

Abstract
Background
Freshwater unionoidean bivalves, and species representing two marine bivalve orders (Mytiloida and Veneroida), exhibit a mode of mtDNA inheritance involving distinct maternal (F) and paternal (M) transmission routes concomitant with highly divergent gender-associated mtDNA genomes. Additionally, male unionoidean bivalves have a ~550 bp 3' coding extension to the cox2 gene (Mcox2e), that is apparently absent from all other metazoan taxa.
Results
Our molecular sequence analyses of MCOX2e indicate that both the primary and secondary structures of the MCOX2e region are evolving much faster than other regions of the F and M COX2-COX1 gene junction. The near N-terminus ~2/3 of the MCOX2e region contains an interspecifically variable number of predicted transmembrane helices (TMH) and interhelical loops (IHL) whereas the C-terminus ~1/3 is relatively conserved and hydrophilic while containing conserved functional motifs. MCOX2e displays an overall pattern of purifying selection that leads to the preservation of TMH/IHL and C-terminus tail sub-regions. However, 14 amino acid positions in the MCOX2e TMH/IHL sub-region might be targeted by diversifying selection, each representing a site where there exists interspecific variation for the constituent amino acids residing in a TMH or IHL.
Conclusion
Our results indicate that Mcox2e is unique to unionoidean bivalves, likely the result of a single insertion event that took place over 65 MYA and that MCOX2e is functional. The predicted TMH number, length and position variability likely stems from substitution-based processes rather than the typically implicated insertion/deletion events. MCOX2e has relatively high rates of primary and secondary structure evolution, with some amino acid residues potentially subjected to site-specific positive selection, yet an overall pattern of purifying selection leading to the preservation of the TMH/IHL and hydrophilic C-terminus tail subregions. The more conserved C-terminus tail (relative to the TMH/IHL sub-region of MCOX2e) is likely biologically active because it contains functional motifs. The rapid evolution of primary and secondary structure in MCOX2e, combined with the action of both positive and purifying selection, provide supporting evidence for the hypothesis that MCOX2e has a novel reproductive function within unionoidean bivalves. All tolled, our data indicate that unionoidean bivalve MCOX2 is the first reported chimeric animal mtDNA-encoded protein.
Background
Cytochrome c oxidase (COX) is a multimeric enzyme that is located in the inner mitochondrial membrane of eukaryotes, belongs to the terminal enzymatic complex (IV) of the respiratory chain and facilitates the transfer of electrons from cytochrome c to molecular oxygen [1]. The three mitochondrially-encoded subunits of COX (COX1, COX2, COX3) typically possess conserved primary and secondary structures, including a relatively invariant number of transmembrane helices (TMHs) per subunit [2-4], with COX1 and COX2 containing highly conserved catalytic sites. Unique among the mtDNA-encoded COX subunits, COX2 has two N-terminus TMHs embedded in the inner mitochondrial membrane while the C-terminus half of the protein, containing the CuA center catalytic site, is located in the intermembrane space. These two distinct regions of COX2 are referred to as the COX2_TM and COX2 Pfam domains, respectively. Most mitochondrial protein coding genes have been shown to evolve under purifying selection [5-9]. However, a few recent studies have detected a signature of positive selection in COX2 lineages and/or sites [10-12]. Nevertheless, the typically conserved pattern of COX2 domains is violated in one category of animal mitochondrial genomes, namely those paternally transmitted in unionoidean bivalves.
Freshwater unionoidean bivalves, as well as representatives of two marine bivalve orders (Mytiloida and Veneroida), exhibit doubly uniparental inheritance (DUI) of mtDNA, which involves distinct maternal (F) and paternal (M) transmission routes concomitant with highly divergent gender-associated mtDNA genomes [13-20]. For a general review of DUI, see [21]. Female bivalves transmit their mitochondria (carrying F mtDNA) to sons and daughters, as in standard maternal inheritance, but males are believed to effectively transmit their mitochondria (via sperm carrying M mtDNA) to only sons (e.g., [22] but see [23]). In the latter, F mtDNA predominates in the somatic tissues while principally M mtDNA is found in the testes. Thus, this genetic system yields homoplasmic female and heteroplasmic male individuals. Intra- and inter-specific comparisons suggest that the M genome is evolving more rapidly than the F genome [16,17,24-27]. The F and M mitochondrial genomes of unionoidean bivalves form reciprocally monophyletic groups [13-16,19], are highly divergent [28] and fossil evidence suggests that the F/M divergence occurred >200 MYA [29].
Recent studies revealed that cox2 from the male-transmitted genomes of unionoidean bivalves has a 3' coding extension that typically yields an ~80% increase in gene length relative to the female-transmitted cox2 genes [13,14]. Because of the pattern of nucleotide substitution and evidence of transcription, it was hypothesized that the MCOX2 extension (MCOX2e) is functional, rapidly evolving and subject to relaxed purging selection [13,14]. It has been demonstrated that the extended Mcox2 gene is translated, most heavily expressed in testes and the protein product is localized to sperm mitochondria [30]. These findings are consistent with the predictions of a male-transmission route for the M genome and functional significance of the extended Mcox2 gene. Furthermore, a secondary structure analysis indicated that the MCOX2e domain has multiple transmembrane helices (TMHs) which suggests a membrane-bound location for this region [30]. These previous studies indicate that the COX2 protein coded by unionoidean bivalve M genomes has a novel third domain (MCOX2e) at its C-terminus.
To obtain further insights into the molecular patterns of primary and secondary structure evolution, and the processes directing these changes, we compared patterns of nucleotide and amino acid substitutions in Mcox2e with those in the other portions of the cox2 and cox1 gene junction region (Fig. 1) from both the F and M genomes among 21 unionoidean bivalve species (Unionoidea: Unionidae: Ambleminae). Our molecular sequence analyses of the MCOX2e region indicate (1) relatively high rates of primary and secondary structure evolution, (2) potential instances of site-specific positive selection and (3) an overall pattern of purifying selection leading to the preservation of the TMH/IHL and C-terminus tail sub-regions of MCOX2e. The pattern of amino acid substitutions indicated that, despite the relatively high degree of sequence divergence observed in some cases, most of the changes did not drastically alter the biochemical properties of the involved amino acid sites. Therefore, the general structure of the MCOX2e region has been preserved since these sequences diverged from a common ancestor > 65 MYA.
Figure 1.

Schematic of the F and Mcox2-cox1 gene junction regions in Venustaconcha ellipsiformis.
Results
In the 21 bivalve species examined, we obtained comparable sequences of the following lengths: 672 bp of Fcox1, 279 bp of Fcox2, 651 bp of Mcox1, and 279 bp of Mcox2h (the region of Mcox2 that is homologous with Fcox2). Mcox2e ranged from 543 (Inversidens and Amblema) to 561 (Ptychobranchus) bp in length (181–187 amino acids; Fig. 2; see Additional file 1). The Mcox2e region contains indels within nucleotide positions 7–135. The first six nucleotides and those from position 136–558 aligned unambiguously and were used in alignment-based analyses containing Mcox2e, while those from positions 7–135 were removed prior to all alignment-based analyses. Thus, slight length variation occurred either within the first 45 amino acid positions, or at the C-terminus end (i.e., the two Quadrula MCOX2e sequences were one amino acid shorter than the others due to the deletion of the terminal residue). No indels were observed between amino acid positions 46 and 186 (constant amino acid positions Y1 and Y2; Fig. 2), and 11 constant amino acid positions were observed (Fig. 2; see Additional file 1). Pairwise amino acid and nucleotide distances (based on Poisson-corrected and Tamura-Nei-corrected models, respectively) are given in Additional file 2. The average corrected amino acid and nucleotide distances were 0.143 and 0.203 for the MCOX2h+COX1 region, 0.013 and 0.137 for the FCOX2+COX1 region, and 0.710 and 0.524 for the MCOX2e region.
Figure 2.

Schematic representation of 21 unionoidean bivalve MCOX2 C-terminus extensions and their evolutionary relationships. A Bayesian Inference estimate of phylogeny (with posterior probabilities [via Mr. Bayes], maximum parsimony bootstrap percentages [when >50%; via PAUP*], and a "+" to indicate significant maximum likelihood ancestral state reconstructions [via Mesquite] given at the tree nodes), based on analyses of 2310 unambiguously alignable protein coding nucleotides from the F and Mcox2-cox1 gene junction regions, and a maximum likelihood optimization of TMH number are presented. The black horizontal lines are proportional to extension length and the red rectangles represent predicted TMH locations and lengths (via ConPredII). The 11 constant amino acid positions are indicated by single letter codes; the eight HyPhy and six codeml sites potentially inferred to be under positive selection are indicated by blue and orange asterisks, respectively (also see Table 2; see Additional file 1).
Database searching
TBLASTX searches of the NCBI database failed to find sequences with significant similarity to the newly characterized MCOX2 extensions. Even when a relaxed search was conducted (cut-off expectation value of E = 1), no significant matches were found outside of mitochondrial genomes from unionoidean bivalves, indicating that this extension is indeed a unique feature of the M mitochondrial genomes of these animals [13,14]. Likewise, PSI-BLAST [31] searches using the deduced amino acid sequences of MCOX2e resulted in convergence (no new matches found) being achieved at the 3rd iteration, and no additional homologs outside of those described above were identified.
