

Abstract
We characterized HIV-1 reverse transcriptase (RT) variants either with or without the (-)-2′,3′-deoxy-3′-thiacytidine-resistant M184I mutation isolated from a single HIV-1 infected patient. First, unlike variants with wild-type M184, M184I RT variants displayed significantly reduced DNA polymerase activity at low dNTP concentrations, which is indicative of reduced dNTP binding affinity. Second, the M184I variant displayed a ∼10- to 13-fold reduction in dNTP binding affinity, compared with the Met-184 variant. However, the kpol values of these two RTs were similar. Third, unlike HIV-1 vectors with wild-type RT, the HIV-1 vector harboring M184I RT failed to transduce cell types containing low dNTP concentrations, such as human macrophage, likely due to the reduced DNA polymerization activity of the M184I RT under low cellular dNTP concentration conditions. Finally, we compared the binary complex structures of wild-type and M184I RTs. The Ile mutation at position 184 with a longer and more rigid β-branched side chain, which was previously known to alter the RT-template interaction, also appears to deform the shape of the dNTP binding pocket. This can restrict ground state dNTP binding and lead to inefficient DNA synthesis particularly at low dNTP concentrations, ultimately contributing to viral replication failure in macrophage and instability in vivo of the M184I mutation.
(-)-2′,3′-Deoxy-3′-thiacytidine (3TC),3 a deoxycytidine analog reverse transcriptase (RT) inhibitor, has been routinely included in the anti-HIV-1 drug regime (1, 2). During 3TC therapy, two amino acid substitutions at position 184 of RT are sequentially selected, conferring viral resistance to this drug. The M184I mutation is detected earlier, and eventually replaced with the M184V mutation. One initial explanation for the delayed selection of the M184V mutation is the requirement of two nucleotide mutations to develop the final M184V mutation from the wild-type methionine codon (ATG). In contrast, the M184I mutation requires only a single nucleotide mutation from the Met codon, which may result in its early selection during 3TC therapy (3–7).
Structural analysis later provided a functional explanation for the instability in vivo of the M184I mutation. The M184I mutation alters the RT interaction with the template nucleotide more significantly than the M184V mutation, leading to more severely decreased processivity, compared with the M184V RT (8). Indeed, the distributive DNA synthesis catalyzed by the M184I RT has been a major explanation for the instability in vivo of this mutation and its rapid transition to the M184V mutation. The long β-branched side chains of these two mutations efficiently block the entry of 3TCTP to the active site (8, 9). Interestingly, pre-steady-state kinetic studies of the M184V mutant demonstrated that the M184V mutation only slightly (1- to 2-fold) affects Kd (dNTP binding affinity), but not kpol (conformational change/chemical catalysis) steps of HIV-1 RT with normal dNTP substrates, implying that the Val mutation does not significantly affect the structural architecture of the HIV-1 RT dNTP binding pocket (10–12). In addition, the M184I RT showed a greater fold increase of enzyme fidelity than the M184V RT, even though the effects of the mutations on fidelity are relatively small (13–15). It was also reported that the Met-184 mutations alter the mutation spectrum of HIV-1 RT (13).
The cellular dNTP concentrations of the two HIV-1 target cells, activated/dividing CD4+ T cells and terminally differentiated/non-dividing macrophage, were recently determined. Macrophage contain very low dNTP concentrations (20∼40 nm) compared with activated CD4+ T cells (∼4 μm) (16). The extremely low availability of dNTP in macrophage raised a question about how HIV-1 RT is able to polymerize DNA in such a low dNTP environment. To address this issue, we recently examined HIV-1 RT for its dNTP concentration-dependent DNA synthesis profile and kinetic parameters such as Kd and kpol and compared these with the same parameters for the RT of MuLV, which replicates only in cells displaying a mitotic index with high cellular dNTP concentrations (i.e. dividing cells). Indeed, HIV-1 RT efficiently synthesizes DNA even at low dNTP concentrations corresponding to macrophage as well as in high dNTP concentrations found in activated CD4+ T cells. Conversely, MuLV RT failed to polymerize DNA at the same low dNTP concentrations but showed activity only at high dNTP concentrations found in dividing cells (∼5 μm dNTP) (16). Furthermore, HIV-1 RT showed between 6 and 150 times tighter dNTP binding affinity than MuLV RT, suggesting that the high dNTP binding affinity of HIV-1 RT enables it to polymerize DNA efficiently at low dNTP concentrations (17). This unique, efficient dNTP synthesis nature was also found in RTs of other lentiviruses such as SIV and feline immunodeficiency virus. In addition, other non-lentiviral RTs such as avian myeloblastosis virus RT and feline leukemia virus RT remain active only at high dNTP concentrations found in dividing cells (16, 18–21). We also observed that two HIV-1 RT mutants, Q151N and V148I, which have reduced dNTP binding affinity, showed high RT activity only at high dNTP concentrations, similar to MuLV RT and other oncoretroviral RTs (16, 22, 23). Importantly, HIV-1 variants harboring these dNTP binding RT mutants failed to infect macrophage even though these mutant viruses still efficiently infect cell types with high cellular dNTP concentrations (16). An HIV-1 vector containing the Q151N RT was unable to transduce normal dividing cells such as human lung fibroblasts (HLFs), but efficiently transduced cancer cell lines containing unusually elevated dNTP concentrations due to their uncontrolled cell cycle (24). The failure of the Q151N HIV-1 vector to transduce HLF was rescued by deoxynucleoside (dN) treatment, which greatly elevates cellular dNTP concentration (24). These findings suggest that the dNTP interaction mechanism of HIV-1 RT and the dNTP availability of virus target cells can contribute to cell type specific replication and infection of retroviruses.
Here, using M184I HIV-1 RT variants isolated from an infected patient, we found that the M184I mutation reduces the dNTP binding affinity of HIV-1 RT, which leads to decreased RT activity, particularly at low in vivo cellular dNTP concentrations. This significantly altered dNTP binding affinity, which was not observed with the M184V HIV-1 RT mutant, may contribute to the in vivo instability and rapid replacement of the M184I mutation with the M184V mutation during 3TC therapy.
