

Summary
B lymphocytes perform somatic hypermutation (SHM) and class switch recombination (CSR) of the immunoglobulin locus to generate an antibody repertoire diverse in both affinity and function. These somatic diversification processes are catalyzed by activation-induced cytidine deaminase (AID), a potent DNA mutator whose expression and function are highly regulated. Here we show that AID is regulated at the post-transcriptional level by a lymphocyte-specific microRNA, miR-155. We find that miR-155 is upregulated in murine B lymphocytes undergoing CSR, and furthermore targets a conserved site in the AID 3′untranslated region. Disruption of this target site in vivo results in quantitative and temporal deregulation of AID expression, accompanied by functional consequences for CSR and affinity maturation. Thus, miR-155, which has recently been shown to play important roles in regulating the germinal center reaction, does so in part by directly downmodulating AID expression.
Introduction
Vertebrates are able to produce a vast repertoire of antibody molecules to combat infection. The number of antibody specificities available during a human lifetime is estimated to exceed 109, far greater than coding capacity of the genome. Instead, the size of the antibody repertoire results from gene diversification processes that occur in antibody-producing B lymphocytes.
During B cell development, antibody genes are assembled by DNA rearrangement (V(D)J recombination) to produce the primary repertoire of antibody specificities (about 105–106 different specificities). However, this repertoire is neither large enough nor specific enough include high-affinity antibodies against the full range of antigens an animal may encounter. Thus, the generation of antibody diversity depends largely on processes that follow V(D)J recombination.
Two main processes drive the generation of the secondary antibody repertoire: somatic hypermutation (or SHM, where mutations are introduced in and around the productively-rearranged V(D)J segment) and gene conversion (or GCV, where nonfunctional pseudogenes are used as templates for diversification). Both SHM and GCV are triggered by activation-induced cytidine deaminase (AID), an enzyme believed to function directly as a DNA mutator. In addition to these two processes, AID-mediated deamination initiates class switch recombination (CSR) a reaction which does not contribute to diversification of antibody specificity, but replaces the default Ig constant region (Cμ) with a downstream constant region (Cγ Cε, or Cα) to alter the effector capacity of the antibody while retaining the specificity for antigen (Teng and Papavasiliou, 2007).
A potent mutator like AID must be stringently regulated in the cell. Indeed, it has been proposed that AID is regulated at the level of transcription (Dedeoglu et al., 2004; Gonda et al., 2003; Sayegh et al., 2003) (R. Casellas, manuscript submitted), by intracellular compartmentalization and trafficking (Ito et al., 2004; McBride et al., 2004), by post-translational modification (Basu et al., 2005; Basu et al., 2007; McBride et al., 2006; Pasqualucci et al., 2006), and by interaction with specific cofactors (Chaudhuri et al., 2004; MacDuff et al., 2006). In addition to these more traditional modes of regulation, we show here that AID is also subject to post-transcriptional regulation by a specific microRNA, miR-155.
mir-155 belongs to a recently identified class of non-coding 21–23 nt RNAs that function as post-transcriptional modulators of gene expression. Encoded by diverse metazoan genomes, miRNAs target their cognate messenger RNAs for degradation or translational repression (Bartel, 2004; Bartel and Chen, 2004). Their functional influence is apparent in a host of biological processes, ranging from pancreatic insulin secretion (Poy et al., 2004) to B and T cell development (Zhou et al., 2007).
miR-155 is processed from the non-coding RNA known as BIC, which serves as its primary miRNA precursor. BIC was originally identified as a common site for insertion of proviral DNA in avian leukosis virus-induced lymphomas (Tam et al., 1997). A role for miR-155 in germinal center development and function has recently been proposed, based on studies using in vivo knock-out and knock-in approaches to analyze the consequences of deletion or ectopic expression of miR-155, which revealed B cell autonomous defects in affinity maturation (Rodriguez et al., 2007; Thai et al., 2007). However, miR-155 has been shown to target over 60 different genes in B cells (Vigorito et al., 2007), and it is unclear which of those genes would be directly targeted by this microRNA to produce the observed phenotype. Furthermore, induction of miR-155 expression has been found in association with the activated phenotype of several immune cell types, including T lymphocytes, dendritic cells, and macrophages (O’Connell et al., 2007; Rodriguez et al., 2007; Thai et al., 2007), suggesting synergistic action of miR-155 on numerous target genes in multiple cell types.
Here we present evidence that AID is a direct target for miR-155 regulation in vivo. Using a bacterial artificial chromosome (BAC) transgenic model of AID expression we show that specific ablation of miR-155:AID interaction deregulates AID protein levels in germinal center B cells and results in ectopic persistence of AID expression in B cells as they exit the germinal centers and enter the peripheral circulation. We also show that lack of AID regulation by miR-155 leads to defective affinity maturation. Hence the central role of miR-155 in control of the germinal center reaction (Thai et al., 2007) is at least in part due to its role in repression of AID expression.
Results
miR-155 is upregulated in B cells undergoing class switch recombination
microRNAs (miRNAs) comprise a class of newly discovered small RNA species that regulate gene expression. To investigate the role these regulators may play in CSR, we cloned and sequenced miRNAs present in primary B cells before and after stimulation with LPS and IL-4, conditions that induce switching from IgM to IgG1. Of the 123 miRNAs that were cloned from these samples (data not shown), we identified one, miR-155, that was upregulated after stimulation (Figure 1a). Importantly, miR-155 was upregulated not only in primary B cells treated with LPS and IL-4, but also in CH12-F3 cells, a murine B cell line which switches from IgM to IgA after treatment with anti-CD40 antibody, IL-4, and TGF-β (Figure 1b). Thus, miR-155 is upregulated during CSR in manner that is not isotype-specific.
Figure 1.

miR-155 is upregulated in B cells undergoing CSR, and targets the 3′UTR of AID. a, Relative cloning frequencies for 10 of the most abundant miRNAs in murine splenic B lymphocytes stimulated with IL-4 and LPS (0, 8, 24, 48, and 72 hours). Color coding scale is shown at bottom, and undetected miRNAs are indicated by black. b, Northern blots for miR-155 from total RNA isolated from murine splenic B cells (left) and the murine B cell line CH12-F3 (right), stimulated in vitro (0–72 hours) to undergo CSR. Ethidium-bromide staining of tRNA bands is shown as a loading control. c, Schematic of AID mRNA from human (Hs), mouse (Mm), dog (Cf), catfish (Ip), and zebrafish (Dr); showing the AID ORF (black boxes), UTRs (black lines), and the predicted miR-155 target site (red). Also shown are sequences of the corresponding miR-155 target sites, with the conserved seed region highlighted in red. d, Base pairing between murine miR-155 (italicized) and its target site in the wt AID 3′UTR. Sequences of the mutated (mut-UTR) and deleted (del-UTR) target site variants are shown underneath. e, CH12-F3 cells stimulated to undergo CSR were transfected with reporter constructs containing firefly luciferase alone (Luc), or fused to the wild-type AID 3′UTR (UTR), del-UTR, or mut-UTR. Data represent mean values of eight independent experiments ± s.e.m. One-way ANOVA F-statistic = 2.95 (P = 0.05).
