

Abstract
The role of positively charged arginines in the fourth transmembrane domain (S4) and a single negatively charged amino acid in the third transmembrane domain (S3) on channel biogenesis and gating of voltage-gated K+ channels (Kv) has been well established. Both intermediate (KCa3.1) and small (KCa2.x) conductance, Ca2+-activated K+ channels have two conserved arginines in S4 and a single conserved glutamic acid in S3, although these channels are voltage-independent. We demonstrate that mutation of any of these charged amino acids in KCa3.1 or KCa2.3 to alanine, glutamine, or charge reversal mutations results in a rapid degradation (<30 min) of total protein, confirming the critical role of these amino acids in channel biogenesis. Mutation of the S4 arginine closest to the cytosolic side of KCa3.1 to histidine resulted in expression at the cell surface. Excised patch clamp experiments revealed that this Arg/His mutation had a dramatically reduced open probability (Po), relative to wild type channels. Additionally, we demonstrate, using a combination of short hairpin RNA, dominant negative, and co-immunoprecipitation studies, that both KCa3.1 and KCa2.3 are translocated out of the endoplasmic reticulum associated with Derlin-1. These misfolded channels are poly-ubiquitylated, recognized by p97, and targeted for proteasomal degradation. Our results suggest that S3 and S4 charged amino acids play an evolutionarily conserved role in the biogenesis and gating of KCa channels. Furthermore, these improperly folded K+ channels are translocated out of the endoplasmic reticulum in a Derlin-1- and p97-dependent fashion, poly-ubiquitylated, and targeted for proteasomal degradation.
Proper folding of K+ channels is critical to both their correct trafficking to the plasma membrane and their eventual function. Recent crystallographic evidence has solidified the notion that the 6-transmembrane (TM)2 domain, voltage-gated K+ channels (Kv) can be thought of as a central pore-forming domain, which is conserved among all K+ channels, and a voltage-sensing domain (S1–S4) (1). This concept of unique domains was confirmed by the demonstration that the voltage-sensing domain could be appended onto KcSA resulting in a voltage-dependent channel (2). Papazian and co-workers (3–7) have studied the structural requirements for the biogenesis of the voltage-sensing domain in Shaker, demonstrating two sets of salt bridges between negatively charged amino acids in S2 and S3 and positively charged amino acids in S4. Specifically, in Shaker Lys-374 in S4 interacts with Asp-316 in S3 and Glu-293 in S2, whereas both Arg-368 and Arg-371 in S4 interact with Glu-283 in S2. Mutation of Lys-374, Asp-316, or Glu-283 results in a destabilized channel and subsequent degradation, whereas mutations in Glu-293, Arg-368, and Arg-371 alter the voltage dependence of gating. Using the KAT1 channel, Sato et al. (8, 9) directly demonstrated that these interactions are required for proper folding and integration into the ER membrane. Thus, these charged amino acids are required to stabilize the fold of the S1–S4 domain such that the channel is efficiently trafficked to the plasma membrane. More recent evidence has confirmed this paradigm in both eag and hERG channels (5, 10). Finally, Horrigan and co-workers (11) demonstrated a role for these salt bridges in the Ca2+- and voltage-dependent K+ channel, BK.
Intermediate (KCa3.1 or IK1) and small (KCa2.x or SK) conductance, Ca2+-activated K+ channels are voltage-independent members of the 6-TM domain superfamily of K+ channels. As such, these channels are evolutionarily older than their voltage-dependent relatives, the Kv channels (12). Despite the lack of voltage dependence, both KCa3.1 and KCa2.x have two conserved arginines in S4 which, upon sequence alignment, correspond to Arg-368 and Lys-374 in Shaker as well as a conserved glutamic acid in S3, which aligns with Asp-316 in Shaker. There are no negatively charged amino acids in S2 of the KCa3.1 and KCa2.x family members. Here we determined whether these S3 and S4 charged amino acids in KCa3.1 and KCa2.x share a conserved function with Kv channels. Namely, are these amino acids required for proper channel biogenesis and gating in a nonvoltage-dependent channel? We demonstrate that mutation of any of these S3 or S4 charged amino acids in KCa3.1 or KCa2.x results in a rapid, proteasome-dependent degradation of the channel. We further demonstrate that this degradation is dependent upon the translocation out of the ER by Derlin-1 after which it is poly-ubiquitylated and shuttled to the proteasome by p97. Finally, we demonstrate that substitution of histidine for one of the S4 arginines results in the plasma membrane expression of a channel with an altered open probability (Po), indicative of a gating change. These results suggest that the fundamental requirements for arginines in S4 and a negative charge in S3 to determine folding of this domain were established prior to the development of voltage dependence.
EXPERIMENTAL PROCEDURES
Molecular Biology—KCa3.1 (hIK1), KCa2.3 (rSK3), and KCa2.2 (rSK2) cDNAs were kindly provided by J. P. Adelman (Vollum Institute, Oregon Health Sciences University). All cDNAs were subcloned into pcDNA3.1(+) (Invitrogen) using the EcoRI and XhoI restriction sites. A hemagglutinin (HA) (YPYDVPDYA) epitope was inserted into KCa3.1 (HA-KCa3.1) between Gly-132 and Ala-133, i.e. the extracellular loop between transmembrane domains S3 and S4 as described previously (13). KCa3.1 and KCa2.2 were tagged with a C-terminal myc epitope (EQKLISEEDL) through PCR amplification as described (13). Derlin-1 was amplified from an ultimate open reading frame clone (Invitrogen) and subcloned into pcDNA3.1(+). A C-terminal HA tag was appended onto Derlin-1 via PCR amplification as described (14). The shRNA targeting Derlin-1 was kindly provided by Dr. Hidde Ploegh (Whitehead Institute). The p97 and p97QQ mutant constructs were generously provided by Dr. Tom Rapoport (Harvard University). All mutations were generated using the Stratagene QuikChange™ site-directed mutagenesis strategy (Stratagene, La Jolla, CA). The fidelity of all constructs utilized in this study was confirmed by sequencing (ABI PRISM 377 automated sequencer, University of Pittsburgh) and subsequent sequence alignment (NCBI BLAST) using GenBank™ accession numbers AF022150, U69884, and U69882.
Cell Culture—Human embryonic kidney (HEK293) cells were obtained from the American Type Culture Collection (Manassas, VA) and cultured in Dulbecco's modified Eagle's medium (Invitrogen) supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin in a humidified 5% CO2, 95% O2 incubator at 37 °C. Cells were transfected using Lipofectamine 2000 (Invitrogen) following the manufacturer's instructions. Stable cell lines were generated for all constructs by subjecting cells to antibiotic selection (1 mg/ml G418). Note that clonal cell lines were not subsequently selected from this stable population to avoid clonal variation.
Electrophysiology; Inside-out Patch Clamp Experiments— The effects of Ca2+, DCEBIO, and clotrimazole on KCa3.1 were assessed with inside-out patch clamp experiments as a functional assay. During patch clamp experiments, the bath solution contained 145 mm potassium gluconate, 5 mm KCl, 2 mm MgCl2, 10 mm HEPES, and 1 mm EGTA (pH adjusted to 7.2 with KOH). Sufficient CaCl2 was added to obtain the desired free Ca2+ concentration (program kindly provided by Dr. Dave Dawson, Oregon Health Sciences University). To obtaina 0 Ca2+ bath solution, EGTA (1 mm) was added without CaCl2 (estimated free Ca2+ <10 nm). The pipette solution was 140 mm potassium gluconate, 5 mm KCl, 1 mm MgCl2, 10 mm HEPES, and 1 mm CaCl2 (pH adjusted to 7.2 with KOH). All experiments were performed at room temperature. All patches were held at a holding potential of -100 mV. Total channel current was determined using Clampfit 8.2 (Axon Instruments).