MATERIALS AND METHODS
Isolation and Cloning of HIV-1 RT Variants from an HIV-1 positive Serum Sample
A serum sample from an HIV-1 infected patient, containing M184I RT variants, was obtained from the Multicenter AIDS Cohort Study (MACS, case number 30329), NIAID, National Institutes of Health. HIV-1 Viral RNA was isolated from the serum samples using the QIAamp Viral RNA Mini kit (Qiagen, Valencia, CA). The isolated viral RNA genomes were reverse transcribed to cDNA using a first strand cDNA synthesis kit (Fermentas, Hanover, MD), which uses M-MuLV as the reverse transcriptase. The primer used for the first strand cDNA synthesis (HIV IN N-terminal reverse primer 5′-CACTAGCCATTGCTCTCC-3′) was designed using complementary regions from the pol regions of strains NL4–3 and BRU of HIV-1 RT. The RT gene was then amplified from cDNA, using pfu DNA polymerase with a mixture of N-T Forward primers (NL43 BRU Forward primer 5′-CCCATTAGTCCTATTGA-3′ and HXB2 Forward primer 5′-CCCATTAGCCCTATTGA-3′) and C-T Reverse primer (5′-CTAAAAATAGTACTTTCCTG-3′). After amplification with pfu polymerase, 3′ A overhangs were added using Taq polymerase. The amplified 1.6-kb product was then cloned directly to TA cloning vector pCR2.1 (Invitrogen), followed by transformation into DH5α (Invitrogen) and plated on 2XYT/Carb/5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside/isopropyl 1-thio-β-d-galactopyranoside plates for blue-white selection. Twenty white colonies were screened by EcoRI digestion (New England Biolabs), and ten isolates positive for RT inserts were sequenced by the ABI system using M13 forward primer (5′-GTAAAACGACGGCCAG-3′). Among the ten RT clones, three RT clones contained the M184I mutation, and these three also contained 3–4 other mutations throughout their genomes. The remaining seven clones contained the wild-type Met residue at the 184 position. Three N-terminal primers containing an NdeI restriction site (shown in bold) were designed for cloning RT inserts into bacterial overexpression vector, pET28a (Novagen, Madison, WI). The primer sequences are as follows: N-T NdeI (bold) CG primer 5′-AAAAAACATATGCCCATTAGCCCTATTGAGAC-3′, N-T NdeI TA primer 5′-AAAAAACATATGCCCATTAGTCCTATTGAAAC-3′, N-T NdeI TG primer 5′-AAAAAACATATGCCCATTAGTCCTATTGAGAC-3′. The C-Terminal reverse primer contains the HindIII site (bold), 5′-AAAAAAAAGCTTTTATAGTACTTTCCTGATTCC-3′.
Construction of the I184M Revertant RT of the M184I Variant
The Ile to Met (I184M) revertant was cloned using overlapping PCR to achieve site-directed mutagenesis. During the first round of PCR, N-T NdeI TG forward and IM reverse primer pairs (to amplify the N-terminal region of RT) and IM forward and C-T HindIII reverse primer pairs (to amplify the C-terminal region of RT) were used. DNA from M184I RT Clone 7 containing the M184I RT mutation was used as the template for this PCR. The second round of PCR was performed using the N-T NdeI TG forward and C-T HindIII reverse primers. The primer sequences for IM forward and reverse primers are: 5′-GATGATTTGTATGTAGGATC-3′ and 5′-GATCCTACATACAAATCATCGAGTATTGATAGATAAC-3′, respectively.
Purification of HIV-1 RT Variants
The RT variants were overexpressed in Escherichia coli BL21 (Novagen) from derivatives of the pET28a-HIV-1 RT plasmid. The HIV-1 RT expression construct encodes full-length HIV-1 p66 fused at the N terminus to a six-histidine tag, which facilitates protein purification using a Ni2+ chelation chromatography according to the manufacturer's protocol (Novagen) as described previously (25, 26). From 1 liter of culture, we were able to purify ∼4 mg of p66/p66 homodimers with >95% purity determined by visual inspection of Coomassie-stained gels using 99% pure bovine serum albumin (Sigma, MO). M184V HIV-1 RT protein was purified from pET28 HIV-1 RT (NL4–3) containing M184V mutation created by site-directed mutagenesis.
dNTP Concentration-dependent Reverse Transcription Assay
To measure the ability of RTs to perform DNA synthesis in the presence of different concentrations of dNTP, template-primer (T·P) was extended using similar activities of RT variants with dNTP concentrations ranging from 5 to 0.05 μm. Briefly, T·P was prepared by annealing a 38-mer RNA template (5′-GCUUGGCUGCAGAAUAUUGCUAGCGGGAAUUCGGCGCG-3′, Dharmacon Research, Chicago, IL) to a 17-mer “A primer” (5′-CGCGCCGAATTCCCGCT-3′; template: primer, 2.5:1) 32P-labeled at the 5′-end by T4 polynucleotide kinase (New England Biolabs) and [γ-32P]ATP (PerkinElmer Life Sciences). Assay mixtures (20 μl) contained 10 nm T·P, all four dNTPs at varying concentrations (5, 1, 0.25, 0.1, and 0.05 μm), 25 mm Tris-HCl (pH 8.0), 100 mm KCl, 2 mm DTT, 5 mm MgCl2, 2 μm (dT)20, and 0.1 mg/ml bovine serum albumin. Reactions were initiated by adding RT activity giving full extension of 50% of labeled primer at 5 μm dNTP incubated at 37 °C for 5 min and terminated by adding 10 μl of 40 mm EDTA and 99% formamide followed by incubation at 95 °C for 5 min. Reactions were resolved on 14% polyacrylamide-urea denaturing gel and then analyzed through phosphorimaging.
3TCTP Sensitivity Assay
Similar to the DNA polymerization assay, 17-mer A primer was 5′-end 32P-labeled using T4 polynucleotide kinase (New England Biolabs) and [γ-32P]ATP (PerkinElmer Life Sciences). T·P complex was prepared by annealing the 38-mer RNA (see above) template to the 17-mer A primer. Assay mixtures (20 μl) contained 10 nm T·P, RT proteins giving similar levels of activity (50% primer extension at 5 μm dNTP), all four dNTPs (5 μm), 3TCTP (Moravek, CA) (0–125 μm), 25 mm Tris-HCl, pH 8.0, 100 mm KCl, 2 mm DTT, 5 mm MgCl2, 2 μm (dT)20, and 0.1 mg/ml bovine serum albumin (New England Biolabs). Reactions were incubated at 37 °C for 5 min and terminated by addition of 10 μl of 40 mm EDTA, 99% formamide. Reaction products were immediately denatured by incubating at 95 °C for 5 min, and reactions were analyzed by electrophoresis on a 14% polyacrylamide-urea denaturing gel followed by phosphorimaging.
Pre-steady state Kinetic Assays
Pre-steady state burst and single turnover experiments were performed to examine the transient kinetics associated with incorporating a single nucleotide on to primer annealed to a template. Because these reactions occur within the millisecond time scale, reactions were performed using a rapid quench-flow machine (Kintek, TX).
Pre-steady state Burst Experiments—Pre-steady state burst experiments were initially performed to determine the concentration of active RT (RT molecules that can bind to and extend T·P) in each protein preparation. For the active site determination we used 23-mer Primer T (5′-CCGAATTCCCGCTAGCAATATTC-3′) annealed to a 38-mer DNA template (Dharmacon Research), 5′-GCTTGGCTGCAGAATATTGCTAGCGGGAATTCGGCGCG-3′. Reactions were carried out by mixing (a) 150 nm of RT preincubated with 300 nm T·P with (b) 800 μm dTTP mixed with 10 mm MgCl2.
Polymerization reactions were quenched with 0.3 m EDTA at time intervals ranging from 5 ms to 2 s. During this period, a fast initial burst occurs followed by a slower steady state due to multiple rounds of extensions. Pre-steady state kinetic data were analyzed using nonlinear regression. Equations were generated from the data with the KaleidaGraph, version 3.51 (Synergy Software, Essex Junction, VT). Data points obtained during the burst experiment were fitted to the burst equation (Equation 1) (27, 28).
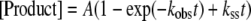 |
(Eq. 1) |
The value A is the amplitude of the burst, which reflects the actual concentration of active enzyme. kobs is the observed first-order rate constant for dNTP incorporation, whereas kss is the observed steady-state rate constant.
