

Abstract
The C-terminal domain (CTD) of bacteriophage Mu immunity repressor (Rep) regulates DNA binding by the N-terminal domain and degradation by ClpXP protease. Five residues at the Rep C terminus (CTD5) can serve as a ClpX recognition motif, but it is dormant unless activated, a state that can be induced by the presence of dominant-negative mutant repressors (Vir). Conversion of Rep to ClpXP-sensitive form was associated with not only increased exposure of CTD5 to solvent but also increased CTD motion or flexibility as measured by fluorescence anisotropy. CTD mutations (V183S, K193S, and V196S) promoting ClpXP resistance without destroying the recognition motif prevented increased CTD motion induced by Vir. Suppression of ClpXP protease resistance conferred by the V196S mutation also correlated with restoration of CTD motion. The temperature-sensitive R47Q mutation present in cis within the DNA-binding domain restored ClpXP protease sensitivity to the V196S mutant, and anisotropy analysis indicated that R47Q allows the V196S CTD to gain increased flexibility when Vir was present. The results indicate that the CTD functions to turn the recognition motif on and off, most likely by modulating flexibility of the domain that harbors the ClpX recognition motif, suggesting a general mechanism by which proteins can regulate their own degradation.
Bacteriophage Mu immunity repressor (Rep)3 establishes and maintains lysogeny by binding to Mu DNA segments (O1, O2, and O3) that act as both operator (1, 2) and transposition enhancer (3, 4), shutting down transposition functions necessary for Mu replication. Certain physiological conditions, such as starvation or entry into stationary phase (S derepression), can promote degradation or inactivation of the repressor, leading to derepression of these transposition functions (5–8). The C-terminal domain (CTD) of Rep not only contains the recognition motif for initiating proteolysis but also modulates association of the N-terminal DNA-binding domain (DBD) with DNA.
In the wild-type repressor (Rep), the CTD is located in close proximity to the DBD (9), a state that we refer to as the closed conformation and that may sterically inhibit interactions of the DBD with DNA. Upon DNA binding, the CTD moves away from the DBD (open conformation). At elevated temperatures, the CTD of temperature-sensitive (ts) repressors, such as the cts62 repressor, which has a R47Q mutation in the DBD, fails to move, and DNA binding is prevented (9). The CTD plays a major role in eliciting the temperature-sensitive DNA binding properties of these mutants; deletions (10) and single amino acid replacements (11) within the CTD can suppress ts mutations to restore stable binding at elevated temperatures. The interconversion of repressor between protease-sensitive and protease-resistant forms also correlates with movement of the CTD; however, the relevant property affected in this interconversion is not the proximity of the CTD to the DBD but rather features such as the exposure of the CTD to solvent (12). Nevertheless, the repressor bound to DNA is not as readily converted to the protease-sensitive form as the unbound form (9, 13).
Although Rep is a substrate of the ClpXP protease (13), it appears to be predominantly in the protease-resistant conformation in purified form. Previously, we determined that the last 5 residues (VKKAV or CTD5) of Rep can serve as a ClpX recognition motif, being able to promote degradation of a heterologous substrate when attached to its C terminus; however, whether the protein is degraded apparently depends upon how this recognition motif is presented (14). Rep can rapidly be degraded when it is heat-denatured (11) or when it is in the presence of dominant negative Vir repressors, which have altered C termini and are highly susceptible to degradation by ClpXP (13, 15–17). The emission spectrum of a fluorescent probe attached to the Rep C terminus has indicated that CTD5 is in a relatively hydrophobic environment (9, 12); however, the presence of Vir induces the Rep CTD5 to enter a more polar environment (9, 12), most likely rendering CTD5 more accessible for recognition by ClpX protein.
ClpX, a member of the AAA+ family of ATPases (18), confers substrate specificity to ClpXP protease by binding the recognition motifs of protein substrates, ultimately unfolding and delivering them to the inner chamber of a double-ringed, serine protease ClpP (19–21). The ClpX subunit consists of an N-terminal zinc-binding domain (residues 1–60) followed by an AAA+ domain. The zinc-binding domain provides a low affinity binding site for ClpX substrates as well as a tethering site that interacts with adaptor proteins, which function to bind ClpX substrates and tether them to ClpX for their delivery to the active site (22–24). The AAA+ domain, on the other hand, functions in direct binding of the substrate and the application of mechanical pulling force starting at the recognition motif to unfold the substrate. ClpX substrates have amino acid sequences that act as degradation tags or recognition motifs, and although ClpX is able to bind internal motifs (25), most are located at either the N or C terminus of the substrate (26–29).
The CTD movement associated with the interconversion between ClpXP-sensitive and -resistant forms of the repressor has suggested an important role in regulating activity of its recognition motif. In addition, cysteine scan mutagenesis has indicated that mutations at 8 individual positions in the CTD (Ile-170 to Val-196 or CTD27; Fig. 1A) confer ClpXP resistance in the presence of Vir (11). Six of these mutations are located outside the CTD5 recognition motif, implying that the CTD may provide more than just a recognition motif. Two of the CTD mutations (V183C and V196C) promoted ClpXP resistance even after heat denaturation (11), raising the question of whether exposure of CTD5 is sufficient to promote degradation of Rep. Furthermore, when CTD5 is attached to the C terminus of green fluorescent protein (GFP), its ability to promote GFP degradation is highly dependent upon the presence of a disordered linker (e.g. the 41 C-terminal residues of the CcdA protein (CcdA41)) between the GFP and CTD5 motif (14). These results suggest that the CTD5 of Rep must be presented in the context of a disordered or flexible domain to trigger Rep degradation.
FIGURE 1.

Wild-type repressor, repressor mutants, and GFP fusion proteins. A, the three domains of the repressor. The DBD participates in DNA binding, and mutations known to cause thermosensitivity of DNA binding (e.g. cts62) are located within this domain (2). The LRD is believed to be required for repressor oligomerization (10). The CTD contains the CTD5 recognition motif and modulates both DNA binding and ClpXP degradation. To attach MIANS to the C terminus of a repressor molecule, we used an engineered repressor protein in which Cys-17 in the repressor DBD is changed to an alanine and a cysteine is added to the C-terminal end of the repressor (RepC197 and VirC192). B, the last 27 residues (CTD27) of Rep and the corresponding sequence of RepV183S, RepK193S, and RepV196S repressors. The CTD5 residues (ClpX recognition motif) are indicated in boldface type. C, GFP fusion proteins. CcdA41 is a 41-residue linker derived from the C-terminal sequence of CcdA protein. In all cases, the mutated residues are marked with an asterisk.
Here we report on the influence of CTD mutations on the presentation of the CTD5 recognition motif as monitored by fluorometric analysis. The results indicate that the exposure of CTD5 is not sufficient for activation and that the conversion of Rep to its ClpXP-sensitive form also correlates with increased flexibility at the CTD5 site. The Rep CTD therefore not only harbors a ClpX recognition motif but also functions in turning the motif on and off by regulating its presentation. This most likely plays a vital role in regulating transposition by allowing Rep to promote its own degradation in response to environmental or physiological cues.