Variance Analysis—Excised patch clamp recordings were subject to variance analysis as described (15). Current records were filtered at 10 kHz and digitized at 20 kHz after which the total current record was divided into 250 episodes and mean current (I) and variance (σ2) were calculated for each episode using Channelab Software (Synaptosoft Inc., Decatur, GA). The number of channels (N) in the patch and single channel amplitude (i) are obtained by fitting the variance (σ2) against mean current (I) distribution to Equation 1,
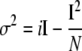 |
(Eq. 1) |
The maximum open probability (Po(max)) can then be calculated using Equation 2,
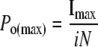 |
(Eq. 2) |
where Imax is the maximum current observed, and i and N are the mean values for single channel amplitude and number of channels calculated from Equation 1. The product of i and N would give the theoretical current at Po = 1; hence, channel Po can be calculated at any given current as a proportion of Po(max) (16–19).
Antibodies—HA (HA.11) and Giantin antibodies were obtained from Covance (HA.11, Richmond, CA); c-myc (clone 9E10) was obtained from Roche Applied Science; α-His, α-ubiquitin, and α-calnexin Ab were obtained from Sigma; α-Derlin-1 Ab was a generous gift from Dr. Tom Rapoport (Harvard University), and α-KCa2.3 Ab was obtained from Chemicon (Temecula, CA).
Proteinase K Digestion—Proteinase K digestion was performed as described previously (20). Briefly, HEK cells stably expressing KCa2.3 were washed three times with ice-cold phosphate-buffered saline and then incubated with 10 mm HEPES, 150 mm NaCl, 2 mm CaCl2, pH 7.4, with or without 200 μg/ml proteinase K (Sigma) at 37 °C for 30 min. Proteinase K digestion was quenched by adding ice-cold phosphate-buffered saline containing 6 mm phenylmethylsulfonyl fluoride and 25 mm EDTA. Cleared lysates were prepared and analyzed by IB.
Chemiluminescence Assay—HEK cells stably expressing the desired channel were subject to a cell surface chemiluminescence assay described previously (20). Nontransfected HEK cells were used for background subtraction, and all mutations were normalized to wild type HA-KCa3.1 cells.
Immunofluorescence (IF)—Immunofluorescent labeling of cell surface and intracellular HA-tagged KCa3.1 was performed as described previously (13, 15, 20, 21). Co-labeling of intracellular HA-KCa3.1 and the ER resident protein, calnexin, was performed sequentially. Nuclei were labeled with Hoechst 33258 (Sigma). Cells were then subjected to laser confocal microscopy using a Leica TCSNT 3 laser 4 PMT system. To ensure maximal X-Y spatial resolution, sections were scanned at 1024 × 1024 pixels, using sequential 2-color image collection to minimize cross-talk between the channels imaged.
Immunoprecipitations (IP) and Immunoblots (IB)—Our IP and IB protocols have been described previously (13, 15, 20, 21). Briefly, cells were lysed in IP buffer (50 mm HEPES, pH 7.4, 150 mm NaCl, 1% v/v Triton X-100, 1 mm EDTA containing Complete™ EDTA-free protease inhibitor mixture mix, Roche Applied Science), and protein concentrations were determined and normalized to achieve equivalent loading. Crude lysates were pre-cleared with protein A/G-Sepharose beads (Sigma) and incubated with the appropriate antibody. Immune complexes were precipitated with protein A/G-Sepharose beads, washed extensively, and resuspended in Laemmli sample buffer. Proteins were resolved by SDS-PAGE (8% gel) and transferred to nitrocellulose for immunoblot analysis. To determine the time constant for channel degradation, HEK cells expressing the channel of interest were incubated in cycloheximide (400 μg/ml) for the indicated times at 37 °C after which they were immediately cooled to 4 °C by washing in ice-cold phosphate-buffered saline and prepared for IB as described (21).
Relative protein levels were quantified by densitometry. For mutations that failed to produce detectable levels of protein upon IB when equivalent amounts of total protein were loaded, we estimated the fold change in protein by the following method. Serial dilutions of total cell lysate expressing either wild type KCa3.1 or KCa2.3 were subject to IB as above, and the smallest detectable amount of channel protein was determined. For KCa3.1, this was a 16-fold decrease, and for KCa2.3 this was a 32-fold decrease. Thus, if we were unable to detect mutated KCa3.1 upon IB, we indicate this as a >16-fold change in channel expression; and if we are unable to detect mutated KCa2.3 upon IB, we indicate this as a >32-fold change in channel expression.
Chemicals—All chemicals were obtained from Sigma, unless otherwise stated. UBEI-41 was obtained from Biogenova Corp. (Ellicott City, MD). DCEBIO was synthesized in the laboratory of R. J. Bridges (University of Pittsburgh), as described previously (22). Both DCEBIO and clotrimazole were made as 10,000-fold stock solutions in Me2SO.
Statistics—All data are presented as means ± S.E., where n indicates the number of experiments. Statistical analysis was performed using a Student's t test. A value of p < 0.05 is considered statistically significant and is reported. All protein biochemical experiments were carried out a minimum of three times on each construct to ensure the voracity of our results. Time constants for protein decay were determined by fitting the data to a single exponential decay function (SigmaPlot 2001).
RESULTS
Papazian and co-workers (3, 4, 6, 7) demonstrated that a series of conserved charged amino acids in S2, S3, and S4 are critical for the biogenesis of Kv channels. Although KCa3.1 is a voltage-independent channel, it has two arginines conserved in S4 (Arg-160 and Arg-166) and a single glutamic acid in S3 (Glu-112). Based upon sequence alignments, these arginines are equivalent to Arg-368 and Lys-374 in Shaker, whereas the glutamic acid in S3 aligns with Asp-316 in Shaker. As an initial test of whether these charged amino acids in KCa3.1 are required for channel biogenesis, each of these was mutated to alanine and total protein expression evaluated by IB. The relative position of these and the other mutated amino acids are shown in Fig. 1A. As is apparent in Fig. 1B, E112A, R160A, and R166A KCa3.1 expressed little or no protein, suggesting these amino acids are fundamentally required for KCa3.1 biogenesis. Similarly, the S2 positively charged lysine (Lys-62) also demonstrates dramatically reduced protein levels, although some K62A was expressed at the cell surface based upon our cell surface chemiluminescence assay (Fig. 1C) and on IF measurements (data not shown). In contrast, mutation of all other charged amino acids between S1 and S5 resulted in detectable protein expression, although some of these were clearly reduced below wild type levels. All IB expressing these mutations were subject to densitometry, and the fold-change, relative to wild type KCa3.1, is shown in Fig. 1B (bottom panel). As we are able to detect a 16-fold change in wild type KCa3.1 protein expression under the conditions of our experiments (see “Experimental Procedures”), we indicate a >16-fold change in channel expression for those mutations where we were unable to detect any protein expression in the majority of blots (indicated by the break shown in the y axis). Given the dramatic decrease in protein expression of the E112A, R160A, and R166A mutations, and their conservation in Kv channels, we focused on determining the degradative fate of these channels.
