

Abstract
Secretogranin II (SgII) belongs to the granin family of prohormones widely distributed in dense-core secretory granules (DCGs) of endocrine, neuroendocrine, and neuronal cells, including sympathoadrenal chromaffin cells. The mechanisms by which secretory proteins, and granins in particular, are sorted into the regulated secretory pathway are unsettled. We designed a strategy based on novel chimeric forms of human SgII fused to fluorescent (green fluorescent protein) or chemiluminescent (embryonic alkaline phosphatase) reporters to identify trafficking determinants mediating DCG targeting of SgII in sympathoadrenal cells. Three-dimensional deconvolution fluorescence microscopy and secretagogue-stimulated release studies demonstrate that SgII chimeras are correctly targeted to DCGs and released by exocytosis in PC12 and primary chromaffin cells. Results from a Golgi-retained mutant form of SgII suggest that sorting of SgII into DCGs depends on a saturable sorting machinery at the trans-Golgi/trans-Golgi network. Truncation analyses reveal the presence of DCG-targeting signals within both the N- and C-terminal regions of SgII, with the putative α-helix-containing SgII-(25-41) and SgII-(334-348) acting as sufficient, independent sorting domains. This study defines sequence features of SgII mediating vesicular targeting in sympathoadrenal cells and suggests a mechanism by which discrete domains of the molecule function in sorting, perhaps by virtue of a particular arrangement in tertiary structure and/or interaction with a specific component of the DCG membrane.
In neuroendocrine cells, newly synthesized secretory proteins are targeted to the constitutive or regulated branches of the secretory pathway (1, 2). Secretory proteins entering the regulated pathway are concentrated into dense-core secretory granules (DCGs)2 prior to release by exocytosis in response to secretagogue. Two basic models have been proposed, namely “sorting-for-entry” and “sorting-by-retention,” and suggest that multimerization/aggregation of a cargo of secretory proteins in a mildly acidic, cation-rich environment is a nexus to both processes (3, 4). Sorting-for-entry proposes that a structural/conformational motif within an aggregate of secretory proteins mediates subsequent association to a protein receptor or a lipid component of the membrane of the trans-Golgi network (TGN). The quest for identifying a consensus sorting signal has proven elusive, and it now appears that such process may rather depend on a variety of motifs (5-7), including hydrophobic disulfide-bonded loop structures (8-12) or amphipathic α-helices (13-16). In the sorting-by-retention model, selective aggregation/condensation occurs in post-TGN immature secretory granules (ISGs), resulting in retention of regulated secretory proteins, whereas nonaggregated proteins are removed into constitutive-like secretory vesicles (3). These two models might not be mutually exclusive. Indeed, sorting-by-retention may perhaps be viewed as an auxiliary condensation mechanism refining the segregation process in a more distal compartment of the secretory pathway, which is the maturing ISG.
The chromogranins/secretogranins (granins) family of regulated secretory proteins are widely distributed in the core of DCGs throughout the neuroendocrine and neuronal systems (17). Index members include chromogranin A (CgA), chromogranin B (CgB), and secretogranin II (SgII). Granins function as prohormones (17), and there is evidence that CgA, and perhaps other granins, mediates the biogenesis of DCGs (18-21). Earlier reports have investigated the presence of specific DCG sorting motifs within CgA and CgB (8, 10-13), such as a conserved N-terminal hydrophobic disulfide-bonded loop (8, 10-13). Although such structure appears necessary and sufficient for sorting of CgB (8, 10), DCG targeting of CgA might require an additional amphipathic α-helix (13). Whether one or many discrete regions in primary structure mediate the vesicular targeting of SgII in the primary sequence of the protein remains undetermined and might not merely be inferred from studies of CgA or CgB trafficking. Indeed, SgII shows little homology with CgA and CgB, with respect to primary structure and genomic organization (17). Most notably, SgII lacks the N-terminal hydrophobic disulfide-bonded loop structure found in CgA and CgB.
We report a strategy in which the expression of novel chimeric forms of SgII fused to GFP or embryonic alkaline phosphatase (EAP) is used to investigate the mechanism by which SgII is targeted to the regulated secretory pathway of sympathoadrenal PC12 cells. We provide evidence that sorting of SgII into DCGs depends on a saturable sorting machinery at the trans-Golgi/TGN, and we find that information necessary for such a process is contained within both the N-terminal and C-terminal regions of SgII, with the α-helix-containing amino acid sequences 25-41 (SgII-(25-41)) and 334-348 (SgII-(334-348)) of the mature protein acting as sufficient, independent sorting signals for trafficking of SgII into the regulated pathway.