Pre-steady state Single Turnover Experiments—To study the incorporation of the four dNTPs (correct), four different 23-mer DNA primers (Integrated DNA Technologies, Coralville, IA) were 5′-end-labeled with [γ-32P]ATP (PerkinElmer Life Sciences) using T4 polynucleotide kinase (New England Biolabs). The primer sequences were as follows: A primer, 5′-CGCGCCGAATTCCCGCTAGCAAT-3′; T primer, 5′-CCGAATTCCCGCTAGCAATATTC-3′; G primer, 5′-CGAATTCCCGCTAGCAATATTCT-3′; C primer, 5′-GCCGAATTCCCGCTAGCAATATT-3′. The 5′-end labeled primers were then individually annealed to a 38-mer DNA template (Dharmacon Research), 5′-GCTTGGCTGCAGAATATTGCTAGCGGGAATTCGGCGCG-3′. Pre-steady state single turnover experiments for the incorporation of correct nucleotide were then measured using excess active RT (200 nm), as determined by the active site titration, onto T·P (50 nm). This ensures single round/single nucleotide extension at the pre-steady-state level, for all four dNTPs when using different primers. For studying the incorporation of incorrect dNTPs (misinsertion), 5′-end 32P-labeled T-primer, was annealed onto a 38-mer DNA template (see above). Incorporation of incorrect dNTP experiments were carried out manually and at longer time points ranging from 30 s to 10 min, with higher concentrations of RT (350 nm). Products were resolved by 14% polyacrylamide-urea denaturing gel electrophoresis and quantified with the Cyclone PhosphorImager (PerkinElmer Life Sciences).
Data from single turnover experiments were fitted to a single exponential equation that measures the rate of dNTP incorporation (kobs) per given dNTP concentration. This was done by fitting the data to the hyperbolic equation (Equation 2).
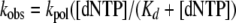 |
(Eq. 2) |
From this equation, we could then identify the kinetic constants for each RT during pre-steady state kinetics: kpol is the maximum rate of dNTP incorporation, and Kd is equilibrium dissociation constant for the interaction of dNTP with the E·DNA complex.
Cells and Cell Lines
Primary human monocyte-derived macrophage were isolated and differentiated as described previously (16) and were maintained in culture in RPMI 1640 (CellGro) supplemented with 10% fetal bovine serum (HyClone). The primary human lung fibroblasts (WI-38) were purchased from ATCC and were cultured in Dulbecco's Modified Eagle's Medium (Invitrogen) supplemented with 10% fetal bovine serum (HyClone) as recommended. The CHME5 cell line, a human fetal microglial cell line transformed by SV40 large T antigen, was a gift from Dr. David Mock (University of Rochester). CHME5 cells were cultured in Dulbecco's modified Eagle's medium from Invitrogen containing 10% fetal bovine serum (HyClone).
Pseudotyped Vector Production and Cell Transduction
The M184I mutation was generated by a PCR-based site-directed mutagenesis in pD3HIV-GFP as previously described (16). Vesicular stomatitis virus a protein-pseudotyped HIV-1 vectors, which express eGFP and all HIV-1 proteins except Env and Nef, were produced as described previously (16), and vectors were normalized by p24 levels determined by the HIV-1 p24 antigen enzyme-linked immunosorbent assay system (Beckman-Coulter). All cell types were transduced with an equal p24 amount (3 × 105 pg) of either wild-type or M184I vectors. HLFs and CHME5 cells were treated with 10 μg/ml Polybrene prior to transduction and 24 h post-transduction, and cells were washed and visualized by fluorescence microscopy (Leica), using eGFP as an indicator of transduction efficiency. Following imaging, cells were trypsinized and fixed in a 0.5% formaldehyde solution for further analysis by FACS. Macrophage were washed after 48 h post transduction and visualized for eGFP expression at 5 days post transduction. For FACS analysis, at day 6, cells were trypsinized, fixed, and analyzed by FACS using the FL-1 channel to detect eGFP expression. The percentage of transduction was determined using the CellQuest program (version 3.3, BD Biosciences).
Determination of Cellular dNTP Concentration
The HIV-1 RT-based dNTP quantitation assay was used to determine cellular dNTP concentrations of WI-38 as previously described (16). About 1 × 106 cells were used to extract dNTP samples, which were applied to the dNTP assay. The dNTP concentrations of macrophage and CHME5 were previously reported.
RESULTS
Isolation of M184I HIV-1 RT Variants—We isolated 10 RTs cloned from serum from a single HIV-1 infected patient containing the M184I RT variants, which was kindly provided by the Multicenter AIDS Cohorts Study (MACS) program. The amplified RT genes from viral RNAs were cloned directly into TA cloning vectors, and 10 isolates positive for RT inserts were sequenced. Among the 10 RT clones, three RT clones contained the M184I mutation. These three RTs also contained 3–4 other mutations (T139A, R172K, H198P, and C162A) throughout their DNA polymerase domain genes. The remaining 7 clones contain the WT Met residue at the 184 position. We observed that none of the 10 RT clones were identical in terms of their nucleotide sequence variations. The amplified RT genes were then cloned to an E. coli overexpression plasmid. All 10 p66/p66 homodimer RT proteins, containing a hexahistidine tag at their N-terminal ends, were purified using Ni2+-charged column chromatography with at least >95% purity as estimated in comparison with purified bovine serum albumin (99%) by SDS-PAGE.
3TC Resistance of the HIV-1 RT Variants—First, we attempted to validate the 3TC resistance of the isolated M184I RT variants using a primer extension assay. A 5′ 32P-labeled 17-mer primer annealed to a 38-mer RNA template was employed in this assay (Fig. 1). RT amounts showing approximately ∼75% full primer extension (“F” in Fig. 1) under standard primer extension conditions (see “Materials and Methods”) in the presence of only dNTPs (5 μm) were determined, and then the reactions were repeated in the presence of both 5 μm dNTPs and two concentrations of 3TCTP (7.5 and 125 μm). This assay qualitatively reveals the 3TCMP incorporation capability of RTs, which results in primer termination specifically at “G” template sequence positions (Fig. 1). In this assay, all seven “Met-184 RT” variants displayed significant early termination of primer extension and reduction of the full-length product to 27–31%, compared with the no 3TCTP control reaction (∼75%), in the presence of 3TCTP due to the 3TCMP incorporation as shown at the positions marked as an asterisk in Fig. 1A with a representative Met-184 Clone 3 RT. In contrast, the three M184I RT variants did not show significant formation of the early termination products, and no reduction of full-length product (70–74%), compared with the no 3TCTP control reaction (∼75%), as shown in Fig. 1B with a representative Clone 7 RT, which indicates lower sensitivity (or increased resistance) to 3TCTP, compared with the Clone 3 RT. Next, to examine the possibility that other RT mutations found in these M184I RT variants affect 3TC resistance, we constructed an I184M revertant RT from an M184I RT variant (Clone 7 RT). This revertant RT contains all the same mutations as the Clone 7 RT variant except the M184I mutation. As shown in Fig. 1C, like other Met-184 RT variants (Clone 3 RT, Fig. 1A), the I184M reversion RT of Clone 7 RT regained sensitivity to 3TCTP (reduction of the full-length product to 28% from 75% of the no 3TCTP reaction), implying that the M184I mutation is solely responsible for the 3TCTP resistance of the Clone 7 M184I RT, and other sequence variations between these RT clones do not significantly contribute to the 3TC resistance. Although there is ∼4–6% less radioactivity in gel C than in gels A and B, this small difference does not change the interpretation of above data because there was ∼250–400% more pausing at the 3TCMP incorporation sites (indicated by an asterisk).
FIGURE 1.

