

Abstract
Structural origin of substrate-enzyme recognition remains incompletely understood. In the model enzyme system of serine protease, canonical anti-parallel β-structure substrate-enzyme complex is the predominant hypothesis for the substrate-enzyme interaction at the atomic level. We used factor Xa (fXa), a key serine protease of the coagulation system, as a model enzyme to test the canonical conformation hypothesis. More than 160 fXa-cleavable substrate phage variants were experimentally selected from three designed substrate phage display libraries. These substrate phage variants were sequenced and their specificities to the model enzyme were quantified with quantitative enzyme-linked immunosorbent assay for substrate phage-enzyme reaction kinetics. At least three substrate-enzyme recognition modes emerged from the experimental data as necessary to account for the sequence-dependent specificity of the model enzyme. Computational molecular models were constructed, with both energetics and pharmacophore criteria, for the substrate-enzyme complexes of several of the representative substrate peptide sequences. In contrast to the canonical conformation hypothesis, the binding modes of the substrates to the model enzyme varied according to the substrate peptide sequence, indicating that an ensemble of binding modes underlay the observed specificity of the model serine protease.
Serine proteases recognize peptide sequences as substrates with a spectrum of specificity (1), for which the structural origin remains largely unclear (2, 3). The enzyme family is one of the most important model families in enzymology as about one-third of proteases (coded in about 2% of human genome) can be classified as serine proteases and many of the serine proteases are involved in important physiological regulation and pathogenic dysfunction (1). Serine proteases hydrolyze peptide bond with nucleophilic Ser195 in the catalytic triad (Asp102–His57–Ser195 in chymotrypsin number) (see Ref. 2, and references therein). The polypeptide binding sites and the binding pockets for the side chains of the peptide substrates in various serine proteases have also been characterized to atomic resolution with x-ray crystallography (see Ref. 4, and references therein). Still, the enzyme-substrate transition state complex structures have been difficult to be elucidated (5), partly due to the rapid hydrolysis of the substrate and partly due to the rapid dissociation of the products from the active site. Hence the structural origin on the substrate specificity of serine proteases has been incompletely resolved, hampering critical understanding toward enzyme-substrate recognition and its implications in inhibitor design and substrate specificity engineering.
Models of enzyme-substrate transition state complexes have been proposed on the basis of the three-dimensional structures of serine protease-inhibitor complexes. Invariably in the models and thus dubbed canonical conformation, the peptide substrate binds to the enzyme in β-strand conformation, anti-parallel to the corresponding β-strand (residues 214–217 in chymotrypsin number) in the serine protease (see Refs. 2–6 for reviews). However, accumulating evidence supporting alternative models suggests that the canonical conformation is not universally applicable to serine protease-substrate complexes (5–8). Using factor Xa as a model system, we asked if the canonical conformation hypothesis can sufficiently account for the enzymatic specificity of the model system and if not, what alternative models are plausible from structural and energetic aspects.
Factor Xa (fXa)3 is situated at the key position of the common pathways of the intrinsic and extrinsic coagulation systems (9), and thus has been an attractive target for antithrombotic drug development. In vivo, factor Xa and factor Va form prothrombinase complexes on the phospholipid membrane surface (10). The membrane-bound prothrombinase recognizes and binds its protein substrate prothrombin through the exosites on the prothrombinase surface (11). Conformational changes of the prothrombinase-prothrombin assembly subsequently enable the active site of fXa to sequentially recognize and cleave the two cleavage sites in the prothrombin to yield activated thrombin as product (12, 13). This two-step substrate recognition mechanism explains the precise specificity and affinity of fXa on its protein substrate (14, 15).
Although both the exosites and the active site in fXa contribute to the protein substrate specificity in vivo (14, 15), the active site of fXa is intrinsically permissive (16). Molecular origins of the active site permissiveness have not been clearly understood. The active site of fXa recognizes substrate sequences (P4–P3–P2–P1): IEGR and IDGR in prothrombin; PQGR in factor VII; and EKGR in the autolysis loop of fXa (8, 17). Phage display experiments identify further that fXa can recognize RGR in the P3–P2–P1 positions of the phage-displayed substrates (18). Positional scanning synthetic combinatorial fluorogenic peptide substrate libraries have identified that fXa can recognize aromatic residues in the P2 position (17) but glycine as the P2 residue still dominates the recognition specificity. In contrast to earlier experimental results, peptide substrates synthesized to replace one residue at a time in the template peptide (VQFRSL from P4 to P2′) show that glycine comes after phenylalanine as the second most favorable residue in the P2 position (19). Little is known as to how the substrate side chains with diverse physicochemical properties can fit into the same peptide binding site as defined by the canonical conformation of the substrate peptide.
To investigate active site-substrate recognition, we re-examined substrate specificity of fXa with substrate phage display selection (18, 20–22) and quantitative ELISA for individual substrate phage-enzyme reaction kinetics (22, 23). In addition, we constructed several representative enzyme-substrate complex structures in atomic details that were consistent simultaneously with the empirical binding energetics and the pharmacophore models derived from 55 high resolution fXa-ligand complex structures in the Protein Data Bank. Together, the substrate phage display experiments and the computational molecular models indicated that an ensemble of substrate-enzyme active site binding modes was necessary to account for the substrate specificity of the active site of fXa. This view is drastically in contrast to the canonical conformation hypothesis for the serine protease active site-substrate recognition.
EXPERIMENTAL PROCEDURES
Library Construction—pCANTAB5E (GE Healthcare) template was constructed as shown in Fig. 1. The DNA libraries coding for substrate peptides were inserted at the site where the TAA stop codons were previously constructed in the template. The TAA stop codons were designed to ensure that only the phagemids carrying inserted degenerate codons would produce pIII fusion protein for phage surface display. The phage display libraries were constructed with the oligonucleotide-directed mutagenesis initially proposed by Kunkel (24). In this work, we followed the Sidhu and Weiss protocol (25). In brief, CJ236 (dut-, ung-, camR) (from New England Biolabs) was transformed with the pCANTAB5E template and a single ampicillin-resistant colony was cultured and recombinant phage was rescued by M13KO7 helper phage (GE Healthcare). Phage particles in the overnight 2xYT supernatant in the presence of ampicillin, kanamycin, and uridine were harvested and precipitated with PEG/NaCl (20% PEG 8000 and 2.5 m NaCl). The dU-single stranded DNA from the phage particles was extracted with the QIAprep spin M13 kit (Qiagen), following the manufacturer's instruction. 35–45 μg of dU-single stranded DNA template was extracted in a typical preparation from 35 ml of culture supernatant.
FIGURE 1.

Recombinant phage variant and library constructions on the basis of pCANTAB5E phagemid.
