the means obtained from mice injected with LPS (261.0±21.8 sec, n=9, Student’s t-test) and mice injected with saline (289.0±9.9 sec, n=8), in spite of a significantly decreased grip force in the same mice injected with LPS (129.3±5.1 g) compared to those injected with saline (163.5±3.7 g, Student’s t-test). We also tested the ability of these mice to support themselves hanging onto a rotating cylinder covered with a metal mesh without falling the 40−cm distance to the countertop. All mice were able to maintain their position on the rotating cylinder, hanging upside down, for a period of 5 min.
4.0. Morphine and grip force responses to LPS
Performance on a Rota-rod reflects coordination but is not absolutely predictive of an animal’s ability to pull on the grip force apparatus. The sensitivity of decreases in grip force to an antinocioceptive compound (Fig. 6) may be a better reflection of whether those decreases are related to nociception. Thirty min after administration of morphine (10 mg/kg i.p.), the reduction in grip force induced by an injection of LPS (5 mg/kg i.p.) 24 hr previously was reversed to a value no different from saline-injected controls, as shown previously (Kehl et al., 2004). Morphine alone had no effect on grip force, consistent with previous studies (Nevins et al., 1993).
Fig. 6. Effect of morphine on the ability of LPS to decrease grip force measurements.
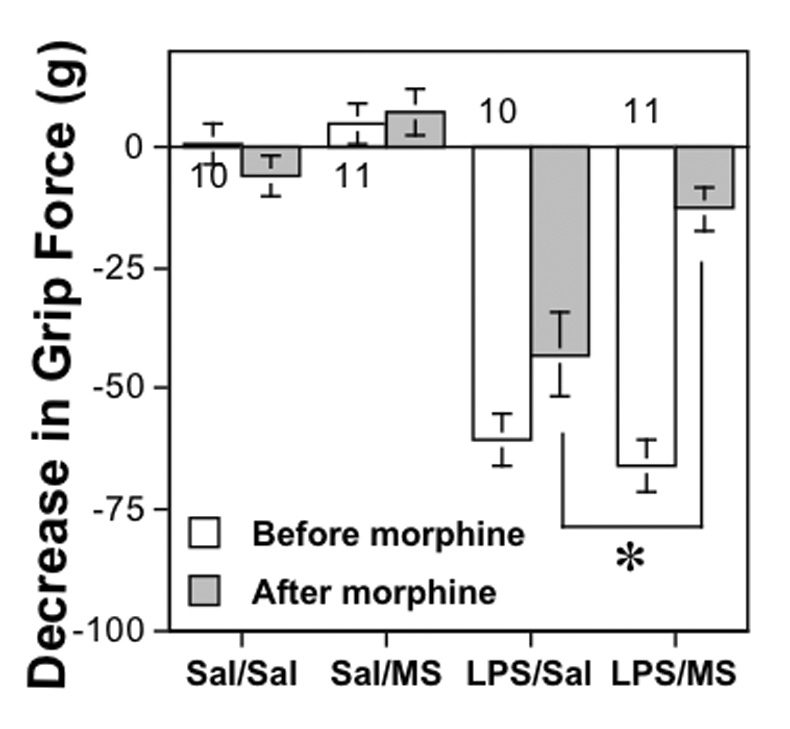
Values represent the mean decrease in grip force responses (mean±SEM) measured 24 hr after LPS (5 mg/kg i.p.) or saline (Sal) and 30 min after morphine (10 mg/kg i.p.) or saline. The asterisk represents a significant difference (P < 0.05) between the effect of morphine sulfate (MS) and that of saline (Sal) on LPS-induced decreases in grip force, as determined using ANOVA followed by PLSD.
4. Discussion
Circulating LPS is most evident following traumatic injury (Buttenschoen et al., 2000), ischemia (Yassin et al., 1998), surgery (Riddington et al., 1996), disc herniation (Sergio et al., 1982) and even head injury (Jiang et al., 1997). Even in healthy individuals, the intestinal lumen is a dense reservoir from which small concentrations transiently pass into the circulation during exercise or stress (Fox et al., 1989; Nakao et al., 1994). Additional sources of endotoxins include the bladder, peritoneal cavity and sinuses. Immune responses to LPS in the circulation include nausea, vomiting, diarrhea, dizziness, fever (Van Leeuwen et al., 1994), muscle aches and pain (Hochstein et al., 1994; Lynn et al., 2003). The tendency for LPS to reduce forelimb grip force responses in mice (Kehl et al., 2004) provides us with a model of the muscular discomfort that patients report during endotoxemia.
While LPS is known to induce illness-like responses, LPS had no significant effect on performance of mice on the Rota-rod. That, together with the ability of morphine to reverse LPS-induced decreases in grip force suggest that the effect of LPS on grip force reflects musculoskeletal hyperalgesia rather than weakness and fatigue. Administered alone, morphine had no effect on grip force in mice, as previously reported (Nevins et al., 1993). The effect of morphine is noteworthy in view of the tendency for LPS to inhibit the antinciceptive effect of morphine (Johnston and Westbrook, 2003).