EXPERIMENTAL PROCEDURES
Cell Culture
Pheochromocytoma PC12 cells and primary chromaffin cells isolated from fresh bovine medulla were cultured as described previously (22, 23). In some experiments, PC12 cells were treated with nerve growth factor (NGF; 2.5 S, 100 ng/ml; Invitrogen) 48 h prior to transfection and further differentiated by NGF for an additional 48 h.
DNA Transfection
Supercoiled plasmid DNA for transfection was grown in Escherichia coli strain DH5α (Invitrogen) and purified on columns (Qiagen). In nucleofection experiments (Amaxa), plasmid DNA was further purified using the MiraCLEAN endotoxin removal kit (MirusBio). Two days before transfection, PC12 cells were split onto either poly-l-lysine (Sigma) plus collagen (Upstate)-coated 15-mm round glass coverslips (Fisher number 1) in 12-well Costar plates or onto poly-l-lysine plus collagen-coated 6- or 12-well Costar plates. Cells were transfected with 1 μg (12-well plate) or 2 μg (6-well plate) of DNA per well using the cationic lipid transfection reagent GenePorter 2 (Genlantis). Five hours after the beginning of the transfection, the culture medium was replaced, and cells were further cultured for 48 h. Nucleofection was performed using 2 × 106 bovine chromaffin cells resuspended in 100 μl of Basic Nucleofector solution for primary neurons, 3 μg of DNA, and program A-33. Immediately after transfection, 500 μl of RPMI 1640 medium was added to the cells, which were allowed to recover 20 min (37 °C, 5% CO2), before plating (6 × 105 cells per well) onto coated glass coverslips in 12-well Costar plates.
Construction of Expression Vectors
pGEM4-hSgII (24) served as a template to amplify full-length human SgII (NCBI NM_003469), including the predicted 30-residue signal peptide (24) (SIG) using primers incorporating an XhoI restriction site followed by a Kozak translation initiation consensus sequence at the 5′ end, and a KpnI restriction site at the 3′ end of the coding sequence. The amplified fragment was purified, digested with XhoI and KpnI, and subcloned in-frame into the same sites of pEGFP-N2 (Clontech), to produce pCMV-SgII-EGFP. SgII domains SIG-SgII-(1-24), SIG-SgII-(1-41), SIG-SgII-(1-76), SIG-SgII-(1-145), and SIG-SgII-(1-301) were obtained by PCR using pCMV-SgII-EGFP as a template and specific primers incorporating a KpnI restriction site at the 3′ end of each domain. After digestion with XhoI and KpnI, each fragment was subcloned in-frame into the same sites of the pEGFP-N2 to create pCMV-SgII181-EGFP, pCMV-SgII253-EGFP, pCMV-SgII304-EGFP, pCMV-SgII409-EGFP, pCMV-SgII616-EGFP, and pCMV-SgII1084-EGFP. pCMV-SgII-EGFP was used to amplify SIG domain flanked by an AgeI site at its 3′ end, and a SgII-(302-587) domain flanked with an AgeI site at its 5′ end. Resulting PCR fragments were digested with either XhoI and AgeI (SIG) or AgeI and KpnI (SgII-(302-587)) and ligated together. The resulting fragment was subcloned in-frame into the XhoI and KpnI sites of pEGFP-N2 to create pCMV-SgIIΔN-EGFP. The C-terminal domains SIG-SgII-(302-333), SIG-SgII-(302-348), SIG-SgII-(302-371), SIG-SgII-(302-402), and SIG-SgII-(302-505) were obtained by PCR using pCMV-SgIIΔN-EGFP as a template and specific primers incorporating KpnI at the 3′ end of each domain. Each fragment was digested with XhoI and KpnI and subcloned in-frame into the same sites of pEGFP-N2 to create pCMV-SgII-(1085-1180)-EGFP, pCMV-SgII-(1085-1225)-EGFP, pCMV-SgII-(1085-1294)-EGFP, pCMV-SgII-(1085-1387)-EGFP, pCMV-SgII-(1085-1588)-EGFP, and pCMV-SgII-(1085-1696)-EGFP. To construct pCMV-SgII-(254-304)-EGFP and pCMV-SgII-(1181-1225)-EGFP, complementary synthetic oligonucleotides of the corresponding sequences, including a KpnI restriction site at the 5′ end and a BamHI at the 3′ end, were annealed and digested with KpnI and BamHI. Each fragment was subcloned into the same sites of pCMV-SgII181-EGFP.