DNA primer fragments encoding for pC-4X, pC-XXGRXX, and pC-XRXRXX libraries were synthesized by IDT (Integrated DNA Technologies). The sequences for the primers were the following: 4X, VCCTGCGGCCGATGCTCCACCTGAMNNMNNMNNMNNGCCGGATCCACTAGAGCCGCC; XXGRXX, VCCTGCGGCCGATGCTCCACCTGAMNNMNNGCGGCCMNNMNNGCCGGATCCACTAGAGCCG; and XRXRXX, VCCTGCGGCCGATGCTCCACCTGAMNNMNNGCGMNNGCGMNNGCCGGATCCACTAGAGCCG, where M = A, C, 50% each; N = A, G, T, C, 25% each. The 5′ end of the primer was phosphorylated with T4 polynucleotide kinase (New England Biolabs) and annealed with the dU-single stranded DNA template before overnight extension and ligation with T7 DNA polymerase (New England Biolabs) and T4 DNA ligase (New England Biolabs). The CCC-double stranded DNA end product was purified with the Qiagen gel extraction kit and electroporated into Escherichia coli ER2738. Typically, 1 μg of dU-single stranded DNA produced 107–108 recombinant phage variants, and 60–80% of the phage variants carried degenerate peptide sequences at the insertion site of the phagemid. The rescued recombinant phage particles were precipitated with 20% PEG/NaCl and resuspended in pure water for the following selection/amplification process.
Substrate Phage Display Selection—We first carried out a pre-selection step with Ni-NTA beads: 1000 μl of phage library (∼1012 colony forming units/ml) in phosphate binding buffer (50 mm NaH2PO4, pH 8, 300 mm NaCl) was immobilized on 100 μl of Ni-NTA-agarose beads (Qiagen) at room temperature for 2 h under constant gentle rocking. The Ni-NTA beads were transferred to a microfilter (Millipore Ultra-free MC, UFC30SV00) and washed 21 times with agarose beads, each with 500 μl of washing buffer (50 mm NaH2PO4, pH 8, 300 mm NaCl, 10 mm imidazole). The phage particles were then eluted from the Ni-NTA beads with elution buffer (50 mm NaH2PO4, pH 8, 300 mm NaCl, 200 mm imidazole). The eluted phage particles were amplified overnight and harvested with PEG/NaCl precipitation. This step was designed to eliminate the phage population carrying the template phagemid.
The Ni-NTA pre-selected phage population was used as input for the fXa selection/amplification cycle as depicted in Fig. 2. Immobilization and wash procedures were carried out as described in the previous paragraph. After the wash procedure, the Ni-NTA beads were rinsed one time with 500 μl of fXa reaction buffer (20 mm Tris-HCl, pH 6.8, 50 mm NaCl, 1 mm CaCl2). 5 units (∼10 μg, purity >90%) of purified bovine fXa (Qiagen) in 1000 μl of fXa reaction buffer were added to the Ni-NTA beads. The optimum pH for bovine fXa activity is 6.5 according to the manufacturer; we modified the reaction buffer to pH 6.8 so as to avoid compromising the His6-Ni-NTA binding while maintaining the optimum activity of fXa. The reaction took place at room temperature for 1.5 h under constant gentle rocking. After the enzymatic reaction, the Ni-NTA beads were washed with 9 × 500 μl of washing buffer and then with 9 × 500 μl of elution buffer (50 mm NaH2PO4, pH 8, 300 mm NaCl, 200 mm imidazole). The washing, reaction, and elution buffers saved from each step of the above process were titered for phage concentration. The histogram in Fig. 3 shows the phage concentration in each of the buffers from the selection cycles.
FIGURE 2.

Flow chart for the substrate phage display selection/amplification cycles. [Im] is the concentration of imidazole in the buffer. W1–21 are 21 times of wash with washing buffer; R and R + fXa are fXa reaction buffer in the absence and presence of fXa, respectively; RW1–9 are 9 times of wash with washing buffer; E1–9 are 9 times of elution with elution buffer.
FIGURE 3.

Titers of phage particles for the washing, reaction, and elution buffers from four selection/amplification cycles with pC-4X library. The collection procedure for the buffers shown in the x axis is depicted in the flow chart of Fig. 2. The x axis label such as W4–6 indicates that the fourth to the sixth washing buffers were pooled together for titer, and the histogram shows the titer in colony forming units/ml.
The phage particles enzymatically cleaved from the Ni-NTA beads were amplified overnight in the host E. coli ER2738, and the rescued phages in the overnight culture were precipitated with 20% PEG/NaCl. The precipitated phages were used as input for the following cycle of selection/amplification. At the end of the last cycle, single colonies of the selected phage particles were cultured for further analysis.
Single Colony Analyses—Single colonies were picked using GENETIX Qpix II colony picker to 96-well deep well culture plate. Each well contained 1 ml of 2xYT, 100 μg of ampicillin, and 5 × 108 colony forming units of helper phage M13KO7. One hour after inoculation, 200 μl of 2xYT, 250 μg/ml kanamycin, 1200 μg/ml ampicillin were added to each well. The culture plates were incubated at 37 °C overnight with vigorously shaking. After being centrifuged at 3000 × g at 4°C for 30 min, 20 μl of supernatant was transferred from each well of the culture plate to the corresponding well in a 96-well His-sorb (Qiagen) plate, for which each well contained 80 μl of fXa reaction buffer and 0.2 units of fXa. The same transfer procedure was repeated for a control 96-well His-sorb plate, where buffer without fXa was added to each well. After 1 h of reaction at room temperature, the His-sorb plates were washed with 3 × 250 μl of PBST (phosphate-buffered saline, pH 7.5, Tween 20, 0.05%) and 1 × 250 μl of PBS. Anti-M13-horseradish peroxidase (1:5000, GE-Amersham Biosciences) in PBS + skim milk (2.5%) was added to each well for 1 h. The anti-M13-horseradish peroxidase was cleared from the His-sorb plates with 3 × 250 μl of PBST and 1 × 250 μl of PBS washing buffer. 100 μl of ABTS substrate (BioFix) was added to each well for 10 min before adding 1% SDS to terminate the chromogenic reaction. The OD of each well was measured at 405 nm. Colonies with control OD405 nm greater than 0.9 were selected for sequencing. Enzymatic kinetics measurement with quantitative ELISA was carried out for the selected variants.
Quantitative ELISA to Measure Enzymatic Kinetics—Enzymatic kinetics measurement was initiated by mixing 20 μl of supernatant phage solution (1010–1011 colony forming units/ml) with 80 μl of fXa solution (0.2 units in reaction buffer) in a well of a low absorbent plate. The reaction was terminated at 0, 10, 20, 30, 40, and 50 min, respectively, by adding 50 μl of 1.5 m guanidine HCl (pH 8.0 in reaction buffer). 100 μl of each terminated reaction mixture was transferred to a His-sorb plate for 1 h binding at room temperature with agitation. The ELISA was developed as described in the previous section. The quantitative measurement of the phage particles capable of binding to the His-sorb surface was calibrated with the standard curve derived from the same plate, where standard pC-Xa phage solution was diluted linearly in a series of 6 wells for a standard curve of OD405 nm versus known phage concentration. The linear range of the standard curves was found between OD405 nm = 0.1 and 2. Hence the supernatant phage concentration was adjusted such that the OD405 nm for the 0-min enzymatic reaction was in the 0.4–1.8 range, which ensured that the substrate phage concentration remained in the linear range of the standard curves throughout the quantitative ELISA measurements. As such, we eliminated the factors that could affect the consistency of the quantitative ELISA through the variance of the His-sorb plate performance and the concentration of the substrate phages outside the linear range of the standard curve.