FIGURE 1.

Mutation of S1–S4 charged amino acids in KCa3.1 alters channel expression and localization. A, schematic of the N terminus to the re-entrant pore loop of KCa3.1 showing relative positions of all charged amino acids mutated in the current study. B, immunoblot showing expression level of all mutations studied. Note that E112A, R160A, R166A, and K62A expressed at very low levels relative to wild type KCa3.1. 20 μg of total protein was loaded per lane. Bottom panel, average expression for the mutations in the top panel expressed as a fold-change. For E112A, R160A, R166A, and K62A expression was too low to be reliably determined and is therefore represented as a change of >16-fold (indicated by the break in the y axis) as this is the maximum change which could be detected for KCa3.1 (see “Experimental Procedures”). The average densitometries are based on three or more IBs for each mutation. C, chemiluminescent detection of cell surface localized KCa3.1 and various mutations. Data are normalized to wild type KCa3.1 and are expressed as relative light units. Note that E45A and R228A fail to express at the cell surface despite being expressed at levels similar to wild type KCa3.1 as evidenced by the IB shown in B. D, E45A KCa3.1 (green, left panel) co-localizes with the ER resident protein, calnexin (red, middle panel). Co-localization is shown as yellow in the overlay (right panel).
Additionally, we observed that both E45A and R228A failed to express at the cell surface (Fig. 1C), although they expressed protein equivalent to wild type (Fig. 1B). We confirmed that E45A failed to exit the ER by immunofluorescent co-localization of E45A KCa3.1 with calnexin, an ER-resident protein (Fig. 1D). In contrast, we saw no co-localization with the Golgi resident protein, Giantin (data not shown). A similar intracellular localization pattern was observed for the Arg-228 mutation (data not shown). A further characterization of these mutations was not carried out in these studies.
We next determined the degradative fate of these mutations in S3 and S4. As shown in Fig. 2A, E112A, R160A, and R166A were insensitive to the lysosomal protease inhibitors, leupeptin and pepstatin, whereas the proteasome inhibitor lactacystin resulted in a dramatic increase in protein expression. A similar result of lactacystin was observed for the K62A mutation (Fig. 2B). It should also be noted that wild type IK1 protein levels were increased by lactacystin, suggesting that some of this channel is targeted for proteasomal degradation.
FIGURE 2.

Immunoblots demonstrating that degradation of S3 and S4 charged amino acid mutations is prevented by lactacystin. A, degradation of E112A, R160A, and R166A KCa3.1 is prevented by the proteasome inhibitor, lactacystin (10 μm), whereas the combination of lysosomal inhibitors, leupeptin (15 μm) and pepstatin (15 μm), had no effect on protein expression. Note that wild type KCa3.1 protein expression is also increased by lactacystin indicating some wild type channel is targeted to the proteasome. B, degradation of the S2 mutation K62A is prevented by lactacystin. C, degradation of E370A, R415A, and R421A KCa2.3 is prevented by the proteasome inhibitor lactacystin, whereas wild type KCa2.3 expression is unaffected. D, S2 mutation K321A in KCa2.3 is expressed at a similar level as wild type KCa2.3 (compare 1st and 3rd lanes). Digestion with proteinase K results in a lower molecular weight band in both wild type and K321A KCa2.3 (2nd and 4th lanes), indicative of these channels being expressed at the plasma membrane. E, degradation of the S4 mutation R266A in KCa2.2 is prevented by lactacystin. A, B, and E, 20 μg of total protein was loaded per lane; C and D, 10 μg of total protein was loaded per lane.
As noted above, each of these transmembrane domain charged amino acids is completely conserved across the KCNN gene family. Thus, we determined whether alanine substitutions would have a similar affect on KCa2.3 and KCa2.2 expression. As shown in Fig. 2C, mutation of Glu-370, Arg-415, or Arg-421 in KCa2.3 (equivalent of Glu-112, Arg-160, and Arg-166 in KCa3.1) resulted in a total loss of protein expression that was sensitive to lactacystin. As we are able to detect a 32-fold decrease in wild type KCa2.3 channel expression (see “Experimental Procedures”), we can conclude that the effect of these mutations is >32-fold. In contrast, the K321A mutation in KCa2.3 (equivalent to K62A in KCa3.1) was expressed at 62 ± 4% (n = 4) of wild type (Fig. 2D). Also, this mutation was clearly expressed at the plasma membrane, based on our proteinase K digestion experiments (Fig. 2D). Based on these results, we conclude that the function of the S2 lysine is not conserved across the gene family and thus was not further studied. Finally, as shown in Fig. 2E, mutation of Arg-266 in KCa2.2 (equivalent of Arg-166 in KCa3.1) to alanine resulted in a loss of protein expression that was lactacystin-dependent. These results indicate that the role of the S4 arginines and S3 glutamic acid is conserved across the KCa2.x and KCa3.1 channels, being required for proper channel biogenesis.
To more thoroughly quantify the effects of these mutations, we determined the time constant for protein degradation of wild type and mutant channels. As shown in Fig. 3, A and C, wild type KCa3.1 was completely degraded over the course of 24 h. The intensity of the protein bands was determined by densitometry, and the data were fit to a single exponential with a time constant of 5.1 ± 0.5 h (n = 3). As shown in Fig. 3, B and C, the R160A mutation was rapidly degraded, having a time constant of 0.4 ± 0.2 h (n = 3). It should be noted that 100 μg of total protein was loaded for each time point shown in B to detect the mutant channel. We also evaluated the effect of the charge-neutralizing mutation, R160Q, in KCa3.1. In this case, the time constant of degradation was shorter still, being 0.2 ± 0.3 h (n = 3). We also determined the time constant for degradation of the E112A and E112Q mutations with these being 0.8 ± 0.1 h (n = 3) and 0.4 ± 0.1 h (n = 3), respectively.
FIGURE 3.

Time course for degradation of wild type and mutant KCa3.1 and KCa2.3 channels. Representative IB showing degradation of wild type KCa3.1 (A), R160A KCa3.1 (B), wild type KCa2.3 (D), and R415 KCa2.3 (E) in the presence of cycloheximide (400 μg/ml) for various periods of time as indicated. For wild type KCa3.1 and KCa2.3, a total of 20 μg of protein was loaded in each lane, whereas 100 μg of total protein was loaded for R160A KCa3.1 and R415A KCa2.3. These time courses for degradation were repeated three times for each construct and the resulting IB digitized, and band intensities for the various time points were determined as a percent change from time 0. The decrease in total protein was fit to an exponential decay function, and the time constant (τ ± S.E.) was determined for each construct. The average intensities and fits are shown in C and F for KCa3.1 and KCa2.3, respectively. Note that the decay of wild type KCa2.3 could not be fit as sufficient protein had not been degraded over a 36-h time period.