Cutaneous hyperalgesia develops following injection of LPS directly into the hindpaw (Kanaan et al., 1996; Safieh-Garabedian et al., 1996; Safieh-Garabedian et al., 1997). Cutaneous hyperalgesia clearly involves secretion of pro-inflammatory cytokines (Ferreira et al., 1988; Maier et al., 1993; Watkins etal., 1994c; Rietschel et al., 1994), eicosanoids (O’Neill and Lewis, 1989; Decker, 1990), and reactive oxygen species (Beasley et al., 1991). These compounds are synthesized peripherally (Nathan, 1987; Jungi et al., 1994; Gardiner et al., 1995; Shapira et al., 1998; Aosasa et al., 2000) but can occur in the brain following systemic injection of LPS (Quan et al., 1999). Hyperalgesia induced by centrally injected LPS may result from induction of cytokines as they, too, are hyperalgesic when injected into the CNS, including TNF, IL-1α and IL-1β (Meller et al., 1994; Ignatowski et al., 1999; Tadano et al., 1999). IL-1β enhances the concentration of nerve growth factor (NGF) (Safieh-Garabedian et al., 1995), a neurotrophin associated with hyperalgesia in rodents (Lewin and Mendell, 1993) and man (Petty et al., 1994). Injected centrally, LPS also upregulates the synthesis of prostanoids (Walker et al., 1996) and activates bradykinin B2 receptors (Walker et al., 1996).
Inhibition of inflammatory mediators by glucocorticoids occurs by interfering with production of inflammatory mediators like the cytokines. Endogenously occurring glucocorticoids, like corticosterone in rodents and cortisol in humans, released from the adrenal cortex in response to LPS, down-regulate the transcription of pro-inflammatory compounds (reviewed by McKay and Cidlowski, 1999). In this fashion, glucocorticoids inhibit the synthesis of IL-1 and TNF-α in macrophages, thereby protecting against the lethal effects of endotoxins. Glucocorticoids are also powerful inhibitors of LPS-induced induction of iNOS (Knowles et al., 1990; Radomski et al., 1990) that catalyzes the synthesis of nitric oxide, a hypotensive and hyperalgesic compound. Removal of the protective influence of endogenously released corticosterone by adrenalectomy enhances the lethal effects of LPS (Bertini et al., 1988). If the movement-induced hyperalgesia caused by LPS were mediated by inflammatory compounds, adrenalectomy would enhance hyperalgesia, similar to its effect on lethality. However, adrenalectomy did not influence movement-induced hyperalgesia compared to that in sham-operated control mice, suggesting adrenal hormones do not provide protection from the hyperalgesic effects of LPS.
Although tolerance to sepsis has been previously attributed by some to glucocorticoid activity, more recently it is thought to occur by an increase in the synthesis of the LPS-binding protein, resulting in an increased clearance of LPS from the circulation (Zweigner et al., 2001). We previously observed decreased circulating LPS coincident with tolerance to movement-induced hyperalgesia (Kehl et al., 2004), suggesting that a similar mechanism underpins the tolerance that develops to both lethality and hyperalgesia. That adrenalectomy failed to alter the development of tolerance to movement-induced hyperalgesia following repeated injection of LPS is consistent with an increased clearance of LPS rather than an effect of glucocorticoids.
If movement-induced hyperalgesia caused by LPS were mediated by inflammatory compounds, pretreatment with potent glucocorticoids like dexamethasone may be expected to attenuate the hyperalgesic effect of LPS, similar to its well-characterized ability to protect against lethality (Geller et al., 1954). However, dexamethasone failed to decrease the effect of LPS on grip force responses, even when administered at a dose that attenuated the enhanced sensitivity of adrenalectomized mice to LPS-induced lethality. The hyperalgesia reflected in the grip force assay may be insensitive to glucocorticoids as decreases in grip force produced by carrageenan, an inflammatory compound, were also not prevented by dexamethasone in rats (Kehl et al., 2000).
Mifepristone antagonizes the interaction of glucocorticoids with their receptors. While mifepristone inhibits both glucocorticoid and progesterone receptors, its protection against lethality is likely due to its ability to inhibit glucocorticoid receptors as endotoxin lethality in mice was diminished by dexamethasone but not progesterone (Lazar et al., 1977). Using this approach, which spares the adrenal but targets the action of hormones derived from the adrenal cortex, we found that mifepristone failed to influence movement-evoked hyperalgesia at a dose that enhanced the lethal effects of LPS. This supports the conclusion that decreases in grip force induced by LPS are not brought about by the same inflammatory mediators as those causing either cutaneous hyperalgesia or lethality.
