

Abstract
Lucina pectinata ctenidia harbor three heme proteins: sulfide-reactive hemoglobin I (HbILp) and the oxygen transporting hemoglobins II and III (HbIILp and HbIIILp) that remain unaffected by the presence of H2S. The mechanisms used by these three proteins for their function, including ligand control, remain unknown. The crystal structure of oxygen-bound HbIILp shows a dimeric oxyHbIILp where oxygen is tightly anchored to the heme through hydrogen bonds with Tyr30(B10) and Gln65(E7). The heme group is buried farther within HbIILp than in HbILp. The proximal His97(F8) is hydrogen bonded to a water molecule, which interacts electrostatically with a propionate group, resulting in a Fe-His vibration at 211 cm-1. The combined effects of the HbIILp small heme pocket, the hydrogen bonding network, the His97 trans-effect, and the orientation of the oxygen molecule confer stability to the oxy-HbIILp complex. Oxidation of HbILp Phe(B10) → Tyr and HbIILp only occurs when the pH is decreased from pH 7.5 to 5.0. Structural and resonance Raman spectroscopy studies suggest that HbIILp oxygen binding and transport to the host bacteria may be regulated by the dynamic displacements of the Gln65(E7) and Tyr30(B10) pair toward the heme to protect it from changes in the heme oxidation state from FeII to FeIII.
Hemoglobins are key proteins in symbiotic relationships between invertebrates and chemoautotrophic bacteria. The clam Lucina pectinata inhabits the sulfide-rich tropical mud and produces three different types of hemoglobin in its gills, first described by Read (1) and characterized by Kraus and Wittenberg (2, 3). Different functionalities have been described for the three hemoglobin variants: hemoglobin I (HbILp)2 is a sulfide-reactive monomeric protein, whereas hemoglobin II and III (HbIILp and HbIIILp) are oxygen transporters. HbIILp and HbIIILp self-associate in a concentration-dependent manner forming a tetramer that remains unaffected in the presence of H2S (2, 3). The mechanisms underlying ligand selection control between HbILp and HbIILp/HbIIILp remain unknown. The structure of HbILp has been solved with different ligands bound at the distal position: water (Protein Data Bank code 1FLP) (4), cyanide (PDB code 1BOB) (5), and hydrosulfuric acid (PDB code 1MOH) (6). The primary structure of HbIILp was determined by Edman degradation (7) and from its cDNA sequence (8). HbILp only shares 32% of its amino acid sequence with HbIILp, but both have a conserved glutamine in the E7 position, and a phenylalanine and a tyrosine in the B10 position for HbILp and HbIILp, respectively. The Tyr(B10) and Gln(E7) distal heme pocket residues of HbIILp are also found in other hemoglobins (8, 9). Hemoglobins from Ascaris suum (HbAsc) (Tyr(B10) and Gln(E7)) (10) and Mycobacterium tuberculosis hemoglobin N (Tyr(B10) and Leu(E7)) (11, 12) show high oxygen affinity and a very slow release of the bound oxygen. The three different hemoglobins from the clam Scapharca inaequivalvis (HbSi) show a very different heme pocket (Met(B10) and His(E7)) (13). HbIIA, Si and HbIIB, Si, assemble to form a heterotetramer (like HbIILp and HbIIILp), whereas HbI, Si forms homodimers (like HbIILp). The different types of hemoglobin from this clam bind oxygen cooperatively. HbIIB, Si exhibits a Hill coefficient for oxygen binding of 2.1 when in a heterotetramer, whereas the homodimer has a Hill coefficient of 1.5 (14). Interestingly, even though the S. inaequivalvis hemoglobins and L. pectinata HbIILp and HbIIILp share ≥40% amino acid similarity, the Lucina hemoglobins do not behave cooperatively (2).
Early biophysical studies (15) compared the structure and functionality of HbIILp and hemoglobin from A. suum because both heme proteins have heme pockets with Tyr(B10) and Gln(E7). The low oxygen off rates were attributed to differences in the hydrogen bonding network of both oxy-protein complexes. Additional studies using UV-visible spectroscopy and pH titration of HbIILp and a HbILp Phe(B10) → Tyr mutant have revealed bands at 486, 541, 577, and 603 nm for both proteins at neutral conditions (3, 9). At basic pH values, the barrier for the reaction increases as the tyrosine adopts a closed conformation and the heme (FeIII) hydroxyl complex replaces the met-aquo species, which suggested the existence of an open and closed conformation due to the interactions between Tyr(B10) and the heme iron. The presence of these conformers were confirmed by resonance Raman spectroscopy showing that, in a neutral environment, met-aquo HbIILp was present as a mixture of coordination and spin states with values for the v2 mode at 1558 cm-1 (6C HS) and 1580 cm-1 (6C LS) and for the v3 mode at 1479 cm-1 (6C HS), 1492 cm-1 (5C), and 1503 cm-1 (6C LS) (9). The infrared spectra of the complex HbIILpCO also showed the presence of the A3 (closed) and A0 (open) conformers at 1924 and 1964 cm-1, respectively (9). We proposed that in the open conformation Tyr(B10) swings away from the heme iron, whereas in the closed conformation Tyr(B10) is closer to and may interact with the ligand. Similarly, the reactions between hydrogen peroxide and both HbIILp and the HbILp Phe(B10) → Tyr mutant showed that Tyr(B10) tailors, in two very distinct ways, the reactivity of compounds I and II ferryl species (9). First, increasing the reaction pH from 4.9 to 7.5, and then to 11.2, the second order rate constant for HbIILp decreases from 141.60 to 77.78 m-1 s-1, and to 2.96 m-1 s-1, respectively. This pH dependence is associated with the disruption of the heme-tyrosine (603 nm) protein moiety, which controls access of H2O2 to the heme protein active center, thus regulating the formation of the ferryl species. Second, the existence of a hydrogen bonding network between the heme pocket amino acids (i.e. Tyr(B10)) and ferryl compound I created a much faster path than 3.0 × 10-2 s-1 for the decay of compound I to compound II. Moreover, the presence of Tyr(B10) in HbIILp and the HbILpPhe(B10) → Tyr mutant appears to afford a more stable O2 adduct in the oxygenated HbILp. The contribution of Tyr(B10) to the stability of the HbIILp and HbILp Phe(B10) → Tyr heme pocket against peroxide attack has recently been shown to be partially due to the presence of hydrogen bonding between the ferryl moiety and the heme pocket amino acids, including Tyr(B10), which ultimately enhances the removal of peroxide by the peroxidative cycle (16, 17). In addition, the close proximity of Tyr(B10) with Gln(E7) to the heme iron contributes largely to the distal control of NO binding, thus providing a model for the design of future oxidative stable oxygen hemoglobins with little or no vasoactivity (16).
Here, we report detailed structural data of HbIILp obtained by x-ray crystallography, providing information about the heme pocket, distal amino acids, and their interaction with the dioxygen molecule. Our results, supported by resonance Raman measurements, suggest a mechanism where HbIILp selects oxygen by tailoring the hydrogen bonding environment between the Tyr(B10) and Gln(E7) and heme-O2 moiety, stabilizing the heme (FeII) oxidation state.