Although wild type KCa3.1 has a relatively short half-life, we demonstrate that the time constant for KCa2.3 degradation is significantly greater than 36 h (Fig. 3, D and F), the longest time point evaluated. Similar to our results above, mutation of Arg-415 to alanine in KCa2.3 dramatically reduced the time constant for degradation to 0.3 ± 0.3 h (n = 3; Fig. 3, E and F). Mutation of Glu-370 to alanine (E370A) reduced the time constant for degradation to 0.4 ± 0.3 h (n = 3). Finally, we determined the time constant for degradation of wild type KCa2.2 to be 16.4 ± 0.9 h (n = 3; data not shown). We were unable to determine a time constant for degradation of the R266A KCa2.2 mutation as too little protein could be detected even after loading 100 μg of total protein into the gel. This is consistent with the observation shown in Fig. 2E where, even in the presence of lactacystin, the expression of R266A is significantly less than wild type KCa2.2 (21 ± 5%, n = 3). This is in contrast to the results observed with lactacystin on KCa3.1 and KCa2.3 mutations (Fig. 2, A and C) and suggests that this mutation in KCa2.2 is even more severe.
Combinations of charge reversal mutations in S2, S3, and S4 have been shown to correct the folding of Kv channels (6, 7, 10), and thus we determined the effect of charge reversal mutations in KCa3.1. As shown in Fig. 4A, the E112R, R160E, and R166E mutations failed to express detectable protein and are thus reduced at least 16-fold. As before, lactacystin increased protein expression of these mutant channels, indicative of proteasomal degradation. Mutating the glutamic acid in S3 (E112R), in combination with the arginines in S4, had no effect on the degradation of KCa3.1 as shown in Fig. 4B. Similarly, mutating the lysine in S2 to glutamic acid (K62E) in combination with charge reversal mutations at Glu-112, Arg-160, and Arg-166 did not alter the degradative fate of KCa3.1 (data not shown). Thus, in contrast to the results from Kv channels (6, 7, 10), we were unable to correct the folding of KCa3.1 by reversing the charges of transmembrane domain amino acids.
FIGURE 4.

Charge reversal mutations do not rescue protein expression of KCa3.1. A, IB showing that the charge reversal mutations, E112R, R160E, and R166E in KCa3.1 are rapidly degraded in a lactacystin-dependent manner. B, combinations of S3 and S4 charge reversal mutations similarly fail to express protein in the absence of lactacystin-dependent proteasome inhibition. 20 μg of total protein was loaded per lane in all experiments.
Finally, we mutated the arginines in S4 to histidines in KCa3.1 (R160H, R166H) based on the premise that the histidine may serve as a conditional positive charge in the acidic trafficking compartments. This would then allow KCa3.1 to traffic to the plasma membrane where it could be functionally studied. As shown in Fig. 5A, both the R160H and R166H mutations expressed reduced channel in the absence of lactacystin, averaging 10 ± 4 and 27 ± 12% (n = 4) of wild type, respectively. However, based on both cell surface chemiluminescence (Fig. 5C) and immunofluorescence labeling of the extracellular HA tag (Fig. 5D), a small amount of R166H is expressed at the cell surface, although no functional channels were observed during patch clamp studies. We previously demonstrated that chloroquine can enhance the expression of a subset of KCa3.1 mutations that are very poorly expressed at the cell surface (21). Thus, we evaluated the effect of chloroquine (100 μm) on cell surface expression of R160H and R166H KCa3.1. As shown in Fig. 5B, chloroquine increased total protein expression of both wild type (2-fold) and R166H (3.3-fold) KCa3.1, while having no effect on R160H KCa3.1 (1.2-fold). We confirmed that chloroquine increased cell surface expression of R166H by both chemiluminescence (Fig. 5C) and IF (Fig. 5D).
FIGURE 5.

R166H KCa3.1 is expressed at the cell surface in a chloroquine-dependent manner. A, IB demonstrating both R160H and R166H are expressed at low levels and that the proteasome inhibitor lactacystin increases protein expression. B, exposure of the cells to chloroquine (100 μm) for 16 h increases expression of both wild type and R166H KCa3.1. 20 μg of total protein was loaded per lane in A and B. C, chemiluminescent detection of cell surface localized wild type, R160H, and R166H KCa3.1. Data are normalized to wild type KCa3.1 and are expressed as relative light units. R160H was not significantly different from background. R166H was expressed at low levels on the cell surface, and this was dramatically increased in the presence of chloroquine. D, immunofluorescent labeling of plasma membrane localized wild type, R160H, and R166H KCa3.1. Labeling was performed using an HA Ab directed against an extracellular epitope as detailed under “Experimental Procedures.” Wild type KCa3.1 was highly expressed at the cell surface (1st panel, green label). No R160H could be detected at the cell surface (2nd panel), consistent with the chemiluminescent data. A small amount of R166H could be detected (3rd panel, green label) and this was increased by exposure to chloroquine (Chlor.) for 16 h (4th panel). Nuclei are labeled with Hoechst (blue).
To evaluate the functional consequences of charge neutralization at the Arg-166 position, we performed inside-out patch clamp studies, in combination with variance analysis, to evaluate R166H. For these studies, the patch was excised into saturating Ca2+ (10 μm) followed by 0 Ca2+ to confirm expression of KCa3.1. After returning to 10 μm Ca2+, the channels were challenged with the KCa opener, DCEBIO (30 μm (22)) followed by clotrimazole (10 μm) to inhibit the channels. As shown in Fig. 6A, for one patch from a cell expressing wild type KCa3.1 incubated overnight in chloroquine, DCEBIO induced an increase in current of wild type KCa3.1 with a subsequent inhibition by clotrimazole as reported previously (22). This DCEBIO-induced increase in current averaged 2.3 ± 0.2-fold (n = 5), which is not different from cells not exposed to chloroquine (2.5 ± 0.1-fold; n = 4). Variance analysis of this current recording demonstrates a Po(max) of 0.67 (Fig. 6B) and a single channel current amplitude (i) of 3.1 pA, consistent with what we have reported previously using this technique (15). In five recordings this Po(max) averaged 0.62 ± 0.07 with a single channel amplitude (i) of 3.3 ± 0.1 pA. Based on this, the Po for wild type channels in the presence of 10 μm Ca2+ averaged 0.26 ± 0.05, similar to what we reported previously (15). As shown in Fig. 6C for a cell expressing the R166H mutation and exposed to chloroquine overnight, DCEBIO induced a dramatic increase in current in an inside-out patch, averaging 8.5 ± 0.9-fold in six patches. This result immediately suggests that either the R166H mutation results in a channel with a significantly increased Po(max) or that the Po in the presence of 10 μm Ca2+ is reduced compared with wild type KCa3.1. Variance analysis confirmed that the Po(max) for these experiments was not significantly higher than that for wild type KCa3.1, averaging 0.59 ± 0.03 with a single channel amplitude of 3.4 ± 0.1 pA. Based on this, the Po in the presence of 10 μm Ca2+ can be calculated as 0.07 ± 0.01 for R166H KCa3.1, a value 3.7-fold lower than wild type KCa3.1. This result suggests that a positively charged amino acid is required at this position in S4 to maintain both the functional properties of KCa3.1 as well as the biogenesis of the channel.
FIGURE 6.

