

Abstract
Cerebral vasospasm (CV) remains a significant cause of delayed neurological deficit and ischemic damage after subarachnoid hemorrhage (SAH), despite intensive research effort. The current lack of an effective therapeutic approach is somewhat due to our lack of understanding regarding the mechanism by which this pathological constriction develops. Recent evidence implicates bilirubin oxidation products (BOXes) in the etiology of CV after SAH: BOXes are found in cerebrospinal fluid from SAH patients with symptomatic or angiographically visible vasospasm (CSFV) but not in CSF from SAH patients with no vasospasm (CSFC). We have previously published research suggesting that the etiology of CV comprises two components: a physiological stimulation to constrict and a pathological failure to relax. Both these components are elicited by CSFV, but not CSFC, and BOXes synthesized in the laboratory potentiate physiological constriction in arterial smooth muscle in vitro, and elicit contraction in pial arteries in vivo. In this paper, we will present our results concerning the action of BOXes on arterial smooth muscle constriction, compared with CSFV. We will also present evidence implicating temporal changes in PKC isoforms and Rho expression in both BOXes- and CSFV-elicited smooth muscle responses.
Keywords: Subarachnoid Hemorrhage, Cerebral Vasospasm, Protein Kinase C, Rho, Vascular Smooth Muscle, Bilirubin, Contractile Function
2. INTRODUCTION
Worldwide, subarachnoid hemorrhage (SAH) and its sequel, cerebral vasospasm (CV), kill or seriously debilitate an estimated 1.2 million people, of both genders, and all ages and ethnic groups annually (1). In the United States alone, complications from SAH, including increased length of hospital stay, increased treatment intensity level, requirement for long-term care and intensive rehabilitation are estimated to have cost 500 million dollars in 2000 (2). The pathological constriction of cerebral vessels that occurs during CV may lead to ischemia, infarction or death (3, 4) and the 3–10 days between the initial hemorrhage and onset of CV potentially affords the clinician a therapeutic window (5). Unfortunately, despite considerable research efforts, the etiology of CV is still unknown, and appears to be a complex combination of events (3, 6–10) Our research indicates that the intractable vasoconstriction seen in CV after SAH is a combination of a physiological protein kinase C- mediated contraction, followed by a pathological failure to relax, mediated by Rho-mediated inhibition of myosin light chain phosphatase (9, 11). Bilirubin oxidation products (BOXes) may be responsible for one or both of these events (12–14).
The literature records a bewildering array of molecules that may be implicated in the etiology of CV after SAH. They include endothelin (15), hemoglobin (7, 16), bilirubin and it’s oxidation products (17–19), arachidonic acid and it’s peroxidized metabolites (12, 20–23), and immune system molecules (24). However, almost all of these molecules are present both in CSFV and CSFC, so they cannot be the only determinant of vasospasm. There is also the question of time-course: the majority of these agents is present acutely and probably plays a role in short-term vasoconstriction to minimize hemorrhage, but delayed CV does not occur until 3–10 days after the SAH.
2.1. Bilirubin Oxidation Products
Bilirubin appears in SAH CSF after around 12 hours post-hemorrhage, but the levels do not peak until days later. We have shown that bilirubin levels are higher in CSFV than CSFC, and that this bilirubin production peaks at 3–8 days, a time course startlingly similar to that of the onset of vasospasm (5). BOXes have been detected in the CSF of vasospastic patients and BOXes synthesized in the laboratory have been shown to be vasoactive both in vivo and in vitro (17, 19). The mixture of BOXes seen in the synthesized product (BOX A and BOX B, see Figure 1) are also seen in CSFV18. In order to have BOXes production, in addition to an elevated bilirubin concentration, an oxidizing environment must also be present (12). An oxidizing environment may result from macrophage infiltration (25), which elevates peroxide concentrations, and leads to non-specific peroxidation of CSF components, as well as induction of lipooxygenases that produce hydroxy-eicosatetranoic acid (HETE) molecules from arachidonic acid (23).
Figure 1.
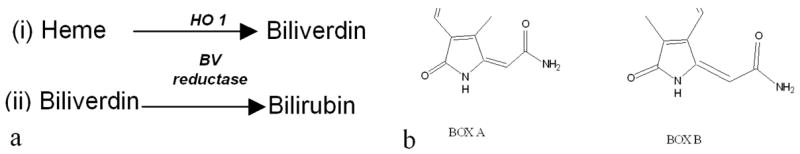
a. The conversion of heme to bilirubin involves two enzymes. Heme Oxygenase 1 (1.14.99.3) and Biliverdin reductase (1.3.1.24). These reactions occur in the CSF, the enzymes are found in the brain tissue. b. Structures of bilirubin oxidation products (BOXes) that are found in CSFV (but not CSFC) and are likely to be formed in the CSF in the presence of bilirubin and an oxidizing environment. These molecules are regularly synthesized in large quantities in our laboratory.
3. MATERIALS AND METHODS
3.1. Materials
All chemicals were obtained from Acros Organics, (Morris Plains, New Jersey) unless otherwise specified.
3.2. BOXes synthesis
BOXes were synthesized using the methods described in Kranc et al (19). The entire procedure was performed protected from light. Reagent grade bilirubin was completely dissolved in 5 M NaOH for 24 hours. The alkaline solution was then neutralized using 10 M HCl. H2O2 was added to a final concentration of 10% and the mixture was incubated at room temperature for 48 hours. The resulting solution was freeze-dried, and extracted into chloroform to facilitate desalting. The chloroform was evaporated under N2 (g), and the resulting powder resuspended in 0.9% saline for use in experiments.
3.2.1. BOXes quantification
BOXes were quantified using HPLC analysis on a Waters Chromatography System consisting of a 2790 Separations Module, and 2487 Variable Wavelength Detector running Millennium 32 Data Analysis Software. Separation is achieved on a Symmetry C18, 5 micrometer × 4.6 × 150 mm, Column. The eluent was monitored at 310 nm and the column temperature held at 35°C with a flow rate of 1.0 ml/min. The mobile phase consists of acetonitrile/water with a gradient of 5% to 50% acetonitrile.
3.3. Tissue preparation
Large White pigs weighing approximately 200 pounds were the source of arteries for this study. The condition of the vessels was maintained thus: the vessels were removed from the carcass within 30 minutes of slaughter, conveyed to the laboratory in ice cold PSS and dissected free of adventitia. The vessels are stored in PSS (Physiological saline solution, 118 mM NaCl; 25 mM NaHCO3; 5.7 mM KCl; 1.2 mM MgCl2; 2.4 mM CaCl2 0.75 mM NaH2PO4 and 11 mM Glucose, pH 7.4) (26) for up to 72 hours, with 12 hourly solution change (27). For each day’s experiments, a control is run concurrently to account for any functional change.
