

Abstract
Microglial neuroinflammatory responses affect the onset and progression of Parkinson’s disease (PD). We posit that such neuroinflammatory responses are, in part, mediated by microglial interactions with nitrated and aggregated α-synuclein (α-syn) released from Lewy bodies as a consequence of dopaminergic neuronal degeneration. As disease progresses, secretions from α-syn activated microglia can engage neighboring glial cells in a cycle of autocrine and paracrine amplification of neurotoxic immune products. Such pathogenic processes affect the balance between a microglial neurotrophic and neurotoxic signature. We now report that microglia secrete both neurotoxic and neuroprotective factors following exposure to nitrated α-syn (N-α-syn). Proteomic [surface enhanced laser desorption-time of flight (SELDI-TOF), 1D SDS electrophoresis, and liquid chromatography-tandem mass spectrometry] and limited metabolomic profiling demonstrated that N-α-syn activated microglia secrete inflammatory, regulatory, redox-active, enzymes, and cytoskeletal proteins. Increased extracellular glutamate and cysteine, dimininshed intracellular glutathione and secreted exosomal proteins were also demonstrated. Increased redox active proteins suggest regulatory microglial responses to N-α-syn. These were linked to discontinuous cystatin expression, cathepsin activity, and NF-κB activation. Inhibition of cathepsin B attenuated, in part, N-α-syn-microglial neurotoxicity. These data support multifaceted microglia functions in PD-associated neurodegeneration.
Keywords: alpha synuclein, microglia, Parkinson’s disease, proteomics, glutamate, neuroinflammation
INTRODUCTION
Activated microglia are linked to Parkinson’s disease (PD) pathobiology (McGeer et al., 1988; Hald et al., 2007; Whitton, 2007; Wilms et al., 2007; Yuan et al., 2007). The primary mediators of neuroinflammatory responses in PD are activated microglia. How such microglial activation can be regulated could present diagnostic and therapeutic options for PD (Hermanowicz, 2007; Klegeris et al., 2007; Lipton et al., 2007; Reynolds et al., 2007a). This is of importance as PD remains the second most common neurodegenerative disorder among the elderly and will increase in incidence and prevalence as the baby boomer generation ages (Khandhar and Marks, 2007). PD is characterized by progressive loss of dopaminergic neurons in the substantia nigra pars compacta (SNpc) and their projections to the caudate-putamen of the basal ganglia (BG). A pathological hallmark of disease is the presence of fibrillar α-synuclein (α-syn) inclusions known as Lewy bodies (LB) in the SN that are associated with degenerating neurons (Hodaie et al., 2007). Although the etiology of PD remains unknown, a large body of evidence links inflammation, mitochondrial dysfunction, oxidative stress, and diminished neurotrophic support to disease (Przedborski, 2005; Zhang et al., 2005; Mandemakers et al., 2007).
Activated microglia are closely associated with dying or damaged dopaminergic (DA) neurons (McGeer et al., 1988; Czlonkowska et al., 1996). A link between microglial secretory activities and neurodegeneration is made through the plethora of neurotoxic factors they produce following activation including tumor necrosis factor alpha (TNF-α, reactive oxygen species (ROS), interferons, excitatory amino acids, interleukin (IL)-1β, IL-6, nitric oxide (NO), and leukotrienes (Rogove and Tsirka, 1998; Wu et al., 2002). There is compelling evidence that the release of aggregated and nitrated α-syn (N-α-syn) from dying or damaged SN dopaminergic neurons serves, in part, to provoke a neuroinflammatory response (Zhang et al., 2005; Reynolds et al., 2007b; Thomas et al., 2007) that, left uncontrolled, contributes to neurodegenerative activities and the tempo of disease.
The mechanism of microglia-mediated DA neurotoxicity is linked to the generation of oxidative insult from microglia. DA neurons, in particular, possess reduced antioxidant capacity as a result of low intracellular glutathione, which renders DA neurons vulnerable to oxidative stress relative to other cell types (Loeffler et al., 1994). Ongoing investigations have identified cellular properties including ROS production, morphological transformation, inflammatory cytokine secretion, nuclear factor-kappa B (NF-κB) activation, and a proteome characteristic of an inflammatory response that accompany microglial stimulation with N-α-syn and DA degeneration (Reynolds et al., 2007b; Thomas et al., 2007). Nonetheless, PD progresses slowly over the span of several years to decades suggesting that in addition to neuroinflammation, a compensatory regulatory mechanism is operative for disease (Przedborski, 2005).
Activated microglia possess dual roles for neural repair and disease that are dependent upon specific environmental cues, degree of injury, stage of disease, and brain regions involved. In our previous works, we demonstrated that stimulation of microglia with sciatic and optic nerve fragments, in addition to inducing expression of pro-inflammatory cytokines, incites a neuroprotective phenotype by upregulation of several signal transducer and activator of transcription (STAT) genes, cytoskeletal proteins, lysosomal proteins, and immunoregulatory proteins, and enhanced expression of neurotrophins including brain derived neurotrophic factor (BDNF) and glial derived neurotrophic factor (GDNF) (Glanzer et al., 2007). Based on those observations, we reasoned that microglial response to N-α-syn might also induce underlying compensatory or protective mechanisms to circumvent the injurious effects. Herein, we demonstrate a dual profile for both a toxic and trophic microglial cell in response to N-α-syn. This profile indicates modulation of the glutamate-glutamine cycle and an upregulation of cytoskeletal proteins, regulatory and redox-active proteins. Most importantly, high levels of cysteine were produced in parallel to reduced cathepsin and high cystatin levels. We demonstrate herein that microglia acquire a neurotoxic phenotype after aggregated N-α-syn stimulation. However, a tandem compensatory response through regulation of cysteine secretion, cystatin expression, cathepsin activity, and NF-κB activation was observed. These data provide a yet undefined regulatory role for microglia in PD pathobiology.
MATERIALS AND METHODS
Purification, nitration, and aggregation of recombinant mouse α-syn
Purification, nitration, and aggregation of recombinant mouse α-syn were performed as previously described (Reynolds et al., 2007b; Thomas et al., 2007). Protein concentration was determined from the dry weight of the lyophilized protein.
Isolation, cultivation, and N-α-syn activation of murine microglia
Microglia from C57BL/6J mice neonates (1–2 days old) were prepared using previously described techniques (Dobrenis, 1998). All animal procedures were in accordance with National Institutes of Health guidelines and were approved by the Institutional Animal Care and Use Committee of the University of Nebraska Medical Center. Brains were removed and placed in Hanks’ balanced salt solution (HBSS) at 4°C. The mixed glial cells were cultured for 7 days in complete Dulbecco’s modified Eagle’s medium (DMEM) containing 10% FBS (fetal bovine serum), 10 μg/ml gentamicin and 2 lg/ml macrophage colony stimulating factor (MCSF; a generous gift of Wyeth, Inc., Cambridge, MA). To obtain homogenous microglial cell populations, culture flasks were gently shaken and nonadherent microglia were transferred to new flasks. The flasks were incubated for 30 min to allow the microglia to adhere, and loose cells removed by washing with DMEM. Microglia were plated at 2 × 106 cells/well in 6-well plates in complete DMEM. The adherent microglia obtained using this method was assessed for purity by immunocytochemical analysis for CD11b positive cells and by morphological examination. As previously reported, the microglial cell population was > 98% CD11b+ (Enose, 2005). One week following re-plating, cells were stimulated with 100 nM of aggregated N-α-syn/well or no stimulation for 4 h and 24 h. Media were replaced with serum free DMEM without phenol red or other additives (Invitrogen/GIBCO) and incubated for 24 h in a 37°C, 5% CO2 incubator.
Surface Enhanced Laser Desorption Ionization-time of Flight (SELDI-TOF)
Protein profiling of culture supernatants was performed using SELDI-TOF ProteinChip® assays (Enose et al., 2005; Kadiu et al., 2007) (Ciphergen Biosystems, Fremont, CA). The normal phase NP20 protein chip was selected to profile culture supernatants for low and high abundant proteins based on the non-discriminating binding affinity of its hydrophobic surface to proteins regardless of their chemical structure. An aliquot of culture supernatant (2 μg of protein) was applied onto each spot and air-dried. To each air-dried spot, 0.5 μl of 50% sinapic acid (SPA) was added and air-dried. The 50% SPA was prepared as a saturated solution in solvent containing 30% acetonitrile (ACN), 15% isopropanol, 0.5% trifluoroacetic acid, and 0.05% Triton X-100. SELDI-TOF spectra were generated on supernatants collected from three separate microglia cultures. Supernatants collected at each time point were run in triplicate, on three protein chips at different spots to control for instrumental and experimental variability. Molecular mass/charge (m/z) ratios of laser beam ionized proteins were measured in a ProteinChip® PBS II reader. The ProteinChip® Reader was externally calibrated for each analysis using the four standard proteins: bovine insulin (5,733.6 Da), cytochrome C (12,230.9 Da), superoxide dismutase (SOD) (15,591.4 Da), and beta-lactoglobulin (18,363.3 Da). Acquired spectra were analyzed using ProteinChip® software 3.2 (Ciphergen Biosystems). The ProteinChip® analyses were performed in three independent experiments, and the data set from each microglia comprised a minimum of 8 spectra. All spectra were combined in one file and normalized to total ion current. Peaks were automatically detected using the Biomarker Wizard of ProteinChip® software 3.2. Peak detection parameters were first pass signal/noise (S/N) ratio = 5, second pass S/N ratio = 2, mass tolerance = 0.5%, and estimated peaks were included in completion of clustering.
Protein identification by LC-MS/MS
Following in gel tryptic digestion and column purification, dried peptide samples from cell supernatant fluids were reconstituted in 0.1% formic acid/HPLC-grade water, detected on a ProteomeX LCQ™ DECA XP Plus mass spectrometer (ThermoElectron, Inc. Waltham, MA), and identified using BioWorks 3.1SR software. Proteins identified by peptides having a Unified Score (BioWorks 3.1SR, ThermoElectron, Inc. Waltham, MA) > 3000 were marked for further analysis (Enose et al., 2005; Kadiu et al., 2007).
Extracellular supernatant fractionation
Culture supernatants were concentrated using Centriplus™ centrifugal filter devices (Millipore, Billerica, MA) and dialyzed against MiliQ water using Cellu·Sep® H1 cellulose membranes (Membrane Filtration Products). Samples of culture supernatants were fractionated using 1D SDS-PAGE. Each 200 μg sample was diluted with NuPAGE® loading buffer and separated using NuPAGE® Novex 10% Bis-Tris (Invitrogen) gel. After electrophoresis, the gels were stained with Coomassie Brilliant blue G-colloidal concentrate (Sigma-Aldrich, St. Louis, MO).
Nuclear/Cytosol Fractionation
Cell lysates were prepared from N-α-syn-stimulated and control microglia. Cells were rinsed 3X with PBS, following gentle scraping, cells were collected following centrifugation at 600×g for 5 min. Cytosol and nuclear fractions were prepared using the Nuclear/Cytosol Fractionation Kit (BioVision; Mountain View, CA) according to manufacturer’s instructions.