EXPERIMENTAL PROCEDURES
Wild-type HbILp and HbIILp Sample Preparations—Proteins were isolated and purified as described with minor modifications (9). HbILp was separated from HbIILp/HbIIILp using a Hi Load 26/60 Superdex 200 gel filtration column (AKTA FPLC, Amersham Biosciences). HbILp was further purified by cation exchange chromatography using DEAE Sephadex Fast Flow equilibrated with 25 mm ammonium bicarbonate buffer, pH 8.3. HbIILp was purified from the HbIILp/HbIIILp fraction by ion exchange chromatography with a HiPrep 16/10 Q FF column equilibrated with 10 mm triethanolamine/acetate buffer at pH 8.3 and eluted with a gradient of sodium chloride concentration from 0 to 180 mm. The purity of the proteins was confirmed by SDS-PAGE.
Recombinant HbILp and Site-directed Mutant Preparation—The mutants from L. pectinata HbI (HbILpPhe(B10) → Tyr, HbILpGln(E7)Val, HbILpGln(E7) → Asn, and HbILpGln(E7) → His) were obtained by introducing single amino acid substitution using the QuikChange Mutagenesis kit (Stratagene, La Jolla) into the HbILp coding region cloned into the pET28(a+) vector (18). The HbILp mutants were expressed in Escherichiacoli Bli5 cells as described (18). Dark red cell pellets were lysed and centrifuged to separate the soluble from the insoluble fractions. The soluble fraction was equilibrated with CO and purified in Co2+ affinity columns (Talon, Invitrogen) followed by size exclusion chromatography (AKTA FPLC, Amersham Biosciences) (9). The purified HbILPPhe(B10) → Tyr showed UV-visible spectra typical of an oxyHbIILP. Met-aquo and CO HbILPPhe(B10) → Tyr derivatives were obtained in a similar manner as the HbIILP complexes (18).
HbIILp Sequence Analysis—Amino acid sequences were obtained from the NCBI protein sequence data base. The accession numbers for L. pectinata hemoglobins are HbI AAG01380, HbII AAO89499, and HbIII AAB28352. Data base searches for sequences with high sequence similarity was performed using the HbIILP sequence and PSI-BLAST (19). Multiple sequence alignments were performed with ClustalW (20). Sequence alignments were used to assess the variation at specific positions and the resulting differences in the overall structure and distal heme cavity.
Crystallization and Data Collection—HbIILP was crystallized as previously reported (21). In brief, lyophilized HbIILP was dissolved in 50 mm BisTris propane pH 7.0 buffer, 0.5 mm EDTA in a final concentration of 30 mg/ml. The crystals were grown by the counter-diffusion technique with a three-chamber configuration (22) using ammonium sulfate as precipitant. X-ray diffraction intensity data were collected at the BM-16 station of the European Synchrotron Radiation Facility (ESRF) using a 0.97-Å wavelength in a Mar CCD-165 detector. Data were indexed, integrated, and scaled with HKL2000 suite (23) at a resolution limit of 1.93 Å.
Structure Solution and Refinement—The structure of HbIILP was solved by molecular replacement methods as reported in Ref. 21. In brief, coordinates from HbILp (PDB code 1EBT) without the water molecule present in the distal position of the heme iron were used as a search model and the molecular replacement solution was found using the CNS suite (24). Refinement was conducted using CNS and REFMAC5 software from CCP4 (25). After several cycles of restrained refinement, manual model building against the electron density maps was conducted with the program COOT (26). Once all the residues were replaced by the HbIILP sequence, the R-factor and R-free decreased to 0.30 and 0.36, respectively. Additional refinement was carried out with REFMAC5 using the TLS parameters (27). The inclusion of TLS parameters in the refinement process improved the R-factor and R-free to a final value of 0.17 and 0.19, respectively. Water molecules were placed in electron density difference maps using the ARP/wARP version 5.0 program from the CCP4 suite. All the structures were checked using the refinement with MolProbity (28), and before deposition using PROCHECK (29). Details on data collection and structure refinement are summarized in Table 1. The secondary structure was tested with iMolTalk (30), whereas β-turn geometry was calculated using the program PROMOTIF version 2.0 (31). Superposition and root mean square deviations of the structures were performed using the CCP4 program LSQKAB (32). Protein interfaces in the crystal were characterized using the PISA server (33). Distances between amino acids were calculated using the program CONTACT from CCP4 (25). The accessible surface areas values were computed with NACCESS (34) with a probe radius of 1.4 Å and a slice width of 0.05 Å. Protein cavities were computed using the CASTp server (35). The coordinates and structure factors of L. pectinata HbII were deposited at the RCSB PDB with entry code 2OLP.
TABLE 1.
Data collection and refinement statistics of HbIILp-O2 structure
Statistical values for the highest resolution shell, 1.96 - 1.93 Å for data collection and 1.98 - 1.93 Å for refinement, are given in parentheses.
| Data collection | |
| Wavelength (Å) | 0.977 |
| Temperature (K) | 100 |
| Space group | P42212 |
| a = b, c (Å) | 73.92, 152.35 |
| α, β, γ (°) | 90 |
| Monomers per asymmetric unit | 2 |
| Resolution (Å) | 20.0-1.93 |
| No. of observed reflections | 338,327 |
| Redundancy | 10.5 (10.6) |
| Completeness (%) | 99.6 (100.0) |
| Rmergea (%) | 5.0 (33.6) |
| Average I/σ(II) | 40.0 (7.0) |
| Refinement | |
| R value (%) | 16.5 (17.5) |
| Rfree value (%) | 19.3 (22.5) |
| No. of reflections in working set | 30,675 (2158) |
| No. of reflections in test set | 1,643 (137) |
| No. of solvent molecules | 283 |
| Average B factor (Å2) | 30.42 |
| Root mean square deviations bond length (Å) | 0.013 |
| Root mean square deviations bond angles (°) | 1.841 |
| Ramachandran plot | |
| Most favored regions (%) | 94.8 |
| Allowed regions (%) | 5.2 |
| General allowed regions (%) | 0.0 |
| Disallowed regions (%) | 0.0 |
Rmerge = ∑hkl∑i|〈I〉 - Ii|/∑hkl∑i (Ii.
Potassium Ferricyanide Titration and Resonance Raman Spectroscopy—Deoxyhemoglobin was obtained by adding sodium dithionite to the purified protein samples, followed by a purification step in a Hi-Trap desalting column (FPLC, GE Healthcare). Oxyhemoglobin complexes were obtained by flushing the deoxy derivatives with oxygen. The initial concentration for the oxyHbLP species was 3.40 μm, at pH 7.5. The titration of oxyHbILP was initiated with the addition of 15-μl aliquots of a 10% potassium ferricyanide solution (2) up to a total volume of 150 μl to obtain the met-aquo HbILP. Full oxidation of HbIILP and the HbILPPhe(B10) → Tyr mutant proteins required a decrease of pH from 7.5 to 5.0, because the above conditions were not adequate. The resonance Raman measurements were made by focusing the output of a krypton ion laser at 413.1 nm (Spectra Physics) to a ∼30-μm spot on a rotating cell to prevent photodamaging. The laser power was set at 10 milliwatts using a CCD back-illuminated detector (800 × 2000 pixels) coupled to a modified Spex 1401 centered at 410 nm. Three spectra were collected, each composed of 60 accumulations of 10 s. Hemoglobin concentration was ∼100 μm.