3.5. Isometric force measurements
Strips (1 mm wide) of porcine carotid arteries were mounted between two wire clips and immersed in a water jacketed chamber containing PSS aerated with 95% O2 and 5% CO2 at 37 degrees C. The tissue was allowed to equilibrate for 1 hour and then stretched to the optimum length. The tissue was twice exposed to 80 mM KCl PSS to determine maximal tension. Tissue viability was assessed by observation of the Herlihy hop (28) which indicates normal contractile function, and a consistent response to the KCl precontractions. The agents of interest were added to the bath at appropriate concentrations, and the tissue allowed to reach a maximal response.
3.6. Western blotting
For western blot analysis, tissue is dissected free of all adventitia, connective tissue and gently rubbed to remove endothelial cells. After the tissue had been stretched to the LO and treated with the agent under investigation it was snap frozen in liquid nitrogen. The tissue was minced in ice-cold homogenization buffer (50 mM Tris-HCl, 5 mM ethylene diamine tetraacetic acid (EDTA), 10 mM ethyleneglycol-bis (aminoethylether)-N-N’ tetraacetic acid (EGTA), and a protease inhibitor cocktail containing 1 mM phenylmethylsulfoxide (PMSF), 5 mM dithiothreitol, 10 mM benzamidine, 25 mg/ml leupeptin). The suspension was then centrifuged at 100,000 × g for 30 minutes at 4 degrees C. The resultant pellet contains the membrane fraction of the tissue, and the supernatant, the cytosol. The cytosolic and membrane fractions were assayed for protein using the BCA protein assay (Pierce®) and then diluted to an appropriate concentration using sample buffer (62.5 mM Tris-HCl, 2% SDS, 10% glycerol and 8 mM dithiothreitol). Samples in sample buffer were boiled for 5 minutes immediately prior to gel loading. SDS-PAGE was performed for 1 hour at 150 V and 20 degrees C in running buffer consisting of 25 mM Tris-base, 0.2 M glycine and 0.1% SDS, at pH 8.3. The resolved proteins were electrophoretically transferred to a nitrocellulose membrane in transfer buffer 20% methanol, 25mM Tris-HCl and 192 mM glycine (pH 8.3) for 90 minutes at 120 mA. After transfer, the membrane was blocked in 0.5% non-fat milk in Tris-buffered saline containing 150 mM NaCl, 20 mM Tris-HCl (pH 7.5) for 60 minutes. They were then incubated overnight at 4°C with a chosen rabbit-derived primary antibody (Sigma, St. Louis, MO, USA) diluted 1:500 v/v in Tris-buffered saline containing 3% horse serum. The membranes were washed (3 times for 5 minutes each time) in Tris-buffered saline, and incubated for 7 hours with 2 micrograms/ml peroxidase-labeled goat-anti-rabbit antibodies, diluted as described for the primary antibodies. The membrane was then washed as before. Enhanced chemiluminescence will be used to develop the signal, and where available, isoform-specific peptides (Sigma, St. Louis, MO, USA) were used to confirm the specificity of the antibodies (10).
3.7. Phosphatase activity assay
The Malachite green phosphatase assay kit from Upstate (Charlottesville, VA, Cat # 17–351) was used to obtain this data. The assay is based on the detection of phosphate as it is released by purified PP1 (Cat # 14–165) from an enzyme-specific synthetic phosphopeptide. The liberated phosphate forms a molybdate:malachite green:phosphate reaction complex which is detected colorimetrically (29, 30). BOXes or okadaic acid were added to the reaction mixture to a final concentration of 20 micromolar (BOXes, equivalent to that seen in CSFV) or 1 nM (OA, potent phosphatase inhibition).
3.7. Human cerebrospinal fluid
Bags of CSF are collected via a cisternal drain as part of standard of care at the University Hospital (Cincinnati, Ohio). The University of Cincinnati IRB has approved use of this CSF for research purposes. No additional risk to the patients is posed by collection of CSF. In order to add CSF to the organ bath at the appropriate concentration to elicit force generation (1:1) (31), we must remove the protein. This is because the tissue must be supplied with O2, and bubbling a proteinous solution leads to unacceptable foaming and aerosol of biohazardous material. We have shown previously that removing protein from the CSF using perchloric acid (PCA) extraction does not significantly reduce the biological activity of CSFV (27). PCA is added to a known volume of CSF so that the final PCA concentration (v/v) is 12%. This solution is centrifuged for 10 minutes at 2000 g at 4 degrees C, and the supernatant immediately neutralized with 10 M NaOH on ice. The resulting solution is then is freeze-dried in the dark, as we have observed that the biological activity of the CSFV is lost if it is exposed to light (32) The weighed powder is reconstituted in the organ bath so that the final concentration in the bath is equivalent to undiluted CSF. CSFC was used as a control for CSFV as it contains all the biological “contaminants” (hemoglobin, bilirubin, proteins) as CSFV, and yet came from patients who did not develop vasospasm.
3.8. Statistics
Significance was determined using ANOVA. P values equal to or less than 0.05 were considered significant.
4. RESULTS
4.1. Potentiation of vascular smooth muscle contraction
In arterial smooth muscle, PKC has been shown to play a role in the sensitization of the contractile apparatus to calcium (33–36). Thus, PKC activation alone does not elicit contraction, but it can intensify an already existing contractile signal. This effect can be seen with BOXes in the porcine carotid artery in vitro. Figure 2 shows that incubation with BOXes, while not eliciting tension in itself, leads to significant potentiation of the contractile response of the artery to phenylephrine. While a small, but significant potentiation is seen with 1 micromolar phorbol myristic acid, a potent PKC activator (37), the potentiation by Okadaic acid, an inhibitor of protein phosphatase (38) is even greater. BOXes, at 20 micromolar, a concentration seen in CSF from vasospastic patients (38) reduces the EC50 from 1.23 +/− 0.09 (control) by almost 10-fold to 0.14 +/− 0.008 micromolar, which is more than PMA, suggesting that more than just PKC activation is involved.
Figure 2.