EXPERIMENTAL PROCEDURES
Bacterial Strains, Plasmids, and Proteins—Expression vectors pHS502 (Rep), pHS504 (Vir3060), pGTN206 (VirC17A, where the single cysteine at residue 17 has been replaced with an alanine), pGTN204 (RepC197, which has an engineered C-terminal cysteine at residue 197 and the C17A alteration), and pGTN211 (VirC192; engineered C-terminal cysteine at residue 192 and the C17A alteration) were used to express the indicated proteins, which were purified as previously described (11–13). The QuikChange™ site-directed mutagenesis kit (Stratagene) was used to introduce single amino acid replacements in the repressor coding sequence. ClpX (30) and ClpP (31) were purified as previously described.
GFP (pHS783), GFP with the CcdA41 linker (GFP-CcdA41; pHS776), and GFP with CTD5 attached to the C terminus via the CcdA41 linker (GFP-CcdA41-CTD5; pHS779) were expressed from the indicated plasmids and purified as previously described (14). Mutations were introduced into the CTD5 coding sequence of GFP-CcdA41-CTD5 by replacing the NheI to HindIII fragment of pHS779, the NheI site located just upstream of the CTD5 coding sequence and HindIII located downstream of the stop codon. Complementary oligonucleotides were designed to contain the mutated CTD5 sequences and to have cohesive NheI and HindIII half-sites for ligation into these sites. When required, a codon for a C-terminal cysteine residue was added to provide a binding site for the fluorescent probe.
All repressor proteins used in this work have the C17A alteration and an added cysteine residue at the C terminus with the following exceptions. First, wild-type repressor retaining Cys-17 was used when attaching the fluorescent probe to this residue within the DBD. Second, Vir that does not have the C17A alteration or added cysteine residue was used in all repressor degradation assays. Third, no additional cysteine was present for the V196C mutant, in which the C-terminal valine was changed to a cysteine residue. Finally, where indicated, properties of repressor proteins with or without the cysteine alterations were compared as control. Neither the C17A nor the added C-terminal cysteine residue have any effect on DNA binding and protease sensitivity of Rep and Vir (supplemental Fig. S1, A–C), as previously described (9, 12). The repressor protein with the C17A mutation and the added C-terminal cysteine (i.e. a unique cysteine at the C-terminal end) therefore provides an appropriate background to examine the effect of single amino acid replacements, such as V196C and V196S. These single mutations confer ClpXP protease resistance when introduced not only into the wild-type repressor but also into the repressor with a unique cysteine at the C-terminal end (supplemental Fig. S1D). Moreover, a cysteine residue added to the C-terminal end of CTD5 does not affect its function as the ClpX recognition motif, indicated by its activity attached to the C terminus of the heterologous protein GFP-CcdA41 (supplemental Fig. S1E).
ClpX and ClpP protein concentrations were determined by the method of Bradford (32), using bovine serum albumin standard and dye reagent purchased from Bio-Rad. Concentrations of all repressor proteins and GFP fusion proteins were determined using calculated molar extinction coefficients, as previously described (33). All concentrations are expressed in equivalents of protein monomers.
ClpXP Protease Assays—Protein degradation assays were all conducted at 37 °C as previously described (14). ClpX and ClpP were present in reaction mixtures at final concentrations of 0.9 and 1.8 μm, respectively, unless otherwise indicated. All gels, stained with Coomassie Blue R-250, were photographed with an Eastman Kodak Co. Gel Logic 100 digital camera. The level of protein degradation was determined from the image using the associated Kodak Molecular Imaging Software, setting the level of protein substrate present at 0 min as 100%.
In assays monitoring degradation of GFP fusion proteins, GFP fluorescence was monitored in 160-μl reaction mixtures at an excitation wavelength of 488 nm and emission wavelength of 509 nm with constant mixing and slit widths set to 3 nm. All degradation assays were performed at least four times, and representative results are shown.
HindIII Protection Assay—Binding of repressor to the Mu operator in pGG215, which bears a mini-Mu element (34), was measured by an assay previously described (2) with the following modifications. Reaction mixtures (30 μl) contained 20 μg/ml pGG215 with varying repressor concentrations (0–8 μm) in 10 mm Tris-HCl (pH 7.9), 50 mm NaCl, 10 mm magnesium chloride, 1 mm dithiothreitol and were incubated at 30 °C and 42 °C for 15 min prior to HindIII digestion and agarose gel electrophoresis (11). All DNA binding assays were performed at least three times, and representative results are shown.
Fluorescence Spectra Determination—Fluorescence measurements were made using a Photon Technology International QuantaMaster spectrofluorometer and FeliX32 software. All analyses were conducted in 160 μl of Buffer A with a path length of 0.4 cm. Samples were mixed and maintained at constant temperature during measurements, using a magnetic stirrer and a circulating water bath.
To measure 2-(4′-maleimidylanilino)naphthalene-6-sulfonic acid (MIANS; Molecular Probes) fluorescence, each reaction mixture contained either a repressor protein that has a single cysteine residue or a GFP fusion protein that has a cysteine introduced at its C terminus (1.5 μm) and 4.5 μm cysteineless VirC17A where indicated. Attachment of MIANS to the proteins and quantification of bound MIANS were performed as described previously (9). A 3-fold excess of MIANS (4.5 μm) was added to each reaction mixture in Buffer A, which was then incubated at 37 °C unless otherwise indicated. Over 90% of the repressor proteins were bound by MIANS after 10 min of incubation, and fluorescence measurements were made at this time. All reaction mixtures containing multiple repressor proteins were incubated for 10 min at 37 °C prior to the addition of MIANS. MIANS fluorescence was monitored at an excitation wavelength of 330 nm unless otherwise stated with slit widths set to 3 nm.
Fluorescence resonance energy transfer from the two tryptophan residues of the Rep DBD (Trp-16 and Trp-44) to MIANS attached to repressor C terminus was measured as previously described (9). Reaction mixtures contained 0.7 μm repressor protein with 7.6 μm MIANS and 6.2 μg/ml pGG215 as indicated. The emission spectrum fluorescence was monitored at an excitation wavelength of 295 nm at either 30 or 42 °C (slit widths 2 nm). All spectra were obtained in at least three independent determinations, and representative spectra are shown.
Steady-state Fluorescence Anisotropy Measurements—Fluorescence anisotropy measurements were performed using the Photon Technology International spectrofluorometer. The FeliX32 software used with the Photon Technology International spectrofluorometer provides automatic polarizer control and calculation of anisotropy, taking into account the G-factor, the correction factor for differential sensitivity to vertically and horizontally polarized light (35).
Measurements were made in Buffer A containing 1.5 μm repressor protein, 4.5 μm MIANS, and when indicated, 4.5 μm cysteineless VirC17A (160-μl reaction mixture with constant stirring). Readings were made at 25 °C with excitation and emission wavelengths of 330 and 420 nm, respectively (slit widths 3 nm). The anisotropy values were generated over 60 s in each of two consecutive readings, which were averaged. The final reported value is the average of at least three separate determinations made in this way, expressing the error as the S.D.