A search of the Pfam database (release 21) for entries whose architecture contains the COX2 domain results in 10705 sequences, of which 9511 are eukaryotic (6978 are partial sequences). Most of the remaining 9511 sequences (8442 or 88.8%) exhibit an architecture with a conserved transmembrane domain (Pfam A domain COX2_TM; avg. size: 76.7 residues) 5' of the COX2 domain. A COX2_TM domain is identified for 71 sequences but the model's significance is below the threshold for inclusion in the sequence architecture. Approximately 97% (972) of the remaining 998 sequences are incomplete with less than 39 residues upstream of the COX2 domain, leaving 26 sequences with greater than 39 residues upstream of COX2 and no predicted COX2_TM domain. We analyzed these sequences with the TMH prediction algorithm ConPred II and identified for 13 sequences two TMHs 5' of the COX2 domain. The cytochrome c oxidase subunit II locus is duplicated in the F genome of the bivalve Venerupis phillipinarium and one copy lacks any 5' TMHs (pfam_acc: Q8WF43), but a COX2_TM domain is present in the second copy. The remaining 12 sequences consist of species from the taxa Alveolata and Viridiplantae and do not exhibit upstream THMs when examined with ConPred II.
In contrast, 13 (0.015%) of the 9513 eukaryotic sequences contain TMHs downstream of the COX2 domain. This result is not an artifact of the large number of incomplete sequences in the Pfam database as only two (0.079%) of the 2532 complete eukaryotic COX2 sequences have TMHs 3' of the COX2 domain. These 13 sequences have between one and five TMHs and are all from the male-transmitted, mitochondrial genomes of unionoidean bivalves and represent the previously identified MCOX2e region [13,14].
Phylogenetic analyses
The majority rule consensus tree from the Bayesian analysis of the concatenated M and Fcox2-cox1 nucleotide sequences (including Mcox2e) using GTR+G+I is shown in Figure 2. A Bayesian analysis that omitted Mcox2e (still using GTR+I+G) produced essentially the same results, the only difference being a three-clade polytomy near the root that was resolved in Figure 2. The species relationships indicated by our gene phylogeny (Figure 2) are very similar to those displayed in the most comprehensive published study of amblemine bivalve phylogeny [32] but our Figure 2 displays generally higher nodal support values.
Transmembrane helix (TMH) prediction and tree-based TMH number optimization
The number and positions of the predicted MCOX2e TMHs are shown in a phylogenetic context in Figure 2 with the number of TMHs ranging from three to five. Two TMH gain events and two TMH loss events are suggested from an examination of character state transitions of nodes with significant ancestral character state reconstructions (Fig. 2; significance indicated by a "+" at the nodes). When considering the inclusion or omission of the Mcox2e region in our phylogenetic analyses, the estimation of ancestral TMH number by ML optimization did not change for the nodes that were significant in Figure 2. All tree topology constraint analyses rejected the hypothesis of monophyly for each of the three groups of sequences possessing the same number of predicted TMHs (p << 0.001; Table 1). In addition to the indicated multiple changes in the number of TMHs, Figure 2 also suggests that the lengths of MCOX2e's interhelical loop regions can change without an associated TMH number transition (e.g., compare the Cyrtonaias and Glebula extensions).
Table 1.
Topology test results.
| Parsimony-based tests: | Test | ||||||
|---|---|---|---|---|---|---|---|
| Tree | Length | Difference | KH | Templeton | Winning sites | ||
| Unconstrained | 2832 | Best | |||||
| Taxa with 3 helices constrained | 2987 | 155 | p < 0.0001 | p < 0.0001 | p < 0.0001 | ||
| Taxa with 4 helices constrained | 3251 | 419 | p < 0.0001 | p < 0.0001 | p < 0.0001 | ||
| Taxa with 5 helices constrained | 3175 | 343 | p < 0.0001 | p < 0.0001 | p < 0.0001 | ||
| All taxa constrained | 3418 | 586 | p < 0.0001 | p < 0.0001 | p < 0.0001 | ||
| Likelihood-based tests: | Test | ||||||
| Tree | -ln L | Difference | AU | KH | SH | WKH | WSH |
| Unconstrained | - 13059.96460 | Best | |||||
| Taxa with 3 helices constrained | - 13286.28131 | 226.31671 | p = 7e-10 | p = 0 | p = 0 | p = 0 | p = 0 |
| Taxa with 4 helices constrained | - 14055.57046 | 995.60586 | p = 1e-08 | p = 0 | p = 0 | p = 0 | p = 0 |
| Taxa with 5 helices constrained | - 13764.70315 | 704.73856 | p = 2e-05 | p = 0 | p = 0 | p = 0 | p = 0 |
| All taxa constrained | - 14218.22053 | 1158.25593 | p = 6e-44 | p = 0 | p = 0 | p = 0 | p = 0 |
Results of the parsimony-based Kishino-Hasegawa (KH), Templeton (Wilcoxon signed-ranks) and winning sites (sign) tests calculated using PAUP*, and the likelihood-based approximately unbiased (AU), Kishino-Hasegawa (KH), Shimodiara-Hasegawa (SH), weighted Kishino-Hasegawa (WKH), and weighted Shimodiara-Hasegawa (WSH) tests calculated using CONSEL. The phylogenetic trees compared were the best topology from the unconstrained Bayesian analysis versus analyses where the species with 3, 4, and 5 helices were individually constrained to be monophyletic, and an analysis where all species with equal numbers of helices were simultaneously constrained to be monophyletic.
Properties of the data: (a) Conservation of Mcox2e sequences
Collectively, the 21 MCOX2 extensions display 11 constant amino acid positions (Fig. 2; see Additional file 1), which is a significantly greater number than that expected by chance (as determined by simulation with the program evolver). Of the 5000 data-sets (each 21 sequences, 143 amino acids in length) simulated using the conservative mtREV24 model of substitution, only 2.3% had 11 or greater constant amino acid positions. Using the less conservative Poisson model of substitution, 97% of sequences do not have any conserved positions and the remaining 3% had either only 1 or 2 conserved positions. Within the bounds of these constant positions, no indels are required for the alignment of a contiguous block of 141 amino acids, but slight length variation is observed outside of the Y1 and Y2 positions (Fig. 2; see Additional file 1). The N- and C-termini of each of the 21 extensions is relatively hydrophilic while the interior ~2/3 is relatively hydrophobic (Fig. 3). The predicted C-terminus tail sub-region (i.e., the amino acid residues downstream of the last predicted TMH) is generally more conserved in its primary structure than is the N-terminus/TMH/IHL sub-region of the extensions (Fig. 4), and contains conserved N-glycosylation (in 20 of the 21 tail regions) and casein kinase II phosphorylation motifs (in 20 of the 21 tail regions) (see Additional file 1).
Figure 3.

Kyte-Doolittle plots of three MCOX2 amino acid sequences. ConPred II-generated Kyte-Doolittle plots of three representative MCOX2 amino acid sequences (one each representing species with three, four and five TMHs respectively) showing the hydrophobic nature of the TMH region of MCOX2e. The vertical dashed line demarcates the boundary between MCOX2 homologous and extension regions.
Figure 4.
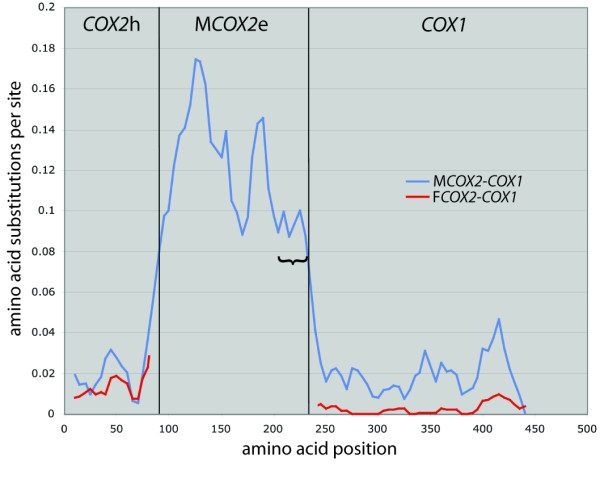
Sliding window plot of amino acid substitutions per site. Sliding window plot of ML-estimated amino acid substitutions per site in the unionoidean bivalve F and MCOX2-COX1 gene junction regions (representing a total of 42 junction regions encoded by 21 F and 21 M genomes and based on a window width of 20 amino acids using five amino acid increments). Regarding the MCOX2 extension region, only the unambiguously alignable (without indels) 143 amino acid positions were utilized in the evaluation of substitution rates. The bracket denotes the C-terminus tail region of MCOX2e.
Properties of the data: (b) Estimates of amino acid substitution rates and positive selection
The hypervariability of the MCOX2 extensions is confirmed by a sliding-window plot (Fig. 4) which illustrates that the overall amino acid substitution rate in the extension is approximately six times greater than that of the MCOX2h+MCOX1 region, and more than 20 times greater than the FCOX2+FCOX1 region. These are likely to be conservative estimates because the most variable portion of the MCOX2e region (i.e., the unalignable N-terminus) was omitted from this analysis. The substitution rate is especially high in the relatively hydrophobic region containing the TMHs and IHLs (see Figs. 2, 3 and 4). Even though the relatively hydrophilic C-terminus tail region is the most conserved portion of the extension, the substitution rate in this region is at least double that of any portion of the MCOX2h+MCOX1 region and at least three times greater than any portion of the FCOX2+FCOX1 region.