The concentration of the substrate phage in the reaction mixture was on the order of ∼10-13 m, which was much lower than the lower limit of Km of fXa, 10-6 m as estimated by Bianchini et al. (19). For [S] << Km, the reaction rate is governed by the approximated Michealis-Menten equation,
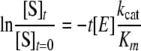 |
(Eq. 1) |
where the hydrolysis kinetics is a pseudo-first order reaction with the reaction constant linearly proportional to kcat/Km, provided that the enzyme concentration [E] is constant. Moreover, the pseudo-first order reaction constant is not affected by the concentration of the substrate when [S] << Km. Based on the Michealis-Menten mechanism, we plotted ln{[S]t/[S]t = 0} versus time (in minute) to derive the slope -kobs.
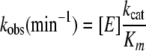 |
(Eq. 2) |
The concentration of the substrate phage [S] in the reaction mixture that was not cleaved by fXa was measured with the quantitative ELISA described above.
Cleavage Site Determination with Mass Spectrometry—Peptides were synthesized with a automated solid phase Fmoc (N-(9-fluorenyl)methoxycarbonyl) peptide synthesizer. Enzymatic reaction was carried out in a 10-μl reaction buffer mixture containing 50 μg of peptide and 4 units of fXa at room temperature for 1 h. The reaction mixtures before and after enzymatic reaction were analyzed with high pressure liquid chromatography to monitor the reaction completeness. The reaction product was analyzed with mass spectrometry to determine the cleavage site in the peptide sequences. The reaction mixture was diluted 30 times in 50% acetonitrile, 0.1% trifluoroacetate, and CHCA (α-cyano-4-hydroxycinnamic acid), 5 mg/ml, before spotting on the MALDI-TOF sample plate for mass spectrometry (Bruker Ultraflex II MALDI-TOF).
Molecular Modeling of Transition State Complex Structure—Bovine fXa structure has been determined with x-ray crystallography (Protein Data Bank code 1KIG) (26). Human fXa is highly homologous to bovine fXa (sequence identity 83%, positive 91%). The constitutive residues of the substrate recognition sites (S2′–S4) are 100% identical between the two proteins. Superimposing the 54 human fXa structures and the bovine fXa structure in the PDB revealed that the structures of human and bovine fXa are almost identical around the active site except that the side chain structures of Gln192, Glu97, Tyr99, and Phe174 vary among human fXa structures, depending on the ligand binding to the substrate recognition sites (see Fig. 5a). During the following docking procedure, the side chain structures of these four residues remained flexible, modeled with the built-in rotamer libraries of the GOLD 3.2 package (27, 28). The partial charges of the atoms in the enzyme were assigned based on the Kollmann all-atom charge set (29) in the SYBYL 7.3 package.
FIGURE 5.

Computational molecular modeling of fXa-substrate transition state complex structures. Pharmacophore models in a were derived from known enzyme-ligand structures. The catalytic triad is colored in red. The S1 and S4 substrate recognition sites are colored in green and magenta, respectively. Pharmacophore centers are labeled D, H-bond donor; A, H-bond acceptor; P, positive ionizable; H, hydrophobic features. The pharmacophore features in the aryl-binding site are D4, P4, H4-1, and H4-2. The pharmacophore features in the S1 site are H1, P1, and D1. A3 and D3 are the H-bonding acceptor and donor pairs for Gly216 backbone. D0, A0, and H0 are features corresponding to the binding pocket above the S1 site. Superimposition of the fXa structures are shown in stick models below the molecular surface in this panel. Only the relevant residues near the active sites are shown. The best 5 complex structure models for each of the substrate peptide sequences: (b) GPQGR (kobs × 1000 = 70.2 (min-1)), (c) GIEGR (kobs × 1000 = 30.8 (min-1)), (d) GYRGR (kobs × 1000 = 25.7 (min-1)), (e) GPSGR (kobs × 1000 = 20.5(min-1)), (f) GLLQR (kobs × 1000 = 13.6 (min-1)), (g) GAQFR (kobs × 1000 = 8.7 (min-1)), and (h) GPRAR (kobs × 1000 = 8.1 (min-1)) are shown in panels b–h. The corresponding kobs are derived from Fig. 4. In each of the panels, the ranks of the models are color coded: the best model is in pure white for the carbon atoms in the substrate peptide, the second to fifth ranked substrate models are shown with increasing grayness for the carbon atoms. The protein structure displayed in each of the panels is the protein model from the top ranked complex structure.
Initial substrate peptide structures were built with SYBYL 7.3 package (Tripos Associates, St. Louis, MO), with partial charges assignment also based on the Kollmann all-atom charge set (29). N and C terminus were blocked with the methyl group. The transition state complex was modeled by constraining the substrate to the enzyme active site with covalent bonding between the P1 carbonyl carbon and Ser195 OG and with hydrogen bonding between side chains of Arg P1 and Asp189. GOLD 3.2 (27, 28) was used for the substrate-enzyme docking. For effective usage of computational resources, the docking calculations were confined in an active site-centered 15-Å radius sphere, which enclosed all possible substrate structures under the applied constraints. For each substrate docking, a maximum of 2000 independent genetic algorithm runs were set with early termination conditions when 10 top ranked conformations (scored with GOLDSCORE (27, 28) in GOLD 3.2) output from each of the genetic algorithm runs first appeared within 1.5 Å root mean square deviation. All sp3 torsion angles in the substrates were allowed to rotate freely during docking. All default parameters were used for the GOLD 3.2 genetic algorithm, except that the number of operations was doubled (200%) for more exhaustive search of the conformational space. The final 100 top GOLDSCORE-ranked substrate-protein complexes for each substrate were selected for the pharmacophore evaluation process below. The docking processes were carried out in a 40-CPU (Intel Xeon CPU 3.00 GHz) Linux cluster. A single substrate docking took on the order of 10 central processor unit days (CPU days).