3TCTP sensitivity of HIV-1 RT variants. A 32P-labeled 17-mer A primer (P) annealed to 38-mer RNA template (T) was extended by Met-184 Clone 3 RT (A), M184I Clone 7 RT (B), and I184M revertant of Clone 7 RT (C), showing ∼75% of primer extension with 5 μm dNTPs at 37 °C for 5 min, and the same reactions were performed in the presence of 5 μm of only dNTPs, and repeated in the presence of both 5 μm dNTPs and two concentrations of 3TCTP (7. 5 and 125 μm). The asterisks indicate termination sites induced by the 3TCTMP incorporation opposite to the “G” sequences of the template. The circle indicates RT. F, 38-nucleotide long full-length product. S, unextended 17-mer primer.
[dNTP]-dependent DNA Polymerization Activity of the RT Protein Variants—Next, we compared the RNA-dependent DNA polymerase activity of the 10 RT clones at dNTP concentrations observed in HIV-1 target cells. A 17-mer 32P5′-end-labeled primer annealed to a 38-mer RNA template was also employed in this assay (Fig. 2). First, RT amounts showing ∼50% primer extension at 5 μm dNTP were determined, and the identical reactions were repeated with decreasing dNTP concentrations (1, 0.25, 0.1, and 0.05 μm). All of the seven RT variants containing Met at position 184 displayed efficient primer extension even at 0.05 μm dNTP (see Clone 3 RT of Fig. 2A as a representative clone of the Met-184 RT variants). In contrast, the M184I RT variants showed significantly reduced RT activity at lower concentrations such as 0.25, 0.1, and 0.05 μm dNTP, as shown by Clone 7 RT in Fig. 2A as a representative clone of the M184I RT variants. The RT variants containing M184I displayed reduced polymerization activity at the low physiological dNTP concentrations where the Met-184 RT variants still efficiently synthesize DNA.
FIGURE 2.

dNTP concentration-dependent reverse transcription activity of HIV-1 RT variants. A, a 32P-labeled 17-mer A primer (P) annealed to a 38-mer RNA template (T) was extended by HIV-1 RT variants, Met-184 Clone 3 RT, M184I Clone 7 RT, and M184V RT showing ∼50% of primer extension with 5 μm dNTPs at 37 °C for 5 min, and the reactions were repeated with decreasing dNTP concentrations (lanes 1–5: 5, 1, 0.25, 0.1, and 0.05 μm). B, the % of the fully extended product (F) in each lane was calculated, and the % extensions at lower dNTP concentrations were normalized with that at 5 μm (lane 1) for each RT protein. Then, the ratios of the normalized % extensions between Met-184 Clone 3 RT and the other two RTs (open circles, M184I Clone 7 RT; black diamond, M184V RT) at each dNTP concentration were plotted and standard deviations in triplicates were <15%.
We also employed M184V HIV-1 RT protein in this test. As shown in Fig. 2B, M184V RT exhibited higher than 50% of the Met-184 Clone 3 RT activity even at low dNTP concentration, whereas the M184I Clone 7 RT showed more severe activity reduction at these low concentrations compared with the Met-184 Clone 3 RT as well as the M184V RT protein. Note that all 10 RT variants showed similar specific activity at the highest dNTP concentration tested (5 μm), and the I184M reversion also displayed efficient primer extension in both high and low dNTP concentration. In addition, the I184M revertant gained the Met-184 RT-like high activity at the low dNTP concentration (data not shown).
Pre-steady-state Kinetic Analysis of M184I and Met-184 RT Variants—Because two previously characterized RT mutations, Q151N and V148I, that specifically reduce the dNTP binding affinity, also render reduced RT activity specifically at low dNTP concentrations (16, 23, 29), we examined the Kd and kpol values of the M184I-containing RT variants, using pre-steady-state kinetic analysis. For this kinetic analysis, we chose one of the three M184I clones (Clone 7 RT) and one Met-184 RT clone (Clone 3 RT) as well as the I184M reversion RT protein of the M184I Clone 7 RT. First, we determined the active concentrations of the three HIV-1 RT proteins on a 32P-labeled 23-mer (T-primer), annealed to a 38-mer DNA template (T-primer-template) as described previously (22). We measured product formation when 800 μm dTTP was mixed with RT (150 nm, protein concentration as determined with a Bradford assay), prebound onto the T·P (300 nm T·P). There was an initial burst of product formation due to dTTP incorporation onto the prebound RT·T·P complex (pre-steady-state kinetics), which was followed by a slower and linear phase of product formation corresponding to the steady state kinetics associated with multiple rounds of DNA polymerization (Fig. 3). Our active site titration analysis revealed that the active site concentrations of RT variants Met-184 clone 3, M184I clone 7, and I184M revertant proteins were all ∼30–50% (I184M RT data not shown). The slope of the burst phase is termed kobs (rate of DNA polymerization during the pre-steady state), and the slope of the second phase is termed kss (rate of DNA polymerization during the steady state). The pre-steady state rates of dTTP incorporation onto the T·P(kobs) for the Met-184 Clone 3, M184I Clone 7, and I184M revertant RT were 39 ± 17, 68.8 ± 16, and 168 ± 21 s-1, respectively, and their rates during the steady state were 8.1 × 10-1 ± 6 × 10-1, 3.7 × 10-1 ± 8 × 10-2, and 7 × 10-2 ± 2 × 10-2 s-1, respectively (Fig. 3).
FIGURE 3.

Active site titration for RT variants. Pre-steady and steady-state kinetics of Met-184 Clone 3 RT (A) and M184I clone 7 RT (B) incorporating correct dTTP onto the 32P-labeled 23-mer T primer annealed to the 38-mer template were analyzed. Reactions were carried out at the indicated times by mixing together a solution of RT (150 nm, protein concentration) prebound to T·P (300 nm) to a second solution with 800 μm dTTP under rapid quench conditions (see “Materials and Methods”). The data were fit into the burst equation as indicated by the solid line, which provides a measure of the active concentration of RT (A), the observed first order rate constant for the burst phase (kobs) and the first order rate constant for the linear phase (kss) for Met-184 Clone 3 and M184I clone 7 RT. The pre-steady-state rates of dTTP incorporation onto the T·P(kobs) for the Met-184 clone 3 and M184I clone 7 RTs were 39 ± 17 and 68.8 ± 16, respectively, and their rates during the steady state were 8.1 × 10-1 ± 6 × 10-1 s-1 and 3.7 × 10-1 ± 8 × 10-2, respectively.
Next, we performed single turnover experiments (200 nm active RT and 50 nm T·Ps) to obtain an actual measurement of the dNTP incorporation rate at different dNTP concentrations during the pre-steady state. By measuring reaction rate (kobs) as a function of dNTP concentration (Fig. 4 for dTTP incorporation), we were able to measure the kinetic parameters Kd, or the binding affinity of RT to the incoming nucleotide substrate, and kpol, the maximum rate of dNTP incorporation (conformational change and chemical catalysis). The pre-steady state kinetic data for RT variants are summarized in Table 1. The dNTP binding affinities (Kd) of M184I RT Clone 7 RT variant for dNTPs were 50–86 μm, and the chemical catalysis (kpol) values were 51–124 s-1. For the wild-type-like Met-184 Clone 3 RT variant, Kd values were comparatively lower, 4.2–9.5 μm, indicating high affinity for the incoming dNTP substrate, however the kpol values, which were 52–88 s-1, were similar to those of M184I RT Clone 7 variant. These kinetic values of the Met-184 Clone 3 RT are very similar to those of an HIV-1 strain HXB2 RT that was previously characterized (17). This kinetic study indicates that M184I RT Clone 7 has 10- to 13-fold lower dNTP binding affinity for the incoming dNTPs, compared with that of Met-184 Clone 3, while these two RTs have similar incorporation capability (kpol). The pattern of the kinetic difference observed between Clone 3 and Clone 7 RTs is identical with that observed between WT HXB2 RT and Q151N (and V148I) dNTP binding HIV-1 RT mutants (22, 29).