miR-155 can target the 3′UTR of AID
We applied several miRNA target prediction algorithms (PicTar (Krek et al., 2005), miRanda (Griffiths-Jones et al., 2006; John et al., 2004), and TargetScan (Grimson et al., 2007; Lewis et al., 2003)) to identify putative targets of miR-155 with relevance to either SHM or CSR. AID was consistently predicted as one such target. Depending on the algorithm used, a number of additional miRNA binding sites in the AID mRNA were identified (not shown); however these miRNAs were not detected in switching B cells by cloning or by Northern analysis. Furthermore, the stringent prediction criteria of most recent version of the TargetScan algorithm (Grimson et al., 2007) identified miR-155 as the sole miRNA target site in the AID 3′UTR. The 3′UTRs of AID mRNA diverge significantly in sequence and length between various species (Figure 1c). However, they coincide strikingly at an 8 nt motif corresponding to the predicted miR-155 target site seed region. To test the possibility of post-transcriptional AID regulation by miR-155, we created reporter constructs containing the 3′UTR of AID downstream of a firefly luciferase reporter gene (Luc-UTR), along with variants harboring deletion (Luc-del-UTR) or mutation (Luc-mut-UTR) of the miR-155 target site (Figure 1d). These constructs were transiently transfected into CH12-F3 cells stimulated to undergo CSR (and hence induced to express endogenous miR-155). We observed repression of luciferase activity by ~50% in cells transfected with Luc-UTR compared to cells transfected with a luciferase-only construct (Figure 1e). This repression was alleviated upon disruption of the miR-155 target site by deletion or mutation. These data indicate that the single target site in the 3′UTR of the AID mRNA renders it susceptible to repression by physiological levels of miR-155 expressed during CSR.
Mutation of the AID miR-155 target site results in deregulated AID expression and higher levels of CSR in splenic B cells stimulated in vitro
To study the behavior of in vivo AID expression in response to miR-155, we took advantage of a recently generated transgenic AID-GFP indicator mouse strain (Crouch et al., 2007). These mice carry between ~2–10 copies of a 75 kilobase-long BAC which contains the entire AID locus as well as its two adjacent gene loci (Mfap5 and Apobec-1). The AID locus in this BAC has been modified such that a GFP gene is inserted downstream of the coding portion of the final AID exon. The transgenic AID-GFP fusion protein expressed from this BAC was previously shown to replicate endogenous patterns of AID expression (Crouch et al., 2007). The AID-GFP gene product also retains catalytic activity, as it can rescue in vitro CSR in AID−/− B lymphocytes (Figure S1). Based on this published AID-GFP BAC, we created a second transgenic construct in which the miR-155 target site seed region was mutated to disrupt binding to miR-155 (AID-GFP-Mut) (Figure 2a). This second construct was used to generate five independent founder lines carrying the AID-GFP-Mut BAC transgene (founders carried varying copy numbers of the transgene, ranging from ~5 up to ~20).
Figure 2.

Mutation of the AID miR-155 target site results in deregulated AID-GFP expression and increased CSR efficiency in vitro. a, The transgenic AID-GFP locus (which is located in the approximate center of a ~75 kbp transgene) is diagrammed, with the five coding exons of AID denoted by black boxes. The hybrid exon 5 includes the GFP gene (hatched box) inserted between the final coding portion of AID and the 3′UTR (white box). The mutated miR-155 target site is marked by an asterisk. Base pairing between miR-155 (italicized) and the wt and mutated UTRs are shown below. b, AID-GFP mRNA expression was monitored by quantitative PCR in representative AID-GFP control and AID-GFP-Mut mice. Data are normalized to Ku70 mRNA expression, and the scale is set to 1 for d0. c, AID-GFP protein expression was monitored by FACS up to four days after in vitro stimulation of splenic B lymphocytes with IL-4 and LPS. Percentages of GFP+ B220+ cells are indicated in the upper right quadrants. Cells from representative AID-GFP control (Tg copy number ~4–5) and AID-GFP-Mut (Tg copy number ~4–5) are shown. d, Median GFP fluorescence intensities, expressed in logarithmic units, are shown for the two samples in (b). e, Splenic B lymphocytes from wt (n=4); AID+/−, AID-GFP control (n=5); AID−/−, AID-GFP control (n=2); AID+/−, AID-GFP-Mut (n=6), or AID−/−, AID-GFP-Mut (n=3) mice were stimulated in vitro for three days with LPS (to induce CSR to IgG3), LPS and IL-4 (to induce CSR to IgG1), or LPS and IFNγ (to induce CSR to IgG2a). Note – AID−/−, AID-GFP control samples did not switch robustly to IgG1 or IgG3, though IgG1+ and IgG3+ populations were detected by FACS analysis(data not shown) – but they did show quantifiable levels of CSR to IgG2a. t-tests, *P <0.05, **P <0.01.
Splenic B lymphocytes from progeny of these transgenic founders were analyzed for AID-GFP expression in response to IL-4 and LPS stimulation in vitro. The dynamics of AID-GFP expression differed considerably between the AID-GFP control and AID-GFP-Mut mice. The controls exhibited a gradual increase of low intensity AID-GFP fluorescence with time of stimulation (as also observed by (Crouch et al., 2007), mirroring the expression profile of endogenous AID. In contrast, cells from AID-GFP-Mut mice showed a rapid induction of AID-GFP, peaking early around day 3, then reaching a plateau (Figure 2b, 2c). The median intensity of AID-GFP fluorescence was also higher in the AID-GFP-Mut mice, suggesting more abundant AID-GFP protein, compared to controls (Figure 2d).
These disparities were not due to position effects due to differential integration of each BAC transgene as a) large BACs such as this are not susceptible to overexpression due to random insertion in active expression sites (Gong et al., 2003; Hatten and Heintz, 2005) and b) we have screened multiple independent AID-GFP-Mut founder lines with identical results (Figure S2). In addition, these differences were not due to AID-GFP overexpression due to copy number variation between the two BAC transgenics, as we have screened animals with similar copy numbers to yield identical results (Figure S2). Therefore, our results demonstrate that in vivo disruption of the AID miR-155 target perturbs the quantitative and temporal expression characteristics of AID.
To assess the functional consequences of miR-155 target site disruption, the AID-GFP control and AID-GFP-Mut mice were bred onto an AID−/− background to extinguish the contribution of endogenous AID. We first compared in vitro CSR efficiency by stimulating splenic B lymphocytes from these mice using LPS (to induce CSR to IgG3), LPS and IL-4 (to induce CSR to IgG1), or LPS and IFNγ (to induce CSR to IgG2a). AID−/− B cells perform negligible in vitro CSR (data not shown). We observed increased levels of CSR in the AID-GFP-Mut mice compared to AID-GFP control or wild type mice (Figure 2e). This was the case regardless of whether these BAC transgenics were deficient or heterozygous for AID in the endogenous locus (Figure 2e). These results indicate that miR-155 directly regulates AID expression in stimulated B lymphocytes.