Fifty-five complex-based pharmacophore models were generated automatically from the 55 fXa-ligand complex structures currently deposited in PDB (1FAX, 1KIG, 1XKA, 1EZQ, 1F0R, 1F0S, 1FJS, 1G2L, 1G2M, 1KSN, 1IOE, 1IQE, 1IQF, 1IQG, 1IQH, 1IQI, 1IQJ, 1IQK, 1IQL, 1IQM, 1IQN, 1KYE, 1LPG, 1LPK, 1LPZ, 1LQD, 1MQ5, 1MQ6, 1NFU, 1NFW, 1NFX, 1NFY, 1V3X, 1WU1, 2BOK, 2BQ6, 1Z6E, 2BMG, 2BOH, 2BQ7, 2BQW, 2CJI, 2D1J, 2FZZ, 2G00, 2J2U, 2J34, 2J38, 2J4I, 2H9E, 2J94, 2J95, 2UWL, 2UWO, and 2UWP) with the LigandScout program (30). These pharmacophore models were read into the Catalyst (31) and the UPGMA programs (Unweighted Pair Group Method with Arithmetic mean) in Discovery Studio 1.7 (Accelrys) for further visual and clustering analyses to remove redundant pharmacophore features. The resulting non-redundant pharmacophore after clustering analysis was composed of two hydrogen bond acceptors, four hydrogen bond donors, two positive ionizables, and four hydrophobic features (see Fig. 5a). This non-redundant pharmacophore model was used to evaluate the 100 top GOLDSCORE-ranked complexes derived from the docking process above with the empirical Fit score shown below (31),
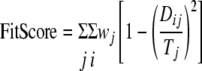 |
(Eq. 3) |
Dij is the distance between the center of the pharmacophore feature i in the ligand and the center of the pharmacophore feature j in the complex-based pharmacophore model as described above. The pharmacophore features in the ligand were generated automatically with the Catalyst program. Only the matching pairs of features within the tolerance distance Tj contributed to the sum of the empirical score. The tolerance radius Tj was set to 5 Å independent to the feature type j, and the weight wj was set to 1 in this work. Equation 3 provided a semi-quantitative evaluation for the consistency of the docked ligand structures with the experimentally derived complex-based pharmacophore model. Because the computational models were constructed with uncertainties in conformational energy calculation, pharmacophore scoring function, and conformational sampling limit, the resulting models can only be regarded as qualitatively adequate in providing molecular level information that could not be arrived at otherwise.
RESULTS
Substrate Phage Display—To establish the substrate phage display system, we first constructed a positive control recombinant phage pC-Xa (see Fig. 1) with a 7-residue flexible linker between the His6 tag and the fXa cutting site. The flexible linker extended the substrate sequence (IEGR) away from the Ni-NTA surface for fXa accessibility. We established this design by carrying out enzymatic reaction on the surface of His-sorb plate pre-adsorbed with the pC-Xa phages. We also found that the titer before and after adding fXa to a control recombinant phage did not change within experimental error, indicating that fXa did not affect the infectivity of the phage particles.
The construction of the pC-4X library generated 1.4 × 109 independent variants. Randomly selecting 24 single colonies for sequencing, we found that 15 colonies contained phagemids with correct insertion and the rest contained template phagemid. Due to the stop codons on the template phagemid, the host cells carrying the template phagemid were not effective in producing any phage particles with displayed protein. Thus, the complexity for the pC-4X phage-displayed library was estimated to be 8.7 × 108 (1.4 × 109 × 15/24). With the same procedure, the complexity for the pC-XXGRXX and pC-XRXRXX libraries were determined to be 6.4 × 108 and 1.6 × 108, respectively. The degenerate codons were analyzed and showed that at the position of N (AGTC with 25% each) the composition of each nucleotide varied from 10 to 40% and at the position of M (AC with 50% each) the composition of the two nucleotides varied from 30 to 70%, no serious bias of the degenerate codon distribution was obvious for the three phage libraries. Thus we constructed phage display libraries with more than 108 phagemid variants, which were more than enough to cover 1.6 × 105 (204) possible peptide sequences.
pC-4X Library—The pC-4X library (see Fig. 1) was applied to the selection procedure depicted in Fig. 2 for 4 cycles of selection/amplification. Fig. 3 shows the phage titers in the washing buffers, reaction buffers, and elution buffers saved from each cycle. The wash of the Ni-NTA beads after the enzymatic reaction released the enzymatically cleaved phage particles (RW1–3 of cycle 1 in Fig. 3). Still, a large portion of the phage particles remained intact bound to the washed Ni-NTA beads (see E1–3 of cycle 1 in Fig. 3). As the cycles progressed, the fraction of the cleavable phage particles increased. In the last cycle, more than half of the phage population on the Ni-NTA beads was able to be released because of the fXa reaction. The progressive increase of the portion of the cleavable phage particles indicated that the selection/amplification cycle was effective in enriching the phage particles displaying substrate peptides for fXa.
We had a concern that some library members with consecutive His in the substrate sequences might be selected by Ni-NTA beads in the above selection cycles, even though the more competitive His6 tag remained the predominant binding moiety to the Ni-NTA-coated surface. This possibility was investigated by sequencing random phage variants in the elution buffers (E1–3, see Fig. 2). No overpopulated His-biased sequences were found, suggesting that our substrate phage display system did not bias the distribution of His in fXa-cleavable sequences.
After the last selection/amplification cycle with the pC-4X library, 96 single colonies of the host E. coli cells infected with the selected phage particles were picked for single colony analysis. Of the 96 single colonies, 31 phage variants satisfied the criteria (see “Experimental Procedures”) for enzymatic kinetics measurement with quantitative ELISA. The IEGR phage variant was not among the 31 selected phage variants but the enzymatic kinetics measurement was carried out for comparison in Fig. 4, which shows the plots of ln{[S]t/[S]t = 0} against time for the variants. The kobs and the substrate sequence displayed on the phage variants are also summarized in the figure. The linearity of the plots in Fig. 4, is in agreement with the approximated Michealis-Menten equation shown in Equations 1 and 2 within experimental error.
FIGURE 4.

Enzymatic reaction kinetics with quantitative ELISA. 32 phage displayed substrate variants are shown in plots of ln{[S]t/[S]t = 0} versus time in minutes. The slopes of the linear plots are shown as kobs. The linearity of the plots are characterized with two indexes: R2 is the coefficient of determination; C is the correlation coefficient.
Two types of sequence patterns clearly emerged from the analysis of the active substrate sequences shown in Fig. 4: the most apparent pattern was XXGR, followed by X(R/Q)XR. Both sequence motifs accounted for 20 of 29 observed active substrate sequences with kobs > 0 (see Fig. 4). Table 1 shows the position of the hydrolyzed peptide bond for a set of representative substrate sequences found in Fig. 4. The cleavage sites shown in Table 1 indicated that G and R in the XXGR sequence motif were the P2 and P1 residues, respectively; the R/Q in the X(R/Q)XR sequence motif was the P3 residue and the C-terminal R was the P1 residue. Following the model of fXa-dansyl-Glu-Gly-Arg methylene inhibitor proposed by Padmanabhan et al. (32), we hypothesized that the XXGR substrates were recognized by fXa through the canonical conformation (32). As such, the P3 side chain was expected to protrude from the protein surface into the solvent and the specificity of the P3 side chain was not particularly confined by the few surrounding residues (Gln192, Glu217, Glu146, Arg222, Glu97, Ser195, and His57 within 10 Å from the Cα of the P3 residue). In contrast, alternative hypothesis for the binding modes of the X(R/Q)XR substrate sequences was more consistent with the specificity of the P3 side chain.
TABLE 1.