FIGURE 4.

Pre-steady state dTTP incorporation by Met-184 clone 3 (A) and M184I clone 7 (B) RTs. The 32P-labeled 23-mer T primer annealed to the 38-mer template (50 nm) was extended with excess RT (200 nm, active site concentration) for single round of dTTP incorporation at five different dTTP concentrations. These data were used for the determination of Kd and kpol values of the in vivo RTs illustrated in Table 1.
TABLE 1.
Pre-steady-state parameters of HIV-1 Clone 3 and 7 RT variants
|
DNTP substrates
|
HIV-1 RTs
|
Kinetic parameters (-fold
differences)a
|
||
|---|---|---|---|---|
| Kd | kpol | kpol/Kd | ||
| μm | s–1 | μm–1 s–1 | ||
| M184 | 7.2 ± 3.2 | 57.5 ± 9.4 | 8 | |
| dGTP | Clone 3 RT | (×1) | (×1) | (×1) |
| M184I | 64 ± 5.7 | 51 ± 8 | 0.8 | |
| Clone 7 RT | (×9) | (×0.8) | (×0.1) | |
| M184 | 4.8 ± 1.8 | 52 ± 5.8 | 10.8 | |
| dATP | Clone 3 RT | (×1) | (×1) | (×1) |
| M184I | 50 ± 4.5 | 84 ± 2 | 1.7 | |
| Clone 7 RT | (×10) | (×1.6) | (×0.15) | |
| M184 | 9.5 ± 2.1 | 88 ± 7.5 | 9.3 | |
| dCTP | Clone 3 RT | (×1) | (×1) | (×1) |
| M184I | 86 ± 9.7 | 124 ± 39 | 1.4 | |
| Clone 7 RT | (×9) | (×1.4) | (×0.16) | |
| M184 | 4.2 ± 2.2 | 63 ± 12 | 15 | |
| dTTP | Clone 3 RT | (×1) | (×1) | (×1) |
| M184I | 56 ± 7.5 | 56 ± 3 | 1 | |
| Clone 7 RT | (×13) | (×0.8) | (×0.06) | |
| I184M | 6.7 ± 2.2 | 79 ± 8 | 11.8 | |
| Revertant RTb | (×1.5) | (×1.25) | (×0.8) | |
The -fold differences are shown for M184I Clone 7 RT relative to M184 Clone 3 RT
The I184M revertant is the same as M184I Clone 7 RT except for the Ile-184 residue
We also performed single turnover kinetics for dTTP incorporation for the I184M revertant on the same T·P. This revertant was cloned using the same background as M184I RT, and the only difference between the two RTs is at position 184. As shown in Table 1, similar to Met-184 clone 3 RT, the I184M revertant showed a higher dNTP binding affinity for the incoming dNTP substrate, 6.7 μm, compared with 56 μm for the M184I Clone 3 RT variant. The kpol values were also similar to that of Met-184 Clone 3 RT and M184I Clone 7 RTs, indicating that the reduced Kd of M184I RT is likely due to the mutation of Met to Ile at position 184 of Clone 7 HIV-1 RT.
Cellular dNTP Concentration-dependent Infectivity of HIV-1 Harboring M184I RT Mutant—We previously reported that human macrophages, which are a key HIV-1 non-dividing target cell type, contain extremely low dNTP concentrations (20–40 nm), compared with other dividing cell types (16). Therefore, based on our results with the dNTP concentration-dependent polymerization assay, we hypothesized that an HIV-1 vector containing the M184I mutant RT showing reduced DNA synthesis at low dNTP concentrations may not be able to support reverse transcription and, therefore, fail to infect macrophage. To test this hypothesis, we employed an HIV-1 vector system expressing eGFP and three cell types containing different levels of dNTP concentrations: primary human macrophage, WI-38 HLFs, and human transformed CHME5 cells. Our dNTP quantitation assay showed that HLF and CHME5 cells have ∼200–400 nm and 3–4 μm dNTPs, respectively (Fig. 5B). We transduced these three cell types with an equal p24 level of wild-type and M184I vectors, and the transduction efficiency was determined by fluorescent microscopy and FACS analysis for GFP expression of transduced cells. As shown in Fig. 5 (A and B), wild-type HIV-1 vector efficiently transduced all three cell types. In contrast, the vector containing the M184I mutant failed to transduce macrophage even though this mutant vector was still capable of transducing HLFs and CHME5 cells containing higher levels of dNTP concentrations than macrophage. This result supports the idea that the M184I RT mutant will fail to support efficient proviral DNA synthesis in macrophage containing very low dNTP concentrations, as demonstrated in the biochemical experiments described in Fig. 2A. Interestingly, the M184I vector showed ∼50% of wild-type vector transduction efficiency in HLF, whereas this mutant vector showed 100% of the wild-type vector level of transduction efficiency in CHME5 cells containing higher dNTP concentration than HLFs. The ∼50% transduction efficiency of M184I vector in HLF suggests that the M184I mutant RT still cannot fully support reverse transcription at the dNTP concentrations found in HLF (200∼400 nm), which are lower than those found in CHME5 cells. These data are consistent with the biochemical data described in Fig. 2B showing that M184I RT displays only 50% of Met-184 Clone 3 RT at 0.25 μm dNTP. More interestingly, it was reported that, unlike M184I virus, M184V virus is able to infect macrophages (30, 31), and this observation is consistent with our biochemical data illustrated in Fig. 2A that M184V RT still maintains RT activity at least 50% of the wild-type RT activity even at low dNTP concentrations found in macrophages. Overall, these vector data are consistent with previous reports that M184I HIV-1 variants isolated during 3TC therapy displays greatly reduced infectivity in human macrophage (32, 33).
FIGURE 5.

Transduction of HIV-1 vectors containing wild-type and M184I RTs in human cell types. A, human macrophage, HLFs, and CHME5 cells were transduced by an equal p24 amount (3 × 105 pg) of wild-type and M184I HIV-1 vectors. eGFP expression of transduced cells was analyzed after 6 days for macrophage and after 24 h post-transduction for HLF and CHME5 cells by fluorescence microscopy. Both bright field (BF) and dark field (GFP) images are shown. B, the average transduction efficiency of duplicated experiments, as determined by FACS for GFP expression, and the transduction efficiency ratios between wild-type and M184I mutant vectors as well as cellular dNTP concentrations of each cell type are summarized. The dNTP concentration of macrophage and CHME5 cells was previously reported (16, 24).