Western blot assays
A total of 20 μg of each sample was loaded onto 4–12% Bis-Tris NuPAGE Novex gels (Invitrogen) and transferred onto PVDF membranes (BioRad, Hercules, CA). Primary antibodies to calmodulin (1:1000) and 14-3-3 σ (1:200) (Millipore), biliverdin reductase (1:5000), thioredoxin (1:2000), β-actin (1:5000), ferritin light chain (1:1000), galectin 3 (1:1000) (Abcam, Cambridge, MA), NF-κB p65 and p50 (1:200) (Santa Cruz Biotechnology, Inc., Santa Cruz, CA), cystatin B (1:500), and cathepsin B (1:1000) (R & D Systems) were used for analyses. Blots were probed with the respective horseradish peroxidase-conjugated secondary antibodies (1:5000; Invitrogen) and detected using SuperSignal West Pico Chemiluminescent substrate (Pierce Biotechnology, Inc). The intensity of protein bands was quantified using ImageJ and normalized to Gapdh (1:5000, Santa Cruz Biotechnology, Inc.).
Metabolite assays
Microglia were cultured with and without N-α-syn in media without added glutamine for 2, 4, 8, and 24 h during stimulation. For analysis of extracellular metabolites, culture supernatants were collected at each time point and mixed with equal volumes of metaphosphoric acid solution (16.8 mg/ml HPO3, 2 mg/ml EDTA and 9 mg/ml NaCl). For analysis of intracellular metabolites, cells were washed three times with ice cold PBS, maintained on ice in PBS, and detached with a tissue culture scraper. To measure protein concentration, an aliquot of the cell suspension was mixed with an equal volume of lysis buffer (0.1 M sodium phosphate, pH 7.4, containing 0.1% Triton-X100, 10 μl/ml protease inhibitor cocktail (Sigma), 25 μg/ml tosyllysine chloromethylketone and 5 μg/ml phenylmethylsulfonyl fluoride). Samples were stored at −80°C until further use. Data are representative of three independent experiments performed in triplicate.
High Performance Liquid Chromatography (HPLC) analyses
For analysis of metabolites, the metaphosphoric acid fixed samples of cells or culture supernatant were thawed, vortexed, and clarified by centrifugation at 14000 × g for 10 min at 4°C. Thiol metabolites in protein free extracts were derivatized with monoiodoacetic acid (7 mM) followed by mixing with an equal volume of 2,4-dinitrofluorobenzene solution (1.5% v/v in absolute ethanol) and analyzed by HPLC using u-Bondapak-NH2 300×3.9 mm column (Waters) with a methanol-acetate gradient as described previously (Mosharov et al., 2000). The concentration of metabolites in the samples was determined using a standard curve generated for each metabolite of interest. Results were normalized to protein concentration in each sample. The protein concentration in samples was measured using the Bradford reagent (Bio-Rad; Hercules, CA) with bovine serum albumin as standard.
Intracellular Glutathione (GSH)
Microglia were cultured with and without N-α-syn for 24 h in media without added glutamine. Cells were washed three times with ice cold PBS, and detached with a tissue culture scraper. Cell suspensions were collected in triplicate and assayed for GSH, GSSG, and total glutathione levels with the Biovision Glutathione Assay Kit (Biovision, Mountain View, CA) according to manufacturer’s protocol. Briefly, cells were homogenized in 100 μl assay buffer, and glutathione was stabilized with 6N perchloric acid. For assay, a standard curve was performed with GSH standard. Samples were prepared according to protocol for determination of GSH, GSSG, and total glutathione. Reducing Agent Mix (Biovision) was added to convert GSSG to GSH. An o-phthalaldehyde probe was added to the samples for 40 min. Samples were then assessed in a 96 well fluorometer plate using a SpectraMAX GEMINI (Molecular Devices, Sunnyvale, CA) at excitation/emission of 340/450 nm.
Cathepsin B activity
Microglia were seeded onto sterile glass coverslips at 105 cells per well and allowed to adhere for 24 h prior to stimulation with aggregated N-α-syn. Cathepsin B activity was determined using the CV-Cathepsin B Detection Kit (BIOMOL International LP). For measurement of cathepsin B activity, arginine conjugated cresyl violet [CV-(RR)2, a red fluorogenic substrate when unconjugated] was added to the culture media and allowed to incubate for 60 min in a 37°C, 5% CO2 incubator. The attached ArgArg group is a substrate for cathepsin B cleavage. Hoechst stain was added at 0.5% v/v and incubated with cells for an additional 5 min to stain nuclei. Fluorescence intensity of unconjugated CV was determined at 550 nm for excitation and 610 nm for emission of 3 wells/10 fields per well/experimental group. Mean Fluorescence intensity of unconjugated CV was normalized to the mean intensities of Hoechst staining at 480 nm for excitation and 540 nm for emission for each sample.
Cathepsin B inhibition and neurotoxicity
MES 23.5 cells, kindly provided by Dr. Stanley Appel (Methodist Hospital; Houston, TX), were cultured in 75-cm2 flasks in DMEM/F12 with 15 mM HEPES (Invitrogen) containing N2 supplement (Invitrogen), 100 U/ml of penicillin, 100 μg/ml streptomycin, and 5% FBS. MES23.5 cells is a cell line derived using somatic cell fusion of rat embryonic mesencephalon cells and the murine neuroblastoma-glioma cell line N18TG2 that produce dopamine and express tyrosine hydroxylase (Crawford et al, 1992). Cells were grown to 80% confluence then plated at a density of 10 × 104 cells on sterile glass coverslips. Microglia were plated in 6 well plates and incubated for 1 h with or without 1 μM of cathepsin B inhibitors, CA-074 [cell impermeable] or CA-074 Me [cell permeable] (Sigma), in a 37°C, 5% CO2 incubator to inhibit residual cathepsin B expression (Gan et al., 2004). N-α-syn (100 nM) was then added directly to this media for 24 h. Unstimulated microglia and N-α-syn stimulation alone served as controls. Following stimulation, supernatants were removed and added to cultures of MES23.5 cells for an additional 24 h. MES 23.5 cell viability was assessed using the Live/Dead cytotoxicity assay (Invitrogen) as previously described (Reynolds et al., 2007b). Live cells were distinguished by the uptake of calcein AM to acquire a green fluorescence [excitation/emission (ex/em) 495/515 nm], while dead cells acquired a red fluorescence (ex/em 495/635 nm) due to the uptake of ethidium homodimer-1 (EthD-1). Cell enumerations were performed using fluorescence microscopy (200× magnification, n= 6 wells per group, 3 frames per well).
Statistical analyses
All values are expressed as means ± SEM. Differences among means were analyzed by one-way ANOVA followed by Bonferroni post-hoc testing for pair-wise comparison.
RESULTS
N-α-syn-activated microglia secretions and neuroinflammatory responses
In previous studies we demonstrated that a temporal pattern of microglial activation occurs following stimulation of microglia with N-α-syn, and consists of increased expression of factors attributed to inflammatory responses, oxidative stress and significant neurocytotoxicity (Zhang et al., 2005; Reynolds et al., 2007b; Thomas et al., 2007). The secretion of potentially neurotoxic compounds characterizes the progression of an activated microglial phenotype to a neurotoxic phenotype, while secretion of trophic factors that support neuronal survival and cell-cell communication characterizes a quiescent microglial phenotype. To analyze the secretory profile induced upon N-α-syn stimulation, supernatants from microglia stimulated for 4 h with N-α-syn were collected at 8, 16, and 24 h post-stimulation. Supernatants were first screened for low and high abundant proteins using SELDI-TOF analysis. In these experiments, spectra were generated from cell supernatants collected from control and stimulated cultures at each time-point in three separate experiments using a NP20 Protein Chip. Signal to noise ratios of 5 and 2 for first and second passes respectively and 0.5% mass tolerance (Enose et al., 2005). Representative spectra of culture supernatants obtained from microglia simulated with aggregated N-α-syn revealed that microglial secretory constituents were significantly altered compared to those of unstimulated microglial controls (Fig. 1A), and consisted of several peaks coinciding with molecular masses of calcium regulatory proteins (calcyclin, 10,051; calmodulin, 16,706 Da; calvasculin, 11,721 Da), redox-active proteins (thioredoxin, 11,544 Da), and TNF-α (17, 907 Da). In contrast, secretory profiles of microglia cultured in the presence of unaggregated N-α-syn revealed similar profiles to unstimulated control microglia (data not shown). These results determined that microglial secretory products were significantly changed by stimulation with N-α-syn, and encouraged us to pursue 1D SDS PAGE methods to isolate and identify differentially secreted proteins upon stimulation. Use of 1D SDS PAGE followed by LC-MS/MS analysis has been shown by our previous works to be reliable in identifying differentially expressed proteins (Enose et al., 2005; Glanzer et al., 2007; Ciborowski et al., 2007) and identification of N-α-syn following immunopreciptiation (Reynolds et al., 2007b; Benner et al., in press) with confirmatory western blot analyses. LC-MS/MS analyses of SDS-PAGE fractions identified 30 proteins common to supernatants from both stimulated and control microglia (data not shown). Analysis of proteins secreted from N-α-syn-stimulated microglia by LC-MS/MS identified those proteins detected by SELDI-TOF. Microglial expressed proteins were considered using criteria that at least two peptides from a protein were detected with a unified score (BioWorks, 3.1SR) greater than 3000 as previously described (Glanzer et al., 2007). Proteins were identified as either greater or lower in abundance based on the number of peptides detected by LC-MS/MS, or the presence of a protein identified with high confidence in culture supernatants of one group but not in culture supernatants of the other. Altogether, LC-MS/MS analysis revealed 40 increased and 34 decreased in abundance within culture supernatants when compared to unstimulated controls (Table 1); seven of which were validated by western blot analysis (Fig. 1C) including decreased secretion of actin, galectin 3, and 14-3-3 sigma along with increased expression of biliverdin reductase, calmodulin, ferritin light chain, and thioredoxin in culture supernatants of N-α-syn-stimulated microglia. Sixteen proteins found in greater abundance in supernatants of aggregated N-α-syn-stimulated microglia were classified as being involved in both cellular activation and regulation. For example, regulators of oxidative stress including thioredoxin, biliverdin reductase, and ferritin light chain were abundantly secreted by stimulated microglia. Also found in these supernatants were cell-morphogenesis proteins L-plastin, actin-related protein 3 homolog B (ARP3) and adenylyl cyclase-associated protein 1 (CAP1); the lysosomal proteins, α-N-acetylglucosaminidase (NAGLU) and N-acetylgalactosamine-6-sulfate sulfatase (GALNS); and the calcium binding proteins EF-hand domain-containing protein 2 (EFHD2, swiprosin1) and nucleobindin (Islam et al., 2006). Proteins less abundant in supernatant fluids from N-α-syn-stimulated microglial supernatants including calcyclin, β-actin, histone H4, triose-phosphate isomerase, phosphoglycerate mutase 1, cathepsin S, 14-3-3σ, and ubiquitin were reported to be associated with exosomal vesicles, whereas only three of the proteins (ferritin light chain, CAP1, and L-plastin) that were more abundant in N-α-syn-stimulated microglial supernatants have been associated with exosomes (Thery et al., 2001; Wubbolts et al., 2003; Pisitkun et al., 2004; Potolicchio et al., 2005; Faure et al., 2006).
Figure 1.