The scissile site in the representative synthesized substrate peptides as determined by MASS The down arrow indicates the location of the scissile peptide bond.
| Substrate peptide sequence |
|---|
| SGSGIEGR ↓ SGGASA |
| SGSGPRAR ↓ SGGASA |
| SGSGYRGR ↓ SGGASA |
| SGSGLQGR ↓ SGGASA |
| SGSGLRWR ↓ SGGASA |
pC-XXGRXX Library—To further investigate the hypothesis for the substrate binding mode of the XXGR sequences, we constructed the pC-XXGRXX library to select for fXa substrate sequences, using the procedure that had been applied on the pC-4X library as shown above. The library was designed to focus on the side chain requirements of the P3 residues, so as to test the hypothesis that the XXGR substrate sequences were recognized by fXa through the canonical conformation. Three cycles of selection/amplification as depicted in Fig. 2 were carried out and the kinetics measurements for selected phage variants are listed in Table 2. Of the total 75 (statistics with more than 50 sequences was considered to be adequate (33)) substrate sequences with kobs > 0, all but three amino acid types (Ala, His, and Thr) appeared at the P3 position, indicating that the P3 side chain requirements were quite tolerant in the XXGR substrate motif. This was in agreement with the implication derived from the canonical conformation hypothesis for the substrate binding mode. Further examination of the distribution of the P3 residues revealed that Arg and Gln were significantly overpopulated (24 and 18 for Arg and Gln, respectively of a total of 75 substrate sequences). This Arg/Gln-enriched distribution was again in agreement with the previous observation in the pC-4X library experiment as shown in Fig. 4, and warranted an alternative hypothesis to account for the strong preference of the Arg and Gln residues at the P3 position.
TABLE 2.
Enzymatic kinetics results for selected variants in the pC-XXGRXX library kobs is in the unit of 1/min. R2 and C are defined in the legend to Fig. 4.
| Sequence | kobs × 1000 | R2 | C |
|---|---|---|---|
| WRGRTA | 44.859 | 0.993 | 0.996 |
| LDGRHP | 40.849 | 0.965 | 0.982 |
| QLGRTT | 35.245 | 0.959 | 0.979 |
| PRGRVF | 32.286 | 0.996 | 0.998 |
| SRGRAW | 29.449 | 0.805 | 0.897 |
| QMGRSW | 28.681 | 0.790 | 0.888 |
| FRGRTY | 27.925 | 0.903 | 0.950 |
| LRGRTY | 26.365 | 0.962 | 0.981 |
| GGGRSG | 22.741 | 0.966 | 0.983 |
| lRGRVG | 22.309 | 0.942 | 0.970 |
| MQGRSS | 22.154 | 0.748 | 0.864 |
| LRGRVY | 21.742 | 0.946 | 0.972 |
| LRGRAN | 19.780 | 0.745 | 0.863 |
| LSGRSY | 19.750 | 0.892 | 0.944 |
| ARGRVA | 18.628 | 0.977 | 0.988 |
| YRGRLY | 18.620 | 0.954 | 0.976 |
| YRGRFE | 18.152 | 0.922 | 0.960 |
| CRGRDA | 17.796 | 0.864 | 0.929 |
| LRGRML | 17.119 | 0.972 | 0.985 |
| IQGRMQ | 16.718 | 0.697 | 0.834 |
| PPGRSG | 16.650 | 0.916 | 0.957 |
| YRGRMI | 16.391 | 0.965 | 0.982 |
| VRGRSE | 16.261 | 0.842 | 0.917 |
| TLGRRP | 15.819 | 0.959 | 0.979 |
| LRGRML | 15.741 | 0.985 | 0.992 |
| LQGRMY | 15.645 | 0.953 | 0.976 |
| WQGRNL | 15.107 | 0.705 | 0.839 |
| IQGRLF | 14.332 | 0.919 | 0.958 |
| VSGRMQ | 13.913 | 0.894 | 0.945 |
| QFGRSS | 13.610 | 0.976 | 0.987 |
| SRGRDI | 13.221 | 0.868 | 0.931 |
| PRGRIG | 13.047 | 0.939 | 0.968 |
| SRGRID | 12.469 | 0.770 | 0.877 |
| YWGRFQ | 12.384 | 0.852 | 0.923 |
| QKGRSN | 12.309 | 0.874 | 0.934 |
| SYGRIQ | 12.259 | 0.622 | 0.788 |
| MRGRVN | 11.914 | 0.958 | 0.978 |
| MQGRTL | 11.672 | 0.987 | 0.993 |
| QQGRVL | 11.076 | 0.865 | 0.930 |
| LLGRQY | 10.808 | 0.594 | 0.770 |
| QQGRSE | 10.086 | 0.940 | 0.969 |
| PLGRTQ | 9.914 | 0.926 | 0.962 |
| MQGRAI | 8.689 | 0.795 | 0.891 |
| TLGRFQ | 8.526 | 0.776 | 0.880 |
| IFGRSQ | 8.518 | 0.661 | 0.812 |
| SYGRQQ | 8.469 | 0.981 | 0.990 |
| GQGRWS | 8.259 | 0.656 | 0.809 |
| FRGRLY | 7.917 | 0.852 | 0.922 |
| SQGRSF | 7.883 | 0.611 | 0.781 |
| LEGRQL | 7.833 | 0.647 | 0.804 |
| MQGRNS | 7.410 | 0.952 | 0.975 |
| SQGRTS | 7.263 | 0.836 | 0.914 |
| AQGRSS | 7.211 | 0.620 | 0.787 |
| LVGRSQ | 7.114 | 0.930 | 0.964 |
| YQGRNA | 6.910 | 0.870 | 0.932 |
| YFGRTQ | 6.723 | 0.848 | 0.921 |
| QLGRGA | 6.718 | 0.919 | 0.958 |
| QLGRTT | 6.346 | 0.954 | 0.976 |
| MQGRNS | 6.321 | 0.842 | 0.917 |
| TRGRFL | 6.290 | 0.943 | 0.971 |
| LIGRMQ | 6.251 | 0.519 | 0.720 |
| CRGRYF | 5.792 | 0.746 | 0.863 |
| NLGRQS | 5.419 | 0.758 | 0.870 |
| SNGRSS | 5.361 | 0.745 | 0.863 |
| QSGRFH | 5.020 | 0.141 | 0.375 |
| QYGRYN | 4.592 | 0.839 | 0.916 |
| NVGRVQ | 3.797 | 0.113 | 0.335 |
| LCGRQL | 3.721 | 0.414 | 0.643 |
| LQGRIS | 3.514 | 0.702 | 0.837 |
| TQGRSS | 2.996 | 0.638 | 0.798 |
| DLGRWD | 2.919 | 0.115 | 0.338 |
| SFGRAQ | 2.217 | 0.200 | 0.446 |
| SVGRAQ | 1.422 | 0.664 | 0.815 |
| VRGRFW | 0.824 | 0.644 | 0.802 |
| QNGRLF | 0.817 | 0.149 | 0.386 |
The alternative hypothesis required that the P3 side chain was enclosed in a binding pocket such that the lining residues of the binding pocket interacted strongly with the side chain of the P3 residue, explaining the distinct specificity imposed on the P3 residue of the substrate. The only binding pocket available for the P3 side chain near the substrate binding site of fXa is the aryl-binding site defined by Tyr99, Phe174, and Trp215 (see Fig. 5). A large body of experimental evidence (8, 34–37) has indicated that the fXa aryl-binding site is particularly favorable for binding the Arg side chain. The binding involves three distinct energetic terms: 1) cation-π interactions between the aromatic rings and the positive charge on Arg (38, 39); 2) hydrophobic interactions due to the aliphatic part of the Arg side chain; 3) the electrostatic interaction between Arg and Glu97 adjacent to the aryl-binding site. Thus, the hypothesis for the binding mode of the XRXR substrate sequence was that the P3 Arg side chain inserted into the aryl-binding pocket and the P1 Arg remained in the conventional S1 binding site forming a salt bridge with Asp189. This substrate binding model is substantially deviated from the canonical conformation.