Many effects of LPS, such as fever, inhibition of water and food intake and reduced locomotor activity, depend on elevated concentrations of TNF-α. These effects occur rapidly after injection of LPS (within hours) and coincide with increases in immune mediators (Nava et al., 1996; Nava et al., 1997a; Nava et al., 1997b; Nava and Caputi, 1999; Nava and Carta, 2000). In contrast, LPS-induced muscle hyperalgesia measured using grip force is not maximal until 24 hr after injection, much longer than the documented elevations in cytokines. The difference in time-course of LPS-induced increases in TNF-α and movement-evoked hyperalgesia suggest that TNF-α does not sustain skeletal muscular hyperalgesia nor initiate downstream mediators responsible for hyperalgesia. Therefore different and as yet unidentified compounds, that are insensitive to glucocorticoids, initiate and maintain the important phenomenon of movement-evoked hyperalgesia and require further study.
Increases in nociceptive sensitivity detected by the grip force assay reflect hyperalgesia common to diverse models of inflammatory muscle hyperalgesia in rats (Kehl et al, 2000; Wacnik et al., 2003) and cancer pain in mice (Kehl et al., 2003; Wacnik et al., 2003). Movement-induced hyperalgesias are associated with several clinical conditions that are known to increase circulating LPS, including irritable bowel syndrome (IBS; Barbara et al., 1997; Nazar and Smishchuk, 1998), inflammatory bowel diseases such as Crohn’s disease and ulcerative colitis (Hotta et al., 1986; Monajemi et al., 1996; Caradonna et al., 2000), chronic fatigue syndrome (Linde et al., 1992; McGregor et al., 1996), interstitial cystitis (Jasmin et al., 1998), hepatitis C (Liehr, 1981) as well as motor vehicle accidents, work-related injuries, surgeries (Riddington et al., 1996) and other infectious diseases (Sergio et al., 1982; Riddington et al, 1996; Buttenschoen et al., 2000). Patients with IBS and other chronic inflammatory disorders of the gastrointestinal tract often experience chronic widespread pain (Tobin and Kimmey, 2001). Because endotoxins cause muscle pain, conditions such as fibromyalgia, which frequently co-exist with inflammatory conditions such as Crohn’s disease (Sperber et al., 1999), IBS (Sivri et al., 1996; Barton et al., 1999; Sperber et al., 1999) and interstitial cystitis (IC) (Jasmin et al., 1998), may develop secondary to processes that allow increased concentrations of endotoxins into the circulation. It is noteworthy that trauma, surgery and infection, conditions that increase circulating endotoxins, are anecdotal events commonly reported to coincide with the onset of fibromyalgia. Thus, LPS-induced decreases in grip force may be a suitable model for these conditions. A compromised ability to develop tolerance to LPS may promote chronically hyperalgesic conditions that result from endotoxins. Because of the high incidence of movement-evoked hyperalgesia, it is important to determine the mechanism by which skeletal muscular hyperalgesia develops.
The present data indicate that this movement-evoked hyperalgesia is not brought about by the same inflammatory mediators as those responsible for the lethal or cutaneous hyperalgesic effects of LPS. This is based on the insensitivity of LPS-induced decreases in grip force responses to changes in glucocorticoids compared to the high sensitivity of LPS-induced lethality to these same manipulations. One clinical implication of this is that the severity of widespread hyperalgesia is not relieved by treatments that prevent shock, so muscle hyperalgesia cannot be used as a barometer of the severity of endotoxemia. A distinction between the mechanisms responsible for cutaneous and muscle hyperalgesia is important as the latter is a common complaint associated with endotoxemic conditions.
5. Summary
Many inflammatory mediators are known to be increased in response to LPS and have been shown to be responsible for LPS-induced cutaneous hyperalgesia and septic shock. However, glucocorticoid-sensitive inflammatory pathways are not responsible for the delayed onset of musculoskeletal hyperalgesia after LPS.