Anisotropy measurements were taken at various glycerol concentrations (0–60% w/v). Viscosities of glycerol solutions were obtained from measurements summarized in a standard table that outlines the physical properties of aqueous glycerol solutions (36). Samples were equilibrated at 25 °C for 3–5 min before anisotropy readings were taken. The data were plotted as 1/A versus T/η (Perrin plot), where A is the anisotropy of the protein in solution, T is the temperature in Kelvin, and η is the viscosity of the solvent (in centipoises). The linear plot, determined by calculating the least squares fit, was extrapolated to T/η = 0 to obtain the y intercept (1/Ao), where Ao is the apparent anisotropy of the fluorescent probe when extrapolated to infinite viscosity (37).
The measured anisotropy A at 0% glycerol (η = 0.89 centipoises) and the extrapolated Ao value were used to calculate the rotational correlation time (τc) of MIANS attached to each protein using the Perrin equation (37).
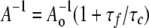 |
(Eq. 1) |
The fluorescence lifetime of the fluorophore MIANS (τf) has previously been determined to be 6 ns (38). To obtain consistent A-1o values (y intercept of the Perrin plot) for calculation of τc, outliers at the highest and lowest solution viscosities were omitted from the linear regression analysis when they deviated significantly from the core majority values (r2 ≥ 0.9). However, all data points are included in Perrin plots shown in the figures.
In this study, we examined decreases in the rotational correlation time τc associated with increased local motion in a protein domain. τc measured as described above would reflect all types of motion of the protein, not only local motion at the attachment site but the overall rotation of the protein as a whole. To verify that the high mobility reflected by low τc values (∼1 ns) is indeed due to local motion, we estimated the correlation time reflecting only the overall rotation of the whole protein (τcWHOLE) at 0.89 centipoises (viscosity of water at 25 °C), using Equation 2 (39).
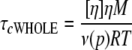 |
(Eq. 2) |
where M is the molecular mass (∼21 kDa for Mu repressor), [η] is the intrinsic viscosity of the protein solution, R is the gas constant, T is the temperature in Kelvin, and ν(p) is the Simha factor. The Simha factor is a coefficient that depends on the molecular shape of the protein of interest. For the purpose of calculating correlation times, Mu repressor was assumed to be a spherical molecule, and a Simha factor of 2.5 was used (40). For intrinsic viscosity, we used the value 3 ml/g, an average of experimental values calculated for various proteins (37). Using these values, we calculated a correlation time (τcWHOLE) of 9 ns for Mu repressor as monomer. Even after considering other possible values for ν(p) and [η] (37), our calculations indicated that τcWHOLE only varied from 8.6 to 10.3 ns. More importantly, correlation times are longer for larger molecules or complexes, which would have more restricted motion, and Mu repressor is known to exist in hexameric and even higher order oligomeric forms in solution (41, 42). Such oligomeric complexes would make up a complex of at least 130 kDa; therefore, the τcWHOLE of the repressor protein is more likely 54 ns or more. Correlation times for MIANS bound to GFP fusion proteins, when calculated in a similar fashion for the protein monomer, yielded estimates of τcWHOLE of 9.6–13.0 ns.
RESULTS
Design and Properties of Engineered Repressor Proteins Used to Examine CTD Movement—Several single amino acid replacements within the repressor CTD confer resistance to degradation by ClpXP even when Vir is present (11). To determine how these mutations affect CTD movement and recognition of the CTD5 motif by ClpX, a fluorescent probe (MIANS) was attached to purified repressor proteins via an engineered C-terminal cysteine (residue 197 in Rep and residue 192 in Vir; Fig. 1A). The single cysteine of Mu repressor at residue 17 was also replaced with an alanine (C17A) such that the C-terminal residue is the sole cysteine on the protein. The C17A mutation and the added C-terminal cysteine residue do not affect properties of repressor as DNA-binding protein or ClpXP substrate (supplemental Fig. S1, A–C).
Mutants with single amino acid changes in the CTD were previously constructed by replacement with cysteine residues (11); however, since we wished to have a single cysteine at the C terminus of mutant repressor proteins, we constructed mutant proteins (RepV183S, RepK193S, and RepV196S; Fig. 1B) in which the amino acid replacement was made with serine instead of cysteine. Analogous to the properties of the corresponding cysteine replacement mutants, all three serine replacement mutants were resistant to ClpXP in the presence of Vir (Fig. 2, B–D).
FIGURE 2.

V183S, K193S, and V196S mutations affect the sensitivity of the repressor to ClpXP. Susceptibility of the indicated repressor proteins (2 μm) to degradation by ClpXP was tested in the presence and absence of Vir (2 μm). A, Rep; B, RepV183S; C, RepK193S; D, RepV196S; E, cts62RepV196S. ClpP levels remain constant throughout the reaction and therefore serve as loading control.
We have previously determined that Rep is by itself a ClpXP substrate that is degraded with a relatively high Michaelis constant (Km); Vir not only has a much lower Km but induces Rep to have a comparably low Km, thus inducing its rapid degradation (13). In contrast, RepV183S, RepK193S, and RepV196S were all found to be ClpXP-resistant. For example, at very high ClpXP concentrations and prolonged incubation time, we are able to detect Rep degradation in the presence of Vir. Under these conditions, neither RepV183S and RepV196S were degraded even when Vir was present (supplemental Fig. S2A).
Like RepV196C (11), RepV196S protected Vir from degradation (Fig. 2D, lane 4), whereas RepV183S and RepK193S did not (Fig. 2, B and C, lane 4). Moreover, the cts62 mutation (R47Q) present in cis to V196S acts as a suppressor, allowing Vir-induced degradation of the cts62RepV196S by ClpXP (Fig. 2E, lane 4). On the other hand, the cts62 mutation present in cis to either V183S or K193S did not promote Vir-induced degradation of the mutant repressor proteins (Supplemental Fig. S2B).
The K193S and V196S Mutations Do Not Inactivate CTD5 as a ClpX Recognition Motif—Since both the K193S and V196S substitutions are within the CTD5 sequence that makes up the ClpX recognition motif, we wished to determine whether they inactivate the ClpX recognition motif. Vir can promote degradation of cts62RepV196S (Fig. 2E, lane 4), suggesting that CTD5 V196S is still a functional recognition motif. However, to further investigate whether amino acid replacements within the CTD5 motif destroys its potential to act as a ClpX recognition motif, the wild-type and mutant CTD5 segments were attached to the C terminus of the heterologous GFP-CcdA41 protein. (Numbering of the 5 residues of CTD5 will correspond to their position on the full-length repressor protein, even when it is attached to a heterologous protein.)
The GFP-CcdA41-CTD5 V196C and the GFP-CcdA41-CTD5 V196S proteins were degraded at even a higher initial rate (744 ± 49 and 616 ± 15 fmol s-1, respectively) than the same protein that has wild-type CTD5 (224 ± 24 fmol s-1) (Fig. 3, A and B), indicating that the V196C and V196S mutations do not destroy CTD5 as a ClpX recognition motif. Although GFP-CcdA41-CTD5 K193S was not degraded as rapidly (103 ± 18 fmol s-1) as the same GFP substrate with the wild-type CTD5 tag, it was clearly a ClpXP substrate, evident when compared with GFP-CcdA41-CTD5 A195D, which is resistant to ClpXP (Fig. 3, A and B). The ClpX recognition signal is generally inactivated by replacement of one of the last two C-terminal residues with an aspartate (26). Thus, the K193S, V196S, and V196C mutations promote ClpXP resistance of the repressor protein without destroying the potential of CTD5 to act as a ClpX recognition motif, suggesting that these mutations affect the presentation of CTD5 as an active recognition motif in the context of the repressor protein.