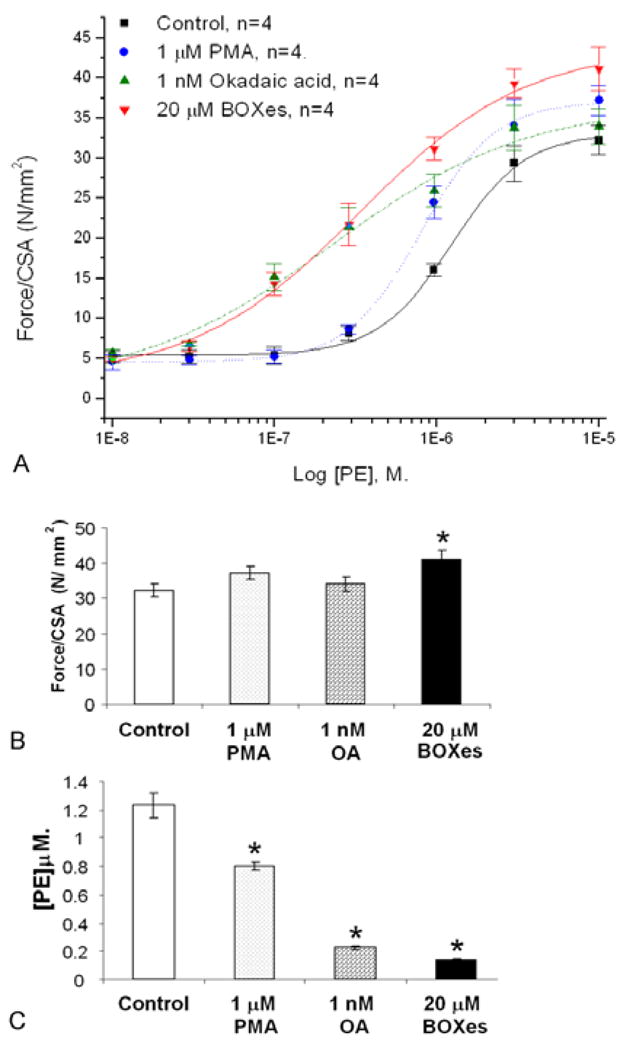
Potentiation of phenylephrine (PE)-induced contraction in porcine carotid artery rings by BOXes. Panel A shows the significant right-shift in the concentration-response curve to cumulative doses of PE, also seen initially with Okadaic acid, a PP inhibitor and to a lesser extent with Phorbol myristic acid, a PKC activator. Panel B compares the maximum contraction obtained in response to PE in the tissues from Panel A. BOXes potentiated the contraction slightly, but significantly (N=4, P<0.05). Panel C shows that PMA significantly reduces the EC50 of PE, and OA, even more so compared to control. BOXes, however, reduced the EC50 almost 10-fold. Errors are s.d.
4.2. Translocation of PKC isoforms, and Rho A
Previously published work from our laboratory has shown that CSF from patients with vasospasm contains a substance that inhibits myosin light chain phosphatase (9). Initiation of contraction in smooth muscle is via myosin light chain kinase. MLCK is activated by the binding of calcium-calmodulin (Ca2+-CaM), and subsequently phosphorylates ser 19 of the regulatory light chains of myosin (MLC20). Phosphorylation of MLC20 activates the intrinsic ATPase activity of the myosin heavy chain, and thus allows cross bridge interaction and contraction. In order for the energy-efficient “latch” state to be entered, MLC20 must be dephosphorylated by myosin light chain phosphatase (MLCP), a type 1 protein phosphatase (PP1) (39). The latch state is a physiological event which enables smooth muscles to maintain tension at low energy cost, and it responds to physiological vasodilatory signals (40). Dephosphorylation must also occur to allow relaxation. Vasospasm is not energy efficient (not latch), and does not respond to physiological or pharmacological dilatory signals (41). Inhibition of MLCP leads to a failure to relax.
We utilized a commercially available kit to assess the effect of synthesized BOXes on purified PP1 in vitro. Figure 3 shows that 20 micromolar BOXes had no significant effect on the activity of the enzyme. The inhibition seen with CSFV was very significant, but the assay was carried out on an enzyme from an intact artery, whereas this assay was in a purified enzyme in isolation from upstream signaling regulation. Thus, we added Rho A to the proteins of interest for our next set of investigations.
Figure 3.
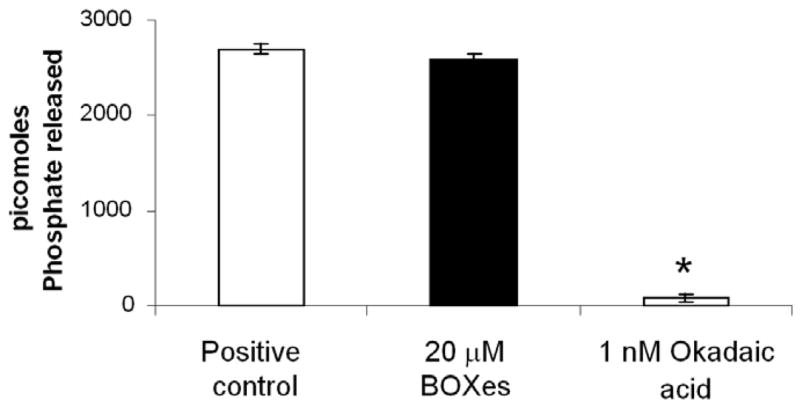
BOXes do not appear to have inhibitory effects on purified protein phosphatase I. The positive control contained no inhibitory agents, and 1 nM Okadaic acid, a known potent PP1 inhibitor at this concentration, acted as a negative control. N=5, errors are s.d.
4.3. BOXes and phosphatase activity
Increased myosin phosphorylation has been reported in the canine double hemorrhage model (7, 42, 43) (44). These increases are reported to be due to mobilization of Rho, which activates rho kinase (ROK). ROK phosphorylates and inactivates MLCP (7, 45–47). Rho is activated by a number of agonists including thromboxane A2 and other lipid signaling molecules (48). Figure 4 is a simplified diagram showing putative mechanisms of control of MLCP by Rho A and PKC. There are additional PKC-mediated mechanisms, discussed later.
Figure 4.