Variance analysis of wild type and R166H KCa3.1. A, following patch excision of wild type KCa3.1, channels were sequentially activated by 10 μm free Ca2+ and DCEBIO (30 μm) and subsequently inhibited by clotrimazole (10 μm). Current traces have been data reduced for display. B, plot of variance (σ2) against mean current (I) for the current record shown in A. The data were fitted to Equation 1 yielding an N of 230 channels and a single channel amplitude (i) of 3.0 pA. These data allowed us to calculate a Po(max) of 0.67 for this recording using Equation 2 and a Po in the presence of 10 μm Ca2+ of 0.23. C, inside-out recording of R166H KCa3.1 demonstrating a large stimulation of current with DCEBIO. D, plot of variance (σ2) against mean current (I) for the current record shown in C. The data were fitted to Equation 1 yielding an N of 270 channels and a single channel amplitude (i) of 3.4 pA. These data allowed us to calculate a Po(max) of 0.62 for this recording using Equation 2 and a Po in the presence of 10 μm Ca2+ of 0.05. Both the wild type and R166H KCa3.1 expressing cells were exposed to chloroquine overnight.
Shaker channels expressing mutations in Asp-316 (S3) and Lys-374 (S4) are primarily degraded in the proteasome, as assessed by lactacystin dependence (4). Our results on KCa3.1 and KCa2.x clearly demonstrate that mutations in charged amino acids in S3 and S4 are degraded in the proteasome. Recent results indicate that misfolded proteins destined for proteasomal degradation are translocated out of the ER in a Derlin-1-dependent fashion, after which they are poly-ubiquitinated and then dislocated into the cytosol by the p97/Cdc48-Ufdl-Npl4 complex (23–25). To determine whether KCa3.1 is translocated out of the ER and targeted to the proteasome by this pathway, we initially determined whether knockdown of Derlin-1 would alter protein expression of wild type and E112A KCa3.1. As shown in Fig. 7A, transfecting a short hairpin RNA (shRNA) targeted against Derlin-1 (shDerlin-1) decreased expression of endogenous Derlin-1 an average of 62 ± 8% in six experiments on KCa3.1 and KCa2.3. This decrease in Derlin-1 expression increased wild type KCa3.1 expression 5.7 ± 0.5-fold and increased E112A expression to 4.1 ± 0.1-fold above wild type KCa3.1 levels (n = 3). This is consistent with the idea that Derlin-1 interacts with KCa3.1 to remove it from the ER. To confirm that Derlin-1 and KCa3.1 are in the same protein complex, we performed co-IP studies on cells co-transfected with Myc-tagged KCa3.1 and HA-tagged Derlin-1. As shown in Fig. 7B, both wild type KCa3.1 and the E112A mutant immunoprecipitated Derlin-1 (lanes 3 and 7), whereas no HA-tagged Derlin-1 was detected when Myc-tagged KCa3.1 (lane 1) or HA-tagged Derlin-1 (lane 2) were transfected alone or in the presence of an IgG control Ab (lane 4). These results confirm that KCa3.1 is present within the Derlin-1 complex.
FIGURE 7.

KCa3.1 and KCa2.3 are translocated out of the ER by Derlin-1. A, cells were co-transfected with a short hairpin RNA against Derlin-1 (shDerlin-1) and either wild type (lanes 1 and 2) or E112A (lanes 3 and 4) KCa3.1. Expression of KCa3.1 was evaluated by IB (top panel). Knockdown of Derlin-1, using shDerlin-1, was confirmed by IB (bottom panel). The ratio in the absence and presence of shDerlin-1 is indicated. As is apparent in the top panel, knockdown of Derlin-1 resulted in an increase in both wild type and E112A KCa3.1 expression. 20 μg of total protein was loaded per lane in these experiments. B, co-IP of Derlin-1 with either Myc-tagged wild type KCa3.1 (lanes 1–4) or E112A KCa3.1 (lanes 5–8). Cells were co-transfected with either wild type or E112A Myc-tagged KCa3.1 plus empty vector (lanes 1 and 5), HA-tagged Derlin-1 plus empty vector (lanes 2 and 6), or Myc-tagged KCa3.1 plus HA-tagged Derlin-1 (lanes 3 and 4 and 7 and 8). Lysate was subject to immunoprecipitation (IP) using either an anti-Myc Ab (lanes 1–3 and 5–7) or a control IgG (lanes 4 and 8) and subsequently IB for Derlin-1 using an anti-HA Ab. Derlin-1 was detected by IB in lanes 3 and 7, confirming association between KCa3.1 and Derlin-1. Bottom panel, total lysate (20 μg) was probed for Derlin-1 to confirm equivalent expression. No Derlin-1 was detected in lanes 1 and 5 as these were only transfected with Myc-tagged KCa3.1. C, cells were co-transfected with a short hairpin RNA against Derlin-1 (shDerlin-1) and either wild type (1st and 2nd lanes) or E370A (3rd and 4th lanes) KCa2.3. Expression of KCa2.3 was evaluated by IB (top panel). Knockdown of Derlin-1 using shDerlin-1 was confirmed by IB (bottom panel). The ratio in the absence and presence of shDerlin-1 is indicated. As is apparent in the top panel, knockdown of Derlin-1, using shDerlin-1, resulted in an increase in E370A KCa2.3 expression. 20 μg of total protein was loaded per lane. D, co-IP of Derlin-1 with either wild type KCa2.3 (lanes 1–4) or E370A KCa2.3 (lanes 5–8). Cells were co-transfected with either wild type or E370A KCa2.3 plus empty vector (lanes 1 and 5), HA-tagged Derlin-1 plus empty vector (lanes 2 and 6), or KCa2.3 plus HA-tagged Derlin-1 (lanes 3 and 4 and 7 and 8). Lysate was subject to IP using either an anti-HA Ab (lanes 1–3 and 5–7) or a control IgG (lanes 4 and 8) and subsequently IB for KCa2.3. KCa2.3 was detected by IB in lanes 3 and 7, confirming association between KCa2.3 and Derlin-1. Bottom panel, total lysate (20 μg) was probed for KCa2.3 to confirm equivalent expression. No KCa2.3 was detected in lanes 2 and 6 as these were only transfected with HA-tagged Derlin-1. Note that E370A KCa2.3 is expressed at a significantly lower level than wild type KCa2.3 as expected.
We further demonstrate that KCa2.3 is recognized by Derlin-1 in a parallel series of studies. As shown in Fig. 7C, knockdown of Derlin-1, using sh-Derlin-1, resulted in a 4.4 ± 0.9-fold (n = 3) increase in E370A KCa2.3 protein level as expected if Derlin-1 is involved in the translocation of mutant KCa2.3 out of the ER. In contrast, knockdown of Derlin-1 had little effect (0.4 ± 0.1-fold; n = 3) on wild type KCa2.3, consistent with the lack of effect of lactacystin on this channel. Co-IP studies (Fig. 7D) confirmed that KCa2.3 and Derlin-1 are in the same protein complex.