RESULTS
HbIILp Structure and Sequence Analysis—The HbIILP crystallizes in space group P42212, and diffracts x-rays with a resolution better than 2.0 Å, having unit cell parameters a = b = 73.92 Å and c = 152.35 Å, and two molecules forming a dimer in the asymmetric unit and a solvent content of 61% per volume. All the residues are placed in the most favorable region of the Ramachandran plot. HbIILP shows the characteristic globin fold with six α-helices surrounding the central heme pocket, and two minor helical segments between B and E helices (Fig. 1). The distance between the iron atoms in the dimer is 17.8 Å, and the plane orientation of the porphyrin ring is almost perpendicular. This short distance between the heme groups is a characteristic feature of the EF-dimers in other types of molluscan hemoglobin (36). The dimer interface includes 25 residues of each monomer and covers a surface of 845 Å2. At the interface, four hydrogen bonds, namely, Lys95B-Ser46A (2.77 Å), Lys95A-Ser46B (2.98 Å), Lys92B-Gln65A (2.73 Å), and Lys92A-Gln65B (2.66 Å), and one salt bridge Asp82B-Lys63A (3.05 Å) are found. It is noteworthy that the HbIILP structure confirms the existence of one aspartate in position 84 and an acetyl group in the N terminus as previously suggested by comparison of matrix-assisted laser desorption ionization-mass spectrometry results and the cDNA-derived amino acid sequences (8).
FIGURE 1.

Structure of the HbII dimer from L. pectinata. Each helix, A to H, is labeled and identified by the colors: red, blue, yellow, magenta, orange, pink, light blue, and salmon, respectively.
The sequence alignments of the hemoglobins of L. pectinata, S. inaequivalvis, and A. suum are shown in Fig. 2. The multiple alignment has three gaps: one between the C and D helices, a second one within the G helix, and the third gap after the G helix. The B10 residue varies from a Phe in HbILP, Tyr in HbIILP, HbIIILP, and HbAsc to a Met in the three types of hemoglobins from S. inaequivalvis, whereas the E7 position contains a His residue in all the S. inaequivalvis hemoglobins and a Gln in the rest of the aligned polypeptides. In the HbIILP structure, the Arg100(F11) and Lys92(F3) form H-bonds with the propionates of the heme group. The Lys92(F3) and Arg100(F11) are conserved in all the hemoglobin sequences, except in HbAsc that are replaced by Glu92 and Asp100, respectively. The replacement of Lys92 by a Phe93 in the HbILP, which cannot form the salt bridge or hydrogen bonds, suggests that Lys92 could be responsible for the HbIILP dimer stability.
FIGURE 2.

Multiple sequence alignment of the three types of hemoglobin from L. pectinata, the bivalve mollusk S. inaequivalvis, and domain one of the nematode A. suum hemoglobin. The corresponding helices as predicted by the program suite iMoltalk applying the STRIDE method are labeled (red squares). Conserved residues are shaded in cyan and residues at position B10 (Tyr30 in L. pectinata), E7 (Gln65 in L. pectinata), Lys92 and Arg100 (L. pectinata) are shaded in yellow. The gaps are labeled in green.
HbIILP is known to form large aggregates at high concentrations (2). Analysis for protein complex assembly using the PISA Server (31) predicted two additional assemblies: a dimer of dimers to form a tetramer, and a dimer of the tetramer to form an octamer, both of low predicted stability. The superposition of the HbIILP dimer with HbI,Si has an overall average root mean square deviation of 2.2 Å (supplemental Fig. 1). The tetrameric HbIIAB,Si in the form of two heterodimers is composed by chains HbIIA,Si and HbIIB,Si (37). The hypothetical HbIILP tetramer in the unit cell is different from that of the S. inaequivalvis hemoglobin.
A channel of water molecules is buried in the HbIILP dimer interface, 40 of them located at a distance <10 Å of the heme groups. They form H-bonds with several residues located between the heme groups in the dimer. This interfacial water has been previously reported in the homodimeric HbI,Si contained in the coelomic fluid erythrocytes from the bivalve mollusk S. inaequivalvis (38) and they have been proposed to play an important role in the allosteric behavior of hemoglobin HbI,Si (39).
The major differences between chains A and B in the L. pectinata HbII dimer are found in the β-turn formed by residues Gly122-Gly123-Leu124-Thr125 with a maximum root mean square deviation of 3.4 Å. The β-turn is a type I in chain A, and a type IV in chain B. This difference in conformation arises from a difference in the environment, the β-turn has several contacts with the symmetry related molecule in chain A, whereas in chain B, it is exposed to the solvent. This β-turn is also one of the major differences between the HbIILP and HbILP structures.
HbIILp Distal and Proximal Heme Pocket—Fig. 3 shows the structure and the hydrogen bonding network of the oxyHbIILP proximal and distal sites. The porphyrin ring adopts a planar configuration with the iron atom in the plane, and the average distance of the pyrrolic nitrogens to the iron is 2.06 Å. The dioxygen forms an angle of 123° with the iron atom in chain A, and 162° in chain B. The heme pocket in chain B is slightly smaller than in chain A and the distance between the OH of the distal tyrosine and the iron atom is shorter (4.84 Å in chain A and 4.53 Å in chain B, but the differences in the volume size of the heme cavity are 912.6 Å3 in chain A and 831.9 Å3 in chain B) might arise from small differences in the position of the atoms as a consequence of the average coordinates position. In the HbIILP heme cavity most of the aromatic residues are conserved when compared with HbILP (for instance, Trp(A11), Phe(B9), Phe(C7), Phe(CD5), Phe(E11), and Trp(H8)), except Tyr(B10), Tyr(G8), and Tyr(G14), which are replaced by phenylalanine in the HbILP sequence, and Phe(E18) that is replaced by tryptophan. Although the HbIILP proximal heme pocket is mainly hydrophobic, it contains one buried water molecule (W9). This water molecule forms a hydrogen bond with the heme propionate group (O2A) within a distance of 2.59 Å and with His97(ND1) within a distance of 3.45 Å (Table 2). It is also part of the water network in the interface of the HbIILP dimer. A water molecule is also present in the heme pocket of the HbILP as part of an identical hydrogen network.
FIGURE 3.

Stereo view of the hydrogen bond network in the oxygen bound HbIILp structure. Residues at a 5-Å distance cut-off are shown. The omit map (contoured at 3 σ) and bond interactions are shown in blue and green lines, respectively. Distances between residues are shown in Table 2.
TABLE 2.