FIGURE 3.

The V196C, V196S, and K193S mutations do not destroy the CTD5 ClpX recognition motif. A, each of the indicated GFP fusion proteins was incubated with ClpXP, and GFP fluorescence was monitored to measure its degradation. B, the initial rate of GFP degradation was determined from data points obtained after the initial decrease in fluorescence that occurs during mixing of GFP substrates with ClpXP. When over 20% of the total GFP was degraded over the 40-min incubation period, we calculated the initial degradation rate of 10% of the GFP substrate. With protease-resistant substrates where there was less than 20% decrease in total GFP fluorescence over 40 min, we determined the apparent initial rates over a 500-s period. The apparent degradation rate of ClpXP-resistant GFP-CcdA41, which was essentially identical to results obtained when it was incubated in the absence of ClpXP, was subtracted from all determined degradation rates. Initial degradation rates are reported as the rate of GFP degradation in the standard 160-μl reaction mixture. They are the average of four determinations, with error expressed as the S.D.
Exposure of CTD5 Alone Is Not Sufficient for Its Activation as a ClpX Recognition Motif in the Repressor—The environmentally sensitive properties of MIANS, which becomes fluorescent only upon being covalently attached to a cysteine residue (43), make it an ideal probe for measuring movement of the repressor C terminus from a hydrophobic to a more hydrophilic environment.
MIANS bound to the C terminus of RepV183S and RepK193S had an emission maximum of 429 and 430 nm, respectively (Fig. 4, B and C), virtually identical to MIANS bound to the C terminus of Rep (Fig. 4A). Analogous to conformational changes it induces in Rep (Fig. 4A), VirC17A caused the C termini of RepV183S and RepK193S to enter a more hydrophilic environment, indicated by the emission maxima of 447 and 450 nm, respectively, of MIANS bound to these proteins (Fig. 4, B and C). On the other hand, the C terminus of RepV196S was already in a relatively hydrophilic environment. The attached MIANS had an emission maximum of 440 nm, and VirC17A was not able to promote an additional red shift (Fig. 4D). As described above, the cts62 mutation suppresses ClpXP resistance elicited by the V196S mutation. Consistent with these results, VirC17A promoted a significant change in the emission spectrum of MIANS attached to the C terminus of cts62RepV196S. The spectrum exhibited a red shift to 449 nm with a significant decrease in fluorescence intensity (Fig. 4D), indicating that VirC17A induces the C terminus to become even more exposed to the aqueous environment.
FIGURE 4.

Exposure of CTD5 is not sufficient to activate it as degradation tag. Shown is fluorescence emission of MIANS attached to the C terminus of Rep (RepC197) (A); RepK193S (B); RepV183S (C); RepV196S and cts62RepV196S (D); GFP, GFP-CTD5, GFP-CcdA41-CTD5, and background (Buffer A) (E); and GFP-CcdA41-CTD5 V196C, GFP-CcdA41-CTD5 K193S, and GFP-CcdA41-CTD5 V196S (F). When present, VirC17A is indicated in parentheses to note that MIANS does not bind to it.
To evaluate whether relative exposure of the CTD5 motif attached to the C terminus of GFP is an important determinant of whether it can promote GFP degradation, we engineered a cysteine at the C-terminal end of each GFP fusion substrate so that MIANS could be attached. Although there are 2 cysteine residues in GFP, they are not accessible for the binding of the fluorophore (44). No detectable MIANS was conjugated to GFP if it did not have an appendage with a C-terminal cysteine (Fig. 4E). Although CTD5 can only promote GFP degradation when the CcdA41 linker is present, MIANS bound to the C terminus of both GFP-CTD5 and GFP-CcdA41-CTD5 exhibited nearly the same emission spectrum with a maximum of 433 nm (Fig. 4E), indicating that the C terminus of the protease-sensitive substrate is not necessarily more exposed to solvent.
When MIANS was bound to GFP-CcdA41-CTD5 K193S, it displayed an emission spectrum similar to GFP-CTD5 with an emission maximum of 430 nm (Fig. 4F). MIANS bound to GFP-CcdA41-CTD5 V196C (the cysteine replacement also serving the role as the MIANS attachment site), and GFP-CcdA41-CTD5 V196S had an emission maximum of 435 and 443 nm, respectively, and a significantly decreased fluorescence intensity (Fig. 4F). These results indicate that the V196C and V196S mutations cause the C terminus of the GFP fusion substrates to enter a more polar environment, most likely due to the replacement of a hydrophobic amino acid with a polar one. However, these mutations cause more rapid degradation of the protein substrate only when the mutant CTD5 is attached to the GFP-CcdA41 substrate and not when it is attached to the repressor. All of these results support the hypothesis that other properties of CTD5 besides its exposure to solvent, most likely properties conferred by the disordered conformation of the CcdA41 linker, may contribute to providing an active recognition motif.
The V196S Mutation Promotes an Open Conformation That Favors a DNA-bound, ClpXP-resistant Form—When bound to DNA, Rep becomes relatively resistant to ClpXP. We therefore determined whether the V196C and V196S mutations induce Rep to assume a conformation analogous to the DNA-bound form.
We first tested whether V196S mutation, like the V196C mutation (11), can suppress in cis the temperature-sensitive DNA binding property of cts62. The cts62RepV196S protein stably binds to Mu operator DNA at both 30 and 42 °C (Fig. 5, B and C), measured in vitro by detecting the protection of one of two HindIII sites located on the Mu DNA substrate plasmid (Fig. 5A). In this assay, repressor binding to Mu DNA protects the HindIII site within the O2 operator region while leaving the other HindIII site located outside the operator unprotected (2, 11, 13). DNA binding properties of cts62RepV183S (Fig. 5C) and cts62RepK193S (data not shown) were however similar to that of cts62Rep, being able to stably bind DNA at 30 °C but not at 42 °C.
FIGURE 5.

The V196S mutation suppresses the thermosensitive DNA binding property conferred by cts62. A, schematic of pGG215 plasmid (34) and the HindIII protection assay. The mini-Mu element harbored on the plasmid is indicated in boldface type. The plasmid has two HindIII sites, one of which is located within the O2 operator region and one located outside of O2. Upon binding of repressor, the HindIII site within the O2 is protected from cleavage. B, reaction mixtures containing cts62Rep or cts62RepV196S and 20 μg/ml pGG215 DNA were incubated at 30 or 42 °C, and the DNA was resolved by agarose gel electrophoresis. The concentration of the repressor protein present in each mixture is shown above each lane (in μm). The two bands obtained from cleavage at both HindIII sites are labeled U to indicate that the HindIII site within O2 is unprotected. The single 7.2 kb band obtained by cleavage only at the HindIII site outside the operator region is labeled P to designate that O2 is protected. The additional slowly migrating band (*) arising at higher concentrations of cts62RepV196S corresponds to plasmid forms that have not been cut at all by HindIII. This arises from nonspecific DNA binding that also protects the HindIII site outside O2. C, quantification of DNA binding by Rep (▪), cts62Rep (□), RepV183S (•), cts62RepV183S (○), RepV196S (▴), and cts62RepV196S (▵) as measured by the HindIII protection analysis. Protection of the O2 HindIII site was quantified as described in the legend to supplemental Fig. S1. Quantification includes the results shown in B plus assays conducted at additional repressor concentrations to cover the full 0–8 μm range. The inset focuses on binding at 0–2 μm repressor concentrations. No detectable binding of cts62RepV183S and cts62Rep could be measured at 42 °C; therefore, the two binding curves overlap at 0% DNA bound.