Following translocation out of the ER, proteins targeted for proteasomal degradation are subject to poly-ubiquitylation. As an initial test to determine whether KCa3.1 and KCa2.3 are being ubiquitylated, we utilized the ubiquitin-activating enzyme-1 (E1) inhibitor UBEI-41. If degradation of these channels is dependent upon poly-ubiquitylation, then inhibition of this initial step of the ubiquitin cascade should increase protein expression. As shown in Fig. 8, inhibition of E1 resulted in an increase in both wild type and E112A KCa3.1 (A) as well as wild type and E370A KCa2.3 (B). These results are consistent with the proposal that poly-ubiquitylation is a prerequisite for channel degradation. To confirm poly-ubiquitylation of both wild type and E112A KCa3.1, we performed co-IP studies. As shown in Fig. 9A, the proteasomal inhibitor MG-132 increased expression of both wild type (2nd lane) and E112A (4th lane) KCa3.1, consistent with our lactacystin results. The inhibition of proteasomal degradation by MG-132 was accompanied by an increase in the total amount of ubiquitylated proteins (Fig. 9B), as expected. As shown in Fig. 9C, following IP of either wild type or E112A KCa3.1, we were able to detect a low level of ubiquitylation (lanes 1 and 3), and this was dramatically increased in the presence of MG-132 (lanes 2 and 4). These results confirm that KCa3.1 is subject to poly-ubiquitylation prior to proteasomal degradation.
FIGURE 8.

Inhibition of the ubiquitin-activating enzyme, E1, increases expression of KCa3.1 and KCa2.3. A, cells expressing either wild type (1st and 2nd lanes) or E112A (3rd and 4th lanes) KCa3.1 were exposed to the E1 inhibitor, UBEI-41 (50 μm) (lanes 2 and 4), overnight, and protein expression was determined by IB. Inhibition of E1 resulted in a large increase in channel expression. 20 μg of total protein was loaded in each lane. B, cells expressing either wild type (1st and 2nd lanes) or E370A (3rd and 4th lanes) KCa2.3 were exposed to the E1 inhibitor, UBEI-41 (50 μm) (2nd and 4th lanes) overnight and protein expression determined by IB. Inhibition of E1 resulted in a large increase in channel expression. 10 μg of total protein was loaded in each lane.
FIGURE 9.

Both wild type and E112A KCa3.1 are poly-ubiquitylated. A, cells were transfected with either Myc-tagged wild type or E112A KCa3.1 and exposed to MG-132 overnight. Both wild type (1st and 2nd lanes) and E112A (3rd and 4th lanes) KCa3.1 expression is increased in the presence of MG-132. B, total cell lysate was probed for ubiquitin in the absence and presence of MG-132. As is apparent, MG-132 increased the total amount of ubiquitylated protein (2nd and 4th lanes). C, co-IP Myc-tagged KCa3.1 with ubiquitin. Cells were transfected with either wild type (lanes 1 and 2) or E112A (lanes 3–5) KCa3.1; the lysates were subjected to IP with either an anti-Myc Ab (lanes 1–4) or a control IgG (lane 5) and subsequently IB for endogenous ubiquitin. Both wild type and E112A KCa3.1 showed levels of ubiquitylation above background (IgG control, lane 5), which was increased following inhibition of proteasomal degradation by MG-132.
Subsequent to Derlin-1 translocation out of the ER and polyubiquitylation, p97, a member of the AAA ATPase family of proteins, shuttles these poly-ubiquitylated proteins to the proteasome (23–25). Thus, a p97 in which the ATPase function has been mutated should not be able to target KCa3.1 to the proteasome, resulting in an increased protein expression similar to the effect of lactacystin. To test this hypothesis, we transfected wild type or E112A KCa3.1 with either wild type p97 or the ATPase mutant p97QQ and determined KCa3.1 protein expression. As shown in Fig. 10A, co-transfecting the p97QQ ATPase mutant with KCa3.1 resulted in a 2.0 ± 0.5-fold (n = 3) increase in protein expression compared with transfection of the wild type p97. Both p97QQ and lactacystin caused a similar increase in E112A expression, averaging 1.2 ± 0.2-fold and 1.4 ± 0.1-fold above wild type KCa3.1 expression levels (Fig. 10A). Similarly, p97QQ increased E370A KCa2.3 protein expression an average of 2.1 ± 0.1-fold (n = 3), whereas it had no effect on wild type KCa2.3 expression (Fig. 10B). These results suggest that p97 is required for the dislocation of KCa3.1 and KCa2.3 from Derlin-1 and is likely involved in the shuttling of these channels to the proteasome for degradation.
FIGURE 10.

Effect of p97 on KCa3.1 and KCa2.3 protein expression. A, cells were co-transfected with wild type or E112A KCa3.1 and either His-tagged wild type p97 (1st and 3rd lanes) or the ATPase mutant p97QQ (2nd and 4th lanes). Expression of KCa3.1 was then evaluated by IB (top panel). p97QQ dramatically increased expression of both wild type and E112A KCa3.1. Lactacystin was used as a control to confirm increased expression of E112A KCa3.1 in the absence of p97 (5th lane). The bottom panel confirms equivalent expression of p97 and p97QQ. Note that His-tagged p97 was not expressed in the E112A KCa3.1 expressing cells exposed to lactacystin. B, cells were co-transfected with wild type or E370A KCa2.3 and either His-tagged wild type p97 (1st and 3rd lanes) or the ATPase mutant p97QQ (2nd and 4th lanes). Expression of KCa2.3 was evaluated by IB. p97QQ dramatically increased expression of both wild type and E370A KCa2.3 while having little effect on wild type KCa2.3. In all experiments 20 μg of total protein was loaded per lane.
DISCUSSION
It is generally believed that prokaryotic 2-TM domain channels gave rise to the ligand-gated 6-TM domain K+ channels through the addition of four additional TM domains. These ligand-gated, 6-TM domain channels likely underwent a series of mutations resulting in the voltage-gated K+ channel family (Kv) which then underwent two separate rounds of gene duplication resulting in the voltage-gated Ca2+ and subsequently Na+ channels (12). From the crystal structure data of Kv channels it is clear that the additional four TM domains (S1–S4), which were appended to the central pore to form the 6-TM domain channels, form a unique structural domain (1). The fundamental difference between the Kv channels and KCa3.1/KCa2.x is that these KCa channels exhibit no voltage dependence to their gating. Therefore, the gating of the KCa channels is solely dependent upon the interaction of Ca2+ with calmodulin, which is constitutively bound to the proximal C terminus (26–28). This Ca2+-dependent gating is then modified by various protein kinase-dependent phosphorylation events (29–32).