Heme contacts in HbIILp-O2 structure
|
Residue 1
|
Atom
|
Residue 2
|
Atom
|
Distance (Å)
|
|
|---|---|---|---|---|---|
| Chain A | Chain B | ||||
| HEM | O1A | Arg100 | NH2 | 2.71 | 2.52 |
| W6/W7 | O | 2.62 | 2.57 | ||
| O2A | Arg100 | NH1 | 2.98 | 2.88 | |
| Arg100 | NH2 | 3.47 | 3.34 | ||
| W9/W2 | O | 2.59 | 2.63 | ||
| O1D | Lys92 a | NZ | 2.63 | 2.72 | |
| W43/W5 | O | 2.64 | 2.67 | ||
| O2D | W8/W3 | O | 2.62 | 2.66 | |
| W34/W5 | O | 2.59 | 3.41 | ||
| W43/33 | O | 3.42 | 2.62 | ||
| W57/139 | O | 2.96 | 2.98 | ||
| Fe | His97 | NE2 | 2.08 | 2.04 | |
| Oxy | O1 | 2.94 | 2.85 | ||
| Oxy | O2 | 2.11 | 1.69 | ||
| Tyr30 | OH | Gln65 | NE2 | 3.00 | 3.07 |
| Oxy | O1 | 1.94 | 1.78 | ||
| Oxy | O2 | 2.99 | 2.95 | ||
| His97 | ND1 | Met93 | O | 2.98 | 2.79 |
| W9/W2 | O | 3.45 | 3.41 | ||
| Q65A | OE1 | Lys92a | NZ | 2.73 | 2.66 |
| NE2 | Tyr30 | OH | 3.00 | 3.07 | |
| Oxy | O1 | 3.02 | 3.04 | ||
| Oxy | O2 | 3.43 | 3.51 | ||
Marked contacts are inter-polypeptides chains.
When HbILP and HbIILP are superimposed using the heme group as a reference, the HbILP as well as the proximal histidine show a displacement of ∼1 Å. The orientation of the proximal His97(F15) is roughly perpendicular to the heme plane and the average distance between the His97(NE2) atom and the iron atom is 2.06 Å, which is a slightly shorter distance when compared with the sulfide (PDB code 1MOH), cyanide (PDB codes 1ETB and 1B0B), and water-bound HbILP, where the distances range from 2.13 to 2.31 Å. It has been suggested that within the various kinds of hemoglobin and myoglobin a hydrogen bond between the proximal histidine His(F8) and the side chain of a residue next to it modulates the strength of the His-Fe bond, regulating the ligand affinity (37, 40). In HbIILP, His(F8) has two possible hydrogen bond interactions, the first one with the buried water molecule and the second one from the interaction of the carbonyl group of Met93 and the ND1 from histidine (Table 2). A similar hydrogen bond is formed between the carbonyl of Met93 and the histidine ND1 atom in HbILP.
Heme Oxidation and Resonance Raman Spectra of Oxy Species—HbI titration with potassium ferricyanide required an additional oxidizing agent to complete oxidation to the met-aquo HbILP form, shifting the Söret band from 416 nm (oxyHbILP) to a maximum at 407 nm (supplemental Fig. 2). A pH-dependent titration for oxyHbIILP and the oxyHbILP-Phe(B10) → Tyr mutant required a decrease in pH from 7.5 to 5.5 to completely change the Söret band from 414 to 403 nm, a characteristic of the met-aquo HbIILP species. This result suggests that Tyr in the B10 position is responsible for the pH oxidation control of oxyHbIILP and the oxyHbILPPhe(B10) → Tyr mutant. Similarly, Fig. 4 compares the low frequency resonance Raman spectra of ferrous deoxygenated recombinant HbILP, HbILPPhe(B10) → Tyr, and HbIILP, which provides information on the Fe-His stretching mode (vFeN) and the trans-effect of the proximal histidine on the Fe-O2 mode. The vibrational frequencies (Fig. 4, a and b) for HbILP and the HbILPPhe(B10) → Tyr mutant are located at 219 cm-1, similar to the myoglobin spectrum. However, for HbIILP the vFeN is present at a lower frequency, 211 cm-1 (Fig. 4c), suggesting that the factors controlling the strength of the Fe-N bond are not the same for both sets of hemoproteins. Normal mode assignments were based on the presence or absence of a particular frequency on the spectra for the HbILP-ligand complexes and compared with the values reported in the literature (17, 41). The vFeN for hemoglobin and myoglobin has been found in the resonance Raman spectra between the 217 and 244 cm-1 regions (17, 41, 42). The spectra also show a band at 370 cm-1, which has previously been attributed to the Cβ-Cc-Cd bending motion of the peripheral propionate heme substituents in HbILP. The spectra for HbILP and HbILPPhe(B10) → Tyr also show the v8 band at 347 cm-1. This is characteristic of the combination of the metal-pyrrole stretch and the in-plane substituent bend, and a shoulder at 331 cm-1 that has been assigned to the out-of-plane pyrrole-tilting mode. In contrast, in HbIILP the spectral bands have been displaced to 350 and 310 cm-1, respectively.
FIGURE 4.

Resonance Raman spectra of deoxy samples of (a) HbILp,(b) HbILpPhe(B10) → Tyr, and (c) HbIILp showing the Fe-His stretching mode and trans-effect of the proximal His.
Fig. 5A shows the vinyl region and the Fe-O2 vibration for HbIILP, recombinant HbILP and a series of HbILP mutants. Some differences in their vibrational modes are observed as function of the polarity of the heme pocket and oxygen isotopic substitution. For example, the recombinant HbILP (Fig. 5A, a) shows a relatively strong band at 412 cm-1 with a small shoulder at 421 cm-1. The former corresponds to an in-plane vinylbending mode (δCbCαCβ-vinyl), whereas the latter is attributed to either a contribution from both vinyls or the same vinyl reflecting heterogeneity among heme subunits. Only small intensity changes are observed as a function of amino acid substitution. Similarly, the v8 band is found at 345 cm-1, which is invariant among the different hemeprotein derivatives. The shoulder of the out-of-plane pyrrole-tilting mode at 333 cm-1 is almost absent in the samples. Fig. 5B shows the 16O2-18O2 isotope difference spectra, where the Fe-O2 stretching mode of the heme O2 moiety presents a significant frequency change as a function of the heme pocket amino acids. Thus, the spectrum of recombinant HbILP (Fig. 5B, a) shows a band at 567 cm-1, which was previously attributed to the Fe-O2 stretching mode, and in this work, has been confirmed by the oxygen isotopic substitution. Changing the residue at the E7 position in the HbILPGln(E7) → Val and HbILPGln(E7) → Asn mutants (Fig. 5B, b and c) produced a decrease in the stretching mode to 563 cm-1, whereas changing the residue at the E7 position with the HbILPGln(E7) → His or at the B10 position in HbILPPhe(B10) → Tyr mutants (Fig. 5B, d and e) increased the frequency of the Fe-O normal mode to 569 and 572 cm-1, respectively. For the wild-type HbIILP this frequency is present at 573 cm-1 (Fig. 5B, f), suggesting that the oxygen in the heme O2 moiety is strongly hydrogen bonded to the distal pocket Tyr30(B10) and Gln65(E7), in good agreement with the proposed x-ray structure.
FIGURE 5.

A, resonance Raman spectra of oxy species HbIILp, HbILp, and HbILp mutants. B, oxygen isotopic shift for the Fe-O2 normal mode vibration of the oxyHbIILp, oxyHbILp, and oxyHbILp mutants.