A truncated domain (EAP) of the full-length human secreted EAP (SEAP) gene devoid of 17-amino acid signal peptide of SEAP was obtained as described previously (25). To design SgII domain-EAP fusion chimeras, EGFP was substituted by EAP at the KpnI and NotI cloning sites of each of the SgII domain/EGFP plasmids described above. pCMV-SgII-(254-304)-EAP and pCMV-SgII-(1181-1225)-EAP were obtained by inserting EAP into the BamHI and NotI cloning sites of EGFP. pECFP-Golgi (Clontech) encoding the membrane-anchoring region of glycoprotein β1,4-galactosyltransferase (GalT-CFP) was used to design pECFP-Golgi-SgII. The ochre codon of enhanced CFP (TAA at position 1581-1583) was mutated to a serine codon (TCA) by site-directed mutagenesis (QuikChange, Stratagene) to produce mutated pECFP-Golgi. Next, a human SgII domain lacking SIG was amplified from pCMV-SgII-EGFP using specific primers incorporating a NotI restriction site at the 5′ end and a TAA ochre codon followed by a NotI restriction site at the 3′ end. The amplified fragment was purified, digested with NotI, and subcloned in-frame into the same site of mutated pECFP-Golgi to produce pECFP-Golgi-SgII encoding GalT-CFP-SgII. All the constructs were verified by restriction and nucleotide sequence analysis.
Chimeric Photoprotein Fluorescence and Immunocytochemistry
Transfected cells cultured on poly-l-lysine- and collagen-coated glass coverslips were fixed for 1 h at room temperature with 2% paraformaldehyde in PBS, pH 7.4, permeabilized for 10 min with 0.1% Triton X-100 in PBS, and exposed to 1 μg/ml nucleic acid stain Hoechst 33342 (Molecular Probes) for nuclei visualization. Prior to fixation, NGF-differentiated PC12 cells were incubated for 30 min at 37 °C with 12.5 μm CellTracker Orange CMRA (Molecular Probes) in serum-free medium to visualize cell bodies and neurites and washed for 30 min at 37 °C in complete medium. For immunocytochemistry, fixed cells were permeabilized for 10 min with 0.5% Triton X-100 in PBS, incubated for 5 min with 150 mm glycine in PBS, and exposed for 30 min to 5% fetal bovine serum in PBS. Cells were then incubated for 90 min at room temperature with goat polyclonal anti-CgB (C19, 1:100; Santa Cruz Biotechnology), rabbit polyclonal anti-human SgII (1:2000) (26), or rabbit polyclonal anti-human placental alkaline phosphatase antibody (1:50; Biomeda) in PBS containing 1% bovine serum albumin. Cells were subsequently washed and incubated for 30 min with a Alexa Fluor 594-conjugated donkey anti-goat IgG (1:350; Molecular Probes) or goat anti-rabbit IgG (F(ab′)2 at 1:250; Molecular Probes) together with 1 μg/ml Hoechst 33342 in PBS containing 1% bovine serum albumin. Coverslips were washed with PBS, mounted in buffered Celvol (Celanese), and processed for three-dimensional imaging by deconvolution fluorescence microscopy.
Three-dimensional Imaging by Deconvolution Fluorescence Microscopy
Images were captured on a DeltaVision deconvolution microscopy system operated by SoftWoRx software (Applied Precision), using oil immersion objectives (60 or 100×, N.A. 1.4). The system included a Photometrics CoolSNAP HQ CCD camera mounted on a Nikon TE2000 fitted with a mercury arc lamp light source. Pixel intensities were kept in the linear response range of the digital camera (≤4095). Optical xy sections along the z axis were acquired with increments of 0.2 μm. The fluorescent data sets were deconvolved to generate optical sections and three-dimensional images of the data sets. Adobe Photoshop 7.0 or ImageJ were used for additional processing of the images.