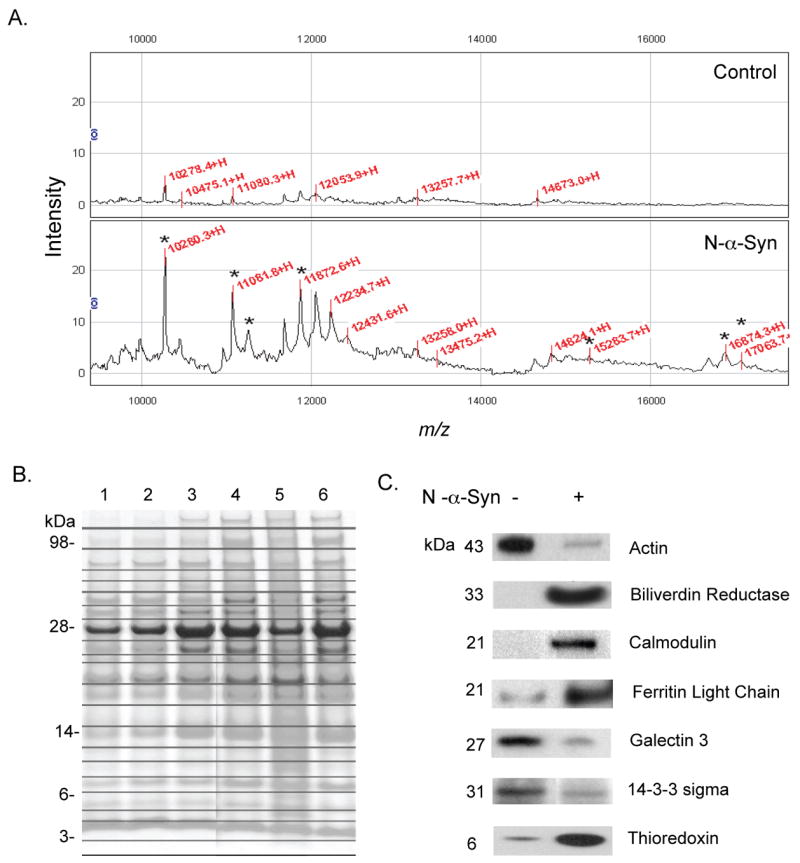
SELDI-TOF, 1D SDS PAGE, and Western blot analyses of supernatant fluids obtained from N-α-syn-activated microglial. Representative SELDI-TOF spectral analysis (region 10–20 kDa) of untreated-control (top panel) and N-α-syn-stimulated microglia (bottom panel) at 16 h post-stimulation, shown in A. Marked by an asterisk are upregulated and uniquely expressed peaks corresponding in molecular weight within 1% of mass tolerance to proteins identified by LC-MS/MS. These include calcyclin (10,051 Da), thioredoxin (11,544 Da), calvasculin (11,721 Da), calmodulin (16,706 Da), and TNF-α (17,907 Da). Bands were excised from 1D SDS PAGE gel, digested by trypsin, and sequenced by LC-MS/MS. Lanes are supernatant fluids obtained from control (unstimulated) microglial = [Lanes 1–3] and supernatants from N-α-syn-activated microglia [Lanes 4–6] collected 8 h, 16 h, and 24 h post-stimulation, respectively. (B). Representative Western blots of supernatant fluids from control and N-α-syn-stimulated microglia 16 h post-stimulation for proteins identified by LC-MS/MS (C).
Table 1.
Secretome of N-α-syn-Stimulated Microglia
| Protein name | Mol. Wt. | PI | Acc. No. | Peptides |
|---|---|---|---|---|
| Proteins with increased abundancee | ||||
| Regulatory | ||||
| SWIPROSIN 1/EF hand domain containing protein 2 (Efhd2) | 26,800 | 5.07 | Q8C845 | 2 |
| Csf1r protein | 109,252 | 5.84 | Q6NXV8 | 5 |
| Adenylyl cyclase-associated protein 1(CAP 1) | 51,444 | 7.3 | P40124 | 2 |
| Calmodulin | 16706 | 4.09 | P62204 | 2 |
| Rho GDP dissociation inhibitor (GDI) alpha | 23407 | 5.12 | Q5XI73 | 2 |
| Nucleobindin | 53425 | 4.90 | Q8BRD3 | 3 |
| Arpc4 protein | 19607 | 8.53 | Q7TPD9 | 2 |
| L-Plastin | 70149 | 5.20 | Q61233 | 3 |
| Calvasculin | 11721 | 5.23 | P07091 | 3 |
| Structural | ||||
| Cofilin-1 | 18428 | 8.26 | P18760 | 3 |
| Lamin A. | 74238 | 6.54 | P48678 | 5 |
| Redox | ||||
| Thioredoxin 1 | 11,544 | 4.8 | P10639 | 2 |
| Ferritin light chain 1 | 20,671 | 5.66 | P29391 | 3 |
| Biliverdin reductase | 33,525 | 6.53 | Q9CY64 | 2 |
| Vesicle amine transport protein (Vat1) | 42,522 | 5.96 | Q5RKP0 | 2 |
| Ferritin heavy chain | 20935 | 5.53 | P09528 | 2 |
| Enzyme | ||||
| Aspartate aminotransferase | 46,100 | 6.75 | P05201 | 2 |
| Naglu | 82,611 | 6.14 | O54752 | 2 |
| N-acetylgalactosamine-6-sulfate sulfatase.- GALNS | 57,673 | 6.13 | Q9JHK9 | 2 |
| Glycogen phosphorylase | 11740 | 6.27 | Q9CZL5 | 2 |
| Phosphoglycerate mutase 1 | 28701 | 6.75 | Q9DBJ1 | 2 |
| Putative membrane-bound dipeptidase-2 | 52664 | 6.6 | Q8C255 | 2 |
| Pyruvate kinase, isozyme M2 | 57756 | 7.42 | P52480 | 8 |
| Transketolase | 60583 | 6.54 | Q9ESA0 | 8 |
| D-dopachrome tautomerase | 12946 | 6.15 | O35215 | 2 |
| Dihydropyrimidinase-related protein 2 | 11507 | 4.79 | Q63826 | 3 |
| Try10-like trypsinogen precursor | 26531 | 4.83 | Q7M754 | 3 |
| Other | ||||
| Fibronectin precursor | 272489 | 5.39 | P11276 | 3 |
| Lyzs protein | 5955 | 8.94 | Q8VE78 | 3 |
| Brain acid soluble protein 1 | 21955 | 4.5 | Q91XV3 | 4 |
| Hypothetical protein | 28630 | 5.13 | Q3U9X3 | 8 |
| Beta-galactoside-binding lectin | 15914 | 9.01 | Q61357 | 4 |
| Complement component 1 | 25992 | 8.84 | Q6DI63 | 3 |
| Elongation factor 2 (EF-2) | 95183 | 6.42 | P58252 | 2 |
| Ezrin (p81) | 69276 | 5.83 | P26040 | 5 |
| Krt2-4 protein | 56283 | 8.32 | P07744 | 3 |
| Monocyte differentiation antigen CD14 | 39204 | 5.08 | P10810 | 2 |
| plasma phospholipid transfer protein | 54453 | 6.17 | P55065 | 12 |
| Radixin | 68601 | 5.85 | P26043 | 5 |
| Stathmin | 17143 | 5.76 | P54227 | 2 |
| Proteins with decreased abundancee | ||||
| Regulatory | ||||
| Ubiquitin | 8,565 | 6.56 | P62991 | 3 |
| Calcyclin | 10,051 | 5.3 | P14069 | 3 |
| 14-3-3 protein sigma | 27,833 | 4.72 | Q9JJ20 | 5 |
| Galectin 3 | 27,384 | 8.5 | P16110 | 9 |
| Structural/cytoskeletal | ||||
| Beta-actin | 41,737 | 5.78 | P60710 | 26 |
| Gamma actin-like protein | 43601 | 5.11 | Q9QZ83 | 11 |
| Nonmuscle heavy chain myosin II-A. | 226226 | 5.54 | Q8VDD5 | 7 |
| Tropomyosin 5 | 32863 | 4.68 | P21107 | 5 |
| Tropomyosin 1 alpha chain | 32681 | 4.69 | P58771 | 2 |
| Profilin-1 | 14826 | 8.5 | P62962 | 5 |
| Tropomyosin 3, gamma | 33149 | 4.73 | Q8K0Z5 | 3 |
| Talin | 269833 | 5.82 | P26039 | 8 |
| Redox | ||||
| Peroxiredoxin 6 | 24739 | 5.72 | O08709 | 2 |
| Enzyme | ||||
| Lysozyme M precursor | 16,689 | 9.11 | P08905 | 2 |
| Phosphoglycerate mutase 1 | 28,701 | 6.75 | Q9DBJ1 | 2 |
| Cathepsin S | 38,707 | 6.51 | Q8BSZ5 | 2 |
| Dipeptidylpeptidase 7 | 56254 | 5.17 | Q8R082 | 3 |
| Phosphoglycerate kinase 1 | 44405 | 7.52 | P09411 | 11 |
| Xaa-Pro dipeptidase | 54898 | 5.5 | Q11136 | 2 |
| Glucose-6-phosphate isomerase | 61718 | 6.19 | P34795 | 4 |
| Glutathione S-transferase Mu 1 | 25839 | 8.14 | P10649 | 2 |
| Mannosidase, beta A, lysosomal | 100831 | 6.82 | Q8K2I4 | 3 |
| Ldh1 protein | 34503 | 8.18 | Q99K20 | 2 |
| Ubiquitin-conjugating enzyme | 17862 | 8.68 | P68037 | 3 |
| Other | ||||
| Beta-2 microglobulin | 13,823 | 7.8 | P01887 | 2 |
| FK506-binding protein 12 | 11,791 | 8.08 | P26883 | 2 |
| Histone H4 | 11,236 | 11.36 | P62806 | 2 |
| Phosphatidylethanolamine-binding protein (PEBP) | 20,699 | 5.19 | P70296 | 2 |
| Extracellular matrix protein | 48359 | 5.72 | Q9Z2R8 | 2 |
| Proteinase inhibitor Spi3 | 42599 | 5.53 | Q60854 | 4 |
| Ribonuclease/angiogenin inhibitor 1 | 49816 | 4.69 | Q91VI7 | 2 |
| Clathrin, heavy polypeptide | 191986 | 5.48 | Q5SXR6 | 4 |
| Histone H2A. I | 14004 | 11.05 | P22752 | 2 |
| Histone H2B F | 13805 | 10.32 | P10853 | 2 |
The CID spectra were compared against those of the EMBL nonredundant protein database by using SEQUEST (ThermoElectron, San Jose, CA). After filtering the results based on cross correlation Xcorr (cutoffs of 2.0 for [M+ H]1+, 2.5 for [M + 2H]2+, and 3.0 for [M+ 3H]3+), peptides with scores greater than 3000 and meeting delta cross-correlation scores ( Cn) > 0.3, and fragment ion numbers > 60% were deemed valid by these SEQUEST criteria thresholds, which have been determined to afford greater than 95% confidence level in peptide identification.
Theoretical molecular mass
Isoelectric point
Accession numbers for UniProt (accessible at http://www.ipr.uniprot.org/search/textSearch.shtml)
Number of peptides identified for each protein selected based on the above mentioned criteria. Proteins were considered if 2 or more peptides were identified.
Proteins were increased or decreased in supernatants of microglia stimulated with N-α-syn for 4 hrs when compared to unstimulated microglia (controls).