Mutation of the miR-155 target site leads to local but not global deregulation of AID in vivo
To study in vivo effects of disrupting the AID miR-155 target site, the transgenic mice were immunized intraperitoneally with nitrophenol conjugated to chicken gamma globulin (NP-CGG). The NP hapten induces a well-characterized immune response (Cumano and Rajewsky, 1985; Furukawa et al., 1999; Taketani et al., 1995) in peripheral lymphoid germinal centers (GC), microenvironments where activated lymphocytes undergo Ig diversification. Eighteen to 21 days after immunization, we evaluated AID-GFP expression by FACS in various B lymphocyte subsets. Mutation of the miR-155 target site did not disrupt global transcriptional control of AID: immature CD93+ B lymphocytes in the bone marrow, along with thymic and peripheral CD4+ or CD8+ T lymphocytes were devoid of AID-GFP (Figure 3). However, AID-GFP was detected in B cell populations associated with activation. GC B lymphocytes (CD95+ B220+) from spleen and intestinal Peyer’s patches expressed AID-GFP in both the control and AID-GFP-Mut mice. The latter, however, showed far more intense GFP fluorescence, indicating a similar overabundance of AID-GFP during the GC reaction, as observed during in vitro CSR (Figure 4a).
Figure 3.
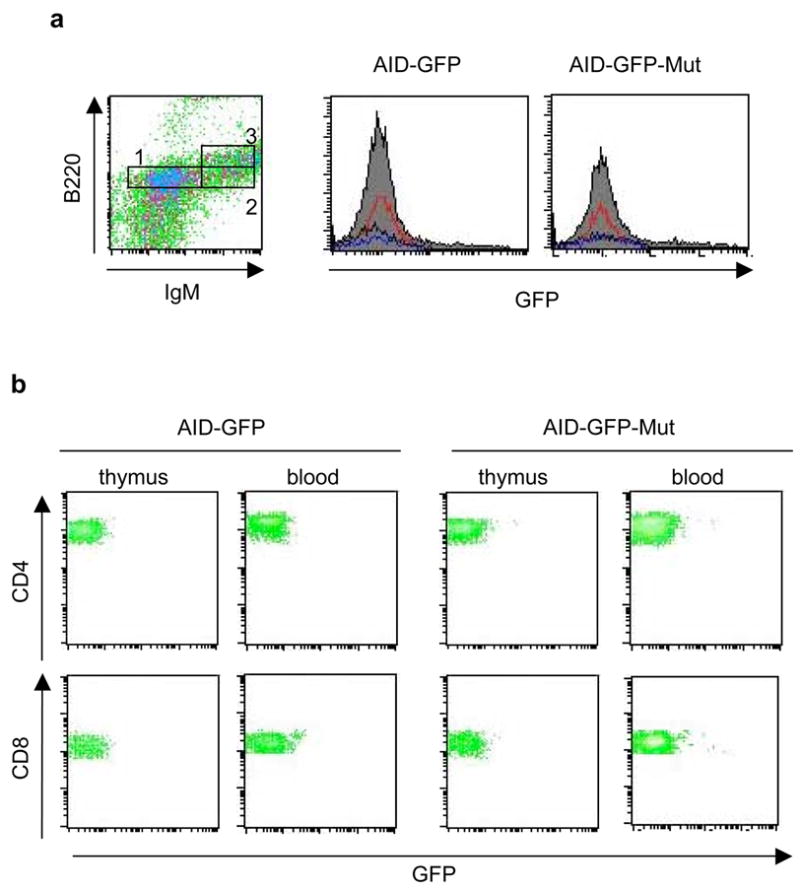
NP-immunized AID-GFP-Mut mice do not express AID-GFP in developing B lymphocytes or in T lymphocytes. FACS analyses are shown for representative AID-GFP and AID-GFP-Mut mice. a, Developing B cells (CD93+) from the bone marrow are subdivided into pro/pre-B cells (box 1 – IgM-, B220low), transitional B cells (box 2 – IgM+, B220low), and recirculating B cells (box 3 – IgM+, B220high). Histograms for GFP fluorescence show pre-pro B cells (red), transitional B cells (blue), and recirculating B cells (black). b, CD8+ and CD4+ T lymphocytes from thymus and peripheral blood are shown.
Figure 4.
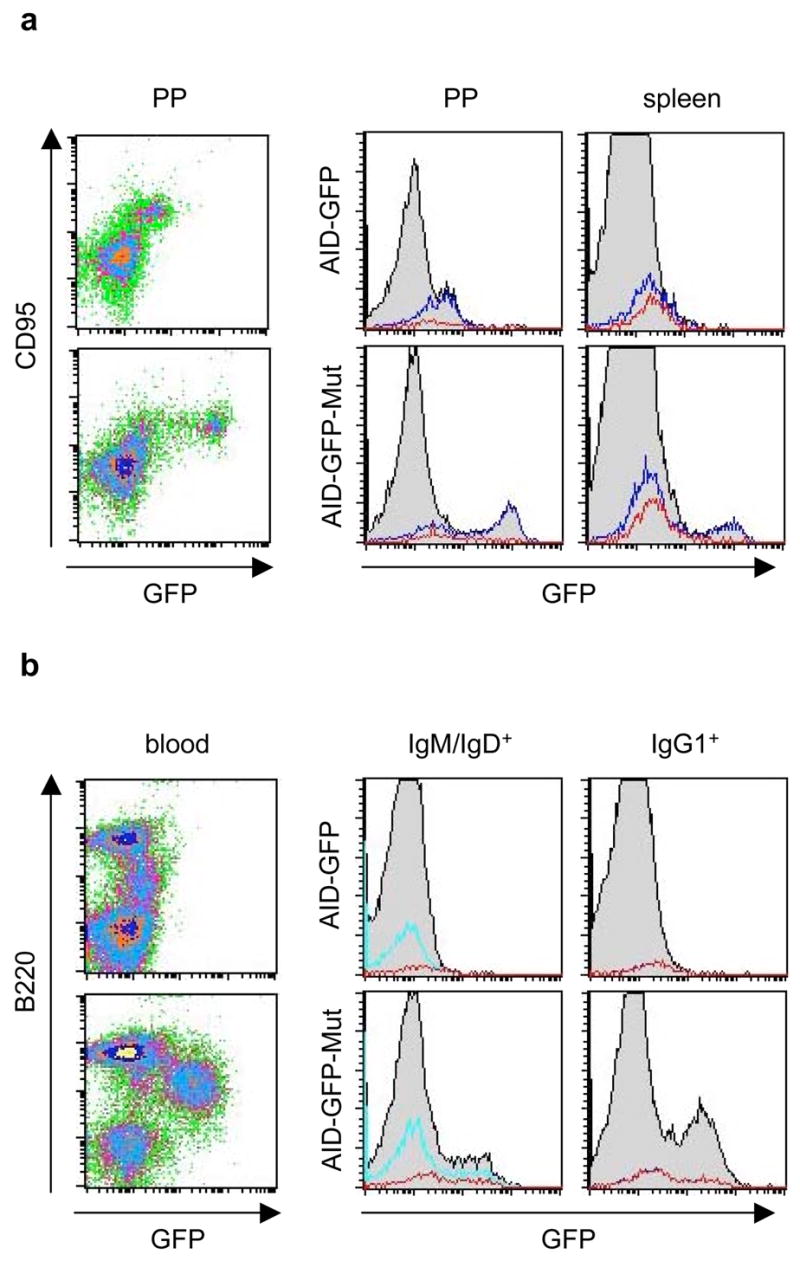
Mutation of the AID miR-155 target site results in deregulated in vivo expression of AID-GFP in NP-CGG immunized mice. a, FACS for AID-GFP expression in GC’s from representative AID-GFP and AID-GFP-Mut mice. Left, GC B cells from Peyer’s Patches (gated on B220+ B cells). Right, histograms show AID-GFP expression in B cell subsets from GC’s of Peyer’s patches (PP) and spleen. Shown in overlay are CD95+ B220+ GC B cells (blue), and the subset of GC B cells that recognize NP (red). b, FACS for AID-GFP expression in peripheral blood from representative AID-GFP and AID-GFP-Mut mice. Left, Total blood lymphocytes are shown. Right, Histograms show AID-GFP expression in IgM/IgD+, B220+ (cyan) and IgG1+, B220+ B cells (blue) subsets. Red overlays indicate the respective subsets of these cells that recognize NP (note – the blue and red overlays in the IgG1+ histogram overlap completely).