Analyses of the Images Data Sets
Quantification of Fluorescence Colocalization—To assess
quantitatively the extent of colocalization between SgII fragment/GFP fusion
proteins, DCG marker CgB, or CFP chimeras, 9-25 optical xy sections
along the z axis of 3-6 cells were analyzed using the pixel-per-pixel
colocalization algorithm of ImageMaster 5.0 imaging software (Photon
Technology International). A scatter plot was generated for each xy
section and represented the intensity of the green channel (x
coordinate, GFP signal) versus the intensity of the red or blue
channel (y coordinate, Alexa Fluor 594 or CFP signal) for each pixel
of the section. Based on the two-dimensional histogram, the software
calculates the Pearson's correlation coefficient (Rp),
which is mathematically related to Pearson's χ2 test, and the
overlap coefficient (Ro). Rp and
Ro are quantitative of the degree of overlap between two
signals S1 and S2. Rp is calculated
according to Equation 1,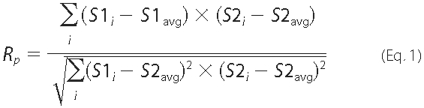
Ro is calculated according to Equation
2,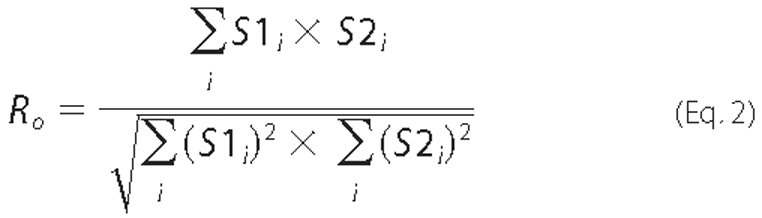
where -1 ≤ Rp ≤1 and 0 ≤ Ro ≤1. A value of 1 stands for complete positive correlation between the S1 and S2. Ro is not sensitive to the differences in pixel intensity between S1 and S2. S1i is signal intensity in the first channel; S1avg is average intensity in the first channel; S2i is signal intensity in the second channel; S2avg is average intensity in the second channel. The averaged Rp and Ro values of each cell were used for statistical analysis of variance by ANOVA with Dunnett's post test. Differences between correlation within each data group of Table 1 were also analyzed by χ2 for a row by column contingency table using pixel counts from a representative xy optical section taken in the mid-cell region. Differences were considered significant when p < 0.05.
TABLE 1.
Quantitative analysis of the colocalization between SgII domain chimeric proteins and markers of the secretory pathway in PC12 cells
Colocalization between endogenous DCG markers SgII and CgB are shown (A). Colocalization between SgII domain/GFP (B) or SgII domain/EAP (C) fusion proteins and endogenous DCG marker CgB is shown. Colocalization between SgII domain-GFP chimeras and trans-Golgi/TGN-retained CFP chimeras GFP is shown (D). xy optical sections were acquired with increments of 0.2 μm along the z axis for the indicated number of cells. The degree of colocalization between fluorescent molecules was measured on a pixel-per-pixel basis for the two wavelength sources by comparing the position of each pixel in each of the 0.2-μm xy image of a given z stack. Rp and Ro were determined using the colocalization algorithm of ImageMaster image processing software, as described under “Experimental Procedures.” Statistical significance of correlation within B, C, and D data sets was also tested by ϰ2 for a 2 × 4 contingency table, using pixel counts from a representative xy optical section taken in the mid-cell region. Group B, c2 = 5925, p < 0.001; group C, ϰ2 = 6548, p < 0.001; group D, c2 = 1642, p < 0.001.
| Pearson's coefficient (Rp) | Overlap coefficient (Ro) | xy optical sections (n) | Cells (n) | |
|---|---|---|---|---|
| A. Endogenous SgII vs. CgB | 0.76 ± 0.02 | 0.75 ± 0.02 | 254 | 17 |
| B. GFP chimera vs. endogenous CgBa | ||||
| SIG-GFP | 0.13 ± 0.04 | 0.17 ± 0.04 | 72 | 4 |
| SIG-SgII-(25-41)-GFP | 0.10 ± 0.05b | 0.17 ± 0.04b | 71 | 5 |
| SIG-SgII-(334-348)-GFP | 0.39 ± 0.04c | 0.44 ± 0.01c | 73 | 4 |
| SIG-SgII-GFP | 0.39 ± 0.05c | 0.41 ± 0.05c | 121 | 6 |
| C. EAP chimera vs. CgBd | ||||
| SIG-EAP | 0.19 ± 0.02 | 0.25 ± 0.02 | 85 | 4 |
| SIG-SgII-(25-41)-EAP | 0.52 ± 0.07c | 0.60 ± 0.06e | 129 | 6 |
| SIG-SgII-(334-348)-EAP | 0.67 ± 0.08e | 0.72 ± 0.06e | 97 | 4 |
| SIG-SgII-EAP | 0.49 ± 0.05c | 0.52 ± 0.05c | 95 | 4 |
| D. GFP chimera vs. CFP chimeraf | ||||
| SIG-SgII-GFP vs. | ||||
| GalT-CFP | 0.15 ± 0.06 | 0.17 ± 0.06 | 43 | 4 |
| GalT-CFP-SgIIa | 0.77 ± 0.03e | 0.77 ± 0.03e | 43 | 4 |
| SIG-GFP vs. | ||||
| GalT-CFP | 0.73 ± 0.07e | 0.74 ± 0.07e | 27 | 3 |
| GalT-CFP-SgII | 0.64 ± 0.03e | 0.65 ± 0.03e | 35 | 3 |
Values are given as the means ± S.E. p values for difference within groups were determined as compared with the lowest value of colocalization coefficient obtained for SIG-GFP versus CgB.