The voltage dependence of Kv channels is conferred by a series of 4–7 positively charged amino acids in S4 as well as critical negatively charged amino acids in S2 and S3 (33, 34). In addition to conferring voltage dependence, Papazian and co-workers (3, 4, 6, 7) have shown that these charged amino acids are essential for channel biogenesis. Namely, key salt bridges are required to maintain the fundamental fold of this S1–S4 domain. Indeed, it has been shown that these electrostatic interactions are required at the initial folding process as the nascent polypeptide chain is being formed prior to entering the ER (8, 9). Although KCa3.1 and KCa2.x channels are voltage-independent, these channels conserve two critical arginines in S4 as well as a glutamic acid in S3. These key charged amino acids are absolutely conserved in all homologues cloned to date, including those in Caenorhabditis elegans, and align with Asp-316, Arg-368, and Lys-374 in Shaker. Of these, Lys-374 and Asp-316 are involved in channel biogenesis, whereas Arg-368 senses membrane voltage during the gating process (6, 7). Here, we demonstrate that mutation of any of these three charged amino acids in S3 or S4 results in the rapid degradation of KCa3.1, KCa2.3, and KCa2.2. Indeed, in the case of the R266A mutation in KCa2.2, the degradation rate was so fast that we could not detect protein regardless of the quantity loaded into the gel. In contrast to the Kv channels, we were unable to directly define the electrostatic interactions involved based on a series of charge reversal mutations. Our results are more in line with those from the BK channel where interacting amino acids were similarly elusive. Ma et al. (11) have demonstrated that Arg-207 and Arg-210 in the BK channel do not sense voltage but alter the stability of the gating conformation. However, well defined salt bridge interactions could not be defined by these authors. Given that both charge neutralization (R/A, R/Q, E/A, and E/Q) and charge reversal (R/E and E/R) mutations similarly resulted in a rapid degradation of the KCa3.1 and KCa2.3 channels, the most parsimonious explanation is that we have disrupted key electrostatic interactions such that folding is abrogated and the channels are targeted for degradation. The observation that S4 arginines, as well as an S3 glutamic acid, share a conserved function between Kv channels and the ligand-gated KCa channels suggests that their role in establishing the proper folding of the S1–S4 domain evolved prior to the establishment of voltage dependence. Thus, we would speculate that the basic structure of the S1–S4 domain is conserved between the Kv and KCa channels and that this was required for voltage dependence to evolve.
In an attempt to evaluate the functional consequences of the S4 arginine mutations in KCa3.1, we mutated Arg-160 and Arg-166 to histidine. We speculated that the histidine would carry sufficient positive charge in the relatively acidic trafficking compartments of the cell to be expressed at the cell surface. As shown in Fig. 5, a small fraction of the R166H channel was expressed at the cell surface, whereas the R160H failed to traffic to the plasma membrane. We have previously found that chloroquine is capable of increasing cell surface expression of mutant channels that express at low levels on the plasma membrane (21). Although we speculate that this is because of an inhibition of endocytosis and hence accumulation of cell surface channel, the exact mechanism is unknown. As shown in Fig. 5, chloroquine increased plasma membrane expression of R166H, thereby allowing its functional evaluation. We demonstrate that the R166H mutation results in a channel with a Po that is reduced ∼4-fold relative to wild type KCa3.1 (Fig. 6). Although this decreased Po is likely caused by a folding defect, as evidenced by the reduced trafficking of R166H (Fig. 5), it clearly demonstrates that this altered folding is communicated to the pore region of the channel such that gating is altered. We would propose then that, similar to Kv channels, S4 arginines in KCa3.1 form critical electrostatic interactions that are required for normal gating. This result further suggests that the basic folding of the S1–S4 domain was established early on and that further mutations allowed for the sensing of the voltage field.
Papazian and co-workers (4) demonstrated that the D316K and K374E mutations in Shaker are degraded in different ways. D316K was degraded in a proteasome-dependent manner, whereas the degradation pathway for K374E remained elusive (4). Here we demonstrate that all of the mutations in either the S3 glutamic acid or S4 arginines in KCa3.1 and KCa2.x are degraded in a lactacystin-dependent manner. Thus, whereas the Shaker mutations were uniquely recognized by distinct degradation pathways, all of the mutations studied herein were degraded in the proteasome. We also demonstrate that overnight exposure to lactacystin results in an increase in wild type KCa3.1 expression, whereas KCa2.3 is insensitive to lactacystin treatment (Fig. 2). However, this should not be interpreted as implying these channels are degraded via different pathways. As the time constant for degradation of KCa3.1 is ∼5 h, whereas it is >36 h for KCa2.3, very little KCa2.3 would have been degraded during the 16-h exposure to lactacystin. We are currently studying the trafficking of plasma membrane localized channels in an attempt to determine why these related channels have such disparate half-lives.
Misfolded proteins are recognized by ER resident chaperones after which some of these proteins are targeted to the ER membrane where they are recognized by the integral membrane protein Derlin-1 and translocated back into the cytosol where they are poly-ubiquitylated (23–25). These poly-ubiquitylated proteins are then recognized by the p97/Cdc48 protein complex and, using the ATPase function of p97, these proteins are dislocated from Derlin-1 and shuttled to the proteasome for degradation (23–25). Our initial studies on the time course for protein degradation of the S3 and S4 charged amino acid mutations as well as their degradation in a lactacystin-dependent manner suggested these channels are misfolded and targeted for proteasomal degradation. We demonstrate that knockdown of Derlin-1, using an shRNA approach, results in an increase in E112A KCa3.1 (Fig. 7A) and E370A KCa2.3 (Fig. 7D) as expected if Derlin-1 associates with this misfolded channel to translocate it out of the ER. We confirmed that both KCa3.1 as well as KCa2.3 are in a protein complex with Derlin-1 using a co-IP approach (Fig. 7, B, C, and E), demonstrating a role of Derlin-1 in the degradation of these channels.
We confirmed that both wild type and E112A KCa3.1 are poly-ubiquitylated prior to being targeted for degradation in two ways. First, we demonstrate that inhibition of E1, the initial step in the ubiquitylation process, increases protein expression of both KCa3.1 and KCa2.3 (Fig. 8), consistent with ubiquitylation being required for channel degradation. Second, we directly confirmed poly-ubiquitylation of KCa3.1 by co-IP (Fig. 9). Finally, we demonstrate a role for p97 in the proteasomal targeting of KCa3.1 and KCa2.3 using the ATPase-deficient p97, p97QQ. Therefore, p97QQ increased protein expression of the S3 glutamic acid mutations in both KCa3.1 and KCa2.3 (Fig. 10) as expected if these channels cannot properly be extracted from Derlin-1 and shuttled to the proteasome for degradation. In total, these results lead us to conclude that KCa3.1 and KCa2.3 are poly-ubiquitylated following translocation from the ER via Derlin-1 and then extracted in a p97-dependent process. Current studies are aimed at elucidating the ubiquitin-protein ligase(s) involved in the ubiquitylation of these channels at the level of the ER. Sun et al. (14) recently demonstrated a similar role for Derlin-1 and p97 in the degradation of ΔF508 cystic fibrosis transmembrane conductance regulator. However, to our knowledge, the role of Derlin-1 and p97 in ER translocation, protein dislocation, and shuttling of K+ channels to the proteasome for degradation has not been determined previously.
Despite the conservation of key charged amino acids in both S3 and S4 between KCa2.x, KCa3.1, and the Kv channels, no role for these amino acids has been described previously in these voltage-independent 6-TM domain channels. In Kv channels, these charged amino acids play two critical roles, they form key electrostatic interactions required for channel biogenesis and sense changes in membrane voltage. We demonstrate this role in channel biogenesis is absolutely conserved in the KCa channels. This suggests that the folding of the S1–S4 domain is likely conserved between KCa and Kv channels at its most fundamental level. Given the requirement for proper folding of this domain in advance of the evolution of voltage dependence, it is perhaps not surprising that similarities would exist in the KCa family. We further demonstrate that misfolding of the S1–S4 domain results in altered channel gating, as assessed by changes in Po. This suggests sufficient linkage between the S1–S4 and pore domains of KCa channels that perturbations in one domain can be transmitted to the other. Of course, it is the transmittal of this conformational change that allows for the voltage dependence of Kv channels. Finally, we demonstrate that misfolded KCa channels are recognized in the ER and translocated out via Derlin-1 before being polyubiquitylated and shuttled to the proteasome by p97. This pathway is likely to be involved in the degradation of a wide array of disease causing K+ channel mutations.