After completing the GC reaction, activated B lymphocytes exit into the periphery and differentiate into plasma or memory cells. These post-GC B lymphocytes have been shown to cease expression of AID (Crouch et al., 2007). Accordingly, the AID- GFP control mice did not express AID-GFP in B cells in the peripheral blood (Figure 4b). In contrast, a GFP+ population of B cells was consistently detected in the blood of immunized AID-GFP-Mut mice (Figure 4b). These appeared to be a heterogeneous population of B lymphocytes (B220intermediate/high), which were mostly IgG1+ but also included IgM/IgD+ non-switched cells. This B lymphocyte population did not appear to arise ectopically as a consequence of AID-GFP overexpression, as a similar B220intermediate (though GFP−) population was also present in the AID-GFP controls. Thus, disruption of the AID miR-155 target site allows for improper persistence of AID-GFP expression beyond the GC compartment. These results confirm once again that the phenotypes we observe cannot be explained by BAC copy number variation, which could be expected to increase protein levels but cannot lead to persistent expression in cells where the gene of interest is normally shut off. Thus our data strongly support the idea that miR-155 controls AID expression levels and specifically, plays a pivotal role in extinguishing AID expression in post-GC B cell populations.
Loss of miR-155 regulation of AID results in impaired affinity maturation
To determine the functional impact of defective AID downregulation, we compared affinity maturation (increased antibody affinity achieved through SHM) of NP-binding IgG1 in sera collected at 8 days and 19 or 21 days after immunization. The ELISA capture substrate was NP conjugated to BSA in different ratios to detect high-affinity (NP3-BSA) and total (NP30-BSA) immunoglobulins (Ig) specific for NP. Affinity maturation was measured as the NP3:NP30 - binding ratio. As expected, we observed an increase in the proportion of high-affinity Ig from day 8 to day19/21 in control AID-GFP mice. However, affinity maturation was significantly impaired in AID-GFP-Mut animals (Figure 5a).-
Figure 5.

AID-GFP-Mut mice show loss of affinity maturation and increased clonal heterogeneity in post-GC B cells. a, Measurement of affinity maturation of NP-specific IgG1 by ELISA. Shown from left to right are average titers of NP3-binding IgG1, NP30-binding IgG1, and NP3:NP30-binding ratios as measured by ELISA in AID-GFP (n=3) and AID-GFP-Mut (n=7) mice ± s.e.m. t-test *P=0.0016. b, Mutation frequencies in VH186.2, JH4 intron, and bcl-6 from splenic GC B cells in NP-immunized AID-GFP and AID-GFP-Mut mice. Mutation frequencies were calculated by dividing the accumulated number of mutations in a given region by the total number of nucleotides sequenced from that region. c, Clonal heterogeneity in the JH4 intron of lymphocytes from peripheral blood of AID-GFP and AID-GFP-Mut mice. We thank Sebastian Fugmann, David Schatz, and Svend Petersen-Mahrt for comments on the manuscript. This work was supported by grants from the Keck foundation, NIH grant CA098495 (FNP), NIH NRSA training grant GM066699 (GT), and the Intramural Research Program of the National Institute of Arthritis and Musculoskeletal and Skin Diseases of the National Institutes of Health (RC).
To ascertain if the observed affinity maturation defect resulted from AID-GFP overexpression in GC’s, we analyzed the mutation load at Ig loci in CD95+ GC B lymphocytes. After sequencing the VH186.2 gene segment which is selected for during the Ig response against NP (Cumano and Rajewsky, 1985) (to assess the mutation profile under selection) and the JH4 intron (to assess the mutation profile in the absence of selection pressure), we found that the frequencies and overall patterns of mutation did not differ significantly between AID-GFP and AID-GFP-Mut mice (Figure 5b). Thus, we considered two possibilities: either the repair mechanisms that process AID-mediated lesions are not overwhelmed by higher lesion loads, or excess AID was not specifically targeted to the Ig locus.
Aside from the Ig genes, a number of other genes, including the oncogene bcl-6, have been shown to hypermutate in the germinal centers (Pasqualucci et al., 2001; Shen et al., 1998). We reasoned that excess AID mutational activity, if not targeted to the Ig genes, could instead be targeted to these other non-Ig loci. To test this hypothesis, we cloned and sequenced bcl-6 from GC B lymphocytes of NP-immunized AID-GFP and AID-GFP-Mut mice. We found that bcl-6 from AID-GFP GC B cells was mutated with a frequency of 0.17×10−3 per base (similar to reported bcl-6 mutation frequency in murine GC B cells from Peyer’s Patches (Muto et al., 2006) (Figure 5b). In contrast, bcl-6 from AID-GFP-Mut GC B lymphocytes mutated about three times as frequently (0.46×10−3 per base) (Figure 5b). This observation supports the hypothesis that excess AID activity within the GC is distributed to non-Ig targets for hypermutation, raising the possibility that loss of miR-155-mediated downregulation of AID could result in higher rates of AID-dependent translocation or lymphomagenesis.