Metabolic response of microglia to N-α-syn stimulation
In addition to activation of signaling pathways that incite neuroinflammatory responses, increased expression and secretion of redox-active proteins suggested that a multifaceted microglial response to N-α-syn may affect PD, and that regulatory mechanisms which counter oxidative stress may accompany the inflammatory response. To assess the latter possibility, we determined the concentrations of metabolites involved in the glutamate-glutamine cycle. We theorized that as a result of stimulation with N-α-syn, changes in the levels of microglial metabolites would occur relative to unstimulated controls (Fig. 2A). Stimulation with N-α-syn resulted in increased secretion of glutamate in supernatants collected at time points measured over a 24 h time period compared to supernatants collected from unstimulated control cells (32.5 ± 0.5 μM versus 21.0 ± 1.0 μM, P< 0.0001 at 8h of stimulation) and continued to rise, so that by 24 h the concentration was 45.5 ± 1.5 μM compared to 26.5 ± 2.5 μM for control (P< 0.0001). Moreover, extracellular cysteine was also found significantly elevated in response to N-α-syn compared to controls (16.6 ± 0.7 μM versus 6.96 ± 0.03 μM, P< 0.0001; 8 h). As expected, increased extracellular cysteine was accompanied by decreased cystine. This was seen at 24 h following exposure to N-α-syn (270.0 μM versus 286.0 ± 3.0 μM in controls, P< 0.0001 at 24 h). Intracellular concentrations of both glutamate and cysteine were decreased in stimulated microglia compared to controls (Fig. 2B).
Figure 2.
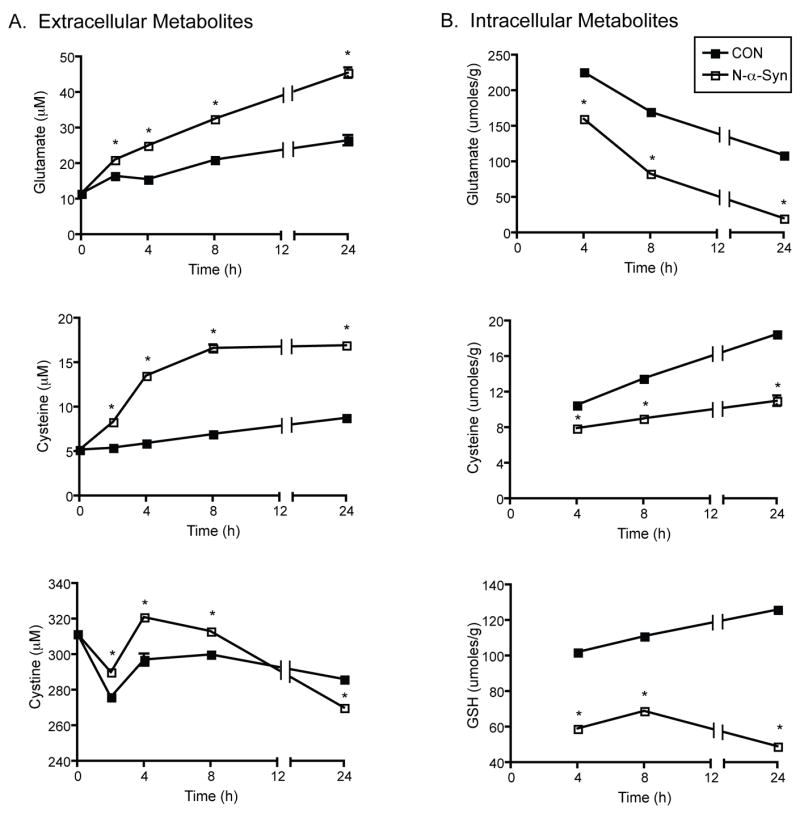
N-α-Syn activated microglial metabolic responses. Microglia were stimulated with N-α-syn over the course of 24h and culture supernatants were collected at specified times following stimulation for analysis of extracellular metabolites, shown in A. Analysis of intracellular metabolites following engagement with N-α-syn is shown in B. Mean metabolite concentrations are presented as values ± SEM, and are representative of three separate experiments, (*P < .001, v. unstimulated control).
Determination of intracellular GSH concentration by HPLC indicated that exposure to N-α-syn resulted in its 2 fold rapid depletion at 2 h to 31.3 ± 0.6 μmoles/g protein compared to controls with 60.0 ± 3.3 μmoles/g protein (data not shown, P< 0.001). During continuous exposure with N-α-syn, glutathione levels rose to nearly pre-stimulatory levels by 8 h (69.0 ± 2 μmoles/g protein versus 111 ± 2 μmoles/g protein), and the levels again dropped by 24h (49 ± 1 μmoles/g protein). In contrast, glutathione levels steadily rose in cell lysates of unstimulated microglia to 126 ± 1 μmoles/g protein (P< 0.0001, Fig. 2B) by 24 h. In addition, the ratio of GSH to oxidized glutathione (GSSG), an indicator of cellular redox status, declined steeply within 2 h in response to N-α-syn stimulation (34.2 ± 1.0 versus 89.4 ± 2.6, P< 0.001), however this ratio gradually rebounded (70.0 ± 2.0 and 65.0 ± 1.0 by 8 and 24 h respectively) but remained at about 2 fold lower throughout the course of stimulation in comparison to unstimulated control (114.0 ± 3.0 and 163.0 ± 1.0 by 8 and 24 h respectively) (data not shown). Investigation of GSH and GSSG levels using the Biovision glutathione assay kit confirmed these results, and revealed that by 24 h the GSH levels were significantly decreased (P< 0.001), and corresponded with decreased GSH/GSSG ratio and total GSH plus GSSG levels compared to levels in unstimulated cell lysates (data not shown).
Cathepsin B activity and N-α-syn microglial activation and cytotoxicity
Microglia have been shown to express a number of proteases, including the cysteine protease cathepsin B, where it may play a role in degradation of matrix proteins and associated signal transduction molecules that can induce neuronal apoptosis (Kingham and Pocock, 2001). To further investigate the microglial response to N-α-syn in regulating neurotoxicity a series of tests were performed to assess the differential expression of cathepsin B, and the cysteine protease inhibitor cystatin B, over the course of stimulation. Western blot analysis revealed a significant increase in expression of cystatin B in microglial cell lysates following 4 hours of N-α-syn stimulation compared to unstimulated controls and decreased expression of cathepsin B (Fig. 3A). However, by 24 h of N-α-syn stimulation, cystatin B expression was decreased, whereas expression of cathepsin B in cell lysates was not significantly changed. Since regulation of cathespin B is primarily post-translational, increased expression of cystatin B would more closely correspond to decreased cathepsin B activity rather than decreased protein expression, therefore we investigated the enzymatic activity of cathespin B before and during stimulation with N-α-syn. Activity of intracellular cathepsin B was significantly decreased in microglia following 4 h of stimulation with N-α-syn compared to basal activity before stimulation, and then enzymatic activity increased significantly over pre-stimulatory level by 24 h (Fig. 3B).
Figure 3.
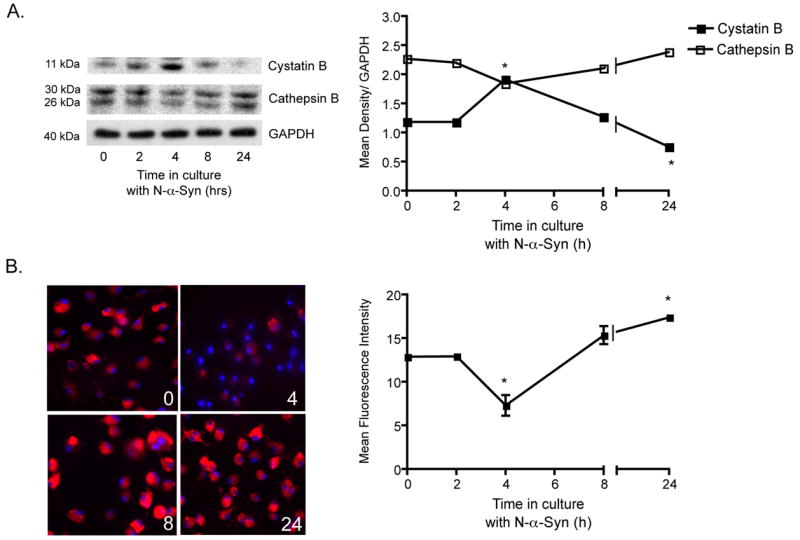
Cathepsin B expression and functional activity following N-α-syn-microglial activation. Comparative analysis of cystatin B and cathepsin B protein expression in microglia at several time points during stimulation with aggregated N-α-syn by western blot, shown in A. Mean density of protein bands (± SEM) were normalized to GAPDH expression on the same blot (*P < 0.05 compared to unstimulated microglia at 0h). (B) Cathepsin B enzymatic activity prior to stimulation (0 h) and at 2 h, 4 h, 8 h, and 24 h after stimulation with N-α-syn. Enzymatic activity is visualized with the red fluorogenic substrate, CV-(RR)2 and nuclei are stained blue with Hoechst dye. Fluorescence intensities were determined by ImageQuant and are presented as mean ± SEM for n = 3 fields for 4 replicates (P < 0.05 compared to mean at 0 h).
We next investigated whether cathepsin B could also contribute to N-α-syn stimulated microglial cytotoxicity. Microglia were stimulated with N-α-syn in the presence of either the selective cathepsin B inhibitor CA-074 or the cell permeable form CA-074 Me for 24 h, when cathepsin B activity was elevated following N-α-syn stimulation alone. The resultant supernatants were then added to cultures of dopaminergic MES23.5 cells (a kind gift from Dr. Stan Appel), for an additional 24 h for cell death measurement (Fig. 4). Supernatants from unstimulated microglia did not induce DA cell death, while N-α-syn stimulation alone resulted in significant cytotoxicity (26% of control, P < 0.001). Inhibition of secreted Cathepsin B by the cell impermeable inhibitor, CA-074, partially protected DA cells against N-α-syn-microglial mediated cell death (53% protection v. N-α-syn alone, P < 0.01), while the cell permeable form, CA-074 Me resulted in heightened DA cell protection (87%, P < 0.001).
Figure 4.
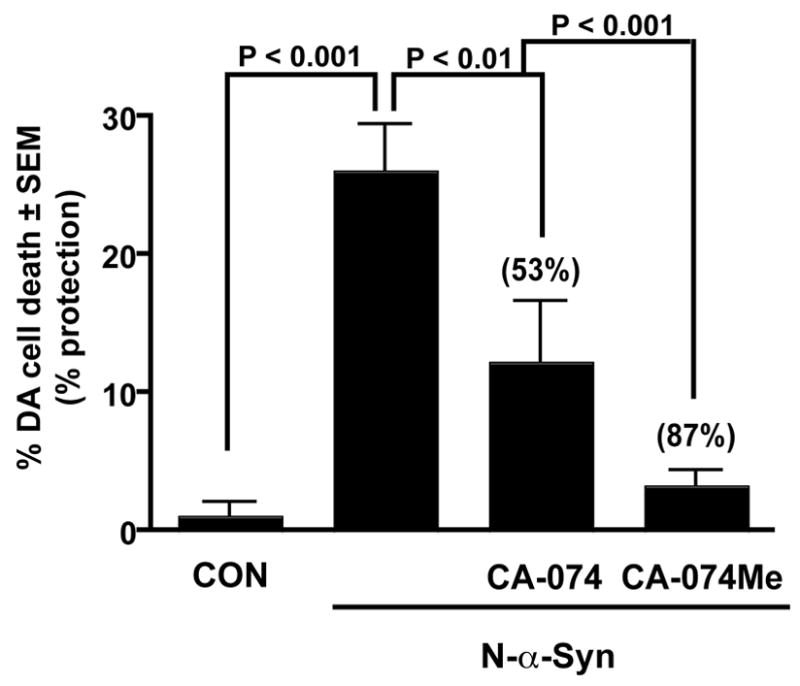
Effect of Cathepsin B inhibition on N-α-syn-mediated cytotoxicity. Supernatants from N-α-syn stimulated microglia induced significant DA cell death. Whereas, inhibition of cathepsin B activity by either the cell impermeable inhibitor CA-074 or the cell-permeable inhibitor CA-074 Me resulted in partial protection from N-α-syn mediated DA cell death, shown. Values are shown as mean dead DA cells ± SEM for n = 6 replicates per treatment paradigm.