Structural Comparison of the Polymerase Active Sites of Wild-type and M184I HIV-1 RTs—Finally, because the pre-steady-state kinetic data described in Table 1 demonstrate the reduced dNTP binding affinity of M184I RT, we attempted to compare the molecular shapes of the polymerase active sites in wild-type and M184I mutant RT proteins. For this, as shown in Fig. 6, we employed three separate structures of HIV-1 RT previously reported: 1) ternary complex of wild-type HIV-1 RT (WT RT·T·P·TTP) (34), 2) binary complex of wild-type HIV-1 RT (WT RT·T·P) (35), and 3) binary complex of M184I HIV-1 RT (MI RT·T·P) (8). We removed the T·Ps in these structures for better visualization of the dNTP binding pocket and the position of residue 184 (gray area of Fig. 6). The ternary complex of WT HIV-1 RT (Fig. 6A with TTP and Fig. 6B after removing TTP) allows us to visualize the surface and shape of the HIV-1 RT dNTP binding pocket. However, because the dNTP substrate targets the dNTP binding pocket of the binary complex (RT·T·P) during the formation of the ternary complex (RT·T·P·dNTP) that is capable of catalyzing the chemical reaction, we compared the molecular shapes of the more relevant binary complexes of the WT (Fig. 6C) and M184I (Fig. 6D) HIV-1 RTs. Unlike the wild-type residue, the Ile mutation indicated by the solid arrow in Fig. 6D exposes its larger/rigid β-branched side chain toward the dNTP binding site. It is plausible that this larger side chain of M184I restricts the entry of dNTP substrates from the solution into the dNTP binding pocket, resulting in the reduction of dNTP binding affinity. Furthermore, it can be clearly observed that the shape of the M184I dNTP binding pocket (Fig. 6D) appears to be widened, compared with that of the WT RT (Fig. 6C, see double-headed dotted arrows). This altered dNTP binding pocket of the M184I mutant may also explain its reduced ground state dNTP binding, compared with the WT RT.
FIGURE 6.

Comparison of the active site structure of wild-type and M184I HIV-1 RTs. A, the active site structure of the wild-type RT ternary complex with TTP (RT·T·P-TTP (34)) and B, the same structure after removing TTP, which allows us to visualize the surface and shape of the dNTP binding site. C, the binary complex of wild-type HIV-1 RT (RT·T·P (35)) and D, M184I RT (8). The residue 184 of each structure was labeled in gray. The larger β-branched side chain of the M184I binary complex (D) was marked with a thick arrow, and the dNTP binding space was marked with a dotted double-headed arrow. The T·Ps in these structures were removed for better visualization of the dNTP binding pocket of each structure.
DISCUSSION
Met-184 in HIV-1 RT is the second residue of the highly conserved YXDD motif that defines the polymerase active site of retroviral reverse transcriptases (36). The YXDD motif makes an unusual type II β-turn that allows the side chains of the four residues to make contacts with the template primer, metal cofactors, and dNTPs (8). Lentiviral RTs, including HIV-1 and SIV RTs, contain Met at the X position in the YXDD motif, whereas RTs of other groups of retroviruses (e.g. MuLV, a γ-retrovirus) mainly contain Val (YVDD) (37, 38). However, two mutations at the Met residue of HIV-1 RT, M184I and M184V, arise during the treatment of the RT inhibitor 3TC (lamivudine) rendering viral resistance to 3TC. Structural analyses suggest that the mechanism of resistance to 3TC of M184V and M184I mutations is a result of the steric hindrance between the β-branched amino acid side chains of the Val and Ile mutations and the unnatural (-) enantiomer sugar structure of 3TC (8, 9).
These 3TC resistance mutations confer several additional enzymatic alterations to HIV-1 RT. One such alteration is reduced viral DNA polymerization (32). Although a less significant difference was observed between viral replication kinetics of wild-type and 3TC resistant HIV-1 viruses in tissue culture infection of a T-cell line, the 3TC mutant viruses displayed delayed replication in the culture with peripheral blood mononuclear cells (32, 33). Gel analysis of reverse transcription products revealed that the 3TC-resistant mutants make shorter DNA products indicating their reduced processivity, compared with the wild-type RT. It is likely that this processivity reduction will be more pronounced under the environments of primary cells, whose dNTP concentrations are much lower than those of the transformed cell lines. In fact, intracellular dNTP levels vary significantly between different cell types and fluctuate during the cell cycle (39). Another enzymatic alteration is that both M184I and M184V showed higher fidelity compared with wild-type RT (13–15, 40).
The dNTP binding and template-primer interaction are two major mechanistic determinants for the processivity of DNA polymerase, which is defined as the number of nucleotides incorporated per single round of the primer extension (41, 42). Under saturating dNTP conditions, the RT·T·P interaction becomes a major factor for the RT processivity. In contrast, with limited dNTP substrates, HIV-1 RT likely stalls more frequently due to kinetic interference throughout the template sequences, and this can promote a higher probability of HIV-1 RT falling from the template, further restricting processive DNA synthesis (43). Even at relatively high dNTP concentrations where wild-type RT efficiently synthesizes DNA, RT mutants with reduced dNTP binding affinity likely exhibit distributive DNA synthesis due to disrupted DNA polymerization kinetics. This was observed with the Q151N- and V148I-reduced dNTP binding mutants of HIV-1 RT (22). We present kinetic evidence that, unlike M184V HIV-1 RT (12), the M184I mutation significantly reduces the binding affinity of HIV-1 RT to normal dNTP substrates without altering the kpol step. Indeed, the accompanying report (48) confirmed that M184I RT displays reduced processive DNA synthesis capability specifically at low dNTP concentrations. Moreover, the processivity defect was eliminated at saturating dNTP concentration, indicating that the effect of the M184I mutation on dNTP binding is the sole cause of the processivity defect.
In this study, we examined M184I HIV-1 RT variants directly isolated from a serum sample from an HIV-1 infected patient containing both M184I and Met-184 RT (wild-type like) variants, provided from the MACS program. The Met-184 RT variants isolated in the same serum sample were used for mechanistic comparison with the M184I RT variants. Due to the typical genetic hypervariable nature of HIV-1, no RT variants cloned from this single serum sample (among 10 clones) were genetically identical, and even among the three M184I RT variants, unique individual and/or combinations of other mutations were observed throughout their RT genetic sequences. The heterogeneous genetic backgrounds of individual RT clones can make it difficult to claim the role of a particular mutation or residue of interest to an overall RT phenotype. To minimize this difficulty, we constructed an I184M revertant RT of a representative M184I RT isolate (Clone 7 RT) and used this RT construct to study the specific enzymatic alterations made by the M184I mutation in these genetically varying RT variants. Only the M184I mutation in Clone 7 RT was reverted to I184 by site-directed mutagenesis. Other mutations within this clone remain unchanged, and the entire RT region of the revertant was sequenced to confirm that other mutations of the original Clone 7 RT remained intact in the revertant. As expected, the I184M reversion RT protein displayed identical enzymatic properties with the Met-184 RT clones (i.e. Clone 3), including 1) similar 3TC sensitivity, 2) high dNTP utilization efficiency and 3) tight dNTP binding affinity. This implies that other mutations contained in the Clone 7 RT variant do not significantly contribute to the biochemical alterations of the M184I mutation observed in this study. Consistent with previous studies, we also observed that the M184I Clone 7 RT variant showed a higher fidelity compared with Met-184 clone 3 RT.4
Finally, we attempted to elucidate how the M184I mutation restricts the dNTP binding of HIV-1 RT by comparing the active site structures of the binary complex form (RT·T·P) of wild-type and M184I HIV-1 RTs, which were previously solved. Previous structural analysis suggested that the two β-branch amino acid mutations (Val and Ile) at position 184 of HIV-1 RT introduce more rigid side chains to the active site, compared with the wild-type Met residue. The rigid side chains of the Val and Ile mutations occupy the space that is normally occupied by the L-form sugar moiety of the 3TCTP, which leads to the restricted binding and resistance to 3TCTP (8, 9). It was also demonstrated that the side chain of the M184I mutation also alters the RT interaction with the template nucleotide, which was used to explain the decreased processivity of the M184I RT (8). Interestingly, however, recent pre-steady-state kinetic analyses of M184V RT demonstrated that, unlike the M184I mutation, the M184V mutation did not significantly affect binding as well as chemical catalysis of HIV-1 RT to normal dNTPs, suggesting that β-branched side chain of the M184V mutation minimally affects the RT interaction with normal dNTP substrates (12).