DISCUSSION
The crystal structure of oxyHbII from L. pectinata has been solved and it shows relevant differences when compared with the ferric HbILP sequence and structure (the oxyHbILP structure has not been resolved yet). The overall hemoglobin fold is conserved in these clam proteins. Some important differences between them are found in the additional residues in the sequence of HbIILP and the flexible β-turn formed by the GGLT residues. The heme cavity is conserved between the HbILP and HbIILP forms, with the exception of the B10 position, which is occupied by Phe and Tyr, respectively. A significant displacement of the heme group is observed in the least square super-imposition of both types of hemoglobins. The heme group is buried farther in HbIILP than in HbILP because of the minor steric hindrance of the Met93 and Phe76 residues that are replaced by Phe92 and Trp75, respectively. Oxygen is tightly bound to the HbIILP heme distal site through simultaneous hydrogen bonds with Tyr(B10) and Gln(E7) (Fig. 3). This last pair of amino acids has been proposed to stabilize the oxygenated form of HbIILP by means of strong and weak hydrogen bonds, respectively (37, 40). HbAsc also has a Tyr(B10) and a Gln(E7), and a high oxygen affinity and unusually slow off rate for oxygen dissociation (15, 41). The HbAsc crystal structure (PDB 1ASH) shows that the distance between the iron to oxygen O1 is 1.90 Å, and between Tyr(B10) (OH) to O2 is 2.73 Å (40). Table 2 presents several distances between the oxygen heme moiety and the amino acids in the distal pocket of HbIILP. For example, the iron to oxygen O1 is 2.94 Å, and from Tyr(B10) (OH) to O2 is 2.99 Å. These longer distances are rather similar to those found in the structure of M. tuberculosis hemoglobin N (12). The orientation of the dioxygen bound to the heme plane is 110°, similar to the value found in the HbIILP structure, but in this case, the oxygen molecule is pointing toward the Tyr(B10) (OH) group. A CASTp calculation resulted in an average volume of 1000 Å3 for the heme cavity in M. tuberculosis hemoglobin, which is slightly larger than the average size of 872 Å3 for HbIILP. Similarly, a large cavity volume was observed in the ferric derivative of the Mb triple mutant (L29F/H64Q/V68F), Mb, and HbILP. The crystal structure reveals the existence of a much greater heme freedom and larger distal cavity volume in HbILP than sperm whale Mb, in both the H2S bound and unbound to the heme group, because of the lack of hydrogen bonding between the heme propionate groups and nearby amino acid residues (43). This dynamic behavior is absent in HbIILP, where the heme group is firmly anchored in place.
Cooperativity and Allosteric Effects—In contrast to a remarkably conserved tetrameric arrangement in vertebrate hemoglobins, invertebrate hemoglobins show a large diversity in quaternary structures (36). The aggregation state of the hemoglobin plays a central role in their cooperative behavior. Hemoglobins from L. pectinata show that HbILP is a monomer in all of the conditions assayed, whereas HbIILP and HbIIILP behave as apparent monomers at low concentrations and aggregates at higher concentrations (2). Lys92 of HbIILP may play an important role in dimer stability, because a Thr that disrupts the possibility to form a bond between chains replaces this residue in the HbILP sequence. Lys92 is conserved in HbIIILP opening up the possibility to form a heterodimer as occurs in S. inaequivalvis hemoglobins. The oligomeric hemoglobins from S. inaequivalvis show cooperativity in oxygen binding. However, a Hill coefficient of 1.1 was obtained for the oxygen binding measurement of HbIILP, regardless of the aggregation state (2, 3). Several mutant forms of the HbI, Si dimer have structurally characterized, suggesting that two residues play an important allosteric role. First, Phe97(F3) is packed tightly in the heme pocket in the deoxy HbI, Si state, and exposed to the dimer interface upon ligand binding (44). The displacement of the bulky Phe97 side chain to the dimer interface displaces several water molecules leaving the interface with less water and a poorer ordered water network. A similar behavior has been described for the heterotetramer of HbIIA, Si and HbIIB, Si in which mutation of the Phe97 to leucine provokes a loss of cooperativity (44). Another residue playing a determinant role is Thr72, and both Phe97 and Thr72 are replaced in HbIILp by methionine and valine, respectively.
The superpositions of the HbIILp structure to the oxygen bound (PDB code 3SDH) and unbound (PDB code 4SDH) hemoglobin states of the S. inaequivalvis show that Asn100 in these hemoglobins is replaced by Leu96 in HbIILp, leading to the loss of a strong hydrogen bond between Asn100 and the heme propionate group. Moreover this propionate moiety of the heme group in S. inaequivalvis shows an important displacement between the oxygen-bound and unbound forms. In particular, the orientation of the unbound form is equivalent to the orientation of the propionate in the oxyHbIILp structure. The interaction with the nearest residues is the same in HbSi, even a water molecule equivalent to the water molecule (W9) bound to His97 (Fig. 3) is found at a distance of 2.52 Å. Interestingly, this water molecule disappears in the oxygen-bound structure of S. inaequivalvis. This observation supports the hypothesis that this water molecule plays an important role in the regulation of the heme group affinity to ligands.
Geometry of the Heme-Oxygen Moiety and Hydrogen Bonding Network—The hydrogen bonding network between the heme bound oxygen and the Gln(E7) and Tyr(B10) of HbIILp is almost identical to the active center of HbAsc. In both cases, it has been proposed that the hydrogen bonding network is responsible for their extremely low oxygen dissociation rates (15, 41). Despite this analogy, the oxygen dissociation rate constant for HbIILp and HbAsc shows a difference of 2 orders of magnitude (0.11 and 0.004 s-1, respectively). This suggests that other factors, like the His97(F8) trans-effect and its orientation, could play an important role in determining this rate difference. The resonance Raman measurements allows a comparison between the Fe-His stretching mode of Mb, HbIILp, and HbAsc, at 220, 211, and 202 cm-1, respectively, whereas for recombinant HbILp and the HbILpPhe(B10) → Tyr mutant, the Fe-N stretching mode is characterized by a band at 219 cm-1 (Fig. 4). The Fe-His normal mode falls into the group of heme proteins that have a neutral proximal histidine, characteristic of those responsible for transporting oxygen, where the proximal histidine environment differs for these three hemeproteins. The strength of the hydrogen bond formed by a nitrogen proton in the proximal histidine and the polarity of the environment are powerful factors for determining the Fe-His stretching frequency in the heme-histidine moiety. This hydrogen bonding network in hemoglobins and myoglobins has also been also related to ligand affinity (37, 