However, these data did not explain the loss of affinity maturation observed in the AID-GFP-Mut mice. We then asked if continued mutation outside of the GC (due to persistent AID-GFP expression in post-GC B cells) could account for this phenotype. To test this possibility, we cloned and sequenced the JH4 intron from post-GC lymphocytes from peripheral blood of AID-GFP and AID-GFP-Mut mice. We found that B lymphocytes from the blood of AID-GFP mice comprised a small number of highly related and affinity-matured clones, with some intraclonal heterogeneity (Figure 5c). This is not surprising, as post-GC B cell clones in the blood have undergone stringent positive selection for those producing high-affinity Ig. In contrast, we found a significantly higher number of unique post-GC B lymphocyte clones in the blood of AID-GFP-Mut animals. Because these clones were so heterogeneous, we could not conclusively ascertain if the mutational load per unique clone was higher in comparison to those from the blood of AID-GFP mice (i.e., whether these cells were continually mutating their JH4 intron). However, in agreement with the serum data, these clones did not appear to have acquired the amino acid substitutions associated with high affinity anti-NP antibodies (notably the W33L substitution and the Y99G substitution (Furukawa et al., 1999)). Thus it did not appear that these clones emerged from the germinal center with high affinity, which would then be lost due to continuing mutation; but rather, that persistent AID expression led to escape from the germinal center of cells bearing low affinity anti-NP antibodies.
Discussion
Although hundreds of miRNAs are present in the mammalian genome, genetic studies addressing their physiological roles are at an early stage. In the present work, we address the function of miR-155 in the context of antibody diversification by 1) identifying this particular miRNA as upregulated in B cells stimulated to undergo CSR, 2) by bioinformatically identifying AID as a putative target, and 3) by genetically mutating the target sequence in the 3′UTR of AID to allow the gene to escape miR-155 control. These experiments are the first to identify a target of a miRNA in vivo by manipulating not the expression of the miRNA itself but rather, by genetically disrupting the association between a miRNA and its putative target gene.
Most genes contain multiple predicted target sites for several different miRNAs. Deletion of just one of these targeting miRNAs can result in protein upregulation in a dose-dependent manner, leading to the hypothesis that multiple microRNAs synergistically fine-tune the expression of a single target gene (Xiao et al., 2007). Surprisingly, the AID 3′UTR is predicted to contain only one miRNA target site, binding miR-155. We have shown that mutation of the seed region of this site leads to dramatic protein overabundance and disruption of proper temporal protein downregulation. We cannot exclude the possibility that the specific point mutations introduced at the miR-155 target site could result in additional miR-155 independent effects (for example, alteration of RNA secondary structure, mRNA stability, or disruption of a binding site for some unknown regulatory factor). However, the previously-reported miR-155 knockout mouse also showed upregulated AID expression (though to a lesser degree than observed in our AID-GFP-Mut mouse, to be addressed below)(Vigorito et al., 2007), and we believe that the phenotypes observed in the AID-GFP-Mut mouse are explained by a model in which miR-155 directly regulates AID expression. In addition, levels of AID expression are stringently regulated in B cells such that mice heterozygous for AID show clear signs of haploinsufficiency (R. Casellas, manuscript submitted). These observations together with our results suggest that, especially when protein levels are already limiting, a single miRNA can contribute far more significantly to control of protein expression than previously appreciated.
Ablating miR-155 control of AID expression leads to increased protein expression in switching cells, which undergo significantly higher levels of CSR (Figure 2e). It also leads to increased protein expression in germinal center cells (Figure 4a). However, increased AID expression does not lead to more SHM in the Ig locus, in accordance with previous work by the Honjo group (Muto et al., 2006), who have shown that transgenic AID overexpression does not lead to an increase in SHM. Our data presented here, and the data published by Muto et al., support the notion of the existence of a limiting factor that targets AID to the Ig locus so that in its presence, excess AID cannot access the locus (though in its absence it can, possibly in a stochastic fashion).
Excess AID in germinal center cells, however, appears to increase hypermutation in other loci such as bcl-6, which are known secondary targets of the SHM apparatus. Excess AID is also likely to increase the rates of chromosomal translocations associated with errant hypermutation (Ramiro et al., 2006; Ramiro et al., 2004). Therefore, lack of miR-155 control of AID expression may be causal for B cell lymphomagenesis. Indeed, Kluiver et al. have recently documented a lack of miR-155 expression in primary cases of B cell Burkitt lymphomas, which constitutively express AID (Kluiver et al., 2006).
In addition to overabundance of AID, we find that lack of miR-155 control leads to persistence of AID expression in post-germinal center B cells, thus effectively marking a novel subset of circulating B cells as recent emigrants from the germinal center. We also find that persistent AID expression is associated with specific defects in affinity maturation. It is possible that persistent AID expression supports ongoing mutation in the Ig locus well after cells exit the germinal center with an affinity-matured antigen receptor, effectively destroying the properly-selected antigen-specific Ig repertoire. However, this scenario is not strongly supported by our data, emphasizing again that specific co-factors may be required to target AID to the Ig locus. Instead, we observe multiple low-affinity B cell clones in the blood of animals where the AID gene is not subject to miR-155 control, suggesting a defect in positive selection of properly matured B cells. Though the mechanism of positive selection and affinity maturation are not well understood, it is thought that B cells cycle between the dark zone of the germinal center, where mutation occurs, to the light zone, where their newly minted receptors are substrates for positive selection. Eventually, high-affinity B cells are thought to emerge after multiple rounds of recycling through the germinal center. Our data would support a scenario where B cells overexpressing AID may not be allowed to “recycle” into a germinal center for proper affinity maturation. We could speculate on the existence of a mechanism for cellular sensing of AID levels that would form a feedback loop between proper AID extinction and GC cycling. However, this would be difficult to experimentally demonstrate as little is presently known about the cellular and molecular parameters governing the mechanism of proper selection and affinity maturation.
The previously described miR-155 knockout mouse models (Rodriguez et al., 2007; Thai et al., 2007) highlighted the immense contribution of miR-155 –mediated regulation to various aspects of vertebrate immunity. The phenotypes therein, specifically – modest increase in AID expression, decreased in vivo CSR to IgG1, and impaired affinity maturation – likely reflect the composite deregulation of at least 60 genes (Vigorito et al., 2007). In contrast to global ablation of miR-155 regulation, we describe here specific disruption of a single miR-155 : target interaction. In our mouse model, we observe significant AID-GFP deregulation, increased in vitro CSR, and impaired affinity maturation – effects which may not be apparent in the more complex context of total miR-155 deficiency.