NF-κB subunit translocation and N-α-syn microglial activation
Activation of the NF-κB pathway leads to the production of inflammatory mediators implicated in inducing neuronal injury (Qin et al., 2005). Indeed, upregulation of NF-κB transcription is induced upon stimulation with N-α-syn activation, as well as being significantly increased in the SN of post-mortem brains from PD patients (Reynolds et al., 2007b). In addition, analysis of the microglial secretome identified many proteins, including for example phosphatidylethanolamine-binding protein, thioredoxin, and FK506-binding protein 12 (FKBP1A) that are downstream of either the NF-κB or mitogen-signaling pathways, suggesting the involvement of NF-κB pathway to initiate not only the inflammatory response, but also regulation of the response. To investigate whether this dual toxic and trophic reaction to N-α-syn by microglia was contingent on activation of the NF-κB pathway, we assessed NF-κB translocation to the nucleus where it can bind DNA and activate transcription of genes encoding proteins involved in an inflammatory response. As early as 1-1.5 h stimulation of microglia with N-α-syn, NF-κB p50 and p65 were increased in the nucleus, and coincided with decreased expression of these subunits within the cytosolic fractions (Fig. 5A). By 4 h stimulation however, the nuclear localization of NF-κB subunit p50 was decreased compared to 1.5 h stimulation, and remained repressed through 8 h. By 24 h of stimulation, the p50 subunit was once again diminished in the cytosol and increased within the nucleus (Fig. 5B). Notably, compared to 0 h, p65 levels in the nucleus were high with correspondingly diminished levels within the cytosolic fraction by 1 h stimulation (Fig. 5C). These data suggest that the microglial compensatory response, albeit temporary, is also reflective of discontinuous activation of the NF-κB pathway and downstream inflammatory cascades.
Figure 5.
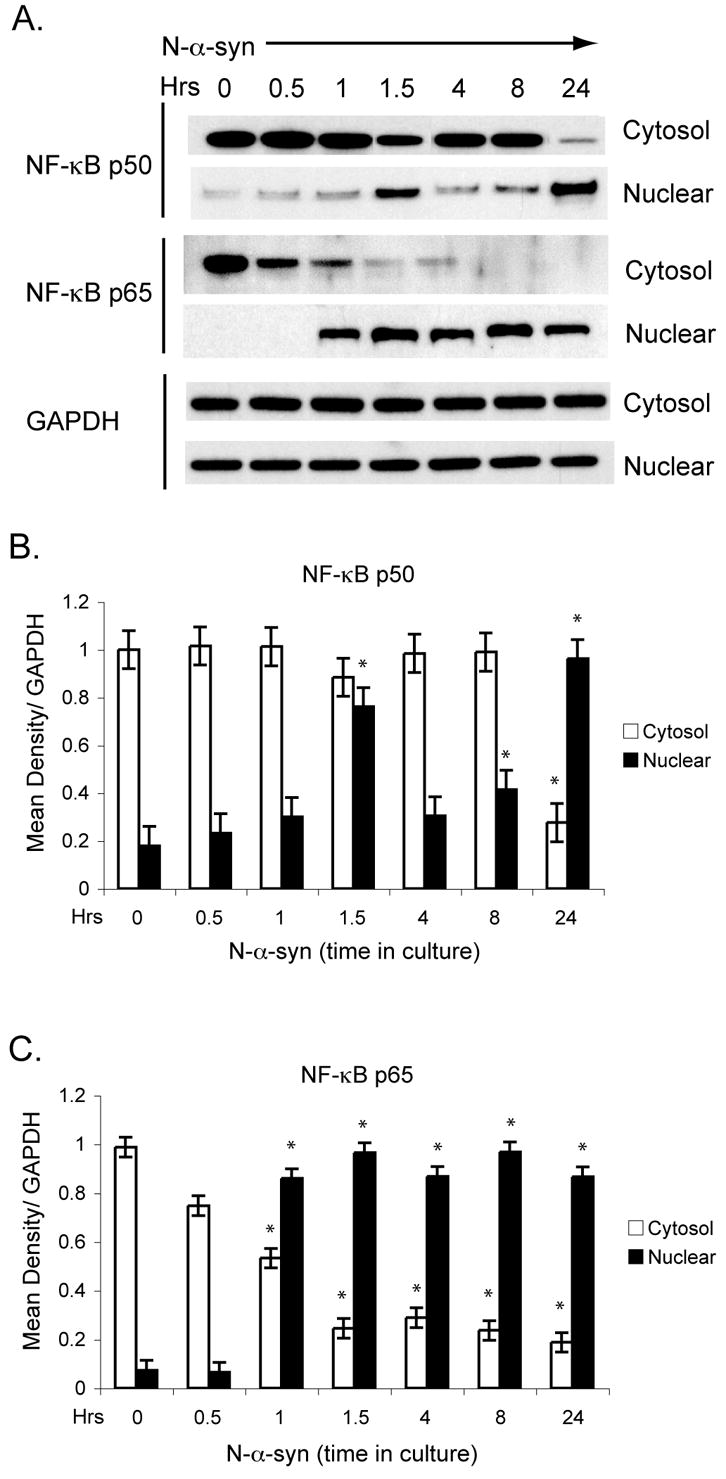
Nuclear translocation of NF-κB subunits. Cytosol (top) and nuclear (bottom) fractions were prepared from microglia stimulated with N-α-syn for subsequent time-points and assessed for expression of NF-κB subunits NFκB1/ p50 and RelA/p65 by western blot (A). Mean density of protein bands for NFκB1/p50 (B) and RelA/p65 (C) by western blot were normalized to GAPDH in the same sample. Values are shown mean ± SEM for n = 3 replicates per time point (*P < 0.01 compared to 0h).
DISCUSSION
Stimulation of microglia with aggregated, N-α-syn results in cellular activation, characterized by morphological transformation and enhanced secretion of ROS (Zhang et al., 2005; Thomas et al., 2007), inflammatory cytokines and chemokines. N-α-syn-mediated microglial activation is further defined by increased NF-κB activation and altered proteome and secretome profiles, which correlate with enhanced N-α-syn-mediated neurotoxicity (Reynolds et al., 2007b). However, in addition to up-regulation of various toxic inflammatory factors, many redox-active and neuroprotective proteins were also elevated as a consequence of microglial activation. These results have led to the hypothesis that microglia have a dual role in the pathogenesis of PD. Indeed, the present report demonstrates that microglia stimulated with N-α-syn not only produce noxious factors that contribute to neuronal cell death, but also secrete a multitude of signals and trophic factors that are potentially neuroprotective.
We demonstrate that N-α-syn microglial stimulation resulted in the secretion of cytoskeletal, enzyme, redox-active, and regulatory proteins. In particular, the cytoskeletal proteins in the N-α-syn-stimulated microglial secretome included vimentin and beta actin whose presence is consistent with cellular activation, morphological transformation, and enhanced cell motility. Aspartate aminotransferase and purine nucleoside phosphorylase are markers for cellular stress (Gonzalez-Flecha et al., 1993; Mochida et al., 1994) and were also secreted. The accumulation of cell morphogenesis proteins L-plastin, ARP3 and CAP1 all support that N-α-syn induced microglial activation (Hubberstey et al., 1996; McCollum et al., 1996; Jones et al., 1998). Production of the polysaccharide modifying lysosomal proteins, NAGLU and GALNS may indicate changes in the secretion of vesicles that are part of enhanced cellular activities which parallel microglial activation (Potolicchio et al., 2005). Several proteins were less abundant in N-α-syn-stimulated microglia. Of these, calcyclin, β-actin, histone H4, triose-phosphate isomerase, phosphoglycerate mutase 1, cathepsin S, 14-3-3σ, and ubiquitin have been reported in exosomes, secretory vesicles common to immune cells (Thery et al., 2001; Wubbolts et al., 2003; Pisitkun et al., 2004; Potolicchio et al., 2005; Faure et al., 2006). In addition PEBP, frount, calvasculin, calcyclin, osteopontin, β-2 microglobulin, commonly contained within secretory vesicles found in supernatants of resting microglial cells (Morozov et al., 1991; Engelkamp et al., 1992; Singh et al., 1993; Terashima et al., 2005), were less abundant in N-α-syn stimulated supernatants. Although exosomal fractionation was not performed, the decreased abundance of normal exosome constituents, coupled with increased abundance of lysosomal or NF-κB-related proteins suggests a secretome characteristic of cell activation. Increased abundance of antioxidants thioredoxin, biliverdin reductase, ferritin light chain, and vat1 was consistent with an upregulation of a response to oxidative stress (Fernando et al., 1992; Baldi et al., 2005; Marchler-Bauer et al., 2005; Liu et al., 2006). At 4 h after stimulation, expression of the antioxidant glutaredoxin-1 was downregulated (Reynolds et al., 2007b) that could result in excessive accumulation of GSSG and subsequently a decline in the ratio of GSH/GSSG rendering the cell less able to cope with excessive ROS production. However, enhanced expression of several redox-active proteins was also observed. Indeed, stimulation resulted in a temporal upregulation of various antioxidants that are capable of combating oxidative stress including superoxide dismutase, peroxiredoxins, cytochrome c oxidase, and thioredoxin-5. The majority of these enzymes act to help the cell cope with oxidative stress through reduction of noxious oxygen free radicals. Moreover, enhancement in enzymatic activity of these proteins was shown to be neuroprotective in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine intoxicated animals (Zhang et al., 2000; Bai et al., 2002; Seo et al., 2006). The major functions of peroxiredoxins include protection against cellular oxidative stress modulation of signaling cascades that utilize hydrogen peroxide as the second messenger, regulation of cell proliferation, reduced NF-κB activation, and possibly protection from apoptosis (Kropotov et al., 2006). Also upregulated was HSP70, a protein chaperone found in the substantia nigra that functions to refold abnormally folded proteins and traffic abnormal proteins to the proteasome (Foster et al., 1995), or to be degraded in the lysosome (Agarraberes and Dice, 2001). Investigations have also reported that HSP70 can reduce the death of dopaminergic neurons expressing mutant N-α-syn in a Drosophila model of PD (Auluck et al., 2002), and can protect against neurodegeneration induced through proteasome inhibition (Ahn and Jeon, 2006). These data, taken together, provide evidence supporting a dual toxic and compensatory microglial response after exposure to N-α-syn.