To determine whether CTD mutations affect the position of the CTD and its movement with respect to the DBD, fluorescence resonance energy transfer analysis was conducted to assess the relative proximity of the C terminus to the DBD. In this analysis, we examined energy transfer from the repressor's two tryptophan residues (Trp-16 and Trp-44), both located in the DBD, to MIANS attached to the C terminus. Tryptophan fluorescence is evident as a single emission peak for cts62Rep at both 30 and 42 °C (Fig. 6, A and B). As previously determined (9), attachment of MIANS at either temperature results in quenching of this peak and the appearance of an additional peak at a longer wavelength that corresponded to MIANS fluorescence, indicating that the C terminus and DBD are in close proximity (Fig. 6, A and B). Binding of cts62Rep to DNA at 30 °C causes energy transfer to be greatly diminished (Fig. 6A), indicating that the repressor has assumed the open conformation. However, at 42 °C cts62Rep fails to bind to DNA, and the high level of energy transfer is observed in both the presence and absence of DNA, indicating that the CTD fails to move away from the DBD and repressor remains in the closed conformation (Fig. 6B). The same results were obtained with both cts62RepV183S (Fig. 6, C and D) and cts62RepK193S (data not shown), indicating that they fail to assume the open conformation at the elevated temperature in the presence of DNA.
FIGURE 6.

The V196S mutation induces the open conformation of the repressor. Fluorescence resonance energy transfer between the tryptophan residues (Trp-16 and Trp-44) in the repressor DBD and MIANS attached to the repressor C terminus was monitored. For each set (A–F), fluorescence of repressor protein with or without MIANS was monitored at an excitation wavelength of 295 nm. Where indicated, pGG215 DNA (6.2 μg/ml) was present. MIANS emission spectrum is denoted by an asterisk.
In contrast, little to no energy transfer was observed with cts62RepV196S at either 30 or 42 °C, regardless of whether DNA was present. The single tryptophan emission peak was essentially unchanged by attachment of MIANS (Fig. 6, E and F), indicating that this protein is in the open conformation even in the absence of DNA. The interpretation that V196S locks the protein in an open conformation is consistent with the finding that RepV196S and cts62RepV196S have a higher affinity for DNA than Rep at both 30 and 42 °C (Fig. 5, B and C). Thus, unlike the V183S and K193S mutations, the V196S mutation prevents the close proximity of the repressor C terminus in the native conformation. RepV196S may generally have properties of repressor proteins in the open conformation, which tend to confer resistance to ClpXP.
Vir Not Only Promotes Exposure of CTD5 but Also Increases Its Local Flexibility—The major question raised by our results so far is what causes the CTD5 motif of Rep to become active. A related question is why proteins such as RepV196S and RepV183S are resistant to ClpXP although they bear a functional ClpX recognition motif that can be exposed to solvent. We previously determined that the CTD5 motif is inactive when attached directly to the heterologous substrate GFP, whereas CTD5 is active when attached to GFP via the disordered CcdA41 linker (14). We therefore considered the possibility that local protein flexibility at the CTD5 recognition motif may be required for CTD5 to be active. To determine local flexibility of the repressor molecules in the protease-resistant and -sensitive forms, we chose to examine anisotropy of a fluorescent probe attached to the C termini of the repressor proteins. Anisotropy measurements were made at varying solution viscosity (Perrin plot analysis) to assess local motion at the attachment site, the level of motion being a measure of local flexibility.
The rotational correlation time reflecting how fast a protein rotates as a whole in solution (τcWHOLE) was calculated to be 9 ns for the repressor monomer (see “Experimental Procedures”). τcWHOLE would be longer for larger molecules or complexes, which would rotate more slowly. Since the repressor is known to assume a hexameric or higher order oligomeric structure in solution (41, 42), the rotational correlation time is most likely 54 ns or greater. However, as further described below, Perrin plot analysis conducted with MIANS attached to the C terminus of wild-type and mutant repressor molecules yielded correlation times (τc) of ∼1 ns when those repressors were highly susceptible to degradation by ClpXP (Table 1). These values are considerably lower than τcWHOLE calculated for the repressor even as a monomer, indicating that the values predominantly reflect high local motion at the C terminus. In general, the differences in slopes of Perrin plots (Tables 1 and 2) reflect differences in the mobility of attached MIANS, with increased steepness reflecting greater motion. When such motion is associated with correlation times (τc) that are considerably less than τcWHOLE, it must reflect increased local mobility at the site of MIANS attachment.
TABLE 1.
Parameters derived from Perrin plots of MIANS-labeled repressor molecules Where indicated, MIANS was attached either to Cys-17 (DBD) or to the C-terminal cysteine (CTD). In all other cases, MIANS was attached to the C-terminal cysteine of the repressor proteins. When present, VirC17A is indicated in parentheses as a reminder that it does not bind MIANS.
| Protein | y intercept (1/Ao) | Slope (centipoises/K × 103) | τc |
|---|---|---|---|
| ns | |||
| Rep | |||
| DBD | 3.8 ± 0.1 | 17 ± 2 | 5.1 ± 0.2 |
| CTD | 3.8 ± 0.2 | 15.8 ± 0.4 | 5.4 ± 0.5 |
| Vir | |||
| DBD | 3.4 ± 0.3 | 6.7 ± 1 | 4.6 ± 0.6 |
| CTD | 3.8 ± 0.1 | 64.0 ± 0.3 | 1.0 ± 0 |
| Rep/(VirC17A) | |||
| DBD | 3.8 ± 0.2 | 7.8 ± 0.5 | 6.5 ± 0.6 |
| CTD | 3.8 ± 0.2 | 69 ± 3 | 1.0 ± 0.1 |
| RepK193S | 3.9 ± 0.3 | 7.0 ± 0.4 | 8.5 ± 1.7 |
| RepK193S/(VirC17A) | 4.0 ± 0.6 | 8.5 ± 0.1 | 8.8 ± 3.8 |
| cts62RepK193S | 4.0 ± 0.2 | 12.0 ± 0.1 | 6.9 ± 1.6 |
| cts62RepK193S/(VirC17A) | 3.5 ± 0.3 | 13.6 ± 0.3 | 7.1 ± 1.7 |
| RepV183S | 3.8 ± 0.2 | 16 ± 2 | 6.9 ± 0.7 |
| RepV183S/(VirC17A) | 4.0 ± 0.2 | 4.7 ± 0.2 | 13 ± 2 |
| cts62RepV183S | 4.4 ± 0.4 | 9.2 ± 0.3 | 7.8 ± 1.8 |
| cts62RepV183S/(VirC17A) | 4.4 ± 0.3 | 4.3 ± 0.1 | 18 ± 7 |
| RepV196S | 3.9 ± 0.2 | 5.6 ± 0.1 | 14.3 ± 0.5 |
| RepV196S/(VirC17A) | 4.4 ± 0.3 | 25.2 ± 0.2 | 7.0 ± 1.0 |
| cts62RepV196S | 4.3 ± 0.2 | 7.2 ± 0.3 | 27 ± 7 |
| cts62RepV196S/(VirC17A) | 4.0 ± 0.2 | 48.4 ± 0.2 | 1.4 ± 0.2 |
TABLE 2.