Codeml (using Bayes Empirical Bayes method using the M8 model) and HyPhy (using Empirical Bayes method and MG94xHKY85x3_4x2_Rates model) analyses of site-specific positive selection identified six and eight (respectively) of the 143 unambiguously alignable MCOX2e amino acid positions (9.8%) as potential targets of positive selection (interpreted here as diversifying selection) (Table 2; Fig. 2; see Additional file 1). However, we should note that different sites were identified by different methods; interestingly, sites identified by different approaches are located close to each other, almost adjacent, in the overall amino acid sequence. Further, none of the individual sites have passed the stringent criterion of being statistically significant at 95% or higher level (based on PP > 0.95 in codeml and Bayes factor > 100 in HyPhy), although one site in each analysis passed a less stringent criterion of > 90% confidence (codeml PP > 0.90; HyPhy Bayes factor > 50). These analyses are suggestive that there is positive selection operating on specific sites in MCOX2e. Therefore, we propose that one or more of these sites may be of potential interest for future studies of MCOX2e, so we are reporting all of the sites with a HyPhy Bayes factor > 20 or a codeml PP > 50. The log-likelihood values and parameter estimates for the four codeml models applied to the M and F COX2+COX1 regions are displayed in Additional file 3. In contrast, none of the MCOX2h+MCOX1 or FCOX2+FCOX1 amino acid positions (Fig. 1) displayed evidence of diversifying selection using either method. Each of these 14 amino acid positions in MCOX2e suggested as potential targets of diversifying selection represent a site where there is interspecific variation for the constituent amino acids residing in a predicted helix vs. inter-helical loop structure (Fig. 2). Overall, these results suggest the possibility of positive selection driving the sequence changes at particular individual sites, although further experimental studies are needed to determine functional significance of these sites.
Table 2.
MCOX2e amino acid sites that may be under positive selection.
| Site | Bayes factor (HyPhy) | Posterior probability (codeml) |
|---|---|---|
| 62 | 39.618 | -------- |
| 67 | -------- | 0.589 |
| 77 | 70.465 | -------- |
| 78 | -------- | 0.743 |
| 82 | 31.017 | -------- |
| 85 | -------- | 0.801 |
| 107 | 29.190 | -------- |
| 112 | 35.985 | -------- |
| 122 | -------- | 0.521 |
| 124 | 28.480 | -------- |
| 128 | -------- | 0.607 |
| 135 | 28.515 | -------- |
| 140 | 24.688 | -------- |
| 152 | -------- | 0.904 |
Bayes factors for the eight sites potentially inferred to be under positive selection by HyPhy and posterior probabilities for the six sites potentially inferred to be under positive selection by the codeml algorithm in PAML. Site numbers correspond to those in Additional file 1.
Properties of the data: (c) Changes in amino acid composition and properties
Because nucleotide composition can (and is known to) influence the amino acid content (e.g., [33]), we compared the overall amino acid composition of MCOX2e to that of the F and M COX counterparts to see whether they differ. Overall amino acid composition was found to be essentially the same between FCOX2+COX1 and MCOX2h+COX1 gene regions (see Fig. 5). Amino acid composition of MCOX2e differs significantly from that observed in FCOX2-COX1 and MCOX2h-COX1 (both comparisons yield significant differences with p < 0.001), primarily due to significantly lower GC-rich codon content. The proportion of AT-rich or neutral codons were not significantly different between the three regions.
Figure 5.

Overall amino acid composition of different COX protein sequences. Data based on average proportions of individual amino acids among different taxa.
The TreeSAAP evaluation of physicochemical changes of 31 amino acid properties for the three gene regions (MCOX2h+MCOX1, MCOX2e and FCOX2+FCOX1) are given in Additional file 4. Following [34], we concentrate on six amino acid properties shown to be correlated with rates of amino acid substitutions: composition of the side chain, polarity, molecular volume, polar requirements, hydropathy, and isoelectric point (Table 3). We also considered alpha-helical properties and turn tendencies, which could affect the formation of TMHs. The hypervariability of MCOX2e is confirmed by both primary and secondary structure analyses (see Figs. 2 and 4). However, the TreeSAAP results indicated that very similar patterns of amino acid substitution exist between the three gene regions, i.e., most amino acid properties are subject to purifying selection on destabilizing changes. Of the 31 properties examined, 30 (96.8%) showed statistically significant signs of purifying selection for the MCOX2h+MCOX1 region, as compared to 27 (87.1%) for MCOX2e, and 23 (74%) for the FCOX2+FCOX1 region. Only two properties (helical contact area and partial specific volume) showed statistically significant signs of positive selection, and only in the FCOX2+FCOX1 region (see Additional file 4). Interestingly, similar trends were detected when only the MCOX2e sequences were considered, although to a somewhat lesser extent (i.e., of 31 amino acid properties, a smaller proportion of properties deviated from neutrality), indicating that purifying selection plays a role in shaping the amino acid composition of MCOX2e. In particular, such properties as beta-structure tendencies, helical contact area, polarity and polar requirements, among others, were found to have a significantly smaller number of observed than expected amino acid substitutions (p < 0.05; see Additional file 4). Moreover, two properties related to the formation of transmembrane helices were either neutral in all three regions or under purifying selection.
Table 3.
Summary of TreeSAAP output.
| M seq | MCOX2-extension only | F seq | ||||
|---|---|---|---|---|---|---|
| Amino Acid Property | 1 -- 3 | 6 -- 8 | 1 -- 3 | 6 -- 8 | 1 -- 3 | 6 -- 8 |
| Composition | neg/destab | |||||
| Hydropathy | neg/destab | neg/destab | neg/stab | neg/destab | ||
| Isoelectric point | neg/stab | neg/stab | ||||
| Molecular volume | pos/stab | neg/destab | pos/stab | |||
| Polar requirement | neg/destab | neg/destab | pos/stab | neg/destab | ||
| Polarity | neg/destab | pos/stab | neg/destab | pos/stab | neg/destab | |
| Alpha-helical tendencies | neg/destab | |||||
| Turn tendencies | neg/destab | neg/destab | neg/destab | |||
The six "most important" TreeSAAP categories according to [34]. Two additional properties (Alpha-helical tendencies and Turn tendencies) are given due to their potential importance to transmembrane helix formation. Absence of selection means neutrality.
Discussion
The MCOX2e region is unique to unionoidean bivalve M genomes
Our TBLASTX, PSI-BLAST and Pfam database searches failed to find any significant matches between the new Mcox2e/MCOX2e sequences analyzed herein and any non-M genome sequences in the current sequence databases. The multiple similarities among the 21 Mcox2e sequences analyzed herein are consistent with the hypothesis that the Mcox2e region was acquired in a single insertion event that took place in a distant common ancestor that lived > 65 MYA [29], rather than by accretionary acquisition. Furthermore, Mcox2e was found in representatives of the genus Margaritifera [14], the hypothesized basal unionoid bivalve lineage [35-38], indicating an origin for the Mcox2e region ≥ 200 MYA. Unfortunately, the original source of the DNA now comprising the Mcox2e region may remain unidentified given the extremely high substitution rate in this region and the relatively large amount of time over which this region has accumulated substitutions.
Another feature novel to unionoidean bivalve M genomes is the presence of the TMH/IHL sub-region of MCOX2e downstream of the COX2 domain. We have shown in the sequences analyzed herein, that there are between three and five such TMHs, and that there has been a minimum of four changes in TMH number (two gains and two losses) during the evolution of MCOX2e (Figure 2) in the 21 amblemine bivalve species analyzed herein. Furthermore, in 20 of the 21 C-terminus tail sub-regions of MCOX2e, both N-glycoslylation and casein kinase II phosphorylation motifs are observed (see Additional file 1) along with this subregion being more conserved in primary structure than the TMH/IHL sub-region (Fig. 4). Thus, while MCOX2e appears volatile with respect to primary structure and the number and position of TMHs/IHLs, purifying selection (TreeSAAP analyses) is apparently acting to preserve the general character of the TMH/IHL and C-terminus tail sub-regions of this unique region.
Is the MCOX2e region functional?
In a study of four freshwater mussel species' (representing three genera) MCOX2 extensions, Curole and Kocher [13] concluded that the "...extension is protein-coding and functional." Their claim was based on three factors: (1) the extension is in frame for each Mcox2 gene and the stop codon is located just upstream of the putative cox1 initiation codon, (2) the relative rates of nucleotide substitution among the three codon positions in the extension are typical for protein coding loci and (3) a polyadenylated transcript of the Mcox2 gene, containing the extension, was detected in testes. Subsequently, Chakrabarti et al. [30] demonstrated that Mcox2e is translated and strongly expressed in testes and sperm mitochondria (as would be expected for a functional, paternally transmitted mitochondrial gene) and they hypothesized "...the C-terminus extension has functional significance for male unionoidean bivalve reproductive success." Our study strongly supports the hypothesis of functionality for the MCOX2e region because the 21 extension sequences in this report (1) are in reading frame with the homologous portion of COX2 and have a 3' stop codon near their putative cox1 initiation codon, (2) show typical relative rates of substitution among the three codon positions, (3) have 11 constant amino acid positions, a number greater than expected by chance, juxtaposed within a relatively larger number of amino acid sites with extremely elevated substitution rates, (4) have at least three predicted transmembrane helices, (5) have conserved functional motifs in the C-terminus tail sub-region, (6) have amino acid compositions indicative of a putative functional state and one that is quite similar to that of the MCOX2h+MCOX1 and FCOX2+FCOX1 regions, (7) have generally conserved hydropathy plots and (8) are evolving largely under the influence of purifying selection (e.g., TreeSAAP analyses), but may potentially be influenced by positive selection at ~10% of sites (as suggested by codeml and HyPhy analyses).
The observed conservation of general MCOX2e features could be due to (1) purifying selection acting on the MCOX2e region per se or (2) purifying selection acting to maintain an ancestral sequestration of the extension so as to prevent it from interfering with vital mitochondrial functions. Because MCOX2 is found in two sub-cellular locations (inner and outer mitochondrial membranes in sperm [39]) vs. the single ancestral location (inner mitochondrial membrane) seems to rule out the second hypothesis. Thus, it is likely that purifying selection is maintaining the general characteristics of a functional MCOX2e region in the face of an extremely high overall amino acid substitution rate. This hypothesis is bolstered by the observation that, similar to the MCOX2h+MCOX1 and FCOX2+FCOX1 regions, two amino acid properties related to transmembrane helix formation (alpha-helical and turn tendencies; Table 3) were found to be evolving under either neutral or purifying selection acting to preserve the physicochemical properties of MCOX2e.