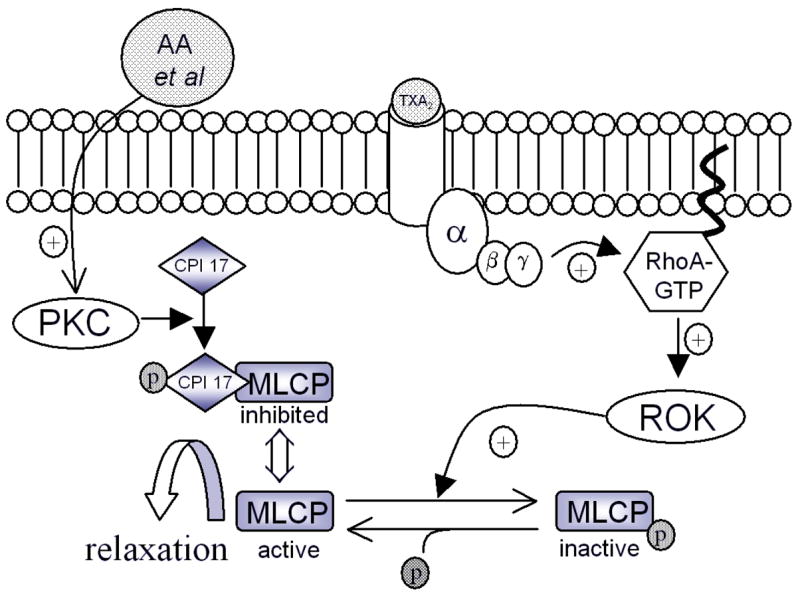
Mechanism of Rho A control of MLCP activity. Inactive Rho A (bound to GDP) is free in the cytosol, and translocates to the membrane when activated by agonists such as TXA2. Rho A then activates Rho kinase (ROK), which phosphorylates and inactivates MLCP. PKC is also thought to inhibit MLCP via PKC-potentiated inhibitor protein (CPI 17) in response to arachidonic acid and other lipid derivatives.
4.4. Translocation of PKC isoforms, and Rho A
PKC isoforms and Rho are translocated to the inside of the cell membrane when activated, and this enables us to monitor their activation by separating membrane and cytosolic fractions for western blot analysis. More than one isoform of PKC has been implicated in the etiology of CV after SAH. Nishizawa et al carried out these studies in a canine double-hemorrhage model, and showed that PKC delta was translocated to the cell membrane during the initiation of contraction in the basilar artery, and PKC alpha during the maintenance phase (10). Figure 4 shows that we see a strikingly similar pattern of translocation in response to both CSFV AND 20 micromolar BOXes. PKC alpha and delta and Rho A are all translocated to the plasma membrane (and we can assume, activated) at 20 minutes of exposure. In contrast to Nishizawa et al, PKC alpha is no longer significantly activated at 3 hours, although perhaps a longer time course may reveal a different pattern. Rho A continued to be significantly activated at 3 hours, although less so than at 20 minutes, when translocation to the membrane was almost double that of control tissue.
In summary, PKC isoforms alpha and delta are both significantly (P < 0.05) translocated after 20 minutes of exposure to either CSFV or 20 micromolar BOXes compared to control. This is no longer occurring at 3 hours, although whether this is a function of the time point chosen remains to be investigated. Rho A is significantly translocated at both 20 minutes, and 3 hours in response to both agents.
5. DISCUSSION
As the effects of BOXes on vascular smooth muscle function and signaling are unraveled, the similarity with the signaling events observed both in vivo and in vitro during cerebral vasospasm after subarachnoid hemorrhage become striking. Prior to the molecular isolation of BOXes from vasospastic CSF, we had reported that CSFV elicited potentiation of the force-O2 consumption relationship in porcine carotid artery (31). The data presented here support the hypothesis that CSFV and BOXes potentiation appears to be mediated through a combination of protein kinase C activation and Rho A-mediated inhibition of myosin light chain phosphatase, as we postulated previously with respect to the etiology of CV (9, 49–51).
5.1. PKC and Rho-mediated calcium sensitization of contraction
Protein kinases C are known to affect smooth muscle contractile function in several ways: in addition to myosin light chain kinase phosphorylation of smooth muscle MLC20, there are PKC specific phosphorylation sites on MLC20 that have been shown to slow cross bridge cycling 2-fold when phosphorylated. While the cycling is slowed, this allows maintenance of contraction in the presence of low calcium (below that which would activate MLCK), and thus the apparatus is sensitized to calcium (52, 53). PKC also controls contraction at the level of the thin filaments (54): unphosphorylated Calponin slows the Vmax of cross bridge cycling (55). When calponin is phosphorylated by PKC, its inhibition is removed as the calponin dissociates (56). PKC also has a role in the Rho kinase pathway (see Figure 5). An endogenous inhibitory protein of MLCP, CPI 17 (PKC-potentiated inhibitor protein) is activated (and thus able to inhibit MLCP) by PKC (57). Yet another role has recently been postulated for PKC, involving MAP kinase (58). In arterial smooth muscle, MAP kinase is involved in upregulation of protein synthesis during hypertrophy and repair (e.g. aneurysm formation, remodeling after CV) (59, 60) as well as increased calcium sensitivity, via MAPK phosphorylation of caldesmon (61). Massett et al reported that PKC phosphorylates and activates MEK (MAP/ERK kinase), which in turn phosphorylates and activates p42/44 MAPK (but not p38 MAPK), thus acutely contributing to the calcium sensitivity of the smooth muscle contractile apparatus (58), and perhaps, chronically, stimulating vascular remodeling to overcome pathological failure of the contractile apparatus in vasospasm.
Figure 5.