Parameters derived from Perrin plots of MIANS-labeled GFP fusion proteins MIANS was attached to the C-terminal cysteine residue.
| Protein | y intercept (1/Ao) | Slope (centipoises/K × 103) | τc |
|---|---|---|---|
| ns | |||
| GFP-CTD5 | 3.9 ± 0.3 | 7.4 ± 0.1 | 5.1 ± 0.6 |
| GFP-CcdA41-CTD5 | 3.9 ± 0 | 31.6 ± 0.9 | 2.2 ± 0.1 |
| GFP-CcdA41-CTD5 K193S | 4.6 ± 0.1 | 31 ± 1 | 2.8 ± 0.2 |
| GFP-CcdA41-CTD5 V196S | 4.4 ± 0.4 | 24 ± 1 | 2.9 ± 0.5 |
| GFP-CcdA41-CTD5 V196C | 4.2 ± 0.2 | 29 ± 1 | 2.2 ± 0.1 |
Perrin plot analyses of MIANS bound to the DBD of Vir and Rep and to the Rep C terminus indicate relatively little motion or flexibility (Table 1 and Fig. 7, B and C). In contrast, MIANS bound to Rep C terminus reported greater motion when VirC17A was also present, analogous to the motion reported by MIANS attached to the Vir C terminus (Table 1 and Fig. 7C). On the other hand, MIANS bound to the Rep DBD (Cys-17) did not report greater motion when VirC17A was present (Table 1 and Fig. 7B) (i.e. the C terminus of Rep, but not the DBD, acquires high flexibility when Vir is present). These results raise the question of whether the high flexibility at the Rep C terminus is a critical feature needed to activate the CTD5 degradation tag.
FIGURE 7.

Vir induces local flexibility of Rep CTD5. Anisotropy of MIANS attached to the indicated proteins was measured at varying glycerol concentrations (0–60%, w/v). The data are shown as a Perrin plots. A, MIANS attached to the C terminus of GFP-CTD5 and GFP-CcdA41-CTD5; B, MIANS attached to Cys-17 (DBD) of Rep and Vir; C, MIANS attached to the C terminus of Rep (RepC197) and Vir (VirC192). When present, VirC17A is indicated in parentheses to note that MIANS does not bind to it.
Whether CTD5 attached to GFP is active as a degradation tag also correlated with local flexibility at CTD5. MIANS bound to the C terminus of the protease-resistant GFP-CTD5 demonstrated relatively little flexibility (Table 2 and Fig. 7A). In contrast, the protease-sensitive GFP-CcdA41-CTD5 displayed significantly higher flexibility at its C terminus (Table 2 and Fig. 7A). These results strongly suggest that local flexibility at the CTD5 attachment site plays a critical role in activating CTD5 as a recognition motif.
CTD Mutations That Confer ClpXP Protease Resistance Decrease Local Flexibility of CTD5—To further examine whether increased local flexibility at CTD5 is associated with its activation, we examined the effect of V183S, K193S, and V196S mutations that confer protease resistance. Perrin plot analysis of MIANS bound to the C terminus of RepV183S, RepK193S, and RepV196S indicated low levels of flexibility even in the presence of VirC17A (Table 1 and Fig. 8, A–C). The presence of the cts62 mutation in cis to the V196S mutation restored high local flexibility at the C terminus induced by Vir (Fig. 8C), consistent with the ability of cts62 to suppress protease resistance conferred by V196S. In contrast, Vir was unable to induce high C-terminal flexibility of cts62RepV183S and cts62RepK193S (Fig. 8, A and B), consistent with our findings that these double mutants remain protease resistant under these conditions. MIANS attached to the C terminus of the protease-sensitive GFP-CcdA41-CTD5 K193S, GFP-CcdA41-CTD5 V196S, and GFP-CcdA41-CTD5 V196C also reported a high degree of flexibility at their C termini (Fig. 8D and Table 2). These results indicate a strong correlation between the acquisition of high flexibility at CTD5 and the conversion of the repressor into a form rapidly degraded by ClpXP protease, implying that the increased flexibility plays a critical role in activating CTD5 as a ClpX recognition motif.
FIGURE 8.

ClpXP resistance of repressor mutants correlates with the inability to acquire increased local flexibility at the C terminus. Anisotropy of MIANS attached to the C terminus of the indicated repressor proteins were measured at varying glycerol concentrations (0–60%, w/v). Data are shown as Perrin plots. A, RepK193S and cts62RepK193S; B, RepV183S and cts62RepV183S; C, RepV196S and cts62RepV196S; D, GFP-CTD5, GFP-CcdA41-CTD5, GFP-CcdA41-CTD5 K193S, GFP-CcdA41-CTD5 V196S, and GFP-CcdA41-CTD5 V196C. When present, VirC17A is indicated in parentheses to note that MIANS does not bind to it.
DISCUSSION
Activation of a ClpX Recognition Motif by Increased Local Flexibility—The CTD of the Mu repressor plays a pivotal role in regulating repressor activity in vivo, being able to modulate both DNA binding and susceptibility to protease digestion. Deletion of the CTD not only suppresses temperature-sensitive mutations in the DBD but also renders the protein resistant to degradation by ClpXP protease (10). When repressor proteins lacking the CTD accumulate, derepression of Mu that normally results under conditions of starvation or stationary phase conditions (S derepression) cannot be triggered (6). We have postulated from these features of CTD deletion mutants that the CTD may function to inactivate the repressor under certain physiological conditions. There are single amino acid replacements both inside and outside of the CTD5 motif, promoting ClpXP resistance in the presence of Vir. This has suggested that at least some of these mutations may alter CTD movement necessary to activate the normally dormant ClpX recognition motif. In this work, we have demonstrated that such single-amino acid replacements can affect movement, positioning, and flexibility of CTD5, preventing its activation as a recognition motif.
The single-amino acid replacement mutants chosen for this study affect CTD movement in different ways. Both the V196C and V196S mutations not only confer protease resistance but also suppress the cts62 (R47Q) mutation in the DBD, allowing stable binding to DNA at elevated temperatures. Although the C terminus of Rep moves away from the DBD upon DNA binding, the C terminus of RepV196C and RepV196S is already positioned away from the DBD in what we call the open conformation. Although the V196C and V196S mutations prevent movement of the CTD into a more polar environment, the CTD is already in a relatively exposed configuration, most likely the result of replacing a nonpolar amino acid with a polar one.
In contrast, the V183S and K193S mutations do not suppress the cts62 mutations, although they confer protease resistance without destroying the CTD5 motif. As with Rep, the C terminus of both RepV183S and RepK193S moves away from the DBD with DNA binding, and CTD movement of both cts62RepV183S and cts62RepK193S is prevented at 42 °C when DNA is added. The C terminus of both RepV183S and K193S also become exposed in response to Vir, but the proteins remain resistant to degradation by ClpXP.