Acknowledgements
This work was supported by NIH grant NS39740 (A.A.L.) funded by the National Institute of Neurological Disorders and Stroke and the National Institute on Arthritis and Musculoskeletal and Skin Diseases.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Jasper MS, Engeland WC. Synchronous ultradian rhythms in adrenocortical secretion detected by microdialysis in awake rats. Am J Physiol. 1991;261:R1257–R1268. doi: 10.1152/ajpregu.1991.261.5.R1257. [DOI] [PubMed] [Google Scholar]
- Thrivikraman KV, Plotsky PM. Absence of glucocorticoid negative feedback to moderate hemorrhage in conscious rats. Am J Physiol. 1993;264:E497–E503. doi: 10.1152/ajpendo.1993.264.4.E497. [DOI] [PubMed] [Google Scholar]
- Aosasa S, Ono S, Mochizuki H, Tsujimoto H, Osada S, Takayama E, Seki S, Hiraide H. Activation of monocytes and endothelial cells depends on the severity of surgical stress. World J Surg. 2000;24(1):10–16. doi: 10.1007/s002689910003. [DOI] [PubMed] [Google Scholar]
- Barbara G, Vallance BA, Collins SM. Persistent intestinal neuromuscular dysfunction after acute nematode infection in mice. Gastroenterology. 1997;113:1224–1232. doi: 10.1053/gast.1997.v113.pm9322517. [DOI] [PubMed] [Google Scholar]
- Barton A, Pal B, Whorwell PJ, Marshal D. Increased prevalence of sicca complex and fibromyalgia in patients with irritable bowel syndrome. Am J Gastroenterol. 1999;94(7):1898–1901. doi: 10.1111/j.1572-0241.1999.01146.x. [DOI] [PubMed] [Google Scholar]
- Beasley D, Schwartz JH, Brenner BM. Interleukin-a induces prolonged L-arginine-dependent cyclic guanosine monophosphate and nitrite production in rat vascular smooth muscle cell. J Clin Invest. 1991;87:602–608. doi: 10.1172/JCI115036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berki T, Pálinkás L, Boldizsár F, Németh P. Glucocorticoid (GC) sensitivity and GC receptor expression differ in thymocyte subpopulations. Int Immunol. 2002;14(5):463–469. doi: 10.1093/intimm/14.5.463. [DOI] [PubMed] [Google Scholar]
- Bertini R, Bianchi M, Ghezzi P. Adrenalectomy sensitizes mice to the lethal effects of interleukin 1 and tumor necrosis factor. J Exp Med. 1988;167:1708–1712. doi: 10.1084/jem.167.5.1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beutler B, Milsark IW, Cerami AC. Passive immunization against cachectin/tumor necrosis factor protects mice from lethal effect of endotoxin. Science. 1985;229(4716):869–871. doi: 10.1126/science.3895437. [DOI] [PubMed] [Google Scholar]
- Buttenschoen K, Berger D, Strecker W, Buttenschoen DC, Stenzel K, Pieper T, Berger HG. Association of endotoxemia and production of antibodies against endotoxins after multiple injuries. J Trauma. 2000;48(5):918–923. doi: 10.1097/00005373-200005000-00017. [DOI] [PubMed] [Google Scholar]
- Cahill CM, Dray A, Coderre TJ. Priming enhances endotoxin-induced thermal hyperalgesia and mechanical allodynia in rats. Brain Res. 1998;808:13–22. doi: 10.1016/s0006-8993(98)00786-0. [DOI] [PubMed] [Google Scholar]
- Caradonna L, Amati L, Magrone T, Pellegrino NM, Jitillo E, Caccavo D. Enteric bacteri, lipopolysaccharides and related cytokines in inflammatory bowel disease: biological and clinical significance. J Endotoxin Res. 2000;6(3):205–214. [PubMed] [Google Scholar]
- Davies E, MacKenzie SM. Extra-adrenal production of corticosteroids. Clin Exp Pharmacol Physiol. 2003;30:437–445. doi: 10.1046/j.1440-1681.2003.03867.x. [DOI] [PubMed] [Google Scholar]
- Decker K. Biologically active products of stimulated liver macrophages (kupffer cells) Eur J Biochem. 1990;192:245–261. doi: 10.1111/j.1432-1033.1990.tb19222.x. [DOI] [PubMed] [Google Scholar]
- Evans GF, Zuckerman SH. Glucocorticoid-dependent and -independent mechanisms involved in lipopolysaccharide tolerance. Eur J Immunol. 1991;21(9):1973–1979. doi: 10.1002/eji.1830210902. [DOI] [PubMed] [Google Scholar]
- Ferreira SH, Lorenzetti BB, Bristow AF, Pooles S. Interleukin-1 beta as a potent hyperalgesic agent antagonized by a tripeptide analogue. Nature. 1988;334:698–700. doi: 10.1038/334698a0. [DOI] [PubMed] [Google Scholar]