As the central catalyst for antibody diversification processes, AID has been shown to be regulated at the level of transcription (Dedeoglu et al., 2004; Gonda et al., 2003; Sayegh et al., 2003) (R. Casellas, manuscript submitted), by nucleo-cytoplasmic trafficking (Ito et al., 2004; McBride et al., 2004), and by phosphorylation (Basu et al., 2005; Basu et al., 2007; McBride et al., 2006; Pasqualucci et al., 2006). Here we reveal an additional miRNA-mediated pathway of AID regulation, which controls AID expression levels in germinal center B cells and ensures proper extinction of AID expression as cells affinity mature and exit the secondary lymphoid organs. Thus, miR-155 plays an important role in the molecular restraint of AID, an enzyme that confers great immunological benefit, but must be tightly regulated to limit its mutagenic potential.
Experimental Procedures
B lymphocyte isolation and cell culture
CD43− naïve splenic B lymphocytes were purified by magnetic separation (MACS, Miltenyi Biotec). Cells were maintained at 0.5 – 1 × 106/ml in standard culture medium, and were treated with 5 ng/mL IL-4 (Sigma) and 25 μg/mL LPS (Sigma) to induce CSR to IgG1; 25 μg/mL LPS to induce CSR to IgG3; or 25 μg/mL LPS and 0.5 μg/mL IFNγ (Sigma) to induce CSR to IgG2a. CH12-F3 cells were maintained in standard culture medium, and were treated with 5 ng/mL IL-4, 0.2 μg/mL anti-CD40 antibody (eBioscience), and 0.1 ng/mL TGF-β (R&D Systems) to induce CSR to IgA. Cell cultures were sampled at various time points for FACS analysis or preparation of RNA.
RNA isolation and small RNA cloning
Total RNA was prepared with Trizol (Invitrogen), and 21–23 nt RNAs were isolated, cloned, sequenced, and catalogued as previously described (Landgraf et al., 2007).
Northern blots
Total RNA (10–20 μg) was run on a 15% TBE-Urea Criterion gel (Bio-rad), and transferred to Hybond N+ nylon membrane (Amersham Biosciences) by semi-dry blotting. Membranes were UV-crosslinked and dried. A miR-155 probe (5′-ACCCCTATCACAATTAGCATTAA) was prepared by T4 polynucleotide kinase labeling with γ32P-ATP. Blots were hybridized in Denhardt’s solution or QuickHyb (Stratagene) at 50 ºC, and were washed twice with 5% SDS, 5x SSC, and twice with 1% SDS, 1x SSC.
Luciferase assays
Reporter constructs were modified from the pRL-TK plasmid (Promega). The renilla luciferase gene was replaced by firefly luciferase(fLuc) to create the Luc construct. The 3′UTR of AID was cloned directly downstream of fLuc to create the Luc-UTR plasmid. This was then altered by Quikchange PCR (Stratagene) to create the Luc-UTR-Del and Luc-UTR Mut constructs. CH12-F3 cells were co-transfected with a reporter construct and pRL-TK by Amaxa nucleofection, and were stimulated for CSR as above. Cells were lysed 48 hours after transfection and the Dual Luciferase Reporter Assay System (Promega) was used to measure firefly and renilla luciferase activities.
CSR rescue by AID-GFP
Replication-deficient retroviruses were generated by transfection of 293T cells with either AID-GFP-pQCXIP or pQCXIP alone, along with pCL-Eco packaging plasmid. Viral stocks were used to transduce naïve splenic B lymphocytes from AID−/− mice, which had been stimulated in culture with IL-4 and LPS.
Transgenic mice
The AID-GFP-Mut BAC was modified from the AID-GFP BAC used to create the previously described AID-GFP reporter mouse (Crouch et al., 2007) (copy number ranging from ~1 to ~10). Mutation of the miR-155 target site was achieved by homologous recombination in bacteria as described previously (Misulovin et al., 2001). Briefly, the AID miR-155 target site plus ~1 kb flanking sequence on either side (for homologous recombination) was amplified by PCR from BAC RP24-68I7 (Genbank AC158651) using the following primers: 5′-GGCGCGCCGGTAAGTCTGCCTGTCTGTCTGCC and 5′-GCGGCCGCGCGTCATTTCCTTGCCACGG. The PCR product was cloned into TOPO-pCR4 (Invitrogen). Point mutations in the miR-155 target site were introduced by Quikchange PCR (Stratagene). The sequence was again amplified with the same primers as above, and cloned into shuttle vector pLD53.SC.AEB (which also contains the RecA and SacB genes). After propagation in PIR2 bacteria (Invitrogen), the construct was electroporated into bacteria carrying the modified BAC RP24-68I7 containing the AID-GFP locus described by Crouch, et al. Cointegrates were selected for in liquid culture in the presence of ampicillin (for the insert-containing shuttle vector) and chloramphenicol (for the BAC), during which RecA-mediated recombination occurred between the BAC and homologous sequences inserted into the shuttle vector. Cultures were plated on chloramphenicol, and the desired recombination event (BAC containing the modified miR-155 target site, with deletion of shuttle vector sequence) was ensured by treating duplicate plates with UV illumination and sucrose to ensure the loss of the shuttle-vector-encoded RecA and SacB, respectively. Generation of the AID-GFP-Mut transgenic founder mice (copy number ranging from ~5 to ~20) was performed by the Rockefeller University Transgenic Services Laboratory, using standard methods. Transgene copy numbers were estimated by comparison to copy number standards in Southern analysis. Integrity of the AID-GFP and AID-GFP-Mut locus was confirmed by sequencing: that is, we sequenced the entire mRNA transcript from AID-GFP and AID-GFP-Mut B cells in vivo, and confirmed that they only differed by the nucleotides we specifically mutated (i.e., we found no additional mutations). Animals were housed and studied in accordance with institutional guidelines.
Quantitative PCR
cDNA was generated from DNAse-I-treated RNA, and AID-GFP expression was monitored by quantitative PCR (5′-GACTTGCGAGATGCATTTCGTATG and 5′-GCTGAACTTGTGGCCGTTTAC). cDNA samples were normalized by also amplifying Ku70 (5′-TGCCCTTTACTGAGAAGGTGAC and 5′-TGCTGCAGGACTGGATTCTC).
Immunization and ELISA
Mice were immunized with 100 μg alum-precipitated NP-CGG (Biosearch Technologies) by intraperitoneal injection. Serum was prepared from peripheral blood collected by retro-orbital bleed at various time points. Serum dilutions were incubated in NP3-BSA or NP30-BSA -coated wells of microtiter plates, and NP-specific Ig were detected by ELISA using reagents from Southern Biotechnologies Clonotyping System-HRP. Affinity maturation was calculated as the ratio of NP3-binding (high affinity anti-NP Ig) to NP30-binding (total anti-NP Ig).
FACS analysis
Cell suspensions from bone marrow, spleen, Peyer’s patches, thymus, and peripheral blood were prepared and stained for FACS using standard procedures. The following reagents were used to stain cells for FACS analysis (all are from BD Biosciences except where indicated): CD93-PE (eBioscience), IgM-APC (Jackson Immunoresearch), CD95-PE-Cy7, B220-PerCP, B220-APC, IgG1-biotin, IgM-biotin, IgD-biotin (eBioscience), CD8-PerCP, CD4-APC, NP-PE (Biosearch Technologies), Streptavidin-PerCP.