pC-XRXRXX Library—To test the hypothesis, we constructed yet another phage library pC-XRXRXX for focused investigation on the side chain requirements of the P2 residue. The rationale was that, as in the proposed binding model in the previous paragraph, the P2 residue had no place to go but to be fitted in the S2 site. In fXa, the S2 site is next to His57 and Tyr99, and unlike many other trypsin-like serine proteases, the space of the S2 site in fXa is limited due to the blockage of the side chain of Tyr99. Thus, we expected that the P2 residue needed to be specific for small amino acids. Table 3 shows the substrate sequences with kobs > 0, selected with the same procedure as described for the derivation of Table 2. As expected, three smallest amino acids: Gly, Ala, and Ser, were the most prevalent P2 residues (20 for Gly, 7 for Ala, and 9 for Ser of total 57 substrate sequences). However, contradicting to the hypothesis above, the P2 residues had a secondary preference for bulky aromatic residues (3 for Trp, 2 for Phe, and 1 for Tyr) and the non-β-branched aliphatic side chain (4 for Leu). The result was in qualitative agreement with that found by Bianchini et al. (19), who have demonstrated that the P2 position for fXa substrate sequences also preferred Phe. It was difficult to reconcile as to why the S2 site simultaneously selected for a group of the smallest amino acid types and a group of the bulkiest amino acid types, except that there might need a third type of binding mode to account for the bulky hydrophobic side chains at the P2 position.
TABLE 3.
Enzymatic kinetics results for selected variants in the pC-XRXRXX library kobs is in the unit of 1/min. R2 and C are defined in the legend to Fig. 4.
| Sequence | kobs × 1000 | R2 | C |
|---|---|---|---|
| DRGRSG | 35.747 | 0.951 | 0.975 |
| SRARNT | 27.385 | 0.824 | 0.907 |
| PRGRDY | 23.696 | 0.922 | 0.960 |
| SRGRVS | 23.420 | 0.976 | 0.988 |
| LRGRIW | 23.211 | 0.961 | 0.980 |
| VRGRDY | 20.860 | 0.996 | 0.997 |
| HRHRLY | 20.354 | 0.974 | 0.986 |
| TRGRTL | 19.477 | 0.969 | 0.984 |
| ARARVQ | 18.234 | 0.865 | 0.929 |
| NRLRSL | 18.009 | 0.663 | 0.814 |
| HRSRGA | 17.514 | 0.995 | 0.997 |
| FRARSS | 16.869 | 0.952 | 0.975 |
| ARGRYF | 16.487 | 0.912 | 0.955 |
| MRGRVL | 16.473 | 0.935 | 0.966 |
| FRGRMS | 15.985 | 0.982 | 0.990 |
| MRGRVL | 15.556 | 0.990 | 0.995 |
| SRFRDN | 14.396 | 0.876 | 0.935 |
| ERGRIM | 13.117 | 0.968 | 0.983 |
| TRGRVW | 12.965 | 0.959 | 0.979 |
| FRPRFY | 12.445 | 0.819 | 0.905 |
| IRGRLS | 11.610 | 0.838 | 0.915 |
| FRARGE | 11.137 | 0.641 | 0.800 |
| NRGRLL | 10.846 | 0.897 | 0.947 |
| LRNRTT | 10.386 | 0.910 | 0.953 |
| GRERTS | 9.799 | 0.712 | 0.843 |
| CRLRSS | 9.719 | 0.759 | 0.871 |
| SRLRFY | 9.276 | 0.749 | 0.865 |
| WRARLY | 9.094 | 0.669 | 0.818 |
| ERGRSQ | 8.810 | 0.413 | 0.642 |
| NRCREG | 8.434 | 0.951 | 0.975 |
| FRARNN | 7.782 | 0.184 | 0.429 |
| WRERVY | 7.359 | 0.884 | 0.940 |
| GRSRVG | 6.947 | 0.700 | 0.836 |
| VRQRLF | 6.813 | 0.953 | 0.976 |
| SRSRIL | 6.368 | 0.817 | 0.903 |
| KRDRSM | 6.283 | 0.389 | 0.623 |
| PRFRFN | 5.978 | 0.518 | 0.719 |
| IRGRDL | 5.825 | 0.830 | 0.911 |
| TRGRLS | 5.767 | 0.859 | 0.926 |
| SRWRVQ | 5.761 | 0.600 | 0.774 |
| MRGRLV | 5.470 | 0.778 | 0.881 |
| MRNRIN | 5.099 | 0.035 | 0.186 |
| SRWRCV | 4.392 | 0.476 | 0.689 |
| SRSRVY | 3.774 | 0.325 | 0.569 |
| NRWRDN | 3.508 | 0.230 | 0.479 |
| YRSRYL | 3.495 | 0.331 | 0.575 |
| IRQRHD | 3.308 | 0.063 | 0.250 |
| LRGRMT | 3.113 | 0.670 | 0.818 |
| LRNRTS | 2.339 | 0.140 | 0.374 |
| GRYRMT | 2.166 | 0.073 | 0.270 |
| SRGRLL | 2.069 | 0.103 | 0.321 |
| NRSRSE | 2.061 | 0.617 | 0.785 |
| GRARDY | 1.613 | 0.157 | 0.395 |
| WRSRNL | 1.607 | 0.027 | 0.164 |
| SRSRVL | 0.876 | 0.109 | 0.329 |
| LRSRPA | 0.514 | 0.015 | 0.124 |
| VRLRYQ | 0.017 | 0.000 | 0.053 |
DISCUSSION
From the experimental data shown in Tables 2 and 3, at least three distinct binding modes for the fXa-substrate recognition were likely: 1) XXGR adapting canonical conformation in enzyme-substrate complex; 2) XR(G/A/S)R with the P3 Arg side chain in the aryl-binding site; and 3) XX(L/W/F/Y)R recognized by fXa through a yet to be defined binding mode. High resolution experimental methods are difficult to determine the proposed fXa-substrate binding modes (5). Alternatively, we resorted to computational molecular modeling to investigate the plausibility of these proposed substrate-enzyme complex structures.