As shown in Fig. 6D, in addition to sterically hindering entry of 3TCTP, the longer and rigid β-branched side chain of the M184I mutation appears to expose its long and rigid side chain toward the dNTP binding pocket, which can restrict the entry and binding of the incoming dNTP substrate (Fig. 6D). It is plausible that the direction and position of the incoming dNTP during entry into the dNTP binding pocket varies depending on the sequence of the template. This is supported by a wide range of dNTP incorporation kinetic variations during DNA synthesis with heterologous template sequences (12, 44, 45). Therefore, the proper space of the dNTP binding pocket of HIV-1 RT likely accommodates efficient entry and uniquely tight binding of dNTP substrates in the active site of HIV-1 RT, and this can facilitate the efficient DNA synthesis rate during reverse transcription with heterologous sequence templates such as the viral RNA genome even with limited dNTP pools found in non-dividing cells (i.e. macrophage). Because the M184V RT showed very similar dNTP binding affinity with WT RT, the interruption of dNTP entry into the dNTP binding pocket by the M184I mutation might not occur in the M184V RT with a shorter β-branched side chain (12). In addition, as displayed in Fig. 6 (C and D), the dNTP binding pocket of the M184I RT seems to be open more widely than that of WT RT, and, together with possible limited entry of dNTP, the deformation of the M184I dNTP binding pocket (Fig. 6D) may also contribute to destabilizing the ground state dNTP binding affinity of Met-184 RT.
Various virological studies have reported the instability of the M184I mutation using both tissue culture and animal models. The M184I mutant virus shows more attenuated viral replication kinetics than M184V virus or WT virus in peripheral blood mononuclear cells even though the M184I virus efficiently grows in established Sup T cell lines (32). The cellular dNTP concentration difference between peripheral blood mononuclear cells and Sup T cells was a suspected reason for the failure of the M184I mutant virus in peripheral blood mononuclear cells. This observation is consistent with our vector data described in Fig. 6. Indeed, the M184I vector, which efficiently transduced both HLF and CHME5 cells containing relatively higher dNTP concentrations, failed to transduce macrophage containing very low dNTP concentrations. Unlike the conditions of pre-steady-state Kd measurements, viral replication occurred in the steady state, where RT executed multiple rounds of DNA synthesis. Therefore, the steady-state DNA synthesis described in Fig. 2 is more relevant for addressing the effects of RT mutations on viral replication kinetics. M184I RT was still highly functional at 1 μm. Because viral replication occurred over a long time period (6–12 h), M184I RT was likely to complete proviral DNA synthesis even at ∼0.3 μm dNTP (HLF) as well as 1–3 μm (CHME5). The virological alteration made by the M184I RT mutation can be explained by our kinetic data that M184I RT has reduced dNTP binding affinity and reduced RT activity at low dNTP concentrations. As discussed above, the more significantly reduced RT processivity of M184I RT was suspected to be responsible for the reduced replication kinetics of the M184I virus. SIV has also been shown to develop very similar 3TC resistance profiles as HIV-1 (46). The pathogenesis of the SIV M184V variant is characterized in a macaque model (SIVmac239) (47). When the M184V mutation was maintained in animals treated with 3TC, the M184V SIV still induced a pathogenesis profile very similar to wild-type SIV. Interestingly, the M184V mutation quickly disappeared if the animals infected with the M184V virus were not treated with 3TC, suggesting that the M184V SIV is less fit than wild-type SIV. However, the pathogenesis in vivo of the M184I virus has not been characterized.
This study presents a line of biochemical evidence that, unlike the M184V mutant, the M184I mutant has reduced dNTP binding affinity, and this kinetic alteration may result from the interruption of the dNTP entry to the dNTP binding pocket. The reduced dNTP binding affinity of M184I RT, together with the altered interaction with template nucleotides, can synergistically restrict processive DNA synthesis under physiological dNTP concentrations, and these alterations caused by the M184I mutation could be mechanistic elements that contribute to the in vivo instability of the M184I mutation and its rapid conversion to M184V.
Acknowledgments
We thank the MACS program for providing the patient samples for this study.
This work was supported by Grants AI49781 (to B. K.) and R25GM64133 (to J. M. S.) from the National Institutes of Health. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: 3TC, (-)-2′,3′-deoxy-3′-thiacytidine; HIV-1, human immunodeficiency virus, type 1; RT, reverse transcriptase; T·P, template-primer complex; p66, 66-kDa HIV-1 RT polypeptide; MACS, Multicenter AIDS Cohort Study; DTT, dithiothreitol; MuLV, murine leukemia virus; SIV, simian immunodeficiency virus; HLF, human lung fibroblast; dN, deoxynucleoside; pol, polymerase; eGFP, enhanced green fluorescent protein; FACS, fluorescence-activated cell sorting; 3TCTP, 2′,3′-Dideoxy-3′-thia-cytidine-5′-triphosphate.
V. K. Jamburuthugoda, J. M. Santos-Velazquez, M. Skasko, D. J. Operario, V. Purohit, P. Chugh, E. A. Szymanski, J. E. Wedekind, R. A. Bambara, and B. Kim, our unpublished data.