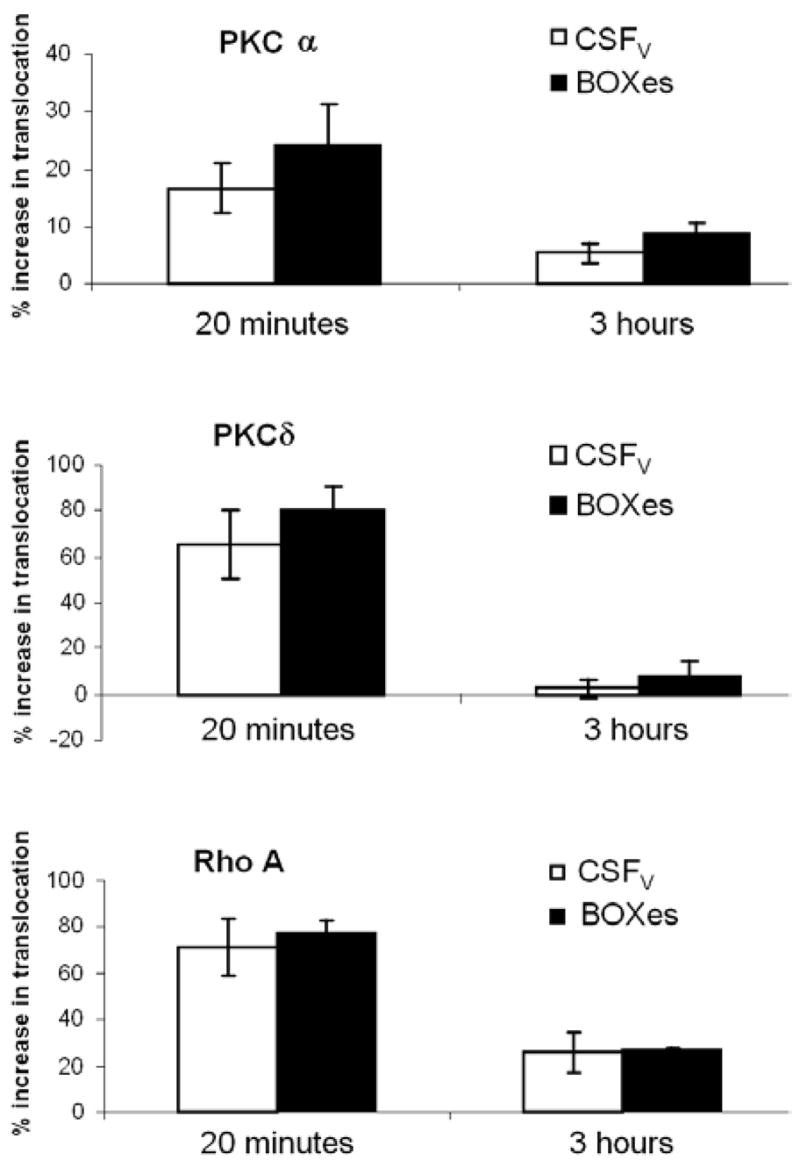
Translocation of PKC alpha, PKC delta or Rho A to the plasma membrane of porcine carotid arteries exposed to CSFV or 20 microM BOXes. Translocation is expressed as percentage of control (CSFC for CSFV and MOPS buffer for BOXes) membrane fraction in the presence of CSFV or BOXes. An early (20 minutes) and later (3 hours) time point were chosen. In all cases, BOXes had a slightly, but not significantly, more pronounced effect than CSFV. The PKC isoforms were both significantly activated at 20 minutes, but much less so at 3 hours, whereas Rho A remained significantly activated at 3 hours. The effects of BOXes and CSFV on these three proteins are strikingly similar. N=5, errors are S.E.M.
Translocation of Rho A at both the early and late time points in this model is not surprising. Rho Kinase is known to be activated via TXA2 binding at TXA2 receptors at the plasma membrane, amongst other agonists (47, 62, 63). Although prostaglandins and eicosanoids are found in both CSFC and CSFV (12, 64–68), we have found that TXA2, specifically, is significantly higher in CSFV than CSFC AND its production is elicited by injection of BOXes into rat brain (69). Rho kinase sensitizes smooth muscle contraction to calcium by phosphorylation, and inhibition of MLCP, leading to increased tension despite lowered calcium (48, 70). Therefore, it is not unexpected that BOXes would have no direct effect on purified protein phosphatase in vitro (Figure 3). The experiments showing inhibition of MLCP by CSFV were carried out in intact tissue, so all levels of the signaling cascade were present (9). This further suggests that BOXes act upstream of MLCP itself, possibly at Rho, perhaps even by increasing a Rho agonist such as TXA2.
In conclusion, both BOXes and CSFV, but not CSFC, elicit early (within 20 minutes) activation of protein kinase C isoforms alpha and delta, and Rho A in arterial smooth muscle. At 3 hours, the PKC alpha and delta translocation has diminished to a level not significantly different than control, but Rho A translocation remains significantly elevated. This is an important set of data, both confirming that our model of CV after SAH corresponds closely to previously reported in vivo models, and that BOXes are very likely to be the main player in a cast of molecules thought to be responsible for the etiology of this enigmatic pathology.
Acknowledgments
This research was supported by an NIH/NINDS award to Gail Pyne-Geithman, D.Phil. (5R01-NS-049428).
References
- 1.Census, Calculated by the author from various epidemiological reports of incidence from around the world from 2000 census data. 2000.
- 2.AHA. American Heart Association: Heart disease and stroke statistics. 2005. [Google Scholar]
- 3.MacDonald RL. Cerebral vasospasm Primer on cerebrovascular diseases. Academic Press; San Diego, CA: 1997. pp. 490–496. [Google Scholar]
- 4.Weir B. The pathophysiology of cerebral vasospasm. Br J Neurosurg. 1995;9:375–390. doi: 10.1080/02688699550041386. [DOI] [PubMed] [Google Scholar]
- 5.Weir B, Grace M, Hansen J, Rothberg C. Time course of vasospasm in man. J Neurosurg. 1978;48:173–178. doi: 10.3171/jns.1978.48.2.0173. [DOI] [PubMed] [Google Scholar]
- 6.Xi G, Reiser G, Keep RF. The role of thrombin and thrombin receptors in ischemic, hemorrhagic and traumatic brain injury: Deleterious or protective? J Neurochem. 2003;84:3–9. doi: 10.1046/j.1471-4159.2003.01268.x. [DOI] [PubMed] [Google Scholar]
- 7.Wickman G, Lan C, Vollrath B. Functional roles of the Rho/Rho kinase pathway and protein kinase C in the regulation of cerebrovascular constriction mediated by hemoglobin: Relevance to subarachnoid hemorrhage and vasospasm. 2003 doi: 10.1161/01.RES.0000066663.12256.B2. [DOI] [PubMed] [Google Scholar]
- 8.Borel CO, McKee A, Parra A, Haglund MM, Solan A, Prabhakar V, Sheng H, Warner DS, Niklason L. Possible role for vascular cell proliferation in cerebral vasospasm after subarachnoid hemorrhage. Stroke. 2003;34:427–433. [PubMed] [Google Scholar]
- 9.Pyne GJ, Cadoux-Hudson TAD, Clark JF. The presence of an extractable substance in the CSF of humans with cerebral vasospasm after subarachnoid haemorrhage that correlates with phosphatase inhibition. Biochim Biophys Acta. 2000;1474:283–290. doi: 10.1016/s0304-4165(00)00030-1. [DOI] [PubMed] [Google Scholar]
- 10.Nishizawa S, Obara K, Nakayama K, Koide M, Yokoyama T, Yokota N, Ohta S. Protein kinase c Delta and alpha are involved in the development of vasospasm after subarachnoid hemorrhage. Eur J Pharmacol. 2000;398:113–119. doi: 10.1016/s0014-2999(00)00311-3. [DOI] [PubMed] [Google Scholar]
- 11.Cadoux-Hudson TAD, Pyne GJ, Clark JF. Subarachnoid haemorrhage-induced cerebral vasospasm: A subcellular perspective on the control of tension. Emerging Therapeutic Targets. 1999;3(3):439–452. [Google Scholar]