Mutational analysis
Genomic DNA was prepared from sorted splenic germinal center B cells and white blood cells from peripheral blood. The following were PCR-amplified with PfuTURBO polymerase (Stratagene): JH4 intron (5′-AGCCTGACATCTGAGGAC and 5′-TAGTGTGGAACATTCCTCAC, annealing temperature 55 ºC for 35 cycles; followed by a nested reaction 5′-CTGACATCTGAGGACTCTGC and 5′-GCTGTCACAGAGGTGGTCCTG, annealing temperature 58 ºC for 35 cycles); VH186.2 (5′-TCTTTACAGTTACTGAGCACACAGGAC and 5′-GGGTCTAGAGGTGTCCCTAGTCCTTCATGACC, annealing temperature 50 ºC for 35 cycles; followed by a nested reaction 5′-CAGTAGCAGGCTTGAGGTCTGGAC and 5′-GGGTCTAGAGGTGTCCCTAGTCCTTCATGACC, annealing temperature 64 ºC for 35 cycles); bcl-6 (5′-GGCCGGACACCAGGTGATTAT and 5′-AGGGAGGGAACTACCGCTGAG, annealing temperature 68 ºC for 35 cycles). PCR products were blunt-end cloned into pSC-B (Stratagene), and sequenced using a standard T3 primer.
Supplementary Material
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- Bartel DP, Chen CZ. Micromanagers of gene expression: the potentially widespread influence of metazoan microRNAs. Nat Rev Genet. 2004;5:396–400. doi: 10.1038/nrg1328. [DOI] [PubMed] [Google Scholar]
- Basu U, Chaudhuri J, Alpert C, Dutt S, Ranganath S, Li G, Schrum JP, Manis JP, Alt FW. The AID antibody diversification enzyme is regulated by protein kinase A phosphorylation. Nature. 2005;438:508–511. doi: 10.1038/nature04255. [DOI] [PubMed] [Google Scholar]
- Basu U, Chaudhuri J, Phan RT, Datta A, Alt FW. Regulation of activation induced deaminase via phosphorylation. Adv Exp Med Biol. 2007;596:129–137. doi: 10.1007/0-387-46530-8_11. [DOI] [PubMed] [Google Scholar]
- Chaudhuri J, Khuong C, Alt FW. Replication protein A interacts with AID to promote deamination of somatic hypermutation targets. Nature. 2004;430:992–998. doi: 10.1038/nature02821. [DOI] [PubMed] [Google Scholar]
- Crouch EE, Li Z, Takizawa M, Fichtner-Feigl S, Gourzi P, Montano C, Feigenbaum L, Wilson P, Janz S, Papavasiliou FN, Casellas R. Regulation of AID expression in the immune response. J Exp Med. 2007;204:1145–1156. doi: 10.1084/jem.20061952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cumano A, Rajewsky K. Structure of primary anti-(4-hydroxy-3-nitrophenyl)acetyl (NP) antibodies in normal and idiotypically suppressed C57BL/6 mice. Eur J Immunol. 1985;15:512–520. doi: 10.1002/eji.1830150517. [DOI] [PubMed] [Google Scholar]
- Dedeoglu F, Horwitz B, Chaudhuri J, Alt FW, Geha RS. Induction of activation-induced cytidine deaminase gene expression by IL-4 and CD40 ligation is dependent on STAT6 and NFkappaB. Int Immunol. 2004;16:395–404. doi: 10.1093/intimm/dxh042. [DOI] [PubMed] [Google Scholar]
- Furukawa K, Akasako-Furukawa A, Shirai H, Nakamura H, Azuma T. Junctional amino acids determine the maturation pathway of an antibody. Immunity. 1999;11:329–338. doi: 10.1016/s1074-7613(00)80108-9. [DOI] [PubMed] [Google Scholar]
- Gonda H, Sugai M, Nambu Y, Katakai T, Agata Y, Mori KJ, Yokota Y, Shimizu A. The balance between Pax5 and Id2 activities is the key to AID gene expression. J Exp Med. 2003;198:1427–1437. doi: 10.1084/jem.20030802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong S, Zheng C, Doughty ML, Losos K, Didkovsky N, Schambra UB, Nowak NJ, Joyner A, Leblanc G, Hatten ME, Heintz N. A gene expression atlas of the central nervous system based on bacterial artificial chromosomes. Nature. 2003;425:917–925. doi: 10.1038/nature02033. [DOI] [PubMed] [Google Scholar]
- Griffiths-Jones S, Grocock RJ, van Dongen S, Bateman A, Enright AJ. miRBase: microRNA sequences, targets and gene nomenclature. Nucleic Acids Res. 2006;34:D140–144. doi: 10.1093/nar/gkj112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimson A, Farh KK, Johnston WK, Garrett-Engele P, Lim LP, Bartel DP. MicroRNA targeting specificity in mammals: determinants beyond seed pairing. Mol Cell. 2007;27:91–105. doi: 10.1016/j.molcel.2007.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatten ME, Heintz N. Large-scale genomic approaches to brain development and circuitry. Annu Rev Neurosci. 2005;28:89–108. doi: 10.1146/annurev.neuro.26.041002.131436. [DOI] [PubMed] [Google Scholar]
- Ito S, Nagaoka H, Shinkura R, Begum N, Muramatsu M, Nakata M, Honjo T. Activation-induced cytidine deaminase shuttles between nucleus and cytoplasm like apolipoprotein B mRNA editing catalytic polypeptide 1. Proc Natl Acad Sci U S A. 2004;101:1975–1980. doi: 10.1073/pnas.0307335101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- John B, Enright AJ, Aravin A, Tuschl T, Sander C, Marks DS. Human MicroRNA targets. PLoS Biol. 2004;2:e363. doi: 10.1371/journal.pbio.0020363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kluiver J, Haralambieva E, de Jong D, Blokzijl T, Jacobs S, Kroesen BJ, Poppema S, van den Berg A. Lack of BIC and microRNA miR-155 expression in primary cases of Burkitt lymphoma. Genes Chromosomes Cancer. 2006;45:147–153. doi: 10.1002/gcc.20273. [DOI] [PubMed] [Google Scholar]
- Krek A, Grun D, Poy MN, Wolf R, Rosenberg L, Epstein EJ, MacMenamin P, da Piedade I, Gunsalus KC, Stoffel M, Rajewsky N. Combinatorial microRNA target predictions. Nat Genet. 2005;37:495–500. doi: 10.1038/ng1536. [DOI] [PubMed] [Google Scholar]
- Landgraf P, Rusu M, Sheridan R, Sewer A, Iovino N, Aravin A, Pfeffer S, Rice A, Kamphorst AO, Landthaler M, et al. A mammalian microRNA expression atlas based on small RNA library sequencing. Cell. 2007;129:1401–1414. doi: 10.1016/j.cell.2007.04.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis BP, Shih IH, Jones-Rhoades MW, Bartel DP, Burge CB. Prediction of mammalian microRNA targets. Cell. 2003;115:787–798. doi: 10.1016/s0092-8674(03)01018-3. [DOI] [PubMed] [Google Scholar]