Within the extension region bounded by the Y1 and Y2 conserved amino acid positions (Fig. 2), the juxtaposition of hypervariability and conservation is consistent with the hypothesis that the predicted TMH number and position variability is due to substitution-based processes rather than the typically implicated duplication/deletion events (e.g., [40]). This hypothesis is supported by "mutating" the MCOX2e sequences in members of sister taxon pairs that have different numbers of estimated helices (Fig. 2), and subsequently employing ConPred II to estimate helix number for the "mutated" sequences. For example, when comparing the Pleurobema and Fusconaia MCOX2e sequences, changing the amino acid at position 138 (see Additional file 1) of Fusconaia's MCOX2e to that in Pleurobema (A to V) resulted in the gain of a 5th helix in Fusconaia's MCOX2e which was in the same position as the 5th helix in Pleurobema. Similarly, a single amino acid substitution (from T to I at position 79; see Additional file 1) in Lemiox's MCOX2e sequence resulted in the gain of a 5th helix in Lemiox which is in the same position as the 3rd helix in Ptychobranchus MCOX2e (Fig. 2). Thus, a single amino acid substitution in MCOX2e can be sufficient to transform an interhelical loop sub-region into a TMH. Furthermore, the suggestion of positively selected sites in the areas of MCOX2e in which some species contain TMHs and others do not (Fig. 2; see Additional file 1) coupled with the possibility of single amino acid substitutions resulting in a change in TMH number, could help explain the volatility in TMH number and position inferred from our MCOX2e sequences. The above results suggest that while the preservation of the TMH/IHL and C-terminus tail subregions is a general feature of MCOX2e evolution, the specific number and positions of TMHs are readily changed.
There are other known genes that exhibit a similar pattern of being composed of a mixture of highly variable and more conserved sites. For example, in mammalian genes of the major histocompatibility complex (MHC), the highly variable antigen recognition sites are subject to strong positive selection [41,42] while other sites evolve under purifying selection. Likewise, different regions of the gamete-recognition protein bindin in sea urchins exhibit signs of both positive and purifying selection [43]. Moreover, the substitution patterns observed within MCOX2e are inconsistent with it being a pseudogene. Generally, pseudogenes experience a relaxation of selection and are gradually degraded through the accumulation of random substitutions [44,45]. While some pseudogenes appear to be relatively conserved (e.g., beta-esterase genes in Drosophila melanogaster; [46,47]), they are unlikely to persist for many millions of years, as is the case with MCOX2e which is shared among taxa that diverged ≥ 200 MYA [29]. Moreover, overall amino acid composition of MCOX2e is quite similar to that of the homologous COX genes analyzed herein (Fig. 5), with the exception of GC-rich codons. However, because the composition of pseudogenes has been shown to strongly correlate with the overall AT composition of the genome [48], similarity of AT-rich and neutral codons' content between MCOX2e and the other COX regions may serve as additional evidence indicating a functional role for MCOX2e.
Significance of rapid evolutionary rate for a functional MCOX2e region?
Both the primary and secondary structures of the MCOX2e region are evolving much faster than in the MCOX2h+MCOX1 and FCOX2+FCOX1 regions. The amino acid substitution rate in the MCOX2e region is ~6× faster than that in the MCOX2h+MCOX1 gene junction region and >20× faster than that in the F gene junction region (Fig. 4). TMH number in the MCOX2e region is evolving much faster than in the typical TM domains of COX proteins (e.g., [2-4]) which generally show no TMH number variation over more than 600 million years of divergence (vs. the ~65 million years of divergence represented by the species in Fig. 2). Furthermore, secondary structure analyses of the two complete Mcox2 sequences available in the GenBank (Anodonta woodiana [GenBank accession no. AB055626] and Inversidens japanensis [GenBank accession no. AB055624]) confirm the presence of two TMHs near the N-terminus of the protein (i.e., the standard metazoan COX2 TMH number and location). The extremely rapid evolution in both primary and secondary structure of the MCOX2e region relative to the other domains of the F and M COX2-COX1 gene junction regions, along with the multiple sub-cellular locations of MCOX2 in sperm mitochondria [39], suggest that MCOX2e has a unique function and consequently a selective regime that is distinct from that of the other mtDNA protein coding domains. The male-specific transmission of M genomes [[24,49], but see [23]], the testes-biased tissue distribution of Mcox2 [50,51], predominant expression of MCOX2 in testes tissue [30], and MCOX2 maximal expression immediately prior to fertilization [39] are all consistent with the hypothesis of a reproductive function for M genomes [52-56]. Relatively rapid rates of evolution are frequently observed for proteins involved in reproduction (e.g., [57-59]). Thus, the rapid evolution of primary and secondary structure in MCOX2e, combined with the action of purifying and possibly site-specific positive selection on this unique domain, provide additional evidence for the reproductive function hypothesis.
Given the published evidence supporting the hypothesis that MCOX2 is localized in the outer mitochondrial membrane of sperm [39], the IHLs and/or C-terminus tail of MCOX2e may serve to "tag" paternal mitochondria in early embryos to facilitate their gender-specific movement [60,61] which is likely a requisite for the maintenance of paternal mitochondria transmission. The impact of positive selection on just a few amino acid sites in the TMH/interhelical loop region of MCOX2e could have a propagating effect on protein function (e.g., paternal mitochondria recognition) by shifting the actual sequence of amino acids that are in the loops (i.e., exposed on the cytoplasmic side of the outer mitochondrial membrane). Positive selection at just a few sites could: (1) lead to switches in the signaling pathway from one to another (i.e., by altering the paternal mitochondria's "tags") and/or (2) diversify the pathway by adding extra recognition sites (i.e., adding new "tags"). Whenever a helix changes position, so does the size and amino acid composition of the two adjacent interhelical loops. A change in helix position could cause a change in the (exposed) loop sequence, even though the overall amino acid sequence in the protein changes by only one amino acid (as in the Fusconaia-Pleurobema and Lemiox-Ptychobranchus MCOX2e comparisons [discussed above]). Adding helices (we see this twice in our gene phylogeny) could expand the number of signaling receptors. The fact that each of the 14 sites in MCOX2e potentially experiencing diversifying selection are sites whose constituent amino acids show interspecific variation as to whether they are within a helix or inter-helical loop, illustrates the potential importance that just a few positively selected sites could have on the evolution of MCOX2 protein function.
Conclusion
Our results indicate that (1) Mcox2e is unique to unionoidean bivalves, (2) MCOX2e is functional and is likely the result of a single insertion event that took place over 65 MYA, (3) the predicted TMH/IHL number, length and position variability likely stems from substitution-based processes rather than the typically implicated insertion/deletion events, (4) MCOX2e has relatively high rates of evolution in its primary and secondary structures, (5) MCOX2e displays evidence suggestive of site-specific positive selection, (6) MCOX2e has an overall pattern of purifying selection that leads to the preservation of the TMH/IHL and hydrophilic C-terminus tail sub-regions, and (7) the more conserved C-terminus tail (relative to the TMH/IHL sub-region of MCOX2e) is likely biologically active because it contains functional motifs. The rapid evolution of primary and secondary structure in MCOX2e, combined with the action of purifying and possibly positive selection, provide supporting evidence for the hypothesis that MCOX2e has a novel reproductive function within unionoidean bivalves. Furthermore, the presence of TMHs on the C-terminus side of the COX2 copper-binding site has never before been observed in metazoan mtDNA. All tolled, our data indicate that unionoidean bivalve MCOX2 is a chimeric animal mitochondrial protein that contains a unique and functional domain of unknown origin.
Methods
Taxa used
We obtained sequences from a thorough cross-section (n = 21 species; Table 4) of the unionoidean bivalve subfamily Ambleminae including 14 genera (16 species) representing the Amblemini and Lampsilini, two genera (two species) from the Pleurobemini, one genus (two species) representing the Quadrulini and one species from the Gonideini. The use of Inversidens japanensis (Gonideini) as the outgroup in our phylogenetic analyses is justified by the results of [19,32]. Paleontological evidence suggests that the sequences analyzed herein diverged from a common ancestor between 65 and 100 MYA [29].
Table 4.
Species and GenBank accession numbers for the taxa used in this study.
Female-transmitted cytochrome c oxidase subunit I (Fcox1); female-transmitted cytochrome c oxidase subunit II (Fcox2); male-transmitted cytochrome c oxidase subunit I (Mcox1); and male-transmitted cytochrome c oxidase subunit II (Mcox2).
DNA Sequencing
The gender of the sampled unionoidean bivalve individuals was determined by microscopical examination of gonadal tissues. Total genomic DNA was isolated from mantle and testes tissues using the Qiagen DNeasy animal kit. The largely M-specific primer pair used in [30] along with those in [62] were used to amplify the Mcox2-cox1 junction region [13] (Fig. 1) from testicular tissue-based DNA isolates. These primers amplified an ~1.7 kbp fragment from the M genomes. A largely F-specific primer pair [19] was used to amplify the corresponding Fcox2-cox1 junction region (Fig. 1) from mantle tissue-based DNA isolates. These primers amplified a ~1.1 kbp fragment from the F genome. PCR reactions consisted of 1× Qiagen PCR buffer, 0.2 mM each dNTP, 0.5 μM each primer, 1 U Qiagen Taq and ~20 ng of template DNA. Reactions using the male-specific primers were cycled at 94°C for 60s, 50°C for 60s, and 72°C for 120s for a total of 40 cycles. Reactions involving the female-specific primer followed the same profile as above but were annealed at 46°C. The above PCR primers ultimately yielded F and Mcox2-cox1 DNA sequences obtained via cycle sequencing with Perkin Elmer AmpliCycle Sequencing Kits. The sequencing primers utilized were identical in sequence to the PCR primers and sequencing template purification was done following [63]. Sequences were visualized using Li-Cor 4200L-2 and 4200S-2 DNA sequencers. Forward and reverse sequencing reads were assembled and verified using AlignIR v2.0 (LI-COR, Inc.) and final sequence alignments were completed manually with MacClade v4.0 [64].