Fig. 5 shows the modeled substrate-fXa transition state complexes for seven representative substrate sequences obtained from Fig. 4, for which at least three binding modes were rationalized as necessary to account for the enzyme-substrate recognition (see above). The corresponding kobs and the enzymatic reaction kinetics of these representative substrate sequences are also shown in Fig. 4 (see also the figure legend of Fig. 5).
Due to the limitation of current energetic scoring systems, it is difficult to single out one substrate-enzyme complex with the lowest conformational free energy among a huge number of conformations derived from an exhaustive search algorithm over a huge conformational space. Hence, in addition to ranking the complex structures with one of the best ranking systems (GOLDSCORE (27, 28)), we further selected from the top 100 GOLDSCORE-ranked complex structures the 5 best models (see Fig. 5, b–h) that scored highest with the semi-quantitative pharmacophore model derived from 55 fXa-ligand complex structures in the PDB (see Equation 3 under “Experimental Procedures”). The chemical properties of these high affinity ligands to fXa have been extensively reviewed (35, 36) as fXa is an important drug target. In panel a of Fig. 5, the centers of the 12 non-redundant complex-based pharmacophore features represented locations of the pharmacophores near the substrate binding site of fXa, where chemical moieties contributing to binding energetics for ligands with high affinities to fXa have been observed in fXa-ligand complex structures. Thus the model structures shown in Fig. 5, b–h, are not only the conformations with the lowest conformational free energy based on the scoring system, these binding substrate structures are also realistically consistent with the space distribution of the energetically favorable ligand chemical moieties observed in high affinity ligand-complex structures.
Only the models of GIEGR consistently have the P4(Ile) side chain inserted into the S4 (aryl-binding) site (see Fig. 5c), in agreement with the canonical conformation hypothesis. Another substrate that shows partial agreement with the canonical conformation hypothesis is the GPSGR (see Fig. 5e), where 3 of 5 models have P4(Pro)-S4 binding conformations. Models for GPQGR (Fig. 5b), GYRGR (Fig. 5d), GLLQR (Fig. 5f), and GPRAR (Fig. 5h) have the binding modes where the S4 (aryl-binding) site prefers to accommodate the P3 side chains. The preference of the Arg and Gln side chains in the aryl-binding site explains the experimental observation of the significantly high occurrence frequencies of Arg and Gln in the P3 position among the cleavable substrate sequences. The binding mode for the GAQFR (Fig. 5g) is striking in that the bulky side chain of the Phe (P2) has no place to go but to fit into the aryl-binding (S4) pocket. Together, the models provide an explanation for the observation that the P2 site accommodated the smallest and the bulkiest amino acid side chains. Nevertheless, due to the limitation of the computational modeling, the findings from molecular modeling can only be regarded as proposition rather than proof.
We found that known substrate sequences from in vivo protein substrates of the coagulation system, such as PQGR in factor VII (see Fig. 4) and I(E/D)GR in prothrombin (see Fig. 4 and Table 2), are among the most active substrate sequences recognized by the fXa active site in vitro. The results indicate that the catalytic site of fXa has been evolved to accomplish the highest specificity toward its substrates in vivo.
Interestingly, the fXa active site recognizes PQGR and I(E/D)GR through different binding modes (see Fig. 5, b and c). The two binding modes are drastically different both in the conformation of the peptide substrate and the active site binding pockets. Moreover, the binding modes shown in Fig. 5 did not suggest that the canonical conformation binding mode correlates with more effective enzymatic kinetics; however, the P2 to S4 binding mode (see Fig. 5g) seems to correlate with less effective enzymatic reactions.
The conformational flexibility of the substrate and the plasticity of catalytic site residues shown in Fig. 5 explain the permissiveness of the active site-substrate recognition shown in Fig. 4 and Tables 2 and 3, and hence imply that serine protease active site-substrate recognitions are intrinsically nonspecific to various extents against peptidyl substrates. System activations requiring high specificity beyond the intrinsic specificity of the involved serine proteases need to rely upon additional enzyme-protein substrate interactions remote from the catalytic site. In this aspect, the exosite-driven mechanism of substrate recognition in the fXa-fVa-catalyzed thrombin formation has provided significant insights (14, 15).
The substrate phage display results have provided a complete mapping of the peptidyl substrate recognitions in the catalytic site of fXa. The binding of Arg/Gln to the aryl-binding site has provided an initial scaffold for designing the peptide-based inhibitor against the active site of fXa. On the other hand, the experimental and modeling results also suggest that difficulties could arise in engineering the catalytic site recognition specificity through mutations of active site residues due to the flexibility of the active site structure and the adaptability of the substrate conformation.
To summarize, substrate phage display selection experiments revealed that the specificity of fXa toward substrate peptide sequences could not be explained with the canonical conformation as the sole binding mode. Molecular modeling results suggest that three binding modes corresponding to the aryl-binding site accommodating the P4, P3, and P2 side chains, respectively, are likely to account for the specificity of fXa toward substrate peptide sequences. Together, the results indicate that the binding mode of the enzyme-substrate complex is sequence-dependent, i.e. the way the sites are occupied in the active site cleft could vary from one substrate to the other, depending on the sequence of the substrate peptide. As such, a sequence-dependent substrate-enzyme recognition model is expected to underlie the experimentally observed fXa specificities toward substrate peptide sequences.
Acknowledgments
The SYBYL and CATALYST computation were conducted at the National Center for High Performance Computing, Taiwan. The DISCOVERY STUDIO 1.7 computation was conducted at the computational center of the Academia Sinica. We thank Dr. Min-Yong Li at the Department of Chemistry, Georgia State University, Atlanta, GA, and Gaokeng Xiao at Shenyang Pharmaceutical University, Shenyang, Liaoning, China, for fruitful discussions. We thank Dr. Thierry Langer at Inteligand for supplying LigandScout to analyze the pharmacophore. We also thank Dr. Wen-Lian Hsu, Ching-Tai Chen, and Ei-Wen Yang at the Institute of Information Science at Academia Sinica for helpful discussions.
This work was supported by National Health Research Institutes Grant NHRI-EX95-9525EI, National Science Council for People's Republic of China Grant NSC 96-2311-B-001-030-MY3, and the Genomics Research Center at Academia Sinica, Taipei, Taiwan. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: fXa, factor Xa; ELISA, enzyme-linked immunosorbent assay; PEG, polyethylene glycol; Ni-NTA, nickel-nitrilotriacetic acid; PBS, phosphate-buffered saline; MALDI-TOF, matrix-assisted laser desorption ionization time-of-flight; dansyl, 5-dimethylaminonaphthalene-1-sulfonyl.