- Fox ES, Thomas P, Broitman SA. Clearance of gut-derived endotoxin by the liver. Release and modification of 3H, 14C-lipopolysaccharide by isolated rat Kupffer cells. Gastroenterology. 1989;96:456–461. doi: 10.1016/0016-5085(89)91571-0. [DOI] [PubMed] [Google Scholar]
- Gardiner KR, Halliday MI, Barclay GR, Milne L, Brown D, Stephens S, Maxwell RJ, Rowlands BJ. Significance of systemic endotoxaemia in inflammatory bowel disease. Gut. 1995;36:897–901. doi: 10.1136/gut.36.6.897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geller P, Merrill ER, Jawetz E. Effects of cortisone and antibiotics on lethal action of endotoxinin in mice. PSEBM. 1954;86:716–719. doi: 10.3181/00379727-86-21211. [DOI] [PubMed] [Google Scholar]
- Hochstein HD, Fitzgerald EA, McMahon FG, Vargas R. Properties of US standard endotoxin (EC-5) in human male volunteers. J Endotoxin Res. 1994;1:52–56. [Google Scholar]
- Hotta T, Yoshida N, Yoshikawa T, Sugino S, Kondo M. Lipopolysaccharide-induced colitis in rabbits. Res Exp Med. 1986;186(1):61–69. doi: 10.1007/BF01851834. [DOI] [PubMed] [Google Scholar]
- Ignatowski TA, Covery WC, Knight PR, Severin CM, Nickola TJ, Spengler RN. Brain-derived TNF-α mediates neuropathic pain. Brain Res. 1999;841:70–77. doi: 10.1016/s0006-8993(99)01782-5. [DOI] [PubMed] [Google Scholar]
- Jasmin L, Jani G, Manz HJ, Rabkin SD. Activation of CNS circuits producing a neurogenic cystitis: evidence for centrally induced peripheral inflammation. J Neurosci. 1998;18(23):10016–10129. doi: 10.1523/JNEUROSCI.18-23-10016.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jasper MS, Engeland WC. Synchronous ultradian rhythms in adrenocortical secretion detected by microdialysis in awake rats. Am J Physiol. 1991;261:R1257–R1268. doi: 10.1152/ajpregu.1991.261.5.R1257. [DOI] [PubMed] [Google Scholar]
- Jiang JX, Tian KL, Chen HS, Zhu PF, Wang ZG. Plasma cytokines and endotoxin levels in patients with severe injury and their relationship with organ damage. Injury. 1997;28(8):509–513. doi: 10.1016/s0020-1383(97)00057-0. [DOI] [PubMed] [Google Scholar]
- Johnston IN, Westbrook RF. Acute and conditioned sickness reduces morphine analgesia. Behavior Brain Res. 2003;142:89–97. doi: 10.1016/s0166-4328(02)00398-4. [DOI] [PubMed] [Google Scholar]
- Jungi TW, Miserez R, Brcic M, Pfister H. Change in sensitivity to lipopolysaccharide during the differentiation of human monocytes to macrophages in vitro. Experientia. 1994;50(2):110–114. doi: 10.1007/BF01984945. [DOI] [PubMed] [Google Scholar]
- Kanaan SA, Saade NE, Haddad JJ, Abdelnoor AM, Atweh SF, Jabbur SJ. Safieh-Garabedian B. Endotoxin-induced local inflammation and hyperalgesia in rats and mice: a new model for inflammatory pain. Pain. 1996;66:373–379. doi: 10.1016/0304-3959(96)03068-0. [DOI] [PubMed] [Google Scholar]
- Kehl LJ, Trempe TM, Hargreaves KM. A new animal model for assessing mechanisms and management of muscle hyperalgesia. Pain. 2000;85(3):333–343. doi: 10.1016/S0304-3959(99)00282-1. [DOI] [PubMed] [Google Scholar]
- Kehl LJ, Hamamoto DT, Wacnik PW, Croft DL, Norsted BD, Wilcox GL, Simone DA. A cannabinoid agonist differentially attenuates deep tissue hyperalgesia in animal models of cancer and inflammatory muscle pain. Pain. 2003;103:175–186. doi: 10.1016/s0304-3959(02)00450-5. [DOI] [PubMed] [Google Scholar]
- Kehl LJ, Kovács KJ, Larson AA. Tolerance develops to the effect of lipopolysaccharides on movement-evoked hyperalgesia when administered chronically by a systemic but not an intrathecal route. Pain. 2004;111:104–115. doi: 10.1016/j.pain.2004.06.014. [DOI] [PubMed] [Google Scholar]
- Knowles RG, Salter M, Brooks SL, Moncada S. Anti-inflammatory glucocorticoids inhibit the induction by endotoxin of nitric oxide synthase in the lung, liver and aorta of the rat. Biochem Biophys Res Commun. 1990;172(3):1042–1048. doi: 10.1016/0006-291x(90)91551-3. [DOI] [PubMed] [Google Scholar]
- Lazar G, Sekiya S, Agarwal MK. Influence of steroid structure in relation to liver metabolism during endotoxin lethality in mice. Exp Cell Biol. 1977;45(3–4):176–183. doi: 10.1159/000162869. [DOI] [PubMed] [Google Scholar]
- Lewin GR, Mendell LM. Nerve growth factor and nociception. Trends Neurosci. 1993;16:353–359. doi: 10.1016/0166-2236(93)90092-z. [DOI] [PubMed] [Google Scholar]