- 12.Pyne GJ, Cadoux-Hudson TA, Clark JF. Platelets play an essential role in the aetiology of cerebral vasospasm after subarachnoid haemorrhage. Med Hypotheses. 2003;60(4):525–30. doi: 10.1016/s0306-9877(02)00452-8. [DOI] [PubMed] [Google Scholar]
- 13.Pyne-Geithman GJ, Morgan CJ, Wagner K, Dulaney EM, Carrozzella J, Kanter DS, Zuccarello M, Clark JF. Bilirubin production and oxidation in CSF of patients with cerebral vasospasm after subarachnoid hemorrhage. J Cereb Blood Flow Metab. 2005;23:23. doi: 10.1038/sj.jcbfm.9600101. [DOI] [PubMed] [Google Scholar]
- 14.Clark JF, Sharp FR. Bilirubin oxidation products (BOXes) and their role in cerebral vasospasm after subarachnoid hemorrhage. J Cereb Blood Flow Metab. 2006;8:8. doi: 10.1038/sj.jcbfm.9600280. [DOI] [PubMed] [Google Scholar]
- 15.Zuccarello M, Romano A, Passalacqua M, Rapoport RM. Endothelin 1 induced endothelin 1 release causes cerebral vasospasm in vivo. J Pharm Pharmacol. 1995;47:702. doi: 10.1111/j.2042-7158.1995.tb05864.x. [DOI] [PubMed] [Google Scholar]
- 16.Buckell M. Demonstration of substances capable of contracting smooth muscle in haematoma fluid from certain cases of ruptured cerebral aneurysm. J Neurol Neurosurg Psych. 1964;27:198–199. doi: 10.1136/jnnp.27.3.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Clark JF, Reilly M, Sharp FR. Oxidation of bilirubin produces compounds that cause prolonged vasospasm of rat cerebral vessels: A contributor to subarachnoid hemorrhage induced cerebral vasospasm. J Cereb Bl Fl Metab. 2002;22:472–478. doi: 10.1097/00004647-200204000-00011. [DOI] [PubMed] [Google Scholar]
- 18.Jia-Pei Miao F, Jer-Fu Lee T. Effects of bilirubin on cerebral artery tone in vitro. J Cereb Bl Fl Metab. 1989;9(5):666–674. doi: 10.1038/jcbfm.1989.94. [DOI] [PubMed] [Google Scholar]
- 19.Kranc KR, Pyne GJ, Tao L, Claridge TDW, Harris DA, Cadoux-Hudson TAD, Turnbull JJ, Schofield CJ, Clark JF. Oxidative degradation of bilirubin produces vasoactive compounds. Eur J Biochem. 2000;267:7094–7101. doi: 10.1046/j.1432-1327.2000.01812.x. [DOI] [PubMed] [Google Scholar]
- 20.Watanabe T, Asano T, Shimizu T, Seyama Y, Takakura K. Participation of lipooxygenase products from arachidonic acid in the pathogenesis of cerebral vasospasm. J Neurochem. 1988;50:1145–1150. doi: 10.1111/j.1471-4159.1988.tb10585.x. [DOI] [PubMed] [Google Scholar]
- 21.Zimpfer U, Hofmann C, Dichmann S, Schopf E, Norgauer J. Synthesis, biological effects and pathophysiological implications of the novel arachidonic acid metabolite 5-oxo-eicosatetranoic acid (review) Int J Mol Med. 1998;2(2):149–153. doi: 10.3892/ijmm.2.2.149. [DOI] [PubMed] [Google Scholar]
- 22.Seifert V, Stolke D, Kaever V, Dietz H. Arachidonic acid metabolism following aneurysm rupture. Surg Neurol. 1987;27:243–252. doi: 10.1016/0090-3019(87)90037-1. [DOI] [PubMed] [Google Scholar]
- 23.Ohta S, Nishihara J, Oka Y, Todo H, Kumon Y, Sakaki S. Possible mechanism to induce protein kinase C-dependent arterial smooth muscle contraction after subarachnoid haemorrhage. Acta Neurochir (Wien) 1995;137:217–225. doi: 10.1007/BF02187196. [DOI] [PubMed] [Google Scholar]
- 24.Vieweg U, Schramm J, Urbach H. Platelet derived growth factor (PGDF-alpha-beta)-like immune reactivity in serum and in cerebral spinal fluid following experimental subarachnoid haemorrhage in dogs. Acta Neurochir. 1999;141:861–866. doi: 10.1007/s007010050388. [DOI] [PubMed] [Google Scholar]
- 25.Oshiro EM, Hoffman PA, Dietsch GN, Watts MC, Pardoll DM, Tamargo RJ. Inhibition of experimental vasospasm with anti-intercellular adhesion molecule-1 monoclonal antibody in rats. Stroke. 1997;28(10):2031–7. doi: 10.1161/01.str.28.10.2031. [DOI] [PubMed] [Google Scholar]
- 26.Krebs EG, Beavo JA. Phosphorylation and dephosphorylation of enzymes. Annu Rev Biochem. 1979;48:923–59. doi: 10.1146/annurev.bi.48.070179.004423. [DOI] [PubMed] [Google Scholar]
- 27.Pyne GJ. Biochemistry. University of Oxford; Oxford (UK): 1999. Vascular smooth muscle oxidative metabolism during cerebral vasospasm after subarachnoid hemorrhage; p. 165. [Google Scholar]
- 28.Herlihy JT, Murphy R. Length-tension relationship of smooth muscle of the hog carotid artery. Circ Res. 1973;33:275–283. doi: 10.1161/01.res.33.3.275. [DOI] [PubMed] [Google Scholar]
- 29.Pinna LA, Donella-Deana A. Phosphorylated synthetic peptides as tools for studying protein phosphatases. Biochim Biophys Acta. 1994;1222(3):415–31. doi: 10.1016/0167-4889(94)90050-7. [DOI] [PubMed] [Google Scholar]
- 30.Van Veldhoven PP, Mannaerts GP. Inorganic and organic phosphate measurements in the nanomolar range. Anal Biochem. 1987;161(1):45–8. doi: 10.1016/0003-2697(87)90649-x. [DOI] [PubMed] [Google Scholar]
- 31.Pyne GJ, Cadoux-Hudson TAD, Clark JF. Cerebrospinal fluid from subarachnoid haemorrhage patients causes excessive oxidative metabolism compared to vascular smooth muscle force generation. Acta Neurochir (Wien) 2001;143:59–63. doi: 10.1007/s007010170139. [DOI] [PubMed] [Google Scholar]