p > 0.05; ANOVA with Dunnett's post test.
p < 0.01; ANOVA with Dunnett's post test.
p values for difference within groups were determined as compared with the lowest value of colocalization coefficient obtained for SIG-EAP versus CgB.
p < 0.001; ANOVA with Dunnett's post test.
p values for difference within groups were determined as compared with the lowest value of colocalization coefficient obtained for SIG-SgII-GFP versus GalT-CFP.
Quantification of Fluorescence Distribution
To assess quantitatively the distribution of SIG-SgII-GFP puncta in PC12 cells coexpressing GalT-CFP-SgII chimera, we analyzed the abundance and size of green fluorescent particles using ImageJ. For each xy section, green puncta that contained above-threshold green pixel values within a specified area range of 0-1.15 μm2 (i.e. a diameter range of 0-600 nm) were outlined and counted, and their diameter was measured. The average diameter was calculated over the total number of identified particles for each condition as follows: SIG-SgII-GFP and GalT-CFP, 2814 counts; SIG-SgII-GFP and GalT-CFP-SgII, 997 counts. A statistical analysis of variance was done by t test.
Chemiluminescence Detection of EAP Secretion Assay
PC12 cells grown onto poly-l-lysine-coated plus collagen-coated 6- or 12-well plates were transiently transfected with an expression plasmid for SgII domain/EAP chimeras or SEAP. Cells were washed twice with calcium saline buffer (CaSB: 150 mm NaCl, 5 mm KCl, 2 mm CaCl2, and 10 mm HEPES, pH 7.4) and subsequently exposed for 15 min to CaSB or barium secretion buffer (BaSB: 150 mm NaCl, 5 mm KCl, 2 mm BaCl2, and 10 mm HEPES, pH 7.4). Supernatants were collected, and cells lysates were prepared by quick freeze/thaw in CaSB containing 0.1% Triton X-100. Detection of EAP enzymatic activity in the culture supernatant and cell lysate was achieved with a high sensitivity chemiluminescence assay (Phospha-Light, Applied Biosystems). The secretion rate of EAP chimeras was calculated as a percentage of the total EAP activity present in the cells before stimulation. Total EAP activity is the sum of the amount released plus the amount remaining in the cells. In the figures, the release of EAP chimeras is expressed as percent EAP activity secretion, or relative to EAP secretion rate in nonstimulating condition (CaSB, mock control).
Secondary Structure Prediction
The secondary structure of mature human SgII was predicted with NNPREDICT (27) and further analyzed with the NPS@ consensus secondary structure prediction program (28) that includes 11 secondary structure prediction algorithms as follows: DPM, DSC, GOR1, GOR3, GOR4, HNNC, PHD, Predator, SIMPA96, SOPM, and SOPMA. Helical wheel projection was accomplished using the PEPWHEEL software of the EMBOSS application package (29).
Presentation of Data and Statistics
Values are given as the mean ± S.E. for at least triplicate determinations. In the figures, data are representative of a typical experiment repeated independently two or more times. The number of independent experiments or analyses (n) performed for the results presented in each figure is as follows: Fig. 2, n > 6; Fig. 3, n ≥ 3; Fig. 4, n ≥ 3; Fig. 5, n > 3; Fig. 6, n ≥ 2; Fig. 7, n ≥ 2; Fig. 8, n ≥ 2; Fig. 9, n≥ 3; Fig. 10, n ≥ 2. Statistical analysis was performed by t test, or ANOVA with Dunnett's or Bonferroni post test, or by χ2 for a row by column contingency table. Differences were considered significant when p < 0.05.