- Liehr H. Endotoxins and the pathogenesis of gastrointestinal diseases. MMW-Munch Med Wochenschr. 1981;123(2):53–56. [PubMed] [Google Scholar]
- Linde A, Andersson B, Svenson SB, Ahrne H, Carlsson M, Forsberg P, Hugo H, Karstrop A, Lenkei R, Lindwall A. Serum levels of lymphokines and soluble cellular receptors in primary Epstein-Barr virus infection and in patients with chronic fatigue syndrome. J Infect Dis. 1992;165(6):994–1000. doi: 10.1093/infdis/165.6.994. [DOI] [PubMed] [Google Scholar]
- Lynn M, Rossignol DP, Wheeler JL, Kao RJ, Perdomo CA, Noveck R, Vargas R, D’Angelo T, Gotzkowsy S, McMahon FG. blocking of responses to endotoxin by E5564 in healthy volunteers with experimental endotoxemia. J Infect Dis. 2003;187:631–639. doi: 10.1086/367990. [DOI] [PubMed] [Google Scholar]
- Maier SF, Wiertelak EP, Martin D, Watkins LR. Interleukin-1 mediates the behavioral hyperalgesia produced by lithium chloride and endotoxin. Brain Res. 1993;623:321–324. doi: 10.1016/0006-8993(93)91446-y. [DOI] [PubMed] [Google Scholar]
- McGregor NR, Dunstan RH, Zerbes M, Butt HL, Roberts TK, Klineberg IJ. Preliminary determination of the association between symptom expression and urinary metabolites in subjects with chronic fatigue syndrome. Biochem Mol Med. 1996;58(1):733–739. doi: 10.1006/bmme.1996.0036. [DOI] [PubMed] [Google Scholar]
- McKay LI, Cidlowski JA. Molecular control of immune/inflammatory responses: interactions between nuclear factor-kappa B and steroid receptor-signaling pathways. Endocrine Reviews. 1999;20(4):435–459. doi: 10.1210/edrv.20.4.0375. [DOI] [PubMed] [Google Scholar]
- Meller ST, Dykstra C, Grzbycki D, Murphy S, Gebhart GF. The possible role of glia in nociceptive processing and hyperalgesia in the spinal cord of the rat. Neuropharmacology. 1994;33:1471–1478. doi: 10.1016/0028-3908(94)90051-5. [DOI] [PubMed] [Google Scholar]
- Monajemi H, Meenan J, Lamping R, Obradov DO, Radema SA, Trown PW, Tytgat GN, Van Deventer SJ. Inflammatory bowel disease is associated with increased mucosal levels of bactericidal/permeability-increasing protein. Gastroenterology. 1996;110(3):733–739. doi: 10.1053/gast.1996.v110.pm8608882. [DOI] [PubMed] [Google Scholar]
- Nakao A, Taki S, Yasui M, Kimura Y, Nonami T, Harada A, Takagi H. The fate of intravenously injected endotoxin in normal rats and in rats with liver failure. Hepatology. 1994;19:1251–1256. [PubMed] [Google Scholar]
- Nathan CF. Secretory products of macrophages. J Clin Invest. 1987;79:319–326. doi: 10.1172/JCI112815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nava F, Calapai G, De Sarro A, Caputi AP. Interleukin-1 receptor antagonist does not reverse lipopolysaccharide-induced inhibition of water intake in rat. Eur J Pharmacol. 1996;309:223–227. doi: 10.1016/0014-2999(96)00352-4. [DOI] [PubMed] [Google Scholar]
- Nava F, Calapai G, Facciola G, Cuzzocrea S, Marciano MC, DeSarro A, Caputi AP. Effects of interleukin-10 on water intake, locomotory activity, and rectal temperature in rat treated with endotoxin. Int J Immunopharmacol. 1997a;19:31–38. doi: 10.1016/s0192-0561(97)00006-4. [DOI] [PubMed] [Google Scholar]
- Nava F, Calapai G, Facciola G, Cuzzocrea S, Giuliani G, DeSarro A, Caputi AP. Melatonin effects on inhibition of thirst and fever induced by lipopolysaccharide in rat. Eur J Pharmacol. 1997b;331:267–274. doi: 10.1016/s0014-2999(97)01049-2. [DOI] [PubMed] [Google Scholar]
- Nava F, Caputi AP. Central effects of cromoglycate sodium salt in rats treated with lipopolysaccharide. Eur J Pharmacol. 1999;367:351–359. doi: 10.1016/s0014-2999(98)00986-8. [DOI] [PubMed] [Google Scholar]
- Nava F, Carta G. Repeated lipopolysaccharide administration produces tolerance to anorexia and fever but not to inhibition of thirst in rat. Int J Immunopharmacol. 2000;22:943–953. doi: 10.1016/s0192-0561(00)00058-8. [DOI] [PubMed] [Google Scholar]
- Nazar PS, Smishchuk IO. The mucosal status of the large intestine in the irritable bowel syndrome and in chronic noninfectious nonspecific colitis. Lik Sprava. 1998;7:111–114. [PubMed] [Google Scholar]