40). The significant differences observed in the heme-ligand dissociation rate constants between Mb and HbAsc have mainly been attributed to the tilted orientation of the His(F8) with regard to the pyrrole rings of the heme (45). Fig. 6 shows an overlay of the oxy heme moiety of HbAsc and HbIILp indicating several structural differences in the orientation of the proximal histidine, the bound oxygen, propionates, and vinyl groups. In particular, the presence of a water molecule between the propionate group of heme and His(F8) in HbIILp suggests that the Fe-His strength and the His-Fe trans-effect may be modulated by this hydrogen bond. Furthermore, the orientation of His(F8), as suggested for HbAsc, can also contribute to the observed Fe-His frequency. The combination of these factors facilitates the strength of the Fe-O2 complex in the distal heme pocket. Moreover, the small distal cavity volume of HbIILp and its hydrogen bonding network with the heme-O2 moiety is consistent with the relationship between the Fe-O2 stretching mode and its oxygen dissociation rate constant (koff) (46). This is supported by data in Table 3, which shows a rough inverse relationship between these two properties for HbIILp, HbILp, and several HbILp mutants. For example, oxyHbIILp and oxyHbILp show Fe-O frequencies at 572 and 567 cm-1 correlated with an oxygen dissociation rate constant (2) of 0.11 and 61 s-1, respectively. Similarly, HbILpGln(E7) → Asp and HbILpGln(E7) → Val mutants show an increase in the dissociation rate constant from 140 s-1 (for HbIr specie) to 375 and 500 s-1, whereas the Fe-O normal mode frequency decreases to 563 cm-1 (46). The faster oxygen dissociation rates suggest that the interactions of the E7 residue in these two mutants are much weaker than in HbILp, thus corroborating that the absence of a hydrogen bond induces a lower Fe-O vibrational frequency. Although no dipolar interaction exists between the residues in the E7 position in these two mutants, the observed frequencies in the HbILp mutants are higher than expected probably due to multipole interactions of the three phenylalanine residues (B10, E11, and CD1) in the distal side of the heme (47). The HbILpGln(E7) → His mutant induces a much stronger polar interaction between this residue and the dioxygen heme complex with a dissociation rate constant (koff) of 3 s-1, suggesting the presence of dipolar interactions with the His(E7) center. This is consistent with the increase in the Fe-O energy and the kinetics studies on Mb, which indicated that the hydrogen bond between His(E7) and the oxygen coordinated to the iron control the koff rate. The Tyr(B10) in the HbILpPhe(B10) → Tyr mutant and in HbIILp is also stabilizing the coordinated dioxygen molecule as evidenced by the increase in the frequency from 567 to 571 cm-1, and to 572 cm-1, respectively, and the decrease in their dissociation rate constants from 61 to 0.6 s-1, and to 0.11 s-1, respectively. As mentioned above, despite similarities in their hydrogen bonding network, HbAsc and HbIILp show significantly different oxygen dissociation rate constants (0.0041 and 0.11 s-1, respectively) (2, 48). Moreover, the HbAsc show a Fe-His normal mode at 202 cm-1, whereas for HbIILp this frequency is present at 211 cm-1. Thus, differences in the His(F8) trans-effect and the orientation of the oxygen molecule in the oxyHbIILp and oxyHbAsc complexes (Fig. 6) must have an important role to explain the observed experimental data. Similarly, this is also supported by studies of CO complexes showing three different conformers at 1912, 1956, and 1965 cm-1 for HbAsc-CO, whereas the HbIILp-CO complex show only the presence of the A3 and A0 conformers at 1924 and 1964 cm-1 in the infrared spectra (15, 46). Therefore, the interplay between the small HbIILp heme pocket structure, the hydrogen bonding network to the proximal and distal heme environments, the His(F8) trans-effect, and the orientation of the oxygen molecule in the oxy complex are responsible for the stability of the oxyHbIILp complex.
FIGURE 6.

Oxygen heme moiety. Least square superposition of HbIILp (yellow) and HbAsc (cyan, PDB code 1ASH). The superposition was carried out taking into account all the residues in the structure (amino acids and prosthetic group). The water molecule forming a hydrogen bond between the propionate group and His97(F8) in HbIILp is also shown. This water molecule is not present in the oxygen bound structure of HbAsc. The oxygen molecule shows opposite orientation in both structures (the oxygen molecule in HbAsc is shown in clear cyan).
TABLE 3.
vFe-O2 and dissociation rate constants (koff) for HbIILp, HbILp, and HbILp mutants
| Heme protein | vFeO2 | koff |
|---|---|---|
| cm−1 | s−1 | |
| HbI (Gln(E7), Phe(B10), Phe(E11)) | 567 | 61.0a |
| HbIr (Gln(E7), Phe(B10), Phe(E11)) | 567 | 140.0 |
| HbIGln(E7) → Val | 563 | 500.0 |
| HbIGln(E7) → Asp | 563 | 375.0 |
| HbIGln(E7) → His | 569 | 3.0 |
| HbIPhe(B10) → Tyr | 571 | 0.60 |
| HbII (Gln(E7), Tyr(B10), Phe(E11)) | 572 | 0.11a |
From Ref. 2.
In truncated hemoglobins (trHbs), the oxy trHb complex shows, in general, a much lower energy for the Fe-O vibrational mode (542-560 cm-1) than Mb (49), HbILp, and HbIILp. Moreover, oxy trHbs tend to present an inverse correlation between the Fe-O frequency and the oxygen dissociation rate, similarly for oxyHbILp and oxyHbIILp there is apparently also a rough inverse relationship with the dissociation constants but their Fe-O frequency is present at higher energy (567-572 cm-1). The fact that trHbs have a hydrogen bonding network interacting with the proximal and distal oxygens of the heme-O2 moiety, whereas HbILp and HbIILp present a hydrogen bonding network interacting only with the distal oxygen of the oxyheme complex may account for the observed trend. Particularly, the Feδ+-O-Oδ- center is highly polar (50) and the formation of hydrogen bonding between heme pocket amino acids and one or both oxygens of the oxyheme complex can generate different moiety resonance structures. Furthermore, because the low frequency Fe-O Raman active vibrational mode arises from the interactions between the stretch and bend motion of Feδ+-O-Oδ-, at equilibrium, an approximate potential leading to the Fe-O vibrational frequency can be expressed as Equation 1,
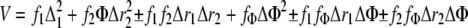 |
(Eq.1) |
where f1, f2, fÔ, r1, r2, andÔ are the force constant and geometries for the Fe-O, O-O, and Fe-O-O atoms, respectively. Similarly, f1 f2, f1 fÔ, and f2 fÔ are the associated interaction force constants, and their signs can be positive or negative (51, 52) depending on the Feδ+-O-Oδ- resonance structures, geometry, and central atom hybridization, which in turn are affected by the hydrogen bonding network present in the oxyhemoglobin species. It is plausible that the combination and magnitude of the sign in these coupling constants may be responsible for the lack of a single correlation between the Fe-O frequency and the hemeproteins oxygen dissociation constant.