- MacDuff DA, Neuberger MS, Harris RS. MDM2 can interact with the C-terminus of AID but it is inessential for antibody diversification in DT40 B cells. Mol Immunol. 2006;43:1099–1108. doi: 10.1016/j.molimm.2005.07.024. [DOI] [PubMed] [Google Scholar]
- McBride KM, Barreto V, Ramiro AR, Stavropoulos P, Nussenzweig MC. Somatic hypermutation is limited by CRM1-dependent nuclear export of activation-induced deaminase. J Exp Med. 2004;199:1235–1244. doi: 10.1084/jem.20040373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBride KM, Gazumyan A, Woo EM, Barreto VM, Robbiani DF, Chait BT, Nussenzweig MC. Regulation of hypermutation by activation-induced cytidine deaminase phosphorylation. Proc Natl Acad Sci U S A. 2006;103:8798–8803. doi: 10.1073/pnas.0603272103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misulovin Z, Yang XW, Yu W, Heintz N, Meffre E. A rapid method for targeted modification and screening of recombinant bacterial artificial chromosome. J Immunol Methods. 2001;257:99–105. doi: 10.1016/s0022-1759(01)00452-5. [DOI] [PubMed] [Google Scholar]
- Muto T, Okazaki IM, Yamada S, Tanaka Y, Kinoshita K, Muramatsu M, Nagaoka H, Honjo T. Negative regulation of activation-induced cytidine deaminase in B cells. Proc Natl Acad Sci U S A. 2006;103:2752–2757. doi: 10.1073/pnas.0510970103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connell RM, Taganov KD, Boldin MP, Cheng G, Baltimore D. MicroRNA-155 is induced during the macrophage inflammatory response. Proc Natl Acad Sci U S A. 2007;104:1604–1609. doi: 10.1073/pnas.0610731104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasqualucci L, Kitaura Y, Gu H, Dalla-Favera R. PKA-mediated phosphorylation regulates the function of activation-induced deaminase (AID) in B cells. Proc Natl Acad Sci U S A. 2006;103:395–400. doi: 10.1073/pnas.0509969103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasqualucci L, Neumeister P, Goossens T, Nanjangud G, Chaganti RS, Kuppers R, Dalla-Favera R. Hypermutation of multiple proto-oncogenes in B-cell diffuse large-cell lymphomas. Nature. 2001;412:341–346. doi: 10.1038/35085588. [DOI] [PubMed] [Google Scholar]
- Poy MN, Eliasson L, Krutzfeldt J, Kuwajima S, Ma X, Macdonald PE, Pfeffer S, Tuschl T, Rajewsky N, Rorsman P, Stoffel M. A pancreatic islet-specific microRNA regulates insulin secretion. Nature. 2004;432:226–230. doi: 10.1038/nature03076. [DOI] [PubMed] [Google Scholar]
- Ramiro AR, Jankovic M, Callen E, Difilippantonio S, Chen HT, McBride KM, Eisenreich TR, Chen J, Dickins RA, Lowe SW, et al. Role of genomic instability and p53 in AID-induced c-myc-Igh translocations. Nature. 2006;440:105–109. doi: 10.1038/nature04495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramiro AR, Jankovic M, Eisenreich T, Difilippantonio S, Chen-Kiang S, Muramatsu M, Honjo T, Nussenzweig A, Nussenzweig MC. AID is required for c-myc/IgH chromosome translocations in vivo. Cell. 2004;118:431–438. doi: 10.1016/j.cell.2004.08.006. [DOI] [PubMed] [Google Scholar]
- Rodriguez A, Vigorito E, Clare S, Warren MV, Couttet P, Soond DR, van Dongen S, Grocock RJ, Das PP, Miska EA, et al. Requirement of bic/microRNA-155 for normal immune function. Science. 2007;316:608–611. doi: 10.1126/science.1139253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayegh CE, Quong MW, Agata Y, Murre C. E-proteins directly regulate expression of activation-induced deaminase in mature B cells. Nat Immunol. 2003;4:586–593. doi: 10.1038/ni923. [DOI] [PubMed] [Google Scholar]
- Shen HM, Peters A, Baron B, Zhu X, Storb U. Mutation of BCL-6 gene in normal B cells by the process of somatic hypermutation of Ig genes. Science. 1998;280:1750–1752. doi: 10.1126/science.280.5370.1750. [DOI] [PubMed] [Google Scholar]
- Taketani M, Naitoh A, Motoyama N, Azuma T. Role of conserved amino acid residues in the complementarity determining regions on hapten-antibody interaction of anti-(4-hydroxy-3-nitrophenyl) acetyl antibodies. Mol Immunol. 1995;32:983–990. doi: 10.1016/0161-5890(95)00057-l. [DOI] [PubMed] [Google Scholar]
- Tam W, Ben-Yehuda D, Hayward WS. bic, a novel gene activated by proviral insertions in avian leukosis virus-induced lymphomas, is likely to function through its noncoding RNA. Mol Cell Biol. 1997;17:1490–1502. doi: 10.1128/mcb.17.3.1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng G, Papavasiliou FN. Immunoglobulin somatic hypermutation. Annu Rev Genet. 2007;41:107–120. doi: 10.1146/annurev.genet.41.110306.130340. [DOI] [PubMed] [Google Scholar]
- Thai TH, Calado DP, Casola S, Ansel KM, Xiao C, Xue Y, Murphy A, Frendewey D, Valenzuela D, Kutok JL, et al. Regulation of the germinal center response by microRNA-155. Science. 2007;316:604–608. doi: 10.1126/science.1141229. [DOI] [PubMed] [Google Scholar]
- Vigorito E, Perks KL, Abreu-Goodger C, Bunting S, Xiang Z, Kohlhaas S, Das PP, Miska EA, Rodriguez A, Bradley A, et al. microRNA-155 Regulates the Generation of Immunoglobulin Class-Switched Plasma Cells. Immunity. 2007 doi: 10.1016/j.immuni.2007.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao C, Calado DP, Galler G, Thai TH, Patterson HC, Wang J, Rajewsky N, Bender TP, Rajewsky K. MiR-150 controls B cell differentiation by targeting the transcription factor c-Myb. Cell. 2007;131:146–159. doi: 10.1016/j.cell.2007.07.021. [DOI] [PubMed] [Google Scholar]
- Zhou B, Wang S, Mayr C, Bartel DP, Lodish HF. miR-150, a microRNA expressed in mature B and T cells, blocks early B cell development when expressed prematurely. Proc Natl Acad Sci U S A. 2007;104:7080–7085. doi: 10.1073/pnas.0702409104. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.