FIGURE 2.

Intracellular distribution of SIG-GFP and SIG-SgII-GFP in PC12 and primary chromaffin cells. Cells were transfected for 48 h with pCMV-SgII181-GFP or pCMV-SgII-GFP and examined by deconvolution microscopy. A series of xy optical sections along the z axis was acquired with increments of 0.2 μm using 60 or 100× oil immersion objectives (1.4 N.A.). The data set was processed to generate three-dimensional (3D)/volume or representative xy section (0.2μm) views of the distribution of the GFP chimeras (green) (A). PC12 cells expressing SIG-GFP or SIG-SgII-GFP were processed for immunocytochemistry using a polyclonal anti-CgB primary and an Alexa Fluor 594-conjugated secondary antibody (B). The distribution of SIG-GFP or SIG-SgII-GFP (green) and the endogenous secretory granule marker CgB (red) was compared in merged images. Yellow regions are indicative of colocalization. Quantification of fluorescence overlap is reported in Table 1. Nuclei were visualized with Hoechst 33342 (blue). Scale bar, 5 μm.
FIGURE 3.

Effect of NGF differentiation on the distribution of SgII domain-GFP chimeras in PC12 cells. NGF-differentiated PC12 cells expressing the indicated GFP fusion protein were labeled with CellTracker Orange CMRA dye (red) prior to fixation to visualize the cell body and neurites. Fluorescence imaging by deconvolution microscopy shows a vesicular distribution of SIG-SgII-GFP, SIG-SgII-(1-41)-GFP, and SIG-SgII-(302-348)-GFP chimeras along and at the tips of processes. Neurite termini are shown in enlarged insets in the deconvolved three-dimensional images. Nuclei were visualized with Hoechst 33342 (blue). The blue intracellular spots (e.g. top right panel) likely represent residual DNA in cell debris, stained by Hoechst 33342. Scale bar, 5 μm.
FIGURE 4.

Trafficking of SIG-EAP and SIG-SgII-EAP chimeras in PC12 cells. A, subcellular distribution of SIG-EAP and SIG-SgII-EAP. PC12 cells transfected with pCMV-SgII181-EAP or pCMV-SgII-EAP were processed for immunocytochemistry using an anti-human placental alkaline phosphatase polyclonal primary and Alexa Fluor 594-conjugated secondary antibodies and analyzed by deconvolution microscopy. B, chemiluminescence detection of secretagogue-evoked secretion of EAP chimeras. Cells transiently expressing SEAP, SIG-EAP, or SIG-SgII-EAP were exposed for 15 min to secretion medium alone (mock) or to 2 mm Ba2+. EAP secretion was calculated relative to total enzymatic activity present in the cells before stimulation. Total enzymatic activity is the sum of the amount released plus the amount remaining in the cells. Release of EAP is expressed either as % EAP activity secretion or relative to enzymatic activity release in the absence of secretagogue. Values are given as the means ± S.E. of triplicate determinations. †, p > 0.05; ***, p < 0.0001 as compared with control (mock), ANOVA with Dunnett's post test. Scale bar, 5 μm.
FIGURE 5.

Dominant negative effect of a GalT-CFP-SgII chimera on DCG targeting of SgII fusion proteins in PC12 cells. SgII fused to the membrane-anchoring region of β1,4-galactosyltransferase (GalT) is retrieved within the trans-Golgi/TGN (A). PC12 cells expressing SIG-GFP together with GalT fused to CFP (GalT-CFP), or GalT-CFP fused to SgII (GalT-CFP-SgII), were processed for deconvolution microscopy. Cells expressing SIG-SgII-GFP together with GalT-CFP or GalT-CFP-SgII were examined by deconvolution microscopy (B and C). Colocalization of GFP (green) and CFP (blue) photoproteins is shown in merged images of three-dimensional (3D) views or representative 0.2 μm xy optical sections (A and B). Cyan regions are indicative of colocalization. Quantification of fluorescence overlap is reported in Table 1. C, quantification of SIG-SgII-GFP fluorescent puncta in PC12 cells expressing GalT-CFP or GalT-CFP-SgII. For each xy section, green puncta that contained above-threshold green pixels values within a specified area range of 0-1.15 μm2 (i.e. a diameter range of 0-600 nm) were outlined and counted, and their diameter was measured (ImageJ). The average diameter was calculated over the total number of identified particles for each condition as follows: SIG-SgII-GFP and GalT-CFP, 2814 counts; SIG-SgII-GFP and GalT-CFP-SgII, 997 counts. Values for diameter and puncta/xy section are given as the means ± S.E. p < 0.05 (*), t test. D, secretagogue-evoked secretion of SIG-EAP and SIG-SgII-EAP chimeras in PC12 cells coexpressing GalT-CFP or GalT-CFP-SgII. Transfected cells were incubated for 15 min in secretion medium alone (mock) or in 2 mm Ba2+. EAP enzymatic activity was assayed in the culture supernatant and cell lysate, and relative secretion was determined as described in the legend of Fig. 4. Values are given as the means ± S.E. of triplicate determinations. †, p > 0.05; ***, p < 0.001, as compared with control (mock), ANOVA with Dunnett's post test. Scale bar, 5 μm.