- Nevins ME, Nash SA. Beardsley PM. Quantitative grip strength assessment as a means of evaluating muscle relaxation in mice. Psychopharmacology. 1993;110:92–96. doi: 10.1007/BF02246955. [DOI] [PubMed] [Google Scholar]
- O’Neill LA, Lewis GP. Interleukin-1 potentiates bradykinin and TNF-alpha-induced PGE2 release. Eur J Pharmacol. 1989;166:131–137. doi: 10.1016/0014-2999(89)90052-6. [DOI] [PubMed] [Google Scholar]
- Petty BG, Cornblath DR, Adornato BT, Chaudhry V, Flexner C, Wachsman M, Sinicropi D, Burton LE, Peroutka SJ. The effect of systemically administered recombinant human nerve growth factor in healthy human subjects. Ann Neurol. 1994;36:244–246. doi: 10.1002/ana.410360221. [DOI] [PubMed] [Google Scholar]
- Philibert D, Teutsch G. RU 486 development. Science. 1990;247:622. doi: 10.1126/science.2300819. [DOI] [PubMed] [Google Scholar]
- Quan N, Stern EL, Whiteside MB, Herkenham M. Induction of pro-inflammatory cytokine mRNAs in the brain after peripheral injection of subseptic doses of lipopolysaccharide in the rat. J Neuroimmunol. 1999;93(1‐2):72–80. doi: 10.1016/s0165-5728(98)00193-3. [DOI] [PubMed] [Google Scholar]
- Radomski MW, Palmer RM, Moncada S. Glucocorticoids inhibit the expression of an inducible, but not the constitutive, nitric oxide synthase in vascular endothelial cells. Proc Natl Acad Sci USA. 1990;87(24):10043–10047. doi: 10.1073/pnas.87.24.10043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raetz CR. Biochemistry of endotoxins. Annu Rev Biochem. 1990;59:129–170. doi: 10.1146/annurev.bi.59.070190.001021. [DOI] [PubMed] [Google Scholar]
- Raghavendra V, Agrewala JN, Kulkarni SK. Melatonin reversal of lipopolysacharides-induced thermal and behavioral hyperalgesia in mice. Eur J Pharmacol. 2000;395:15–21. doi: 10.1016/s0014-2999(00)00196-5. [DOI] [PubMed] [Google Scholar]
- Riddington DW, Venkatesh B, Boivin CM, Bonser RS, Elliott TS, Marshall T, Mountford PJ, Bion JF. Intestinal permeability, gastric intramucosal pH, and systemic endotoxemia in patients undergoing cardiopulmonary bypass. JAMA. 1996;275(13):1007–1012. [PubMed] [Google Scholar]
- Rietschel E, Kirikae T, Schade U, Mamat U, Schimidt G, Loppnow H, Ulmer A, Zahringer U, Seydel U, Padova F, Schrier M, Brade H. Bacterial endotoxin: molecular relationships of structure to activity and function. FASEB J. 1994;8:217–225. doi: 10.1096/fasebj.8.2.8119492. [DOI] [PubMed] [Google Scholar]
- Safieh-Garabedian B, Poole S, Allchorne A, Winter J, Woolf CJ. Contribution of interleukin-1β to the inflammation-induced increase in nerve growth factor levels and inflammatory hyperalgesia. Br J Pharmacol. 1995;115:1265–1275. doi: 10.1111/j.1476-5381.1995.tb15035.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safieh-Garabedian B, Poole S S, Allchorne A, Kanaan S, Saade N, Woolf CJ. Zinc reduces the hyperalgesia and upregulation of NGF and IL-1β produced by peripheral inflammation in the rat. Neuropharmacology. 1996;35(5):599–603. doi: 10.1016/0028-3908(96)84630-2. [DOI] [PubMed] [Google Scholar]
- Safieh-Garabedian B, Kanaan SA, Haddad JJ, Jaoude PA, Jabbur SJ, Saade NE. Involvement of interleukin-1β, nerve growth factor and prostaglandin E2 in endotoxin-induced localized inflammatory hyperalgesia. Br J Pharmacol. 1997;121:1619–1626. doi: 10.1038/sj.bjp.0701313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sergio G, Esposito P, Ferraro L, Insidioso M, Pane P, Zagaria MP. The limulus test of the cerebrospinal fluid in the diagnosis of disc herniation and its possible correlation with the auto-immune pathogenesis of root pain. Ital J Orthop Traumatol. 1982;8(2):199–204. [PubMed] [Google Scholar]
- Shapira L, Champagne C, Van Dyke TE, Amar S. Strain-dependent activation of monocytes and inflammatory macrophages by lipopolysaccharide of Porphyromonas gingivalis. Infect Immun. 1998;66(6):2736–2742. doi: 10.1128/iai.66.6.2736-2742.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sivri A, Cindas A, Dincer F, Sivri B. Bowel dysfunction and irritable bowel syndrome in fibromyalgia patients. Clin Rheumatol. 1996;15(3):283–286. doi: 10.1007/BF02229708. [DOI] [PubMed] [Google Scholar]