The one common feature of all protease-resistant forms of the repressor is that the C terminus is relatively motionless, reflected by high anisotropy of an attached fluorescent probe. Fluorescent probes attached to the C terminus of protease-sensitive Vir or Rep in the presence of Vir report considerable local motion with respect to the rest of the molecule, indicating a high level of flexibility at the site of the recognition motif. RepV196S, RepV183S, and RepK193S all exhibit relatively little motion (lower flexibility) at the C terminus even in the presence of Vir. The cts62 mutation present in cis can only confer protease sensitivity to RepV196S among these three mutants, and accordingly, Vir was able to induce C-terminal motion in cts62RepV196S but not in cts62RepV183S and cts62RepK193S. These results indicate high local motion at CTD5 when it is activated as a recognition motif, and mutations able to prevent this motion effectively prevent its activation by Vir.
Possible Role of Local Flexibility in Activating a Recognition Motif—The enhanced local flexibility at CTD5 could increase the rate of repressor degradation by ClpXP in at least two ways. First, it could facilitate unfolding of the repressor for delivery into the inner chamber of ClpP tetradecamer, where the protease active sites are located. Second, it could enhance the recognition of the ClpX recognition motif by increasing ligand flexibility of CTD5. These two possibilities are not mutually exclusive.
The C-terminal flexibility of protease-sensitive repressor may reflect local protein instability at the site of the recognition motif, enhancing unfolding initiated by ClpX at that site (45, 46). Resistance of ClpXP substrates to unfolding and degradation has been found to be strongly dependent on local protein stability rather than global protein stability (47, 48). These studies suggest that ClpX can lose grip of the protein substrate as it applies mechanical pulling force at the recognition motif if the local protein structure resists unfolding. Moreover, ClpX may also lose grip if the recognition motif is weak such that ClpX binds with low affinity. Destabilization of the local protein structure at the Rep C terminus may allow ClpX to apply a sufficient force pulling at a weak recognition motif to initiate protein unfolding.
Alternatively, increased flexibility of the Rep C terminus may activate a weak or inactive recognition motif such that ClpX binds with high efficiency. One effect would be to increase accessibility of the recognition motif so that it can be bound by ClpX. In addition, increased ligand flexibility of CTD5 could increase its affinity for ClpX. There are examples of ligands that achieve binding diversity and specificity through conformational disorder (49–52). These studies indicate that conformational flexibility can potentially increase binding affinity not only by increasing the diversity of ligand conformations but also by the gain of entropy that results from displacement of water molecules around the ligand as it is bound and by increasing the rate of sampling of ligand conformations.
trans-Targeting of ClpX substrates such as peptides with a C-terminal ssrA tag, UmuD′, and σS subunit of RNA polymerase involves the association of the respective delivery factors SspB (53), UmuD (54), and RssB (55), which provide a tethering motif bound by the zinc-binding domain of ClpX. Current thinking is that anchoring of the substrate to ClpX via this tethering motif orients the recognition motif (the degradation tag) for binding by ClpX AAA+ domain and the application of force that unfolds the substrate. However, our recent work has indicated that Vir-induced degradation of Rep can be catalyzed by ClpX that lacks the zinc-binding domain.4 This indicates that Vir-induced degradation of Rep involves a different type of trans-targeting mechanism. Instead of an adaptor protein providing a tethering motif, the change takes place in the substrate to be degraded such that a dormant degradation tag recognized by the AAA+ domain is activated.
Supplementary Material
Acknowledgments
We thank Olga Tcherkasskaya for discussions and assistance in carrying out the steady-state fluorescence anisotropy and Perrin plot analysis. We thank Dawn A. Defenbaugh for help in purifying the GFP fusion proteins. We thank Jessica Jones for critical reading of the manuscript.
This work was supported by National Institutes of Health Grant GM58265 (to H. N.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1 and S2.
Footnotes
The abbreviations used are: Rep, wild-type Mu repressor; DBD, DNA-binding domain; CTD, C-terminal domain; CcdA41, peptide containing 41 C-terminal residues of F plasmid CcdA41 protein; GFP, green fluorescent protein; MIANS, 2-(4′-maleimidylanilino)naphthalene-6-sulfonic acid; RepC197, repressor with a C17A alteration and a cysteine residue added at the C terminus; Vir, repressor encoded by virulent vir3060 mutant of Mu; VirC192, Vir repressor with a C17A alteration and a cysteine added at the C terminus; VirC17A, cysteineless Vir repressor having a C17A alteration; ts, temperature-sensitive; AAA+, ATPases associated with diverse cellular activities.
Marshall-Batty, K. R., and Nakai, H. (2008) Mol. Microbiol. 67, 920–933.
References
- 1.Krause, H. M., and Higgins, N. P. (1986) J. Biol. Chem. 261 3744-3752 [PubMed] [Google Scholar]
- 2.Vogel, J. L., Li, Z. J., Howe, M. M., Toussaint, A., and Higgins, N. P. (1991) J. Bacteriol. 173 6568-6577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Leung, P. C., Teplow, D. B., and Harshey, R. M. (1989) Nature 338 656-658 [DOI] [PubMed] [Google Scholar]
- 4.Mizuuchi, M., and Mizuuchi, K. (1989) Cell 58 399-408 [DOI] [PubMed] [Google Scholar]
- 5.Lamrani, S., Ranquet, C., Gama, M. J., Nakai, H., Shapiro, J. A., Toussaint, A., and Maenhaut-Michel, G. (1999) Mol. Microbiol. 32 327-343 [DOI] [PubMed] [Google Scholar]
- 6.Ranquet, C., Geiselmann, J., and Toussaint, A. (2001) Proc. Natl. Acad. Sci. U. S. A. 98 10220-10225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shapiro, J. A., and Higgins, N. P. (1989) J. Bacteriol. 171 5975-5986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shapiro, J. A., and Hsu, C. (1989) J. Bacteriol. 171 5963-5974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rai, S. S., O'Handley, D., and Nakai, H. (2001) J. Mol. Biol. 312 311-322 [DOI] [PubMed] [Google Scholar]