- 32.Lyons MA, Shukla R, Zhang K, Pyne GJ, Sing M, Biehle SJ, Clark JF. Increase of metabolic activity and disruption of normal contractile protein distribution by bilirubin oxidation products in vascular smooth-muscle cells. J Neurosurg. 2004;100:505–11. doi: 10.3171/jns.2004.100.3.0505. [DOI] [PubMed] [Google Scholar]
- 33.Ruegg JC. Smooth muscle: PKC-induced Ca2+ sensitization by myosin phosphatase inhibition. J Physiol. 1999;520(1):3. doi: 10.1111/j.1469-7793.1999.t01-1-00003.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Laher I, Bevan JA. Protein kinase C activation selectively augments a stretch induced, calcium dependent tone in vascular smooth muscle. J Pharmacol Exp Ther. 1987;242(2):566–572. [PubMed] [Google Scholar]
- 35.Erdodi F, Rokolya A, Barany M, Barany K. Phosphorylation of the 20,000-da myosin light chain isoforms of arterial smooth muscle by myosin light chain kinase and protein kinase C. J Biochem Biophys. 1988;306(2):583–591. doi: 10.1016/0003-9861(88)90291-3. [DOI] [PubMed] [Google Scholar]
- 36.Haller H, Smallwood JI. Protein kinase C translocation in intact vascular smooth muscle strips. Biochem J. 1990;270:375–381. doi: 10.1042/bj2700375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Castagna M, Takai Y, Kaibuchi K, Sano K, Kikkawa U, Nishizuko Y. Direct activation of calcium-activated, phospholipid dependent protein kinase by tumour promoting phorbol esters. J Biol Chem. 1982;257(13):7847–7851. [PubMed] [Google Scholar]
- 38.Takai A. Okadaic acid. Protein phosphatase inhibition and muscle contractile effects. J Muscle Res Cell Motil. 1988;9(6):563–5. doi: 10.1007/BF01738761. [DOI] [PubMed] [Google Scholar]
- 39.Sellers JR. Unphosphorylated crossbridges and latch: Smooth muscle regulation revisited. J Muscle Res Cell Motil. 1999;20:347–349. doi: 10.1023/a:1005557724418. [DOI] [PubMed] [Google Scholar]
- 40.Murphy RA, Rembold CM, Hai CM. Contraction in smooth muscle. What is latch? Front Smooth Musc Res. 1990;327:39–50. [PubMed] [Google Scholar]
- 41.Varsos V, Liszak T, Han D, Kistler J, Vielma J, Black P, Heros R, Zervas N. Delayed cerebral vasospasm is not reversible by aminophylline, nifedipine, or papaverine in a “Two-hemorrhage” Canine model. Journal of Neurosurgery. 1983;58:11–17. doi: 10.3171/jns.1983.58.1.0011. [DOI] [PubMed] [Google Scholar]
- 42.Sato M, Tani E, Fujikawa H, Kaibuchi K. Involvement of rho-kinase-mediated phosphorylation of myosin light chain in enhancement of cerebral vasospasm. Circ Res. 2000;87(3):195–200. doi: 10.1161/01.res.87.3.195. [DOI] [PubMed] [Google Scholar]
- 43.Butler WE, Peterson JW, Zervas NT, Morgan KG. Intracellular calcium, myosin light chain phosphorylation and contractile force in experimental cerebral vasospasm. Neurosurgery. 1996;38(4):781–788. doi: 10.1227/00006123-199604000-00029. [DOI] [PubMed] [Google Scholar]
- 44.Butler TM, Narayan SR, Mooers SU, Siegman MJ. Rapid turnover of myosin light chain phosphate during cross bridge cycling. Am J Physiol. 1994;267:C1160–1166. doi: 10.1152/ajpcell.1994.267.4.C1160. Cell Physiol 36. [DOI] [PubMed] [Google Scholar]
- 45.Koyama M, Ito M, Feng J, Seko T, Shiraki K, Takase K, Hartshorne DJ, Nakano T. Phosphorylation of CPI-17, an inhibitory phosphoprotein of smooth muscle myosin phosphatase, by Rho-kinase. FEBS Lett. 2000;475:197–200. doi: 10.1016/s0014-5793(00)01654-9. [DOI] [PubMed] [Google Scholar]
- 46.Feng J, Ito M, Ichikawa K, Isaki N, Nishikawa M, Hartshorne DJ, Nakano T. Inhibitory phosphorylation site for rho-associated kinase on smooth muscle myosin phosphatase. J Biol Chem. 1999;274:37385–37390. doi: 10.1074/jbc.274.52.37385. [DOI] [PubMed] [Google Scholar]
- 47.Nobe K, Paul RJ. Distinct pathways of Ca2+ sensitization in porcine coronary artery. Effects of Rho-related kinase and protein kinase C inhibition on force and intracellular ca2+ Circ Res. 2001;88 doi: 10.1161/hh1201.092035. [DOI] [PubMed] [Google Scholar]
- 48.Somlyo AP, Somlyo AV. Signal transduction by G-proteins, Rho-kinase and protein phosphatase to smooth muscle and non-muscle myosin ii. J Physiol. 2000;522(2):177–185. doi: 10.1111/j.1469-7793.2000.t01-2-00177.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pyne GJ, Cadoux-Hudson TAD, Clark JF. Cerebral spinal fluid from sub-arachnoid haemorrhage patients causes excessive oxidative metabolism compared to vascular smooth muscle force generation. Acta Neurochirurgica. 2001;142(12):59–63. doi: 10.1007/s007010170139. [DOI] [PubMed] [Google Scholar]
- 50.Pyne GJ, Cadoux-Hudson TAD, Clark JF. Platelets play an essential role in the aetiology of cerebral vasospasm after subarachnoid haemorrhage. Med Hypoth. 2003;60(4):525–530. doi: 10.1016/s0306-9877(02)00452-8. [DOI] [PubMed] [Google Scholar]