Factors Influencing HbIILp Oxygen Selection—Different roles have been suggested for the hemoglobin variants of L. pectinata (1, 3). HbILp is a sulfide-reactive monomeric protein, whereas HbIILp and HbIIILp are responsible for the oxygen transport and remain unaffected by the presence of H2S. The factors underlying this unique behavior and ligand selection between HbILp and HbIILp/HbIIILp clearly depend on the heme iron oxidation state; however, the mechanism underlying the deoxy heme stability in HbIILp is still unknown. In vitro HbILp and HbIILp bind oxygen with an association rate constant of 100-200 × 106 and 0.39 × 106 m-1 s-1, and hydrogen sulfide with association rate constant values of 226 × 103 and 11.3 × 103 m-1 s-1, respectively (2, 3). This indicates that HbILp binds both ligands much faster than HbIILp. Similar to HbIILp, kinetic analysis of the HbILpPhe(B10) → Tyr mutant with O2 and H2S indicates that in this variant the association constant of both ligands decreases to 6.8 × 106 and 3.37 × 103 m-1 s-1, respectively, when compared with HbILp. Furthermore, the H2S dissociation constant for HbIILp, rHbILp, and the HbILpPhe(B10) → Tyr mutants are 17 × 10-3, 0.04 × 10-3, and 0.06 × 10-3 s-1, respectively, whereas for the oxygen species these values are 0.11, 140, and 0.6 s-1, respectively. Thus, the data indicate that in HbILp the Phe(B10) → Tyr mutation cannot account for the properties of HbIILp-SH2 complex reactivity but it can explain the smaller dissociation constant of oxyHbIILp. Replacing Phe(B10) by Tyr in the HbILpPhe(B10) → Tyr mutant may decrease the size of the HbILp heme cavity, thus causing a reduction in ligand association. Independently of the heme iron oxidation state driving force for the selection of O2 or H2S, the heme cavity of HbIILp is smaller (872 Å3) than the HbILp. Dynamic features in HbILp, Mb, and triple Mb mutant (L29F/H64Q/V68F) revealed a large cavity, suggesting that the larger cavities favor the binding of H2SbyHbILp (43). The reduction of the heme cavity of both HbIILp and HbILpPhe(B10) → Tyr may help to stabilize the FeII-O2 moiety once O2 binds the heme iron center by forming hydrogen bonding interactions with Gln(E7) and Tyr(B10) as evidenced by the resonance Raman and oxygen off rate analyses presented above. Moreover, the reduction of the heme cavities in both proteins, caused in part by Tyr(B10), may help prevent oxidation of the ferrous iron center by impeding access of external water molecules into the distal environment. Indeed, a direct role of Tyr(B10) in preventing oxidation of FeII as well as ligand selection can be observed in the pH titration analysis of HbILp, HbILpPhe(B10) → Tyr, and HbIILp. The data showed that the high affinity of HbIILp and the HbILpPhe(B10) → Tyr mutant for oxygen is pH-dependent. Similarly, a decrease from pH 7.5 to 5.5 was necessary to fully oxidize both HbIILp and the HbILpPhe(B10) → Tyr mutant, suggesting that the tyrosine plays an important role in regulating the oxidation of the heme group. Previous experiments has been suggested a relationship between the pKa values of the ionizable groups associated with the heme and the role of the hydrogen bonding interactions on the heme oxygen dissociation rate (53). Furthermore, for met-aquo HbIILp and HbILpPhe(B10) → Tyr, the UV-visible pH data shows, at neutral conditions, bands at 486, 541, 577, and 603 nm for both proteins. This suggests the existence of an open and closed conformation due to the interactions in the coordination of the Tyr(B10) (OH) and the ligand, to the heme iron. This means that, in the open conformation the Tyr30(B10) swings away from the iron, whereas in the closed conformation remains at very close distance and may interact with the ligand (47).
Overall, the data suggest a model for the in vivo mechanism of the clam L. pectinata where the function of HbIILp to bind and possibly transport oxygen to the host bacteria is regulated by the dynamic displacements of the Gln65(E7) and Tyr30(B10) pair toward the heme to protect it from the change in the heme oxidation state from FeII to FeIII. This suggested mechanism avoids the binding of H2S to HbIILp that disrupts its function of oxygen transport in an environment rich in hydrogen sulfide. In summary, the results from the crystallographic data show that a small heme pocket cavity for HbIILp induces the formation of strong hydrogen bonds between the iron and oxygen molecule. Resonance Raman data supports the existence of a hydrogen bonding network between Gln(E7) and Tyr(B10) that stabilizes the binding of the oxygen to HbIILp complex shown in Fig. 3. This, together with the proximal histidine trans-effect, and the pH dependence of the oxidation state substantiates the role of HbIILp in controlling the oxygen dissociation rate.
Supplementary Material
The atomic coordinates and structure factors (code 2OLP) have been deposited in the Protein Data Bank, Research Collaboratory for structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
This work was supported by National Science Foundation Grant NSF-MCB-0544250 (to J. L. G.), National Institutes of Health NIGMS MBRS-SCORE 2 Grant S06GM08103-34 (to J. L. G.), Grants P20RR016470 and G12RR03051 from the National Center for Research Resources, National Institutes of Health, Grant BIO2006-15517-C02-02 from the Spanish Ministry of Education and Sciences, Project RMN-1344, the Junta de Andalucía (Spain) (J. A. G.), Factoría de Cristalización funded by the program Consolider-Ingenio 2010, and National Institutes of Health Grant HL65465 (to S. R. Y.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1 and S2.
Footnotes
The abbreviations used are: HbLp, L. pectinata hemoglobin; HbAsc, A. suum hemoglobin; HbSi, S. inaequivalvis hemoglobin; HbIr, L. pectinata recombinant HbI; trHb, truncated hemoglobins; FPLC, fast performance liquid chromatography; BisTris, 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol.
References
- 1.Read, K. R. (1965) Comp. Biochem. Physiol. 15 137-157 [DOI] [PubMed] [Google Scholar]
- 2.Kraus, D. W., and Wittenberg, J. B. (1990) J. Biol. Chem. 265 16043-16053 [PubMed] [Google Scholar]
- 3.Kraus, D. W., Wittenberg, J. B., Lu, J. F., and Peisach, J. (1990) J. Biol. Chem. 265 16054-16059 [PubMed] [Google Scholar]
- 4.Rizzi, M., Wittenberg, J. B., Coda, A., Fasano, M., Ascenzi, P., and Bolognesi, M. (1994) J. Mol. Biol. 244 86-99 [DOI] [PubMed] [Google Scholar]
- 5.Bolognesi, M., Rosano, C., Losso, R., Borassi, A., Rizzi, M., Wittenberg, J. B., Boffi, A., and Ascenzi, P. (1999) Biophys. J. 77 1093-1099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rizzi, M., Wittenberg, J. B., Coda, A., Ascenzi, P., and Bolognesi, M. (1996) J. Mol. Biol. 258 1-5 [DOI] [PubMed] [Google Scholar]
- 7.Hockenhull-Johnson, J. D., Stern, M. S., Martin, P., Dass, C., Desidero, D. M., Wittenberg, J. B., Vinogradov, S. N., and Walz, D. A. (1991) J. Protein Chem. 10 609-622 [DOI] [PubMed] [Google Scholar]