- 10.Vogel, J. L., Geuskens, V., Desmet, L., Higgins, N. P., and Toussaint, A. (1996) Genetics 142 661-672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mukhopadhyay, B., Marshall-Batty, K. R., Kim, B. D., O'Handley, D., and Nakai, H. (2003) Mol. Microbiol. 47 171-182 [DOI] [PubMed] [Google Scholar]
- 12.Marshall-Batty, K. R., and Nakai, H. (2003) J. Biol. Chem. 278 1612-1617 [DOI] [PubMed] [Google Scholar]
- 13.Welty, D. J., Jones, J. M., and Nakai, H. (1997) J. Mol. Biol. 272 31-41 [DOI] [PubMed] [Google Scholar]
- 14.Defenbaugh, D. A., and Nakai, H. (2003) J. Biol. Chem. 278 52333-52339 [DOI] [PubMed] [Google Scholar]
- 15.Geuskens, V., Mhammedi-Alaoui, A., Desmet, L., and Toussaint, A. (1992) EMBO J. 11 5121-5127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Laachouch, J. E., Desmet, L., Geuskens, V., Grimaud, R., and Toussaint, A. (1996) EMBO J. 15 437-444 [PMC free article] [PubMed] [Google Scholar]
- 17.Mhammedi-Alaoui, A., Pato, M., Gama, M.-J., and Toussaint, A. (1994) Mol. Microbiol. 11 1109-1116 [DOI] [PubMed] [Google Scholar]
- 18.Neuwald, A. F., Aravind, L., Spouge, J. L., and Koonin, E. V. (1999) Genome Res. 9 27-43 [PubMed] [Google Scholar]
- 19.Grimaud, R., Kessel, M., Beuron, F., Steven, A. C., and Maurizi, M. R. (1998) J. Biol. Chem. 273 12476-12481 [DOI] [PubMed] [Google Scholar]
- 20.Kim, Y. I., Burton, R. E., Burton, B. M., Sauer, R. T., and Baker, T. A. (2000) Mol. Cell 5 639-648 [DOI] [PubMed] [Google Scholar]
- 21.Wang, J., Hartling, J. A., and Flanagan, J. M. (1997) Cell 91 447-456 [DOI] [PubMed] [Google Scholar]
- 22.Bolon, D. N., Wah, D. A., Hersch, G. L., Baker, T. A., and Sauer, R. T. (2004) Mol. Cell 13 443-449 [DOI] [PubMed] [Google Scholar]
- 23.Dougan, D. A., Weber-Ban, E., and Bukau, B. (2003) Mol. Cell 12 373-380 [DOI] [PubMed] [Google Scholar]
- 24.Wojtyra, U. A., Thibault, G., Tuite, A., and Houry, W. A. (2003) J. Biol. Chem. 278 48981-48990 [DOI] [PubMed] [Google Scholar]
- 25.Hoskins, J. R., Yanagihara, K., Mizuuchi, K., and Wickner, S. (2002) Proc. Natl. Acad. Sci. U. S. A. 99 11037-11042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Flynn, J. M., Levchenko, I., Seidel, M., Wickner, S. H., Sauer, R. T., and Baker, T. A. (2001) Proc. Natl. Acad. Sci. U. S. A. 98 10584-10589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Flynn, J. M., Neher, S. B., Kim, Y.-I., Sauer, R. T., and Baker, T. A. (2003) Mol. Cell 11 671-683 [DOI] [PubMed] [Google Scholar]
- 28.Gonciarz-Swiatek, M., Wawrzynow, A., Um, S. J., Learn, B. A., McMacken, R., Kelley, W. L., Georgopoulos, C., Sliekers, O., and Zylicz, M. (1999) J. Biol. Chem. 274 13999-14005 [DOI] [PubMed] [Google Scholar]
- 29.Levchenko, I., Yamauchi, M., and Baker, T. A. (1997) Genes Dev. 11 1561-1572 [DOI] [PubMed] [Google Scholar]
- 30.Kruklitis, R., Welty, D. J., and Nakai, H. (1996) EMBO J. 15 935-944 [PMC free article] [PubMed] [Google Scholar]
- 31.Jones, J. M., Welty, D. J., and Nakai, H. (1998) J. Biol. Chem. 273 459-465 [DOI] [PubMed] [Google Scholar]
- 32.Bradford, M. M. (1976) Anal. Biochem. 72 248-254 [DOI] [PubMed] [Google Scholar]
- 33.Pace, C. N., Vajdos, F., Fee, L., Grimsley, G., and Gray, T. (1995) Protein Sci. 4 2411-2423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Surette, M. G., Buch, S. J., and Chaconas, G. (1987) Cell 49 253-262 [DOI] [PubMed] [Google Scholar]
- 35.Lackowicz, J. R. (1999) Principles of Fluorescence Spectroscopy, 2nd Ed., Kluwer Academic/Plenum Publishers, New York
- 36.Weast, R. C. (1985) CRC Handbook of Chemistry and Physics, 66th Ed., CRC Press, Inc., Boca Raton, FL
- 37.Cantor, C. R., and Schimmel, P. R. (1980) Biophysical Chemistry: Techniques for the Study of Biological Structure and Function, W. H. Freeman and Co., New York
- 38.Stapelfeldt, H., Olsen, C. E., and Skibsted, L. H. (1999) J. Agric. Food Chem. 47 3986-3990 [DOI] [PubMed] [Google Scholar]
- 39.Tcherkasskaya, O., Ptitsyn, O. B., and Knutson, J. R. (2000) Biochemistry 39 1879-1889 [DOI] [PubMed] [Google Scholar]
- 40.Van Holde, K. E. (1971) Physical Biochemistry, Prentice-Hall, Inc., Englewood Cliffs, NJ
- 41.Alazard, R., Ebel, C., Venien-Bryan, V., Mourey, L., Samama, J. P., and Chandler, M. (1998) Eur. J. Biochem. 252 408-415 [DOI] [PubMed] [Google Scholar]
- 42.Rousseau, P., Bétermier, M., Chandler, M., and Alazard, R. (1996) J. Biol. Chem. 271 9739-9745 [DOI] [PubMed] [Google Scholar]
- 43.Haugland, R. P. (2002) Handbook of Fluorescent Probes and Research Products, 9th Ed., Molecular Probes, Inc., Eugene, OR
- 44.Plafker, K., and Macara, I. G. (2002) J. Biol. Chem. 277 30121-30127 [DOI] [PubMed] [Google Scholar]
- 45.Lee, C., Schwartz, M. P., Prakash, S., Iwakura, M., and Matouschek, A. (2001) Mol. Cell 7 627-637 [DOI] [PubMed] [Google Scholar]
- 46.Matouschek, A. (2003) Curr. Opin. Struct. Biol. 13 98-109 [DOI] [PubMed] [Google Scholar]
- 47.Kenniston, J. A., Baker, T. A., Fernandez, J. M., and Sauer, R. T. (2003) Cell 114 511-520 [DOI] [PubMed] [Google Scholar]
- 48.Kenniston, J. A., Burton, R. E., Siddiqui, S. M., Baker, T. A., and Sauer, R. T. (2004) J. Struct. Biol. 146 130-140 [DOI] [PubMed] [Google Scholar]
- 49.Hauer, J. A., Taylor, S. S., and Johnson, D. A. (1999) Biochemistry 38 6774-6780 [DOI] [PubMed] [Google Scholar]
- 50.Jorgensen, C. I., Kallipolitis, B. H., and Valentin-Hansen, P. (1998) Mol. Microbiol. 27 41-50 [DOI] [PubMed] [Google Scholar]
- 51.Kriwacki, R. W., Hengst, L., Tennant, L., Reed, S. I., and Wright, P. E. (1996) Proc. Natl. Acad. Sci. U. S. A. 93 11504-11509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vajda, S., Weng, Z., Rosenfeld, R., and DeLisi, C. (1994) Biochemistry 33 13977-13988 [DOI] [PubMed] [Google Scholar]
- 53.Wah, D. A., Levchenko, I., Baker, T. A., and Sauer, R. T. (2002) Chem. Biol. 9 1237-1245 [DOI] [PubMed] [Google Scholar]
- 54.Neher, S. B., Sauer, R. T., and Baker, T. A. (2003) Proc. Natl. Acad. Sci. U. S. A. 100 13219-13224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhou, Y., Gottesman, S., Hoskins, J. R., Maurizi, M. R., and Wickner, S. (2001) Genes Dev. 15 627-637 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.