References
- 1.Marnett, A. B., and Craik, C. S. (2005) Trends Biotechnol. 23 59-64 [DOI] [PubMed] [Google Scholar]
- 2.Hedstrom, L. (2002) Chem. Rev. 102 4501-4524 [DOI] [PubMed] [Google Scholar]
- 3.Perona, J. J., and Craik, C. S. (1995) Protein Sci. 4 337-360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tyndall, J. D., Nall, T., and Fairlie, D. P. (2005) Chem. Rev. 105 973-999 [DOI] [PubMed] [Google Scholar]
- 5.Ding, X., Rasmussen, B. F., Petsko, G. A., and Ringe, D. (2006) Bioorg. Chem. 34 410-423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Laskowski, M., and Qasim, M. A. (2000) Biochim. Biophys. Acta 1477 324-337 [DOI] [PubMed] [Google Scholar]
- 7.Coombs, G. S., Rao, M. S., Olson, A. J., Dawson, P. E., and Madison, E. L. (1999) J. Biol. Chem. 274 24074-24079 [DOI] [PubMed] [Google Scholar]
- 8.Brandstetter, H., Kuhne, A., Bode, W., Huber, R., von der Saal, W., Wirthensohn, K., and Engh, R. A. (1996) J. Biol. Chem. 271 29988-29992 [DOI] [PubMed] [Google Scholar]
- 9.Hertzberg, M. (1994) Blood Rev. 8 56-62 [DOI] [PubMed] [Google Scholar]
- 10.Krishnaswamy, S., Jones, K. C., and Mann, K. G. (1988) J. Biol. Chem. 263 3823-3834 [PubMed] [Google Scholar]
- 11.Betz, A., and Krishnaswamy, S. (1998) J. Biol. Chem. 273 10709-10718 [DOI] [PubMed] [Google Scholar]
- 12.Bianchini, E. P., Orcutt, S. J., Panizzi, P., Bock, P. E., and Krishnaswamy, S. (2005) Proc. Natl. Acad. Sci. U. S. A. 102 10099-10104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hacisalihoglu, A., Panizzi, P., Bock, P. E., Camire, R. M., and Krishnaswamy, S. (2007) J. Biol. Chem. 282 32974-32982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bock, P. E., Panizzi, P., and Verhamme, I. M. (2007) J. Thromb. Haemostasis 5 Suppl. 1, 81-94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Krishnaswamy, S. (2005) J. Thromb. Haemostasis 3 54-67 [DOI] [PubMed] [Google Scholar]
- 16.Orcutt, S. J., Pietropaolo, C., and Krishnaswamy, S. (2002) J. Biol. Chem. 277 46191-46196 [DOI] [PubMed] [Google Scholar]
- 17.Harris, J. L., Backes, B. J., Leonetti, F., Mahrus, S., Ellman, J. A., and Craik, C. S. (2000) Proc. Natl. Acad. Sci. U. S. A. 97 7754-7759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Matthews, D. J., and Wells, J. A. (1993) Science 260 1113-1117 [DOI] [PubMed] [Google Scholar]
- 19.Bianchini, E. P., Louvain, V. B., Marque, P. E., Juliano, M. A., Juliano, L., and Le Bonniec, B. F. (2002) J. Biol. Chem. 277 20527-20534 [DOI] [PubMed] [Google Scholar]
- 20.Deperthes, D. (2002) Biol. Chem. 383 1107-1112 [DOI] [PubMed] [Google Scholar]
- 21.Ohkubo, S., Miyadera, K., Sugimoto, Y., Matsuo, K., Wierzba, K., and Yamada, Y. (2001) Comb. Chem. High Throughput Screening 4 573-583 [DOI] [PubMed] [Google Scholar]
- 22.Smith, M. M., Shi, L., and Navre, M. (1995) J. Biol. Chem. 270 6440-6449 [DOI] [PubMed] [Google Scholar]
- 23.Sharkov, N. A., Davis, R. M., Reidhaar-Olson, J. F., Navre, M., and Cai, D. (2001) J. Biol. Chem. 276 10788-10793 [DOI] [PubMed] [Google Scholar]
- 24.Kunkel, T. A., Roberts, J. D., and Zakour, R. A. (1987) Methods Enzymol. 154 367-382 [DOI] [PubMed] [Google Scholar]
- 25.Sidhu, S. S., and Weiss, G. A. (2004) in Phage Display (Clackson, T., and Lowman, H. B., eds) 1st Ed., pp. 27-41, Oxford University Press, New York
- 26.Wei, A., Alexander, R. S., Duke, J., Ross, H., Rosenfeld, S. A., and Chang, C. H. (1998) J. Mol. Biol. 283 147-154 [DOI] [PubMed] [Google Scholar]
- 27.Jones, G., Willett, P., Glen, R. C., Leach, A. R., and Taylor, R. (1997) J. Mol. Biol. 267 727-748 [DOI] [PubMed] [Google Scholar]
- 28.Jones, G., Willett, P., and Glen, R. C. (1995) J. Mol. Biol. 245 43-53 [DOI] [PubMed] [Google Scholar]
- 29.Cornell, W. D., Cieplak, P., Bayly, C. I., Gould, I. R., Merz, K. M., Ferguson, D. M., Spellmeyer, D. C., Fox, T., Caldwell, J. W., and Kollman, P. A. (1995) J. Am. Chem. Soc. 117 5179-5197 [Google Scholar]
- 30.Wolber, G., and Langer, T. (2005) J. Chem. Inf. Model 45 160-169 [DOI] [PubMed] [Google Scholar]
- 31.Li, H., Sutter, J., and Hoffmann, R. (1999) in Pharmacophore Perception, Development, and Use in Drug Design (Guner, O. F., ed) 1st Ed., pp. 173-188, International University Line, La Jolla, CA
- 32.Padmanabhan, K., Padmanabhan, K. P., Tulinsky, A., Park, C. H., Bode, W., Huber, R., Blankenship, D. T., Cardin, A. D., and Kisiel, W. (1993) J. Mol. Biol. 232 947-966 [DOI] [PubMed] [Google Scholar]
- 33.Rodi, D. J., Soares, A. S., and Makowski, L. (2002) J. Mol. Biol. 322 1039-1052 [DOI] [PubMed] [Google Scholar]
- 34.Bromfield, K. M., Quinsey, N. S., Duggan, P. J., and Pike, R. N. (2006) Chem. Biol. Drug Des. 68 11-19 [DOI] [PubMed] [Google Scholar]
- 35.Kontogiorgis, C. A., and Hadjipavlou-Litina, D. (2004) Med. Res. Rev. 24 687-747 [DOI] [PubMed] [Google Scholar]
- 36.Maignan, S., and Mikol, V. (2001) Curr. Top. Med. Chem. 1 161-174 [DOI] [PubMed] [Google Scholar]
- 37.Marlowe, C. K., Sinha, U., Gunn, A. C., and Scarborough, R. M. (2000) Bioorg. Med. Chem. Lett. 10 13-16 [DOI] [PubMed] [Google Scholar]
- 38.Gallivan, J. P., and Dougherty, D. A. (1999) Proc. Natl. Acad. Sci. U. S. A. 96 9459-9464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ma, J. C., and Dougherty, D. A. (1997) Chem. Rev. 97 1303-1324 [DOI] [PubMed] [Google Scholar]