Damage and the subsequent selective degeneration of dopamine-producing neurons in the substantia nigra and striatum occurs as a prominent pathobiological feature of PD. Examinations of postmortem brain tissue from PD patients demonstrate that oxidative stress, mitochondrial dysfunction and impaired glucose uptake disrupt neuronal energy metabolism and homeostasis, by, in part, affecting glutamate transporters. Such oxidative and metabolic dysfunctions leading to neuronal vulnerability to excitotoxicity and apoptosis are linked to microglial responses (Przedborski and Ischiropoulos, 2005). Activated microglia express inducible transporters [excitatory amino acid transporters 1 and 2] and enzymes [glutamine synthetase], both major effectors of glutamate metabolism (Chretien et al., 2002; Gras et al., 2006; Beschorner et al., 2007), which evolves during disease progression. Nonetheless, it is the level of uncoupled synaptic glutamate, not related to energy metabolism, that leads to neuronal death (Ramonet et al., 2004). Glutamate is the principle excitatory neurotransmitter in the CNS, is produced and regulated from glia (principally astrocytes and microglia), and its extracellular concentrations increase under oxidative stress conditions. Glutamate-mediated neuronal damage occurs through excitoxic pathways due to hyper-activation of specific transporters or inhibition of cystine uptake resulting in glutathione depletion and oxidative stress (Gras et al., 2006). Microglia can control these responses leading to neuroprotective outcomes through uptake and clearance of glutamate. This may partially compensate for the deleterious state of their activation. It is possible that microglial responses are distinct early and late in the disease course. Early in disease microglial responses are protective and the cells can compensate for changes in glutamate metabolism. In contrast, more chronic microglial activation occurs when compensatory responses are spent. This accompanies neuronal apoptosis and cell loss as it pertains to the role played by macrophages and microglia in HIV-induced neuronal dysfunction (Gras et al., 2006) as well as to PD. Indeed, our own analyses of extracellular metabolites within culture supernatants revealed that microglia secrete several factors that could constitute a dual trophic and toxic response to N-α-syn. For example, extracellular levels of cysteine were enhanced during N-α-syn stimulation. Cysteine can be taken up by adjacent neurons to produce glutathione (Shih et al., 2006) needed to cope with oxidative stress and supports a neuroprotective role for activated microglia. On the other hand, cysteine may also be excitotoxic in large amounts and contribute to neuronal demise (Yeh et al., 2000). In addition, enhanced accumulation of extracellular glutamate was also observed, albeit the concentrations observed in this study are modest compared to what is induced upon macrophage interaction with human immunodeficiency virus-1 (Jiang et al., 2001). Accumulation of extracellular glutamate may also inhibit cystine uptake via the cystine/glutamate antiporter leading to depletion of glutathione (Simantov, 1989; Jiang et al., 2001). Moreover, analysis of intracellular metabolites indicated that N-α-syn stimulation renders cells more prone to oxidative stress as GSH, the GSH/GSSG ratio, and total glutathione were decreased. Also decreased were intracellular glutamate and cysteine, corresponding to the elevated levels of both metabolites in culture fluids. As these metabolites can be taken up by adjacent glia and neurons to be used as precursors for glutathione synthesis (Gras et al., 2006), efflux of these metabolites in low concentrations may constitute a protective rather than toxic response of the activated microglia. Therefore, the microglia could constitute a local, inducible, cell population for glutamate clearance and metabolism (Shaked et al., 2005). How this property is changed during states of chronic activation has yet to be determined, but depletion of intracellular reservoirs of glutathione and its precursors following prolonged activation may contribute to disease. Thus, this property may be critical to neuronal survival and disease outcome in PD-associated neuroinflammation.
Cathepsins consist of intracellular proteases localized within lysosomes that are important for normal protein turnover and proper cell function (Turk et al., 2001), including antigen presentation (Riese and Chapman, 2000) and may aid cell migration (Kawada et al., 1997). Whereas normal levels of cathepsin B are crucial for neuronal viability and brain function, elevated cathepsin B enzymatic activity in serum or in the extracellular matrix is associated with serious pathological conditions such as AD, cancer, stroke, autoimmune disease, and osteoporosis (Buhling et al., 2000; Gerber et al., 2000). Cathepsin B can be translocated to the cytosol, released from lysosomes during oxidative stress (Kagedal et al., 2001) by neurons as well as by activated microglia leading to the degradation of extracellular matrix proteins (Buck et al., 1992) resulting in neuronal apoptosis (Kingham and Pocock, 2001), whereas inhibition of microglial cathepsin B expression diminishes microglial-induced neurotoxicity in vitro (Gan et al., 2004). In the present study, we have investigated cathepsin B activity in N-α-syn-stimulated microglia, and compared it with the expression of the endogenous inhibitors cystatins B and C. Cystatin C expression, although present in culture supernatants, was not altered by stimulation with N-α-syn, in contrast to other stimulators of microglial activation (Chapman et al., 1990; Kingham and Pocock, 2001). In contrast, cystatin B expression was changed, and paralleled decreased cathepsin B enzymatic activity. Gene deletion of cystatin B increased neuronal apoptosis in mice, demonstrating that cystatin B is important for neuronal survival (Brannvall et al., 2003; Riccio et al., 2005). Release of cathespin B may have consequences for neuronal loss by triggering apoptosis or by fueling inflammatory cascades. Therefore the transient downregulation of cathepsin B activity observed in this study is a potential compensatory mechanism to regulate subsequent neurotoxicity. Indeed, inhibition of cathepsin B partially protected from N-α-syn mediated cytotoxicity. A relationship between cathepsin B activity and cysteine regulation is suggested by the observation that microglial-derived glutamate-mediated cytototoxity as well as cathepsin B-mediated toxicity were significantly reduced in response to cathepsin B inhibition (Kingham and Pocock, 2001).
Activation of NF-κB and related signaling pathways predominate in the N-α-syn-mediated microglial inflammatory response. NF-κB consists of dimeric transcription factors (RelA/p65: NFκB1/p50) that regulate cell division, apoptosis, and inflammation. Usually sequestered in the cytoplasm of unstimulated cells by binding to IκB proteins, activating stimuli of NF-κB activate the inhibitor kappa B kinase signalosome to phosphorylate IκB, liberating NF-κB dimmers to translocate to the nucleus and regulate target gene transcription (Nelson et al., 2004). The observation that both subunits (RelA/p65 and NFκB1/p50) are present within the nucleus as early as 1 h of stimulation with sustained presence of RelA/p65 proteins and transient peaks of NFκB1/p50 levels at 90 min and 24 h presents insight into the mechanisms of N-α-syn induction of microglial ROS and mediating neuronal cytotoxicity. The transient decrease in nuclear NFκB1/p50 expression coincides with an increase in transcript levels of Nfkbia observed after 4 h of N-α-syn stimulation. However, transcript levels of Ikbkb was also induced (Reynolds et al., 2007b) and may in turn inhibit NFκBIA from binding NF-κB dimers allowing for renewed translocation to the nucleus by 24 h. Previous work by others also demonstrated an asynchronous oscillation of NF-κB activation following cell stimulation, and suggested that the functional consequences of NF-κB activation may depend on the persistence of these oscillations (Nelson et al., 2004) to maintain NF-κB-dependent gene expression. These results provide further insight into how compensatory responses are overcome by continued stimulation with N-α-syn. Indeed, activation of the NF-κB pathway was also found in nuclear fractions prepared from the SN of PD patients with increased expression of NFκB1/p50 and RelA/p65, relaying that this pathway predominates in the inflammatory processes within the SN of PD patients (Reynolds et al., 2007b).
One function of microglial activation is to signal to resident cells of the CNS the presence of invading organisms, intracellular killing, and leukocyte recruitment. Underlying these functions is the secretion of a plethora of cytokines and chemokines that activate neighboring cells as a defense mechanism against invading pathogens. However, microglial cellular activation is a double-edged sword, as these same factors are also neurotoxic. The ability of a microglial cell to compensate for the devastating consequences of their activation and initiate effective repairs results from the capacity to produce several protective factors including antioxidants, growth factors, and signals to astrocytes that can in-turn secrete neuroprotective factors. However, as microglial activation is associated with a variety of neurodegenerative disorders, this compensatory response elicited by the microglia cell is not sufficient to protect neurons from secondary degeneration under conditions conducive to disease progression. This analysis now provides evidence for a dual destructive and potentially protective role for microglia in affecting disease onset and progression.
Acknowledgments
We thank Dr. E. Benner for providing recombinant mouse α-synuclein and Dr. S. Appel for providing the MES23.5 cell line. The authors also thank Dr. R. Lee Mosley and Robin Taylor for critical reading of the manuscript.
This work was supported by the Frances and Louie Blumkin Foundation, the Community Neuroscience Pride of Nebraska Research Initiative, and the Alan Baer Charitable Trust (to H.E.G.), a University of Nebraska Medical Center Graduate Student Excellence Award and the Patterson Fellowship (to A.R.) and NIH grants 1T32 NS07488 (to A.R. and H.E.G.), and 2R37 NS36136, PO1 NS43985, PO1 MH64570, R01 MH79886 (to H.E.G.) and DK64959 (to RB).
Footnotes
“This article is published in the Journal of Neuroimmune Pharmacology. The original publication is available at www.springerlink.com”.