- Sperber AD, Atzmon Y, Neumann L, Weisberg I, Shalit Y, Abu-Shakrah M, Fich A, Buskila D. Fibromyalgia in the irritable bowel syndrome: studies of prevalence and clinical implications. Am J Gastroenterol. 1999;94(12):3541–3546. doi: 10.1111/j.1572-0241.1999.01643.x. [DOI] [PubMed] [Google Scholar]
- Tadano T, Namioka M, Nakagawasai O, Tan-No K, Matsushima K, Endo Y, Kisara K. Induction of nociceptive responses by intrathecal injection of interleukin-1 in mice. Life Sci. 1999;65(3):255–261. doi: 10.1016/s0024-3205(99)00244-1. [DOI] [PubMed] [Google Scholar]
- Takemura T, Makino S, Takao T, Asaba K, Suemaru S, Hashimoto K. Hypothalamic-pituitary-adrenocortical responses to single vs. repeated endotoxin lipopolysaccharide administration in the rat. Brain Res. 1997;767:181–191. doi: 10.1016/s0006-8993(97)00460-5. [DOI] [PubMed] [Google Scholar]
- Thrivikraman KV, Plotsky PM. Absence of glucocorticoid negative feedback to moderate hemorrhage in conscious rats. Am J Physiol. 1993;264:E497–E503. doi: 10.1152/ajpendo.1993.264.4.E497. [DOI] [PubMed] [Google Scholar]
- Tobin RW, Kimmey MB. Painful Diseases of the Gastrointestinal Tract. In: Loeser JD, editor. Bonica’s Management of Pain. 3rd edition. Philadelphia: Lippincott Williams & Wilkins; 2001. pp. 1269–1292. [Google Scholar]
- Tracey KJ, Beutler B, Lowry SF, Merryweather J, Wolpe S, Milsark IW, Hariri RJ, Fahey TJ, 3rd, Zentella A, Albert JD. Shock and tissue injury induced by recombinant human cachectin. Science. 1986;234(4775):470–474. doi: 10.1126/science.3764421. [DOI] [PubMed] [Google Scholar]
- Van Leeuwen PA, Boermeester MA, Houdijk AP, Ferwerda CC, Cuesta MA, Meyer S, Wesdorp RI. Clinical significance of translocation. Gut. 1994;35 Suppl:S28–S34. doi: 10.1136/gut.35.1_suppl.s28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wacnik PW, Kehl LJ, Trempe TM, Ramnaraine ML, Beitz AJ, Wilcox GL. Tumor implantation in mouse humerus evokes movement-related hyperalgesia exceeding that evoked by intramuscular carrageenan. Pain. 2003;101:175–186. doi: 10.1016/s0304-3959(02)00312-3. [DOI] [PubMed] [Google Scholar]
- Walker K, Dray A, Perkins M. Hyperalgesia in rats following intracerebroventricular administration of endotoxin: effect of bradykinin B1 and B2 receptor antagonist treatment. Pain. 1996;65:211–219. doi: 10.1016/0304-3959(95)00195-6. [DOI] [PubMed] [Google Scholar]
- Watkins LR, Wiertelak EP, Furness LW, Maier SF. Illness-induced hyperalgesia is mediated by spinal neuropeptides and excitatory amino acids. Brain Res. 1994a;664:17–24. doi: 10.1016/0006-8993(94)91948-8. [DOI] [PubMed] [Google Scholar]
- Watkins LR, Wiertelak EP, Goehler LE, Mooney-Heiberger K, Martinez J, Furness L, Smith KP, Maier SF. Neurocircuitry of illness-induced hyperalgesia. Brain Res. 1994b;639:283–299. doi: 10.1016/0006-8993(94)91742-6. [DOI] [PubMed] [Google Scholar]
- Watkins LR, Wiertelak EP, Goehler LE, Smith KP, Martin D, Maier SF. Characterization of cytokine induced hyperalgesia. Brain Res. 1994c;654:15–26. doi: 10.1016/0006-8993(94)91566-0. [DOI] [PubMed] [Google Scholar]
- Wiertelak EP, Furness L, Watkins LR, Maier SF. Illness-induced hyperalgesia is mediated by a spinal NMDA-nitric oxide cascade. Brain Res. 1994a;664:9–16. doi: 10.1016/0006-8993(94)91947-x. [DOI] [PubMed] [Google Scholar]
- Wiertelak EP, Smith KP, Furness L, Mooney-Heiberger K, Mayr T, Maier SF, Watkins LR. Acute and conditioned hyperalgesic responses to illness. Pain. 1994b;56:227–234. doi: 10.1016/0304-3959(94)90098-1. [DOI] [PubMed] [Google Scholar]
- Yassin MM, Dsa AB, Parks TG, Soong CV, Halliday MI, Mccaigue MD, Erwin PJ, Rowlands BJ. Lower limb ischemia-reperfusion injury causes endotoxemia and endogenous antiendotoxin antibody consumption but not bacterial translocation. Br J Surg. 1998;85(6):785–789. doi: 10.1046/j.1365-2168.1998.00717.x. [DOI] [PubMed] [Google Scholar]
- Zweigner J, Gramm H-J, Singer OC, Wegscheider K, Schumann RR. High concentrations of lipopolysaccharide-binding protein in serum of patients with sever sepsis or septic shock inhibit the lipopolysaccharide response in human monocytes. Blood. 2001;98(13):3800–3808. doi: 10.1182/blood.v98.13.3800. [DOI] [PubMed] [Google Scholar]