Database searching
To identify potential homologs, Mcox2e nucleotide sequences were used as queries for TBLASTX [31] searches (six-frames, translated nucleotides for both query and subject) against the non-redundant Genbank database. We searched with a relaxed expectation value of E = 1 to ensure completeness. Putative translated Mcox2e sequences, using the Drosophila mtDNA genetic code, were used in subsequent PSI-BLAST [31] searches of the GenBank with an expectation E-value of 0.5 for inclusion.
To understand better the secondary structure of the cox2 gene we searched the Pfam database (release 21; [65]) for all sequences with an architecture containing the COX2 Pfam A domain. Sequences from Danaus plexippus plexippus (Pfamseq_acc: Q6J977), Tirumala hamata (Q6J937) and Agathis sp. DMA-1998 (Q9T6C7) were excluded because of the large number of ambiguous residues present at the 5' end of the sequences. A Caenorhabditis briggsae sequence (Q60IM7) was excluded because it is a whole genome shotgun entry consisting of a cytochrome c oxidase II periplasmic domain, a NADH:ubiquinone oxidoreductase subunit 5 domain and a NADH-Ubiquinone/plastoquinone domain; thus, it is likely an unannotated clone of the C. briggsae mitochondrial genome. Sequences were split into two groups based on the presence or absence of a COX2_TM Pfam A domain. A small subset of sequences have a putative COX2_TM Pfam A domain, but the model significance is too low for inclusion in the sequence architecture. A large number of sequences have a small number of residues upstream of the COX2 domain. To estimate a minimum cutoff length for COX2_TM prediction, we generated the distribution of upstream sequence length (length 5' of the COX2 domain) for all sequences whose architecture includes the COX2_TM domain. We chose a 1% cutoff level, which corresponds to 39 residues (i.e. 99% of sequences with a COX2_TM~COX2 architecture have an upstream sequence length greater than 39 residues). Sequences with greater than 39 upstream residues and no COX2_TM Pfam A domain were subjected to transmembrane helix prediction using the ConPred II algorithm [66]. Transmembrane helices are considered upstream if the 3' end of the transmembrane helix is upstream of the 5' end of the COX2 Pfam A domain. In addition, we scanned the Pfam annotation for TMHs 3' of the COX2 domain.
Phylogenetic analyses
Our Mcox2 sequences were aligned with unionoidean bivalve Fcox2 nucleotide sequences (which have a uniform length) obtained from the Genbank to determine the boundaries and length of Mcox2e. The 5' end of Mcox2e is designated as the nucleotide of the Mcox2 sequence that aligns with position one in the stop codon of the Fcox2 sequences. The 3' end of Mcox2e is the stop codon for Mcox2. The F and Mcox2-cox1 nucleotide sequences were translated to protein sequences using the Drosophila mtDNA genetic code. All DNA sequences generated herein were submitted to the GenBank database (Table 4).
Phylogenetic trees were reconstructed using Bayesian inference (BI) and maximum parsimony (MP) approaches. Bayesian analyses were conducted using the program MrBayes (v3.1.2; [67,68]). Bayesian searches were run for 10 million generations with 10 search chains and the data were partitioned by gene region and by codon position (five gene regions × three codon positions for the full data-set; four gene regions × three codon positions for the data-set with the Mcox2e sequences removed), saving 10,000 trees (one tree saved every 1000 generations) and using GTR+G+I substitution model [69], as selected by the program Modeltest [70]. To allow each partition to have its own set of parameter estimates, revmat, tratio, statefreq, shape, and pinvar were all unlinked during the analysis. The burn-in was determined by visual inspection of the likelihood score plot obtained as the trees were written to the tree file. In all analyses, stationarity was reached before one million generations, and the first 1000 trees were discarded (i.e., the first million generations) from each analysis as the burn-in. To obtain the most accurate branch length estimates possible, the option prset ratepr = variable was employed as per the recommendations of [71]. Maximum parsimony analyses, using PAUP* (v.4.0b10 [72]), were run on a transformed data-set wherein only transversions were coded for all third codon positions. Reliability of the internal nodes was estimated by bootstrapping the data-set with 100,000 full heuristic replications for the MP analysis using PAUP*. Reliability of the Bayesian topologies was evaluated with the posterior probabilities from the majority-rule consensus tree.
Transmembrane helix (TMH) prediction and optimization
Secondary structure prediction was done using the ConPred II software package [66] that utilizes five TM prediction methods. The "Markov k-state one parameter model" (MK1; [73]), which assumes equally probable forward and backward rates of change, was used for maximum likelihood estimation of the ancestral number of MCOX2e TMHs. Ancestral states were determined using the Bayesian phylogenetic estimate from the 2,310 bp data-set under the GTR+G+I model and MK1 model parameters with the software package Mesquite (v.1.05; [74]). Ancestral character state estimates with a log likelihood two or more units lower than the best state estimate (decision threshold [T] set to T = 2) were rejected [75,76].
Because the best phylogenetic estimates suggested substantial MCOX2e TMH number volatility, the robustness of the ancestral state inferences was evaluated using constraint analyses. Separate BI analyses were conducted on our non-transformed 2,310 bp nucleotide dataset to produce trees in which terminals with identical TMH number were individually constrained to be monophyletic, and a tree in which all terminals of equal helix number were simultaneously constrained to be monophyletic. Differences in topology between these trees and the best tree from an unconstrained analysis were tested with the parsimony-based Kishino-Hasegawa test (KH; [77]), the Templeton test (Wilcoxon signed-ranks test; [78]) and the winning sites test [79] in PAUP*. Differences between these topologies were also tested with the likelihood-based approximately unbiased test (AU; [80]), the Shimodaira-Hasegawa test (SH; [81]), the weighted Shimodaira-Hasegawa test (WSH; [80]), the Kishino-Hasegawa test (KH) and the weighted Kishino-Hasegawa test (KWH) in CONSEL [82].
Properties of the data: (a) Conservation of MCOX2e sequences
To determine whether the observed number of conserved amino acid positions in the MCOX2e region would be expected by chance alone, we generated a null distribution of the number of MCOX2e conserved amino acid sites. Data-sets consisting of 21 sequences each 143 amino acids in length were simulated (5,000 total) with the program evolver (from the PAML package; [83]) using the Mtrev model of substitution [84]. Evolver simulates data sets by generating a root sequence from user defined amino acid frequencies and then evolving this sequence along the user specified tree. Branch lengths used for sequence simulation were estimated by subjecting the extension region's 143 unambiguously aligned amino acids and the topology in Figure 2 to a maximum-likelihood analysis with PAML assuming a constant substitution rate across sites and the mtREV24 substitution matrix. The average amino acid frequencies across the 21 species were used as the starting frequencies for simulation. To reflect the null hypothesis that this region is evolving in the absence of selection the simulation assumed a constant substitution rate across sites. Simulations were done with both the conservative mtREV24 model and the less conservative Poisson model of sequence substitution rates. Each of the 5,000 simulated sequence data-sets was then scanned for conserved positions. The number of conserved positions for each simulated data-set was used to construct a probability density distribution. In addition, PROSITE [85] was used to search for conserved functional motifs in the MCOX2e region.
Properties of the data: (b) Estimates of amino acid substitution rates, hydropathy indices, and positive selection
The ML-based program HyPhy [86] was used to generate sliding window estimates of amino acid substitutions per site in the COX2+COX1 regions (Mtrev substitution model; [87]) using the topology from Figure 2. Kyte-Doolittle hydropathy plots were generated for the sequenced MCOX2 region (MCOX2h+MCOX2e) using ConPred II [66]. The codeml program (PAML; [83]) and HyPhy [86] were used to search for positively selected codons in the M and F COX2-COX1 junction regions. These programs estimate model parameters by numerical optimization of the likelihood function (maximum likelihood) via reference to a user-specified tree topology. We explored four models of codon substitution with codeml in a pairwise fashion. First, a nearly neutral model (M1a) that does not allow for positive selection (ω1 = 1 and 0<ω0<1) was compared with a positive selection model (M2a), which is an extension of model M1a that has an additional (third) site class with ω >1. Differences in log-likelihood values were tested against a X2 distribution. If a majority of sites in a gene have undergone purifying selection or neutral evolution, models M1 and M2 may not be sufficiently sensitive to detect positive selection. Therefore, a similar X2 test is employed with two models (M7 and M8) that assume a β distribution of ω ratios among sites. Unlike the M7 model, the M8 model allows for some sites to have ωs > 1. If the positive selection models are found to be significantly better than the other models, codeml employs the Bayes empirical Bayes method [87] to calculate the posterior probabilities (PP) that sites are experiencing positive selection. Some authors [e.g., [88-90]] report the presence of positive selection with a PP > 0.9, while others [e.g., [91]] feel a more stringent cutoff of PP > 0.95 is more appropriate. The HyPhy analysis utilized the "MG94xHKY85x3_4x2_Rates" model of codon substitution [92] and searched for positively selected sites using the empirical Bayes method [6] following the procedure in [92]. Following [92], Bayes factor values >20 show some evidence for positive selection and Bayes factor values > 50 correspond to "extremely high posterior probabilities" (at least greater than PP = 0.9) [89].
Properties of the data: (c) Changes in amino acid composition and properties
The program TreeSAAP (v.3.2; 93]) was used to measure the influences of selection at the amino acid level using quantitative amino acid properties. Distribution of potential changes in physicochemical amino acid properties was inferred using the topology from Figure 2, and differences between expected and observed changes were evaluated. Positive and negative z-scores indicate positive and negative (purifying) selection, respectively [34,93,94].