- 8.Torres, E., Renta, J. Y., Rodríguez, Y., López-Garriga, J., and Cadilla, C. L. (2003) J. Protein Chem. 22 683-690 [DOI] [PubMed] [Google Scholar]
- 9.Pietri, R., Granell, L., Cruz, A., De Jesús, W., Lewis, A., León, R., Cadilla, C. L., and López Garriga, J. (2005) Biochim. Biophys. Acta 1747 195-203 [DOI] [PubMed] [Google Scholar]
- 10.Yang, J., Kloek, A. P., Goldberg, D. E., and Mathews, F. S. (1995) Proc. Natl. Acad. Sci. U. S. A. 92 4224-4228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Couture, M., Yeh, S., Wittenberg, B. A., Wittenberg, J. B., Ouellet, Y., Rousseau, D. L., and Guertin, M. (1999) Proc. Natl. Acad. Sci. U. S. A. 96 11223-11228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Milani, M., Pesce, A., Ouellet, Y., Ascenzi, P., Guertin, M., and Bolognesi, M., (2001) EMBO J. 20 3902-3909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Condon, P. J., and Royer, W. E., Jr. (1994) J. Biol. Chem. 269 25259-25267 [DOI] [PubMed] [Google Scholar]
- 14.Royer, W. E., Jr., Heard, K. S., Harrington, D. J., and Chiancone, E. (1995) J. Mol. Biol. 253 168-186 [DOI] [PubMed] [Google Scholar]
- 15.Peterson, E. S., Huang, S., Wang, J., Miller, L. M., Vidugiris, G., Kloek, A. P., Goldberg, D. E., Chance, M. R., Wittenberg, J. B., and Friedman, J. M. (1997) Biochemistry 36 13110-13121 [DOI] [PubMed] [Google Scholar]
- 16.De Jesús-Bonilla, W., Jia, Y., Alayash, A. I., and López Garriga, J. (2007) Biochemistry 46 10451-10460 [DOI] [PubMed] [Google Scholar]
- 17.De Jesús-Bonilla, W., Cruz, A. Lewis, A., Cerda, J., Bacelo, D. E., Cadilla, C. L., and López-Garriga, J. (2006) J. Biol. Inorg. Chem. 11 334-342 [DOI] [PubMed] [Google Scholar]
- 18.León, R. G., Munier-Lehmann, H., Barzu, O., Baudin-Creuza, V., Pietri, R., López-Garriga, J., and Cadilla, C. L. (2004) Protein Expr. Purif. 38 184-195 [DOI] [PubMed] [Google Scholar]
- 19.Altschul, S. F., Madden, T. L., Schäffer, A. A., Zhang, J., Zhang, Z., Miller, W., and Lipman, D. J. (1997) Nucleic Acids Res. 25 3389-3402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Thompson, J. D., Higgins, D. G., and Gibson, T. J. (1994) Nucleic Acids Res. 22 4673-4680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gavira, J. A., De Jesús, W., Camara-Artigas, A., Lopez-Garriga, J., and Garcia-Ruiz, J. M. (2006) Acta Crystallogr. Sect. F Struct. Biol. Crystallogr. Commun. 62 196-199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.García-Ruiz, J. M. (2003) Methods Enzymol. 368 130-154 [DOI] [PubMed] [Google Scholar]
- 23.Otwinowski, Z., and Minor, W. (1997) Methods Enzymol. 276 307-326 [DOI] [PubMed] [Google Scholar]
- 24.Brunger, A. T., Adams, P. D., Clore, G. M., Delano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J. S., Kuszewski, J., Nilges, M., Pannu, N. S., Read, R. J., Rice, L. M., Simonson, T., and Warren, G. L. (1998) Acta Crystallogr. Sect. D Biol. Crystallogr. 54 905-921 [DOI] [PubMed] [Google Scholar]
- 25.Bailey, S. (1994) Acta Crystallogr. Sect. D Biol. Crystallogr. 50 760-763 [DOI] [PubMed] [Google Scholar]
- 26.Emsley, P., and Cowtan, K. (2004) Acta Crystallogr. Sect. D Biol. Crystallogr. 60 2126-2132 [DOI] [PubMed] [Google Scholar]
- 27.Winn, M. D., Isupov, M. N., and Murshudov, G. N. (2001) Acta Crystallogr. Sect. D Biol. Crystallogr. 57 122-133 [DOI] [PubMed] [Google Scholar]
- 28.Lovell, S. C., Davis, I. W., Adrendall, W. B., III, de Bakker, P. I., Word, J. M., Prisant, M. G., Richardson, J. S., and Richardson, D. C. (2003) Proteins 50 437-450 [DOI] [PubMed] [Google Scholar]
- 29.Laskowski, R. A., MacArthur, M. W., Moss, D. S., and Thornton, J. M. (1993) J. Appl. Crystallogr. 26 283-291 [Google Scholar]
- 30.Frishman, D., and Argos, P. (1995) Proteins 23 566-579 [DOI] [PubMed] [Google Scholar]
- 31.Hutchinson, E. G., and Thornton, J. M. (1996) Protein Sci. 5 212-220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kabsch, W. (1976) Acta Crystallogr. Sect. A 32 922-923 [Google Scholar]
- 33.Krissinel, E., and Henrick, K. (2005) Comput. Life Sci. Proc. 3695 163-174 [Google Scholar]
- 34.Hubbard, S. J., and Thornton, J. M. (1993) NACCESS, Computer Program, Department of Biochemistry and Molecular Biology, University College, London
- 35.Binkowski, T. A., Naghibzadeh, S., and Liang, J. (2003) Nucleic Acids Res. 31 3352-3355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Royer, W. E., Knapp, J. E., Strand, K., and Heaslet, H. A. (2001) Trends Biochem. Sci 26 297-304 [DOI] [PubMed] [Google Scholar]
- 37.Smerdon, S. J., Krzywda, S., Wilkinson, A. J., Brantley, R. E., Carver, T. E., Hargrove, M. S., and Olson, J. S. (1993) Biochemistry 32 5132-5138 [DOI] [PubMed] [Google Scholar]
- 38.Royer, W. E., Jr., Pardanani, A., Gibson, Q. H., Peterson, E. S., and Friedman, J. M. (1996) Proc. Natl. Acad. Sci. U. S. A. 93 14526-14531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pardanani, A., Gambacurta, A., Ascoli, F., and Royer, W. E. (1998) J. Mol. Biol. 284 729-739 [DOI] [PubMed] [Google Scholar]
- 40.Carver, T. E., Brantley, R. E., Jr., Singleton, E. W., Arduini, R. M., Quillin, M. L., Phillips, G. N., Jr., and Olson, J. S. (1992) J. Biol. Chem. 267 14443-14450 [PubMed] [Google Scholar]
- 41.Cerda, J., Echevarría, Y., Morales, E., and López-Garriga, J. (1999) Biospectroscopy 5 289-301 [Google Scholar]
- 42.Mukai, M., Mills, C. E., Poole, R. K., and Yeh, S. R. (2001) J. Biol. Chem. 276 7272-7277 [DOI] [PubMed] [Google Scholar]
- 43.Fernández-Alberti, S., Bacelo, D. E., Binning, R. C., Echave, J., Chergui, M., and Lopez-Garriga, J., (2006) Biophys. J. 91 1698-1709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhou, Y. Q., Zhou, H. Y., and Karplus, M. (2003) J. Mol. Biol. 326 593-606 [DOI] [PubMed] [Google Scholar]
- 45.Das, K. T., Boffi, A., Chiancone, E., and Rousseau, A. D. (1999) J. Biol. Chem. 274 2916-2919 [DOI] [PubMed] [Google Scholar]
- 46.Pietri, R., Granell, L. B., León, R., Cadilla, C. L., and López-Garriga, J. (2006) Biochim. Biophys. Acta 1764 758-765 [DOI] [PubMed] [Google Scholar]
- 47.Navarro, A., Maldonado, M., González-Lagoa, J., López, R., López-Garriga, J., and Colón, J. (1996) Inorg. Chim. Acta 243 161-166 [Google Scholar]
- 48.Blaxter, M. L., Vanfleteren, J. R., Xia, J., and Moens, L. (1994) J. Biol. Chem. 269 30181-30186 [PubMed] [Google Scholar]
- 49.Lu, C., Egawa, T., Wainwright, L. M., Poole, R. K., and Yeh, S.-R. (2007) J. Biol. Chem. 282 13627-13636 [DOI] [PubMed] [Google Scholar]
- 50.Hirota, S., Li, T., Phillips, G. H., Olson, J. S., Mukai, M., and Kitagawa, T. (1996) J. Am. Chem. Soc. 118 7845-7846 [Google Scholar]
- 51.Coulson, C. A., Duchesne, J., and Manneback, C. (1947) Nature 159 794-795 [PubMed] [Google Scholar]
- 52.Califano, S. (1976) Vibrational States, pp. 95-97, John Wiley & Sons, New York
- 53.Saroff, H. A. (2004) J. Theor. Biol. 229 113-118 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.