References
- Agarraberes FA, Dice JF. A molecular chaperone complex at the lysosomal membrane is required for protein translocation. J Cell Sci. 2001;114:2491–2499. doi: 10.1242/jcs.114.13.2491. [DOI] [PubMed] [Google Scholar]
- Ahn TB, Jeon BS. Protective role of heat shock and heat shock protein 70 in lactacystin-induced cell death both in the rat substantia nigra and PC12 cells. Brain Res. 2006;1087:159–167. doi: 10.1016/j.brainres.2006.02.097. [DOI] [PubMed] [Google Scholar]
- Auluck PK, Chan HY, Trojanowski JQ, Lee VM, Bonini NM. Chaperone suppression of alpha-synuclein toxicity in a Drosophila model for Parkinson's disease. Science. 2002;295:865–868. doi: 10.1126/science.1067389. [DOI] [PubMed] [Google Scholar]
- Bai J, Nakamura H, Hattori I, Tanito M, Yodoi J. Thioredoxin suppresses 1-methyl-4-phenylpyridinium-induced neurotoxicity in rat PC12 cells. Neurosci Lett. 2002;321:81–84. doi: 10.1016/s0304-3940(02)00058-7. [DOI] [PubMed] [Google Scholar]
- Baldi A, Lombardi D, Russo P, Palescandolo E, De Luca A, Santini D, Baldi F, Rossiello L, Dell'Anna ML, Mastrofrancesco A, Maresca V, Flori E, Natali PG, Picardo M, Paggi MG. Ferritin contributes to melanoma progression by modulating cell growth and sensitivity to oxidative stress. Clin Cancer Res. 2005;11:3175–3183. doi: 10.1158/1078-0432.CCR-04-0631. [DOI] [PubMed] [Google Scholar]
- Benner EJ, Banerjee R, Reynolds AD, Sherman S, Pisarev V, Tsiperson V, Nemachek C, Ciborowski P, Przedborski S, Mosley RL, Gendelman HE. Nitrated α-synuclein immunity accelerates degeneration of nigral dopaminergic neurons. PLoS ONE. doi: 10.1371/journal.pone.0001376. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beschorner R, Simon P, Schauer N, Mittelbronn M, Schluesener HJ, Trautmann K, Dietz K, Meyermann R. Reactive astrocytes and activated microglial cells express EAAT1, but not EAAT2, reflecting a neuroprotective potential following ischaemia. Histopathology. 2007;50:897–910. doi: 10.1111/j.1365-2559.2007.02703.x. [DOI] [PubMed] [Google Scholar]
- Brannvall K, Hjelm H, Korhonen L, Lahtinen U, Lehesjoki AE, Lindholm D. Cystatin-B is expressed by neural stem cells and by differentiated neurons and astrocytes. Biochem Biophys Res Commun. 2003;308:369–374. doi: 10.1016/s0006-291x(03)01386-x. [DOI] [PubMed] [Google Scholar]
- Buck MR, Karustis DG, Day NA, Honn KV, Sloane BF. Degradation of extracellular-matrix proteins by human cathepsin B from normal and tumour tissues. Biochem J. 1992;282 ( Pt 1):273–278. doi: 10.1042/bj2820273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buhling F, Fengler A, Brandt W, Welte T, Ansorge S, Nagler DK. Review: novel cysteine proteases of the papain family. Adv Exp Med Biol. 2000;477:241–254. doi: 10.1007/0-306-46826-3_26. [DOI] [PubMed] [Google Scholar]
- Ciborowski P, Kadiu I, Rozek W, Smith L, Bernhadt K, Fladseth M, Ricardo-Dukelow M, Gendelman HE. Investigating the human immunodeficiency virus type 1-infected monocyte-derived macrophage secretome. Virology. 2007;363:198–209. doi: 10.1016/j.virol.2007.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman HA, Jr, Reilly JJ, Jr, Yee R, Grubb A. Identification of cystatin C, a cysteine proteinase inhibitor, as a major secretory product of human alveolar macrophages in vitro. Am Rev Respir Dis. 1990;141:698–705. doi: 10.1164/ajrccm/141.3.698. [DOI] [PubMed] [Google Scholar]
- Chretien F, Vallat-Decouvelaere AV, Bossuet C, Rimaniol AC, Le Grand R, Le Pavec G, Creminon C, Dormont D, Gray F, Gras G. Expression of excitatory amino acid transporter-2 (EAAT-2) and glutamine synthetase (GS) in brain macrophages and microglia of SIVmac251-infected macaques. Neuropathol Appl Neurobiol. 2002;28:410–417. doi: 10.1046/j.1365-2990.2002.00426.x. [DOI] [PubMed] [Google Scholar]
- Crawford GD, Jr, Le WD, Smith RG, Xie WJ, Stefani E, Appel SH. A novel N18TG2 x mesencephalin cell hybrid expresses properties that suggest a dopaminergic cell line of substantia nigra origin. J Neuroscience. 1992;12:3392–3398. doi: 10.1523/JNEUROSCI.12-09-03392.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czlonkowska A, Kohutnicka M, Kurkowska-Jastrzebska I, Czlonkowski A. Microglial reaction in MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) induced Parkinson's disease mice model. Neurodegeneration. 1996;5:137–143. doi: 10.1006/neur.1996.0020. [DOI] [PubMed] [Google Scholar]
- Dobrenis K. Microglia in cell culture and in transplantation therapy for central nervous system disease. Methods. 1998;16:320–344. doi: 10.1006/meth.1998.0688. [DOI] [PubMed] [Google Scholar]
- Engelkamp D, Schafer BW, Erne P, Heizmann CW. S100 alpha, CAPL, and CACY: molecular cloning and expression analysis of three calcium-binding proteins from human heart. Biochemistry. 1992;31:10258–10264. doi: 10.1021/bi00157a012. [DOI] [PubMed] [Google Scholar]
- Enose Y, Destache CJ, Mack AL, Anderson JR, Ullrich F, Ciborowski PS, Gendelman HE. Proteomic fingerprints distinguish microglia, bone marrow, and spleen macrophage populations. Glia. 2005;51:161–172. doi: 10.1002/glia.20193. [DOI] [PubMed] [Google Scholar]
- Faure J, Lachenal G, Court M, Hirrlinger J, Chatellard-Causse C, Blot B, Grange J, Schoehn G, Goldberg Y, Boyer V, Kirchhoff F, Raposo G, Garin J, Sadoul R. Exosomes are released by cultured cortical neurones. Mol Cell Neurosci. 2006;31:642–648. doi: 10.1016/j.mcn.2005.12.003. [DOI] [PubMed] [Google Scholar]
- Fernando MR, Nanri H, Yoshitake S, Nagata-Kuno K, Minakami S. Thioredoxin regenerates proteins inactivated by oxidative stress in endothelial cells. Eur J Biochem. 1992;209:917–922. doi: 10.1111/j.1432-1033.1992.tb17363.x. [DOI] [PubMed] [Google Scholar]
- Foster JA, Rush SJ, Brown IR. Localization of constitutive and hyperthermia-inducible heat shock mRNAs (hsc70 and hsp70) in the rabbit cerebellum and brainstem by non-radioactive in situ hybridization. J Neurosci Res. 1995;41:603–612. doi: 10.1002/jnr.490410506. [DOI] [PubMed] [Google Scholar]
- Gan L, Ye S, Chu A, Anton K, Yi S, Vincent VA, von Schack D, Chin D, Murray J, Lohr S, Patthy L, Gonzalez-Zulueta M, Nikolich K, Urfer R. Identification of cathepsin B as a mediator of neuronal death induced by Abeta-activated microglial cells using a functional genomics approach. J Biol Chem. 2004;279:5565–5572. doi: 10.1074/jbc.M306183200. [DOI] [PubMed] [Google Scholar]
- Gerber A, Welte T, Ansorge S, Buhling F. Expression of cathepsins B and L in human lung epithelial cells is regulated by cytokines. Adv Exp Med Biol. 2000;477:287–292. doi: 10.1007/0-306-46826-3_31. [DOI] [PubMed] [Google Scholar]
- Glanzer JG, Enose Y, Wang T, Kadiu I, Gong N, Rozek W, Liu J, Schlautman JD, Ciborowski PS, Thomas MP, Gendelman HE. Genomic and proteomic microglial profiling: pathways for neuroprotective inflammatory responses following nerve fragment clearance and activation. J Neurochem. 2007;102:627–645. doi: 10.1111/j.1471-4159.2007.04568.x. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Flecha B, Reides C, Cutrin JC, Llesuy SF, Boveris A. Oxidative stress produced by suprahepatic occlusion and reperfusion. Hepatology. 1993;18:881–889. doi: 10.1002/hep.1840180421. [DOI] [PubMed] [Google Scholar]
- Gras G, Porcheray F, Samah B, Leone C. The glutamate-glutamine cycle as an inducible, protective face of macrophage activation. J Leukoc Biol. 2006;80:1067–1075. doi: 10.1189/jlb.0306153. [DOI] [PubMed] [Google Scholar]
- Hald A, Van Beek J, Lotharius J. Inflammation in Parkinson's disease: causative or epiphenomenal? Subcell Biochem. 2007;42:249–279. [PubMed] [Google Scholar]
- Hermanowicz N. Drug therapy for Parkinson's disease. Semin Neurol. 2007;27:97–105. doi: 10.1055/s-2007-971177. [DOI] [PubMed] [Google Scholar]
- Hodaie M, Neimat JS, Lozano AM. The dopaminergic nigrostriatal system and Parkinson's disease: molecular events in development, disease, and cell death, and new therapeutic strategies. Neurosurgery. 2007;60:17–28. 28–30. doi: 10.1227/01.NEU.0000249209.11967.CB. [DOI] [PubMed] [Google Scholar]
- Hubberstey A, Yu G, Loewith R, Lakusta C, Young D. Mammalian CAP interacts with CAP, CAP2, and actin. J Cell Biochem. 1996;61:459–466. doi: 10.1002/(SICI)1097-4644(19960601)61:3%3C459::AID-JCB13%3E3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- Islam A, Adamik B, Hawari FI, Ma G, Rouhani FN, Zhang J, Levine SJ. Extracellular TNFR1 release requires the calcium-dependent formation of a nucleobindin 2-ARTS-1 complex. J Biol Chem. 2006;281:6860–6873. doi: 10.1074/jbc.M509397200. [DOI] [PubMed] [Google Scholar]
- Jiang ZG, Piggee C, Heyes MP, Murphy C, Quearry B, Bauer M, Zheng J, Gendelman HE, Markey SP. Glutamate is a mediator of neurotoxicity in secretions of activated HIV-1-infected macrophages. J Neuroimmunol. 2001;117:97–107. doi: 10.1016/s0165-5728(01)00315-0. [DOI] [PubMed] [Google Scholar]
- Jones SL, Wang J, Turck CW, Brown EJ. A role for the actin-bundling protein L-plastin in the regulation of leukocyte integrin function. Proc Natl Acad Sci U S A. 1998;95:9331–9336. doi: 10.1073/pnas.95.16.9331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadiu I, Ricardo-Dukelow M, Ciborowski P, Gendelman HE. Cytoskeletal protein transformation in HIV-1-infected macrophage giant cells. J Immunol. 2007;178:6404–6415. doi: 10.4049/jimmunol.178.10.6404. [DOI] [PubMed] [Google Scholar]
- Kagedal K, Johansson U, Ollinger K. The lysosomal protease cathepsin D mediates apoptosis induced by oxidative stress. Faseb J. 2001;15:1592–1594. doi: 10.1096/fj.00-0708fje. [DOI] [PubMed] [Google Scholar]
- Kawada A, Hara K, Kominami E, Hiruma M, Akiyama M, Ishibashi A, Abe H, Ichikawa E, Nakamura Y, Watanabe S, Yamamoto T, Umeda T, Nishioka K. Expression of cathepsin D and B in invasion and metastasis of squamous cell carcinoma. Br J Dermatol. 1997;137:361–366. [PubMed] [Google Scholar]
- Khandhar SM, Marks WJ. Epidemiology of Parkinson's disease. Dis Mon. 2007;53:200–205. doi: 10.1016/j.disamonth.2007.02.001. [DOI] [PubMed] [Google Scholar]
- Kingham PJ, Pocock JM. Microglial secreted cathepsin B induces neuronal apoptosis. J Neurochem. 2001;76:1475–1484. doi: 10.1046/j.1471-4159.2001.00146.x. [DOI] [PubMed] [Google Scholar]
- Klegeris A, McGeer EG, McGeer PL. Therapeutic approaches to inflammation in neurodegenerative disease. Curr Opin Neurol. 2007;20:351–357. doi: 10.1097/WCO.0b013e3280adc943. [DOI] [PubMed] [Google Scholar]
- Kropotov A, Gogvadze V, Shupliakov O, Tomilin N, Serikov VB, Tomilin NV, Zhivotovsky B. Peroxiredoxin V is essential for protection against apoptosis in human lung carcinoma cells. Exp Cell Res. 2006;312:2806–2815. doi: 10.1016/j.yexcr.2006.05.006. [DOI] [PubMed] [Google Scholar]