We also compared overall amino acid composition of MCOX2e with that of the male and female COX1 and COX2h proteins. Amino acids were partitioned into three categories: those encoded by GC-rich codons (i.e., FYMINK amino acids, namely, phenylalanine, tyrosine, methionine, isoleucine, asparagine, and lysine), those encoded by AT-rich codons (GARP amino acids, namely, glycine, alanine, arginine, and proline) and those encoded by neutral codons [95]. The proportion of each individual amino acid was estimated for each sequence. A chi-square test was used to determine whether the average amino acid composition significantly differed between different regions.
Abbreviations
BI: Bayesian inference; bp: base pairs; cox: cytochrome c oxidase DNA; COX: cytochrome c oxidase protein; MCOX2h/Mcox2h: homologous portion of COX2/cox2 shared by both M and F genomes; DUI: doubly uniparental inheritance; F: maternally transmitted; M: paternally transmitted; IHL: interhelical loop; MCOX2e/Mcox2e: MCOX2/Mcox2 extension; MP: maximum parsimony; mtDNA: mitochondrial DNA; PP: posterior probability; TMH: transmembrane helix.
Authors' contributions
EGC conducted the Bayesian phylogenetic analyses, the constraint analyses and topology tests and the TMH prediction and ML optimizations. HP carried out the TBLASTX and PSI-BLAST searches, PAML (codeml) and TreeSAAP analyses. JMW did all DNA sequencing. JPC conducted the pfam and evolver (PAML) analyses. WRH carried out the HyPhy and maximum parsimony analyses. WRH, JPC and DTS developed the concept and design of the study. All authors participated in writing the manuscript and read and approved the final version of the manuscript.
Supplementary Material
Amino acid sequences deduced from the complete Mcox2 extension nucleotide sequences of the 21 species used in this study.
Pairwise amino acid and nucleotide distances (based on PC and TN models, respectively).
Codeml log-likelihood values and parameter estimates for four models applied to the M and F COX2+COX1 regions.
Summary of TreeSAAP output showing the 31 amino acid properties of MCOX2h+MCOX1, MCOX2e and FCOX2+FCOX1.
Contributor Information
Eric G Chapman, Email: echapman@kent.edu.
Helen Piontkivska, Email: opiontki@kent.edu.
Jennifer M Walker, Email: jennifwalker@gmail.com.
Donald T Stewart, Email: don.stewart@acadiau.ca.
Jason P Curole, Email: jcurole@usc.edu.
Walter R Hoeh, Email: randy.hoeh@gmail.com.
Acknowledgements
We thank S. Ahlstedt, C. Barnhart, A. Bogan, R. Butler, D. Campbell, A. Christian, M. Gordon, J. Harris, W. Heard, R. Howells, D. Hubbs, J. Jones, K. Kuehnl, B. Lang, B. Posey, B. Sietman and T. Watters for providing specimens, D. Senyo for assistance in the lab and V. Dvornyk for assistance with PAML. We thank A. Bogan, A. Case, V. Dvornyk, M. Gordon, P. Lorch, and K. Mock for commenting on previous versions of this manuscript. This work was supported by grants from the USA's National Science Foundation (DEB-0237175 to W.R.H.) and Canada's Natural Sciences and Engineering Research Council (Discovery Grant 217175 to D.T.S.). Finally, we thank the three anonymous reviewers for their helpful comments and criticisms of this manuscript.
References
- Richter O-MH, Ludwig B. Cytochrome c oxidase – structure, function, and physiology of a redox-driven molecular machine. Rev Physiol Biochem Pharmacol. 2003;147:47–74. doi: 10.1007/s10254-003-0006-0. [DOI] [PubMed] [Google Scholar]
- Castresana J, Lübben M, Saraste M, Higgins DG. Evolution of cytochrome oxidase, an enzyme older than atmospheric oxygen. EMBO J. 1994;13:2516–2525. doi: 10.1002/j.1460-2075.1994.tb06541.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwata S, Ostermeier C, Ludwig B, Michel H. Structure at 2.8 Å resolution of cytochrome c oxidase from Paracoccus denitrificans. Nature. 1995;376:660–669. doi: 10.1038/376660a0. [DOI] [PubMed] [Google Scholar]
- Tsukhihara T, Aoyama H, Yamashita E, Tomizaki T, Yamaguchi H, Shinzawa-Itoh K, Nakashima R, Yaono R, Yoshikawa S. The whole structure of the 13-subunit oxidized cytochrome c oxidase at 2.8 Å. Science. 1996;272:1136–1144. doi: 10.1126/science.272.5265.1136. [DOI] [PubMed] [Google Scholar]
- Nielsen R, Yang Z. Likelihood models for detecting positively selected amino acid sites and applications to the HIV-1 envelope gene. Genetics. 1998;148:929–936. doi: 10.1093/genetics/148.3.929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang ZH, Nielsen R, Goldman R, Pedersen AMK. Codon-substitution models for heterogeneous selection pressure at amino acid sites. Genetics. 2000;155:431–449. doi: 10.1093/genetics/155.1.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerber AS, Loggins R, Kumar S, Dowling TE. Does nonneutral evolution shape observed patterns of DNA variation in animal mitochondrial genomes? Annu Rev Genet. 2001;35:539–566. doi: 10.1146/annurev.genet.35.102401.091106. [DOI] [PubMed] [Google Scholar]
- Rand DM. The units of selection on mitochondrial DNA. Annu Rev Ecol Syst. 2001;32:415–48. [Google Scholar]
- Ballard JWO, Whitlock MC. The incomplete natural history of mitochondria. Molec Ecol. 2004;13:729–744. doi: 10.1046/j.1365-294x.2003.02063.x. [DOI] [PubMed] [Google Scholar]
- Grossman LI, Wildman DE, Schmidt TR, Goodman M. Accelerated evolution of the electron transport chain in anthropoid primates. TREE. 2004;20:578–585. doi: 10.1016/j.tig.2004.09.002. [DOI] [PubMed] [Google Scholar]
- Dalziel AC, Moyes CD, Fredriksson E, Lougheed SC. Molecular evolution of cytochrome c oxidase in high performance fish. J Mol Evol. 2006;62:319–331. doi: 10.1007/s00239-005-0110-7. [DOI] [PubMed] [Google Scholar]
- Rawson PD, Burton RS. Molecular evolution at the cytochrome oxidase subunit 2 gene among divergent populations of the intertidal copepod, Tigriopus californicus. J Mol Evol. 2006;62:753–764. doi: 10.1007/s00239-005-0074-7. [DOI] [PubMed] [Google Scholar]
- Curole JP, Kocher TD. Ancient sex-specific extension of the cytochrome c oxidase II gene in bivalves and the fidelity of doubly-uniparental inheritance. Mol Biol Evol. 2002;19:1323–1328. doi: 10.1093/oxfordjournals.molbev.a004193. [DOI] [PubMed] [Google Scholar]
- Curole JP, Kocher TD. Evolution of a unique mitotype-specific protein-coding extension of the cytochrome c oxidase II gene in freshwater mussels (Bivalvia: Unionoida) J Mol Evol. 2005;61:381–389. doi: 10.1007/s00239-004-0192-7. [DOI] [PubMed] [Google Scholar]
- Hoeh WR, Stewart DT, Sutherland B, Zouros E. Multiple origins of gender-associated mitochondrial DNA lineages in bivalves (Mollusca: Bivalvia) Evolution. 1996;50:2276–2286. doi: 10.1111/j.1558-5646.1996.tb03616.x. [DOI] [PubMed] [Google Scholar]
- Hoeh WR, Stewart DT, Guttman SI. High fidelity of mitochondrial genome transmission under the doubly uniparental mode of inheritance in fresh water mussels (Bivalvia: Unionoidea) Evolution. 2002;56:2252–2261. doi: 10.1111/j.0014-3820.2002.tb00149.x. [DOI] [PubMed] [Google Scholar]
- Liu H-P, Mitton JB, Wu S-K. Paternal mitochondrial DNA differentiation far exceeds maternal mitochondrial DNA and allozyme differentiation in the freshwater mussel, Anodonta grandis grandis. Evolution. 1996;50:952–957. doi: 10.1111/j.1558-5646.1996.tb03907.x. [DOI] [PubMed] [Google Scholar]
- Passamonti M. An unusual case of gender-associated mitochondrial DNA heteroplasmy: the mytilid Musculista senhousia (Mollusca Bivalvia) BMC Evol Biol. 2007;7(Suppl 2):S7. doi: 10.1186/1471-2148-7-S2-S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker JM, Curole JP, Wade DE, Chapman EG, Bogan AE, Watters GT, Hoeh WR. Taxonomic distribution and phylogenetic utility of gender-associated mitochondrial genomes in the Unionoida (Bivalvia) Malacologia. 2006;48:265–282. [Google Scholar]
- Passamonti M, Scali V. Gender-associated mitochondrial DNA heteroplasmy in the venerid clam Tapes phillippinarum (Mollusca: Bivalvia) Curr Genet. 2001;39:117–124. doi: 10.1007/s002940100188. [DOI] [PubMed] [Google Scholar]
- Breton S, Beaupré HD, Stewart DT, Hoeh WH, Blier PU. The unusual system of doubly uniparental inheritance of mtDNA: isn't one enough? Trends Genet. 2007;23:465–474. doi: 10.1016/j.tig.2007.05.011. [DOI] [PubMed] [Google Scholar]