References
- 1.Gulick, R. (1998) AIDS 12 17-22 [Google Scholar]
- 2.Mathe, C., and Gosselin, G. (2006) Antiviral Res. 71 276-281 [DOI] [PubMed] [Google Scholar]
- 3.Boucher, C. A., Cammack, N., Schipper, P., Schuurman, R., Rouse, P., Wainberg, M. A., and Cameron, J. M. (1993) Antimicrob. Agents Chemother. 37 2231-2234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gao, Q., Gu, Z., Parniak, M. A., Cameron, J., Cammack, N., Boucher, C., and Wainberg, M. A. (1993) Antimicrob. Agents Chemother. 37 1390-1392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schinazi, R. F., Lloyd, R. M., Jr., Nguyen, M. H., Cannon, D. L., McMillan, A., Ilksoy, N., Chu, C. K., Liotta, D. C., Bazmi, H. Z., and Mellors, J. W. (1993) Antimicrob. Agents Chemother. 37 875-881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tisdale, M., Kemp, S. D., Parry, N. R., and Larder, B. A. (1993) Proc. Natl. Acad. Sci. U. S. A. 90 5653-5656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Frost, S. D., Nijhuis, M., Schuurman, R., Boucher, C. A., and Brown, A. J. (2000) J. Virol. 74 6262-6268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sarafianos, S. G., Das, K., Clark, A. D., Jr., Ding, J., Boyer, P. L., Hughes, S. H., and Arnold, E. (1999) Proc. Natl. Acad. Sci. U. S. A. 96 10027-10032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gao, H. Q., Boyer, P. L., Sarafianos, S. G., Arnold, E., and Hughes, S. H. (2000) J. Mol. Biol. 300 403-418 [DOI] [PubMed] [Google Scholar]
- 10.Feng, J. Y., and Anderson, K. S. (1999) Biochemistry 38 55-63 [DOI] [PubMed] [Google Scholar]
- 11.Deval, J., White, K. L., Miller, M. D., Parkin, N. T., Courcambeck, J., Halfon, P., Selmi, B., Boretto, J., and Canard, B. (2004) J. Biol. Chem. 279 509-516 [DOI] [PubMed] [Google Scholar]
- 12.Feng, J. Y., and Anderson, K. S. (1999) Biochemistry 38 9440-9448 [DOI] [PubMed] [Google Scholar]
- 13.Rezende, L. F., Drosopoulos, W. C., and Prasad, V. R. (1998) Nucleic Acids Res. 26 3066-3072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Oude Essink, B. B., Back, N. K., and Berkhout, B. (1997) Nucleic Acids Res. 25 3212-3217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hsu, M., Inouye, P., Rezende, L., Richard, N., Li, Z., Prasad, V. R., and Wainberg, M. A. (1997) Nucleic Acids Res. 25 4532-4536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Diamond, T. L., Roshal, M., Jamburuthugoda, V. K., Reynolds, H. M., Merriam, A. R., Lee, K. Y., Balakrishnan, M., Bambara, R. A., Planelles, V., Dewhurst, S., and Kim, B. (2004) J. Biol. Chem. 279 51545-51553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Skasko, M., Weiss, K. K., Reynolds, H. M., Jamburuthugoda, V., Lee, K., and Kim, B. (2005) J. Biol. Chem. 280 12190-12200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Roberts, J. D., Preston, B. D., Johnston, L. A., Soni, A., Loeb, L. A., and Kunkel, T. A. (1989) Mol. Cell. Biol. 9 469-476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bakhanashvili, M., and Hizi, A. (1993) Biochemistry 32 7559-7567 [DOI] [PubMed] [Google Scholar]
- 20.Avidan, O., Meer, M. E., Oz, I., and Hizi, A. (2002) Eur. J. Biochem. 269 859-867 [DOI] [PubMed] [Google Scholar]
- 21.Operario, D. J., Reynolds, H. M., and Kim, B. (2005) Virology 25 106-121 [DOI] [PubMed] [Google Scholar]
- 22.Weiss, K. K., Chen, R., Skasko, M., Reynolds, H. M., Lee, K., Bambara, R. A., Mansky, L. M., and Kim, B. (2004) Biochemistry 43 4490-4500 [DOI] [PubMed] [Google Scholar]
- 23.Diamond, T. L., Souroullas, G., Weiss, K. K., Lee, K. Y., Bambara, R. A., Dewhurst, S., and Kim, B. (2003) J. Biol. Chem. 278 29913-29924 [DOI] [PubMed] [Google Scholar]
- 24.Jamburuthugoda, V. K., Chugh, P., and Kim, B. (2006) J. Biol. Chem. 281 13388-13395 [DOI] [PubMed] [Google Scholar]
- 25.Kim, B. (1997) Methods 12 318-324 [DOI] [PubMed] [Google Scholar]
- 26.Weiss, K. K., Isaacs, S. J., Tran, N. H., Adman, E. T., and Kim, B. (2000) Biochemistry 39 10684-10694 [DOI] [PubMed] [Google Scholar]
- 27.Johnson, K. A. (1993) Annu. Rev. Biochem. 62 685-713 [DOI] [PubMed] [Google Scholar]
- 28.Johnson, K. A. (1995) Methods Enzymol. 249 38-61 [DOI] [PubMed] [Google Scholar]
- 29.Weiss, K. K., Bambara, R. A., and Kim, B. (2002) J. Biol. Chem. 277 22662-22669 [DOI] [PubMed] [Google Scholar]
- 30.Aquaro, S., Svicher, V., Ceccherini-Silberstein, F., Cenci, A., Marcuccilli, F., Giannella, S., Marcon, L., Caliò, R., Balzarini, J., and Perno, C. F. (2005) J. Antimicrob. Chemother. 55 872-878 [DOI] [PubMed] [Google Scholar]
- 31.Perez-Bercoff, D., Wurtzer, S., Compain, S., Benech, H., and Clavel, F. (2007) J. Virol. 81 4540-4550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Back, N. K., Nijhuis, M., Keulen, W., Boucher, C. A., Oude Essink, B. O., van Kuilenburg, A. B., van Gennip, A. H., and Berkhout, B. (1996) EMBO J. 15 4040-4049 [PMC free article] [PubMed] [Google Scholar]
- 33.Back, N. K., and Berkhout, B. (1997) Antimicrob. Agents Chemother. 41 2484-2491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huang, H., Chopra, R., Verdine, G. L., and Harrison, S. C. (1998) Science 282 1669-1675 [DOI] [PubMed] [Google Scholar]
- 35.Ding, J., Das, K., Hsiou, Y., Sarafianos, S. G., Clark, A. D., Jacobo-Molina, A., Tantillo, C., Hughes, S. H., and Arnold, E. (1998) J. Mol. Biol. 284 1095-1111 [DOI] [PubMed] [Google Scholar]
- 36.Larder, B. A., Kemp, S. D., and Darby, G. (1987) Nature 327 716-717 [DOI] [PubMed] [Google Scholar]
- 37.Doolittle, R. F., Feng, D. F., Johnson, M. S., and McClure, M. A. (1989) Q. Rev. Biol. 64 1-30 [DOI] [PubMed] [Google Scholar]
- 38.Poch, O., Sauvaget, I., Delarue, M., and Tordo, N. (1989) EMBO J. 8 3867-3874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Traut, T. W. (1994) Mol. Cell Biochem. 140 1-22 [DOI] [PubMed] [Google Scholar]
- 40.Julias, J. G., Boyer, P. L., McWilliams, M. J., Alvord, W. G., and Hughes, S. H. (2004) Virology 322 13-21 [DOI] [PubMed] [Google Scholar]
- 41.Suo, Z., and Johnson, K. A. (1998) J. Biol. Chem. 273 27257-27267 [Google Scholar]
- 42.Bambara, R. A., Uyemura, D., and Choi, T. (1978) J. Biol. Chem. 253 413-423 [PubMed] [Google Scholar]
- 43.DeStefano, J. J., Mallaber, L. M., Rodriguez-Rodriguez, L., Fay, P. J., and Bambara, R. A. (1992) J. Virol. 66 6370-6378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vaccaro, J. A., Parnell, K. M., Terezakis, S. A., and Anderson, K. S. (2000) Antimicrob. Agents Chemother. 44 217-221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Selmi, B., Boretto, J., Sarfati, S. R., Guerreiro, C., and Canard, B. (2001) J. Biol. Chem. 276 48466-48472 [DOI] [PubMed] [Google Scholar]
- 46.Cherry, E., Slater, M., Salomon, H., Rud, E., and Wainberg, M. A. (1997) Antimicrob. Agents Chemother. 41 2763-2765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Van Rompay, K. K., Matthews, T. B., Higgins, J., Canfield, D. R., Tarara, R. P., Wainberg, M. A., Schinazi, R. F., Pedersen, N. C., and North, T. W. (2002) J. Virol. 76 6083-6092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gao, L., Hanson, M. N., Balakrishnan, M., Boyer, P. L., Roques, B. P., Hughes, S. H., Kim, B., and Bambara, R. A. (2008) J. Biol Chem. 283 9196-9205 [DOI] [PMC free article] [PubMed] [Google Scholar]