- Lipton SA, Gu Z, Nakamura T. Inflammatory mediators leading to protein misfolding and uncompetitive/fast off-rate drug therapy for neurodegenerative disorders. Int Rev Neurobiol. 2007;82:1–27. doi: 10.1016/S0074-7742(07)82001-0. [DOI] [PubMed] [Google Scholar]
- Liu Y, Liu J, Tetzlaff W, Paty DW, Cynader MS. Biliverdin reductase, a major physiologic cytoprotectant, suppresses experimental autoimmune encephalomyelitis. Free Radic Biol Med. 2006;40:960–967. doi: 10.1016/j.freeradbiomed.2005.07.021. [DOI] [PubMed] [Google Scholar]
- Loeffler DA, DeMaggio AJ, Juneau PL, Havaich MK, LeWitt PA. Effects of enhanced striatal dopamine turnover in vivo on glutathione oxidation. Clin Neuropharmacol. 1994;17:370–379. doi: 10.1097/00002826-199408000-00009. [DOI] [PubMed] [Google Scholar]
- Mandemakers W, Morais VA, De Strooper B. A cell biological perspective on mitochondrial dysfunction in Parkinson disease and other neurodegenerative diseases. J Cell Sci. 2007;120:1707–1716. doi: 10.1242/jcs.03443. [DOI] [PubMed] [Google Scholar]
- Marchler-Bauer A, Anderson JB, Cherukuri PF, DeWeese-Scott C, Geer LY, Gwadz M, He S, Hurwitz DI, Jackson JD, Ke Z, Lanczycki CJ, Liebert CA, Liu C, Lu F, Marchler GH, Mullokandov M, Shoemaker BA, Simonyan V, Song JS, Thiessen PA, Yamashita RA, Yin JJ, Zhang D, Bryant SH. CDD: a Conserved Domain Database for protein classification. Nucleic Acids Res. 2005;33:D192–196. doi: 10.1093/nar/gki069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCollum D, Feoktistova A, Morphew M, Balasubramanian M, Gould KL. The Schizosaccharomyces pombe actin-related protein, Arp3, is a component of the cortical actin cytoskeleton and interacts with profilin. Embo J. 1996;15:6438–6446. [PMC free article] [PubMed] [Google Scholar]
- McGeer PL, Itagaki S, Boyes BE, McGeer EG. Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson's and Alzheimer's disease brains. Neurology. 1988;38:1285–1291. doi: 10.1212/wnl.38.8.1285. [DOI] [PubMed] [Google Scholar]
- Mochida S, Arai M, Ohno A, Masaki N, Ogata I, Fujiwara K. Oxidative stress in hepatocytes and stimulatory state of Kupffer cells after reperfusion differ between warm and cold ischemia in rats. Liver. 1994;14:234–240. doi: 10.1111/j.1600-0676.1994.tb00081.x. [DOI] [PubMed] [Google Scholar]
- Morozov VN, Morozova T, Bray P, Hranisavljevic J, Vucelic D. Survey of small molecule and ion binding to beta 2-microglobulin--possible relation to BEN. Kidney Int Suppl. 1991;34:S85–88. [PubMed] [Google Scholar]
- Mosharov E, Cranford MR, Banerjee R. The quantitatively important relationship between homocysteine metabolism and glutathione synthesis by the transsulfuration pathway and its regulation by redox changes. Biochemistry. 2000;39:13005–13011. doi: 10.1021/bi001088w. [DOI] [PubMed] [Google Scholar]
- Nelson DE, Ihekwaba AE, Elliott M, Johnson JR, Gibney CA, Foreman BE, Nelson G, See V, Horton CA, Spiller DG, Edwards SW, McDowell HP, Unitt JF, Sullivan E, Grimley R, Benson N, Broomhead D, Kell DB, White MR. Oscillations in NF-kappaB signaling control the dynamics of gene expression. Science. 2004;306:704–708. doi: 10.1126/science.1099962. [DOI] [PubMed] [Google Scholar]
- Pisitkun T, Shen RF, Knepper MA. Identification and proteomic profiling of exosomes in human urine. Proc Natl Acad Sci U S A. 2004;101:13368–13373. doi: 10.1073/pnas.0403453101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potolicchio I, Carven GJ, Xu X, Stipp C, Riese RJ, Stern LJ, Santambrogio L. Proteomic analysis of microglia-derived exosomes: metabolic role of the aminopeptidase CD13 in neuropeptide catabolism. J Immunol. 2005;175:2237–2243. doi: 10.4049/jimmunol.175.4.2237. [DOI] [PubMed] [Google Scholar]
- Przedborski S. Pathogenesis of nigral cell death in Parkinson's disease. Parkinsonism Relat Disord. 2005;11(Suppl 1):S3–7. doi: 10.1016/j.parkreldis.2004.10.012. [DOI] [PubMed] [Google Scholar]
- Przedborski S, Ischiropoulos H. Reactive oxygen and nitrogen species: weapons of neuronal destruction in models of Parkinson's disease. Antioxid Redox Signal. 2005;7:685–693. doi: 10.1089/ars.2005.7.685. [DOI] [PubMed] [Google Scholar]
- Qin H, Wilson CA, Lee SJ, Zhao X, Benveniste EN. LPS induces CD40 gene expression through the activation of NF-kappaB and STAT-1alpha in macrophages and microglia. Blood. 2005;106:3114–3122. doi: 10.1182/blood-2005-02-0759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramonet D, Rodriguez MJ, Fredriksson K, Bernal F, Mahy N. In vivo neuroprotective adaptation of the glutamate/glutamine cycle to neuronal death. Hippocampus. 2004;14:586–594. doi: 10.1002/hipo.10188. [DOI] [PubMed] [Google Scholar]
- Reynolds A, Laurie C, Mosley RL, Gendelman HE. Oxidative stress and the pathogenesis of neurodegenerative disorders. Int Rev Neurobiol. 2007a;82:297–325. doi: 10.1016/S0074-7742(07)82016-2. [DOI] [PubMed] [Google Scholar]
- Reynolds AD, Glanzer JG, Kadiu I, Ricardo-Dukelow M, Chaudhuri A, Ciborowski P, Cerny R, Gelman B, Thomas MP, Mosley RL, Gendelman HE. Nitrated alpha-synuclein-activated microglial profiling for Parkinson's disease. J Neurochem. 2007b doi: 10.1111/j.1471-4159.2007.05087.x. In press. [DOI] [PubMed] [Google Scholar]
- Riccio M, Santi S, Dembic M, Di Giaimo R, Cipollini E, Costantino-Ceccarini E, Ambrosetti D, Maraldi NM, Melli M. Cell-specific expression of the epm1 (cystatin B) gene in developing rat cerebellum. Neurobiol Dis. 2005;20:104–114. doi: 10.1016/j.nbd.2005.02.012. [DOI] [PubMed] [Google Scholar]
- Riese RJ, Chapman HA. Cathepsins and compartmentalization in antigen presentation. Curr Opin Immunol. 2000;12:107–113. doi: 10.1016/s0952-7915(99)00058-8. [DOI] [PubMed] [Google Scholar]
- Rogove AD, Tsirka SE. Neurotoxic responses by microglia elicited by excitotoxic injury in the mouse hippocampus. Curr Biol. 1998;8:19–25. doi: 10.1016/s0960-9822(98)70016-8. [DOI] [PubMed] [Google Scholar]
- Seo BB, Nakamaru-Ogiso E, Flotte TR, Matsuno-Yagi A, Yagi T. In vivo complementation of complex I by the yeast Ndi1 enzyme. Possible application for treatment of Parkinson disease. J Biol Chem. 2006;281:14250–14255. doi: 10.1074/jbc.M600922200. [DOI] [PubMed] [Google Scholar]
- Shaked I, Tchoresh D, Gersner R, Meiri G, Mordechai S, Xiao X, Hart RP, Schwartz M. Protective autoimmunity: interferon-gamma enables microglia to remove glutamate without evoking inflammatory mediators. J Neurochem. 2005;92:997–1009. doi: 10.1111/j.1471-4159.2004.02954.x. [DOI] [PubMed] [Google Scholar]
- Shih AY, Erb H, Sun X, Toda S, Kalivas PW, Murphy TH. Cystine/glutamate exchange modulates glutathione supply for neuroprotection from oxidative stress and cell proliferation. J Neurosci. 2006;26:10514–10523. doi: 10.1523/JNEUROSCI.3178-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simantov R. Glutamate neurotoxicity in culture depends on the presence of glutamine: implications for the role of glial cells in normal and pathological brain development. J Neurochem. 1989;52:1694–1699. doi: 10.1111/j.1471-4159.1989.tb07246.x. [DOI] [PubMed] [Google Scholar]
- Singh K, Deonarine D, Shanmugam V, Senger DR, Mukherjee AB, Chang PL, Prince CW, Mukherjee BB. Calcium-binding properties of osteopontin derived from non-osteogenic sources. J Biochem (Tokyo) 1993;114:702–707. doi: 10.1093/oxfordjournals.jbchem.a124240. [DOI] [PubMed] [Google Scholar]
- Terashima Y, Onai N, Murai M, Enomoto M, Poonpiriya V, Hamada T, Motomura K, Suwa M, Ezaki T, Haga T, Kanegasaki S, Matsushima K. Pivotal function for cytoplasmic protein FROUNT in CCR2-mediated monocyte chemotaxis. Nat Immunol. 2005;6:827–835. doi: 10.1038/ni1222. [DOI] [PubMed] [Google Scholar]
- Thery C, Boussac M, Veron P, Ricciardi-Castagnoli P, Raposo G, Garin J, Amigorena S. Proteomic analysis of dendritic cell-derived exosomes: a secreted subcellular compartment distinct from apoptotic vesicles. J Immunol. 2001;166:7309–7318. doi: 10.4049/jimmunol.166.12.7309. [DOI] [PubMed] [Google Scholar]
- Thomas MP, Chartrand K, Reynolds A, Vitvitsky V, Banerjee R, Gendelman HE. Ion channel blockade attenuates aggregated alpha synuclein induction of microglial reactive oxygen species: relevance for the pathogenesis of Parkinson's disease. J Neurochem. 2007;100:503–519. doi: 10.1111/j.1471-4159.2006.04315.x. [DOI] [PubMed] [Google Scholar]
- Turk V, Turk B, Turk D. Lysosomal cysteine proteases: facts and opportunities. Embo J. 2001;20:4629–4633. doi: 10.1093/emboj/20.17.4629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitton PS. Inflammation as a causative factor in the aetiology of Parkinson's disease. Br J Pharmacol. 2007;150:963–976. doi: 10.1038/sj.bjp.0707167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilms H, Zecca L, Rosenstiel P, Sievers J, Deuschl G, Lucius R. Inflammation in Parkinson's diseases and other neurodegenerative diseases: cause and therapeutic implications. Curr Pharm Des. 2007;13:1925–1928. doi: 10.2174/138161207780858429. [DOI] [PubMed] [Google Scholar]
- Wu DC, Jackson-Lewis V, Vila M, Tieu K, Teismann P, Vadseth C, Choi DK, Ischiropoulos H, Przedborski S. Blockade of microglial activation is neuroprotective in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson disease. J Neurosci. 2002;22:1763–1771. doi: 10.1523/JNEUROSCI.22-05-01763.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wubbolts R, Leckie RS, Veenhuizen PT, Schwarzmann G, Mobius W, Hoernschemeyer J, Slot JW, Geuze HJ, Stoorvogel W. Proteomic and biochemical analyses of human B cell-derived exosomes. Potential implications for their function and multivesicular body formation. J Biol Chem. 2003;278:10963–10972. doi: 10.1074/jbc.M207550200. [DOI] [PubMed] [Google Scholar]
- Yeh MW, Kaul M, Zheng J, Nottet HS, Thylin M, Gendelman HE, Lipton SA. Cytokine-stimulated, but not HIV-infected, human monocyte-derived macrophages produce neurotoxic levels of l -cysteine. J Immunol. 2000;164:4265–4270. doi: 10.4049/jimmunol.164.8.4265. [DOI] [PubMed] [Google Scholar]
- Yuan H, Zheng JC, Liu P, Zhang SF, Xu JY, Bai LM. Pathogenesis of Parkinson's disease: oxidative stress, environmental impact factors and inflammatory processes. Neurosci Bull. 2007;23:125–130. doi: 10.1007/s12264-007-0018-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Graham DG, Montine TJ, Ho YS. Enhanced N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine toxicity in mice deficient in CuZn-superoxide dismutase or glutathione peroxidase. J Neuropathol Exp Neurol. 2000;59:53–61. doi: 10.1093/jnen/59.1.53. [DOI] [PubMed] [Google Scholar]
- Zhang W, Wang T, Pei Z, Miller DS, Wu X, Block ML, Wilson B, Zhou Y, Hong JS, Zhang J. Aggregated alpha-synuclein activates microglia: a process leading to disease progression in Parkinson's disease. Faseb J. 2005;19:533–542. doi: 10.1096/fj.04-2751com. [DOI] [PubMed] [Google Scholar]