- Sutherland B, Stewart D, Kenchington ER, Zouros E. The fate of paternal mitochondrial DNA in developing female mussels Mytilus edulis: implications for the mechanism of doubly uniparental inheritance of mitochondrial DNA. Genetics. 1998;148:341–347. doi: 10.1093/genetics/148.1.341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obata M, Sano N, Kawamura K, Komaru A. Inheritance of two M type mitochondrial DNA from sperm and unfertilized eggs to offspring in Mytilus galloprovincialis. Develop Growth Differ. 2007;49:335–344. doi: 10.1111/j.1440-169X.2007.00930.x. [DOI] [PubMed] [Google Scholar]
- Skibinski DOF, Gallagher C, Beynon CM. Sex-limited mitochondrial DNA transmission in the marine mussel Mytilus edulis. Genetics. 1994;138:801–809. doi: 10.1093/genetics/138.3.801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawson PD, Hilbish TJ. Evolutionary relationships among the male and female mitochondrial DNA lineages in the Mytilus edulis species complex. Mol Biol Evol. 1995;12:893–901. doi: 10.1093/oxfordjournals.molbev.a040266. [DOI] [PubMed] [Google Scholar]
- Stewart DT, Saavedra C, Stanwood RR, Ball A, Zouros E. Male and female mitochondrial DNA lineages in the Blue mussel (Mytilus edulis) species group. Mol Biol Evol. 1995;12:735–747. doi: 10.1093/oxfordjournals.molbev.a040252. [DOI] [PubMed] [Google Scholar]
- Krebs RA. Combining paternally and maternally inherited mitochondrial DNA for analysis of population structure in mussels. Molec Ecol. 2004;13:1701–1705. doi: 10.1111/j.1365-294X.2004.02133.x. [DOI] [PubMed] [Google Scholar]
- Mizi A, Zouros E, Moschonas N, Rodakis GC. The complete maternal and paternal mitochondrial genomes of the Mediterranean mussel Mytilus galloprovincialis: implications for the doubly uniparental inheritance mode of mtDNA. Mol Biol Evol. 2005;22:952–967. doi: 10.1093/molbev/msi079. [DOI] [PubMed] [Google Scholar]
- Watters GT. In: Ecology and Evolution of the Freshwater Mussels (Unionoida) Bauer G, Wächtler K, editor. Berlin: Springer-Verlag; 2001. The evolution of the Unionacea in North America, and its implications for the worldwide fauna; pp. 281–307. [Google Scholar]
- Chakrabarti R, Walker JM, Stewart DT, Trdan RJ, Vijatraghavan S, Curole J, Hoeh WR. Presence of a unique male-specific extension of C-terminus to the cytochrome c oxidase subunit II protein coded by the male-transmitted mitochondrial genome of Venustaconcha ellipsiformis (Bivalvia: Unionoidea) FEBS Lett. 2006;580:862–866. doi: 10.1016/j.febslet.2005.12.104. [DOI] [PubMed] [Google Scholar]
- Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. http://ncbi.nlm.nih.gov/BLAST [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell DC, Serb JM, Buhay JE, Roe KJ, Minton RL, Lydeard C. Phylogeny of North American amblemines (Bivalvia, Unionoida): prodigious polyphyly proves pervasive across genera. Invert Biol. 2005;124:131–164. [Google Scholar]
- Jermiin LS, Graur D, Lowe RM, Crozier RH. Analysis of directional mutation pressure and nucleotide content in mitochondrial cytochrome b genes. J Mol Evol. 1994;39:160–173. doi: 10.1007/BF00163805. [DOI] [PubMed] [Google Scholar]
- McClellan DA, McCracken KG. Estimating the influence of selection on the variable amino acid sites of the cytochrome b protein functional domains. Mol Biol Evol. 2001;18:917–925. doi: 10.1093/oxfordjournals.molbev.a003892. [DOI] [PubMed] [Google Scholar]
- Heard WH, Guckert RH. A re-evaluation of the recent Unionacea (Pelecypoda) of North America. Malacalogia. 1970;10:333–335. [Google Scholar]
- Graf DL. The Etherioidea revisited: a phylogenetic analysis of hyriid relationships (Mollusca: Bivalvia: Paleoheterodonta: Unionoida) Occas Pap Mus Zool Univ Mich. 2000;729:1–21. [Google Scholar]
- Hoeh WR, Bogan AE, Heard WH. In: Ecological studies, 145: Ecology and evolution of the freshwater mussels Unionoida. Bauer G, Wächlter K, editor. Berlin: Springer-Verlag; 2001. A phylogenetic perspective on the evolution of morphological and reproductive characteristics in the Unionoida; pp. 257–280. [Google Scholar]
- Roe KJ, Hoeh WR. In: Molecular Systematics and Phylogeography of Mollusks. Lydeard C, Lindberg DR, editor. Washington: Smithsonian Books; 2003. Systematics of freshwater mussels (Bivalvia: Unionoida) pp. 91–122. [Google Scholar]
- Chakrabarti R, Walker JM, Chapman EG, Shepardson S, Trdan RJ, Curole JP, Watters GT, Stewart DT, Vijatraghavan S, Hoeh WR. Reproductive function for a c-terminus extended, male-transmitted cytochrome c oxidase subunit II protein expressed in both spermatozoa and eggs. FEBS Lett. 2007;581:5213–5219. doi: 10.1016/j.febslet.2007.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Heijne G. Membrane-protein topology. Nat Rev Mol Cell Biol. 2006;7:909–918. doi: 10.1038/nrm2063. [DOI] [PubMed] [Google Scholar]
- Hughes AL, Nei M. Pattern of nucleotide substitution at major histocompatibility complex class I loci reveals overdominant selection. Nature. 1988;335:167–170. doi: 10.1038/335167a0. [DOI] [PubMed] [Google Scholar]
- Hughes AL, Nei M. Nucleotide substitution at major histocompatibility complex class II loci: evidence for overdominant selection. Proc Natl Acad Sci USA. 1989;86:958–962. doi: 10.1073/pnas.86.3.958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metz EC, Palumbi SR. Positive selection and sequence rearrangements generate extensive polymorphism in the gamete recognition protein bindin. Mol Biol Evol. 1996;13:397–406. doi: 10.1093/oxfordjournals.molbev.a025598. [DOI] [PubMed] [Google Scholar]
- Gojobori T, Li WH, Graur D. Patterns of nucleotide substitution in pseudogenes and functional genes. J Mol Evol. 1982;18:360–369. doi: 10.1007/BF01733904. [DOI] [PubMed] [Google Scholar]
- Li WH, Gojobori T, Nei M. Pseudogenes as a paradigm of neutral evolution. Nature. 1981;292:237–239. doi: 10.1038/292237a0. [DOI] [PubMed] [Google Scholar]
- Balakirev ES, Ayala FJ. Pseudogenes: are they "junk" or functional DNA? Annu Rev Genet. 2003;37:123–151. doi: 10.1146/annurev.genet.37.040103.103949. [DOI] [PubMed] [Google Scholar]
- Balakirev ES, Ayala FJ. Nucleotide variation of the Est-6 gene region in natural populations of Drosophila melanogaster. Genetics. 2003;165:1901–1914. doi: 10.1093/genetics/165.4.1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Echols N, Harrison P, Balasubramanian S, Luscombe NM, Bertone P, Zhang Z, Gerstein M. Comprehensive analysis of amino acid and nucleotide composition in eukaryotic genomes, comparing genes and pseudogenes. Nucleic Acids Res. 2002;30:2515–2523. doi: 10.1093/nar/30.11.2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zouros E, Ball AO, Saavedra C, Freeman KR. An unusual type of mitochondrial DNA inheritance in the blue mussel Mytilus. Proc Natl Acad Sci USA. 1994;91:7463–7467. doi: 10.1073/pnas.91.16.7463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrido-Ramos MA, Stewart DT, Sutherland BW, Zouros E. The distribution of male-transmitted and female-transmitted mitochondrial DNA types in somatic tissues of blue mussels: Implications for the operation of doubly uniparental inheritance of mitochondrial DNA. Genome. 1998;41:818–824. [Google Scholar]
- Dalziel AC, Stewart DT. Tissue-specific expression of male-transmitted mitochondrial DNA and its implications for rates of molecular evolution in Mytilus mussels (Bivalvia: Mytilidae) Genome. 2002;45:348–355. doi: 10.1139/g01-159. [DOI] [PubMed] [Google Scholar]
- Saavedra C, Reyero M-I, Zouros E. Male-dependent doubly uniparental inheritance of mitochondrial DNA and female-dependent sex-ratio in the mussel Mytilus galloprovincialis. Genetics. 1997;145:1073–1082. doi: 10.1093/genetics/145.4.1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zouros E. The exceptional mitochondrial DNA system of the mussel family Mytilidae. Genes Genet Syst. 2000;75:313–318. doi: 10.1266/ggs.75.313. [DOI] [PubMed] [Google Scholar]
- Kenchington E, MacDonald B, Cao L, Tsagkarakis D, Zouros E. Genetics of mother-dependent sex ratio in Blue Mussels (Mytilus spp.) and implications for doubly uniparental inheritance of mitochondrial DNA. Genetics. 2002;161:1579–1588. doi: 10.1093/genetics/161.4.1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeh JA, Zeh DW. Maternal inheritance, sexual conflict and the maladapted male. Trends Genet. 2005;21:281–286. doi: 10.1016/j.tig.2005.03.006. [DOI] [PubMed] [Google Scholar]
- Burt A, Trivers R. Genes in Conflict: The Biology of Selfish Genetic Elements. Cambridge: Belknap Press, Cambridge; 2006. [Google Scholar]