This work was supported by a National Institutes of Health Grant HL083060 (to D. C. D.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: TM, transmembrane; ER, endoplasmic reticulum; IF, immunofluorescence; Ab, antibody; IB, immunoblot; shRNA, short hairpin RNA; HA, hemagglutinin; E1, ubiquitin-activating enzyme; DCEBIO, 5,6-dichloro-1-ethyl-1,3-dihydro-2H-benzimidazol-2-one.
References
- 1.Lee, S.-Y., Lee, A., Chen, J., and MacKinnon, R. (2005) Proc. Natl. Acad. Sci. U. S. A. 102 15441-15446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lu, Z., Klem, A. M., and Ramu, Y. (2001) Nature 413 809-813 [DOI] [PubMed] [Google Scholar]
- 3.Papazian, D. M., Shao, X. M., Seoh, S.-A., Mock, A. F., Huang, Y., and Wainstock, D. H. (1995) Neuron 14 1293-1301 [DOI] [PubMed] [Google Scholar]
- 4.Myers, M. P., Khanna, R., Lee, E. J., and Papazian, D. M. (2004) FEBS Lett. 568 110-116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Silverman, W. R., Roux, B., and Papazian, D. M. (2003) Proc. Natl. Acad. Sci. U. S. A. 100 2935-2940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tiwari-Woodruff, S. K., Lin, M.-C. A., Schulteis, C. T., and Papazian, D. M. (2000) J. Gen. Physiol. 115 123-138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tiwari-Woodruff, S. K., Schulteis, C. T., Mock, A. F., and Papazian, D. M. (1997) Biophys. J. 72 1489-1500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sato, Y., Sakaguchi, M., Goshima, S., Nakamura, T., and Uozumi, N. (2002) Proc. Natl. Acad. Sci. U. S. A. 99 60-65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sato, Y., Sakaguchi, M., Goshima, S., Nakamura, T., and Uozumi, N. (2003) J. Biol. Chem. 278 13227-13234 [DOI] [PubMed] [Google Scholar]
- 10.Zhang, M., Liu, J., Jiang, M., Wu, D. M., Sonawane, K., Guy, H. R., and Tseng, G. N. (2005) J. Membr. Biol. 207 169-181 [DOI] [PubMed] [Google Scholar]
- 11.Ma, Z., Lou, X. J., and Horrigan, F. T. (2006) J. Gen. Physiol. 127 309-328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Anderson, P. A. V., and Greenberg, R. M. (2001) Comp. Biochem. Physiol. B Biochem. Mol. Biol. 129 17-28 [DOI] [PubMed] [Google Scholar]
- 13.Syme, C. A., Hamilton, K. L., Jones, H. M., Gerlach, A. C., Giltinan, L., Papworth, G. D., Watkins, S. C., Bradbury, N. A., and Devor, D. C. (2003) J. Biol. Chem. 278 8476-8486 [DOI] [PubMed] [Google Scholar]
- 14.Sun, F., Zhang, R., Gong, X., Geng, X., Drain, P. F., and Frizzell, R. A. (2006) J. Biol. Chem. 281 36856-36863 [DOI] [PubMed] [Google Scholar]
- 15.Jones, H. M., Bailey, M. A., Baty, C. J., MacGregor, G. G., Syme, C. A., Hamilton, K. L., and Devor, D. C. (2007) Channels 1 80-91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Alvarez, O., Gonzalez, C., and Latorre, R. (2002) Adv. Physiol. Educ. 26 327-341 [DOI] [PubMed] [Google Scholar]
- 17.Sigworth, F. J. (1977) Nature 270 265-267 [DOI] [PubMed] [Google Scholar]
- 18.Sigworth, F. J. (1980) J. Physiol. (Lond.) 307 97-129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Traynelis, S. F., and Jaramillo, F. (1998) Trends Neurosci. 21 137-145 [DOI] [PubMed] [Google Scholar]
- 20.Jones, H. M., Hamilton, K. L., and Devor, D. C. (2005) J. Biol. Chem. 280 37257-37265 [DOI] [PubMed] [Google Scholar]
- 21.Jones, H. M., Hamilton, K. L., Papworth, G. D., Syme, C. A., Watkins, S. C., Bradbury, N. A., and Devor, D. C. (2004) J. Biol. Chem. 279 15531-15540 [DOI] [PubMed] [Google Scholar]
- 22.Singh, S., Syme, C. A., Singh, A. K., Devor, D. C., and Bridges, R. J. (2001) J. Pharmacol. Exp. Ther. 296 600-611 [PubMed] [Google Scholar]
- 23.Jentsch, S., and Rumpf, S. (2007) Trends Biochem. Sci. 32 6-11 [DOI] [PubMed] [Google Scholar]
- 24.Ye, Y. (2006) J. Struct. Biol. 156 29-40 [DOI] [PubMed] [Google Scholar]
- 25.Ye, Y., Meyer, H. H., and Rapoport, T. A. (2001) Nature 414 652-656 [DOI] [PubMed] [Google Scholar]
- 26.Keen, J. E., Khawaled, R., Farrens, D. L., Neelands, T., Rivard, A., Bond, C. T., Janowsky, A., Fakler, B., Adelman, J. P., and Maylie, J. (1999) J. Neurosci. 19 8830-8838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schumacher, M. A., Rivard, A. F., Bachinger, H. P., and Adelman, J. P. (2001) Nature 410 1120-1124 [DOI] [PubMed] [Google Scholar]
- 28.Xia, X. M., Fakler, B., Rivard, A., Wayman, G., Johnson-Pais, T., Keen, J. E., Ishii, T., Hirschberg, B., Bond, C. T., Lutsenko, S., Maylie, J., and Adelman, J. P. (1998) Nature 395 503-507 [DOI] [PubMed] [Google Scholar]
- 29.Bildl, W., Strassmaier, T., Thurm, H., Andersen, J., Eble, S., Oliver, D., Knipper, M., Mann, M., Schulte, U., Adelman, J. P., and Fakler, B. (2004) Neuron 43 847-858 [DOI] [PubMed] [Google Scholar]
- 30.Gerlach, A. C., Gangopadhyay, N. N., and Devor, D. C. (2000) J. Biol. Chem. 275 585-598 [DOI] [PubMed] [Google Scholar]
- 31.Gerlach, A. C., Syme, C. A., Giltinan, L., Adelman, J. P., and Devor, D. C. (2001) J. Biol. Chem. 276 10963-10970 [DOI] [PubMed] [Google Scholar]
- 32.Srivastava, S., Li, Z., Ko, K., Choudhury, P., Albaqumi, M., Johnson, A. K., Yan, Y., Backer, J. M., Unutmaz, D., Coetzee, W. A., and Skolnik, E. Y. (2006) Mol. Cell 24 665-675 [DOI] [PubMed] [Google Scholar]
- 33.Aggarwal, S. K., and MacKinnon, R. (1996) Neuron 16 1169-1177 [DOI] [PubMed] [Google Scholar]
- 34.Seoh, S.-A., Sigg, D., Papazian, D. M., and Bezanilla, F. (1996) Neuron 16 1159-1167 [DOI] [PubMed] [Google Scholar]