- 51.Clark JF, Pyne-Geithman GJ. Vascular smooth muscle function: The physiology and pathology of vasoconstriction. Pathophysiology. 2005;12:35–45. doi: 10.1016/j.pathophys.2005.02.007. [DOI] [PubMed] [Google Scholar]
- 52.Danthuluri NR, Deth NC. Phorbol ester induced contraction of arterial smooth muscle and inhibition of alpha adrenergic response. Biochem Biophys Res Commun. 1984;125(3):1103–1109. doi: 10.1016/0006-291x(84)91397-4. [DOI] [PubMed] [Google Scholar]
- 53.Forder J, Scriabine A, Rasmussen H. Plasma membrane calcium flux, protein kinase C activation and smooth muscle contraction. J Pharmacol Exp Ther. 1985;235(2):267–73. [PubMed] [Google Scholar]
- 54.Nakamura F, Yamamoto J, Naka M, Tanaka T. Identification of the sites and effects of phosphorylation of smooth muscle calponin by protein kinase C. Jpn J Pharmacol. 1992;58(suppl 2):263. [PubMed] [Google Scholar]
- 55.Nishida W, Abe M, Takahashi K, Hiwada K. Do thin filaments of smooth muscle contain calponin? FEBS Lett. 1990;268:165–168. doi: 10.1016/0014-5793(90)80999-y. [DOI] [PubMed] [Google Scholar]
- 56.Naka M, Kureishi Y, Muroga Y, Takahashi K, Ito M, Tanaka T. Modulation of smooth muscle calponin by protein kinase C and calmodulin. Biochem Biophys Res Commun. 1990;171(3):933–937. doi: 10.1016/0006-291x(90)90773-g. [DOI] [PubMed] [Google Scholar]
- 57.Eto M, Ohmori T, Suziki M, Furuya K, Morita F. A novel protein phosphatase-1 inhibitory protein potentiated by protein kinase C. Isolation from porcine aorta media and characterisation. J Biochem. 1995;118:1104–1107. doi: 10.1093/oxfordjournals.jbchem.a124993. [DOI] [PubMed] [Google Scholar]
- 58.Massett MP, Ungvari Z, Csiszar A, Kaley G, Koller A. Different roles of PKC and MAP kinases in arteriolar constrictions to pressure and agonists. Am J Physiol Heart Circ Physiol. 2002;(283):2282–2287. doi: 10.1152/ajpheart.00544.2002. [DOI] [PubMed] [Google Scholar]
- 59.Zhang J. Role of MAPK in cerebral vasospasm. Drug News Perspect. 2001;14(5):261–7. doi: 10.1358/dnp.2001.14.5.858392. [DOI] [PubMed] [Google Scholar]
- 60.Aoki K, Zubkov AY, Tibbs RE, Zhang JH. Role of MAPK in chronic cerebral vasospasm. Life Sci. 2002;70(16):1901–8. doi: 10.1016/s0024-3205(02)01499-6. [DOI] [PubMed] [Google Scholar]
- 61.Yamboliev IA, Gerthoffer WT. Modulatory role of ERK-MAPK-caldesmon pathway in PDGF-stimulated migration of cultured pulmonary artery smc’s. Am J Physiol, Cell Physiol. 2001;(280):1680–1688. doi: 10.1152/ajpcell.2001.280.6.C1680. [DOI] [PubMed] [Google Scholar]
- 62.Fukata Y, Amano M, Kaibuchi K. Rho-rho kinase pathway in smooth muscle contraction and cytoskeletal reorganization of non-muscle cells. TIPS. 2001;22(1):32–39. doi: 10.1016/s0165-6147(00)01596-0. [DOI] [PubMed] [Google Scholar]
- 63.Ito K, Shimomura E, Iwanage T, Shiraishi M, Shindo K, Nakamura J, Nagumo H, Seto M, Sasaki Y, Takuwa Y. Essential role of Rho kinase in the Ca2+ sensitization of prostaglandin f2α-induced contraction of rabbit aortae. J Physiol. 2003;546(3):823–836. doi: 10.1113/jphysiol.2002.030775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.D’Avella D, Germano A, Santoro G, Costa G, Zuccarello M, Caputi AP, Hayes RL, Tomasello F. Effect of experimental subarachnoid hemorrhage on CSF eicosanoids in the rat. J Neurotrauma. 1990;7(3):121–9. doi: 10.1089/neu.1990.7.121. [DOI] [PubMed] [Google Scholar]
- 65.Pickard JD, Walker V, Vile J, Perry S, Smythe PJ, Hunt R. Oral nimodipine reduces prostaglandin and thromboxane production by arteries chronically exposed to a periarterial haematoma and the antifibrinolytic agent tranexamic acid. J Neurol Neurosurg Psychiatry. 1987;50(6):727–31. doi: 10.1136/jnnp.50.6.727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pickard JD, Walker V, Brandt L, Zygmunt S, Smythe J. Effect of intraventricular haemorrhage and rebleeding following subarachnoid haemorrhage on CSFeicosanoids. Acta Neurochir (Wien) 1994;129(3–4):152–7. doi: 10.1007/BF01406495. [DOI] [PubMed] [Google Scholar]
- 67.Romero SD, Chyatte D, Byer DE, Romero JC, Yaksh TL. Measurement of prostaglandins in the BOXes in CV after SAH: smooth muscle signaling cerebrospinal fluid in cat, dog, and man. J Neurochem. 1984;43(6):1642–9. doi: 10.1111/j.1471-4159.1984.tb06090.x. [DOI] [PubMed] [Google Scholar]
- 68.Zuccarello M, Marsch JT, Schmitt G, Woodward J, Anderson DK. Effect of the 21-aminosteroid U74006f on cerebral vasospasm following subarachnoid hemorrhage. J Neurosurg. 1989;71(1):98–104. doi: 10.3171/jns.1989.71.1.0098. [DOI] [PubMed] [Google Scholar]
- 69.Pyne-Geithman GJ, Caudell DN, Harm AL, Chasse LA, Wagner KR, Clark JF. Bilirubin oxidation products elicit significant elevation of thromboxane A2 in rat brain. Arterioscl Thromb and Vasc Biol. 2007;27(6):e49. [Google Scholar]
- 70.Somlyo AV. New roads leading to Ca2+ sensitization. Circ Res. 2002;91:83–84. doi: 10.1161/01.res.0000028341.63905.91. [DOI] [PubMed] [Google Scholar]