- Metz EC, Robles-Sikisaka R, Vacquier VD. Nonsynonymous substitution in abalone sperm fertilization genes exceeds substitution in introns and mitochondrial DNA. Proc Natl Acad Sci USA. 1998;95:10676–10681. doi: 10.1073/pnas.95.18.10676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson WJ, Vacquier VD. The rapid evolution of reproductive proteins. Nat Rev Genet. 2002;3:137–144. doi: 10.1038/nrg733. [DOI] [PubMed] [Google Scholar]
- Clark NL, Aagaard JE, Swanson WJ. Evolution of reproductive proteins from animals and plants. Reproduction. 2006;131:11–22. doi: 10.1530/rep.1.00357. [DOI] [PubMed] [Google Scholar]
- Cao L, Kenchington ELR, Zouros E. Differential segregation patterns of sperm mitochondria in embryos of the blue mussel (Mytilus edulis) Genetics. 2004;166:883–894. doi: 10.1534/genetics.166.2.883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cogswell AT, Kenchington ELR, Zouros E. Segregation of sperm mitochondria in two- and four cell embryos of the blue mussel Mytilus edulis: implications for the mechanism of doubly uniparental inheritance of mitochondrial DNA. Genome. 2006;49:799–807. doi: 10.1139/g06-036. [DOI] [PubMed] [Google Scholar]
- Walker JM, Bogan AE, Bonfiglio EA, Campbell DC, Christian AD, Curole JP, Harris JL, Wojtecki RJ, Hoeh WR. Primers for amplifying the hypervariable, male-transmitted COII-COI junction region in amblemine freshwater mussels (Bivalvia: Unionoidea: Ambleminae) Mol Ecol Notes. 2007;7:489–491. [Google Scholar]
- Folmer O, Black M, Hoeh WR, Lutz R, Vrijenhoek R. DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol Mar Biol Biotechnol. 1994;3:294–299. [PubMed] [Google Scholar]
- Maddison WP, Maddison DR. MacClade: Analysis of Phylogeny and Character Evolution. Sunderland: Sinauer Assoc., Inc; 2000. [DOI] [PubMed] [Google Scholar]
- Bateman A, Coin L, Durbin R, Finn RD, Hollich V, Griffiths-Jones S, Khanna A, Marshall M, Moxon S, Sonnhammer ELL, Studholme DJ, Yeats C, Eddy SR. The Pfam protein families database. Nucleic Acids Res. 2004;32:138–141. doi: 10.1093/nar/gkh121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arai M, Mitsuke H, Ikeda M, Xia JX, Kikuchi T, Satake M, Shimizu T. ConPred II: a consensus prediction method for obtaining transmembrane topology models with high reliability. Nucleic Acids Res. 2004. pp. W390–393. [DOI] [PMC free article] [PubMed]
- Huelsenbeck JP, Ronquist F. MrBayes: Bayesian inference of phylogeny. Bioinformatics. 2003;17:754–755. doi: 10.1093/bioinformatics/17.8.754. [DOI] [PubMed] [Google Scholar]
- Ronquist F, Huelsenbeck JP. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003;19:1572–1574. doi: 10.1093/bioinformatics/btg180. [DOI] [PubMed] [Google Scholar]
- Rodriguez F, Oliver JL, Marin A, Medina JR. The general stochastic model of nucleotide substitution. J Theor Biol. 1990;142:485–501. doi: 10.1016/s0022-5193(05)80104-3. [DOI] [PubMed] [Google Scholar]
- Posada D, Crandall KA. Modeltest: Testing the model of DNA substitution. Bioinformatics. 1998;14:817–818. doi: 10.1093/bioinformatics/14.9.817. [DOI] [PubMed] [Google Scholar]
- Marshall DC, Simon C, Buckley TR. Accurate branch length estimation in partitioned Bayesian analyses requires accommodation of among-partition rate variation and attention to branch length priors. Syst Biol. 2006;55:992–1003. doi: 10.1080/10635150601087641. [DOI] [PubMed] [Google Scholar]
- Swofford DL. PAUP*. Phylogenetic Analysis Using Parsimony (*and Other Methods) Sunderland: Sinauer Associates; 2001. [Google Scholar]
- Lewis PO. A likelihood approach to estimating phylogeny from discrete morphological character data. Syst Biol. 2001;50:913–925. doi: 10.1080/106351501753462876. [DOI] [PubMed] [Google Scholar]
- Maddison WP, Maddison DR. Mesquite: a modular system for evolutionary analysis. Version 1.05. 2003. http://mesquiteproject.org
- Edwards AWF. Likelihood. Cambridge: Cambridge University Press; 1972. [Google Scholar]
- Pagel M. The maximum likelihood approach to reconstructing ancestral character states on phylogenies. Syst Biol. 1999;48:612–622. [Google Scholar]
- Kishino H, Hasegawa M. Evaluation of the maximum likelihood estimate of the evolutionary tree topologies from DNA sequence data, and the branching order of the Hominoidea. J Mol Evol. 1989;29:170–179. doi: 10.1007/BF02100115. [DOI] [PubMed] [Google Scholar]
- Templeton AR. Phylogenetic inference from restriction endonuclease cleavage site maps with particular reference to the humans and apes. Evolution. 1983;37:221–244. doi: 10.1111/j.1558-5646.1983.tb05533.x. [DOI] [PubMed] [Google Scholar]
- Prager EM, Wilson AC. Ancient origin of lactabumin from lysozyme: analysis of DNA and amino acid sequences. J Mol Evol. 1988;27:326–335. doi: 10.1007/BF02101195. [DOI] [PubMed] [Google Scholar]
- Shimodaira H. An approximately unbiased test of phylogenetic tree selection. Syst Biol. 2002;51:492–508. doi: 10.1080/10635150290069913. [DOI] [PubMed] [Google Scholar]
- Shimodaira H, Hasegawa M. Multiple comparisons of log-likelihoods with applications to phylogenetic inference. Mol Biol Evol. 1999;16:1114–1116. [Google Scholar]
- Shimodaira H, Hasegawa H. CONSEL: for assessing the confidence of phylogenetic tree selection. Bioinformatics. 2001;17:1246–1247. doi: 10.1093/bioinformatics/17.12.1246. [DOI] [PubMed] [Google Scholar]
- Yang ZH. PAML: A program package for phylogenetic analysis by maximum likelihood. Comput Appl Biosci. 1997;13:555–556. doi: 10.1093/bioinformatics/13.5.555. [DOI] [PubMed] [Google Scholar]
- Yang ZH, Nielsen R, Hasegawa M. Models of amino acid substitution and applications to mitochondrial protein evolution. Mol Biol Evol. 1998;15:1600–1611. doi: 10.1093/oxfordjournals.molbev.a025888. [DOI] [PubMed] [Google Scholar]
- Bairoch A, Bucher P, Hofmann K. The PROSITE database, its status in 1997. Nucleic Acids Res. 1997;25:217–221. doi: 10.1093/nar/25.1.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosakovsky Pond SL, Frost SDW, Muse SV. HyPhy: hypothesis testing using phylogenies. Bioinformatics. 2005;21:676–679. doi: 10.1093/bioinformatics/bti079. [DOI] [PubMed] [Google Scholar]
- Yang ZH, Wong WSW, Nielsen R. Bayes empirical Bayes inference of amino acid sites under positive selection. Mol Biol Evol. 2005;22:1107–1118. doi: 10.1093/molbev/msi097. [DOI] [PubMed] [Google Scholar]
- Zhu S, Bosmans F, Tytgat J. Adaptive evolution of scorpion sodium channel toxins. J Mol Evol. 2004;58:145–153. doi: 10.1007/s00239-003-2534-2. [DOI] [PubMed] [Google Scholar]
- Kosakovsky Pond SL, Frost SDW. Not so different after all: A comparison of methods for detecting amino acid sites under selection. Mol Biol Evol. 2005;22:1208–1222. doi: 10.1093/molbev/msi105. [DOI] [PubMed] [Google Scholar]
- Xia X, Kumar S. Codon-based detection of positive selection can be biased by heterogenous distribution of polar amino acids along protein sequences. Comput Syst Bioinformatics conf. 2006;4:335–340. [PMC free article] [PubMed] [Google Scholar]
- Anisimova M, Bielawski JP, Yang Z. Accuracy and power of bayes prediction of amino acid sites under positive selection. Mol Biol Evol. 2002;19:950–958. doi: 10.1093/oxfordjournals.molbev.a004152. [DOI] [PubMed] [Google Scholar]
- Kosakovsky Pond SL, Muse SV. Site-to-site variation of synonymous substitution rates. Mol Biol Evol. 2005;22:2375–2385. doi: 10.1093/molbev/msi232. [DOI] [PubMed] [Google Scholar]
- Woolley S, Johnson J, Smith MJ, Crandall KA, McClellan DA. TreeSAAP: Selection on amino acid properties using phylogenetic trees. Bioinformatics. 2003;19:671–672. doi: 10.1093/bioinformatics/btg043. [DOI] [PubMed] [Google Scholar]
- McClellan DA, Whiting DG, Christensen RG, Sailsbery JK. Genetic codes as evolutionary filters: subtle differences in the structure of genetic codes result in significant differences in patterns of nucleotide substitution. J Theoret Biol. 2004;226:393–400. doi: 10.1016/j.jtbi.2003.09.019. [DOI] [PubMed] [Google Scholar]
- Foster PG, Jermiin LS, Hickey DA. Nucleotide composition bias affects amino acid content in proteins coded by animal mitochondria. J Mol Evol. 1997;44:282–288. doi: 10.1007/pl00006145. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Amino acid sequences deduced from the complete Mcox2 extension nucleotide sequences of the 21 species used in this study.
Pairwise amino acid and nucleotide distances (based on PC and TN models, respectively).
Codeml log-likelihood values and parameter estimates for four models applied to the M and F COX2+COX1 regions.
Summary of TreeSAAP output showing the 31 amino acid properties of MCOX2h+MCOX1, MCOX2e and FCOX2+FCOX1.