FIGURE 6.

Subcellular distribution and secretagogue-stimulated release of the N- and C-terminal half-domains of SgII in PC12 cells. A, three-dimensional reconstruction of the subcellular distribution of SIG-SgII-GFP, SIG-SgII-(1-301)-GFP, and SIG-SgII-(302-587)-GFP expressed in PC12 cells. B, secretagogue-evoked secretion of SgII domain-EAP chimeras. PC12 cells expressing SIG-EAP, SIG-SgII-EAP, SIG-SgII-(1-301)-EAP, or SIG-SgII-(302-587)-EAP were exposed for 15 min to secretion medium alone (mock) or to 2 mm Ba2+. The enzymatic activity of EAP chimeras was assayed in the culture supernatant and cell lysate, and relative secretion was determined as described in the legend of Fig. 4. Values are given as the means ± S.E. of triplicate determinations. †, p > 0.05; **, p < 0.01; ***, p < 0.001, as compared with control (mock), ANOVA with Dunnett's post test. Scale bar, 5 μm.
FIGURE 7.

Trafficking of progressively truncated N-terminal domains of SgII fused to GFP or EAP in PC12 cells. Cells transfected with expression plasmids encoding the indicated photoproteins were processed for deconvolution microscopy (A). A transition from a punctate/vesicular distribution (e.g. SIG-SgII-GFP) to a juxtanuclear localization (e.g. SIG-GFP) is seen as the N-terminal domain of SgII is reduced from +41 to +24 residues. B, secretagogue-evoked release of SgII domain-EAP chimeras. PC12 cells expressing the indicated EAP fusion proteins were exposed for 15 min to secretion medium alone (mock) or to 2 mm Ba2+. The enzymatic activity of EAP chimeras was assayed in the culture supernatant and cell lysate, and relative secretion was determined as described in the legend of Fig. 4. Values are given as the means ± S.E. of triplicate determinations. †, p > 0.05; **, p < 0.01; ***, p < 0.001 as compared with control (mock), ANOVA with Dunnett's post test. Scale bar, 5 μm.
FIGURE 8.

The domain SgII-(25-41) is a sufficient sorting signal for the regulated secretory pathway in PC12 cells. SIG-SgII-(25-41)-GFP- or SIG-SgII-(25-41)-EAP-expressing cells were processed for immunocytochemistry and deconvolution microscopy using a polyclonal anti-CgB primary and an Alexa Fluor 594-conjugated secondary antibody. Absence of overlap between SIG-SgII-(25-41)-GFP (green) and CgB (red) is revealed in the merged images (A) and after quantification of fluorescence colocalization (Table 1). SIG-SgII-(25-41)-EAP was stained using a polyclonal anti-human placental alkaline phosphatase primary and an Alexa Fluor 488-conjugated secondary antibodies (B). Yellow regions are indicative of colocalization between SIG-SgII-(25-41)-EAP (green) and CgB (red). Quantification of fluorescence overlap is reported in Table 1. C, regulated secretion of SIG-SgII-(25-41)-EAP. PC12 cells transiently expressing SIG-SgII-EAP or SIG-SgII-(25-41)-EAP were incubated for 15 min in secretion medium alone (mock) or 2 mm Ba2+. The enzymatic activity of EAP chimeras was assayed in the culture supernatant and cell lysate, and relative secretion was determined as described in the legend of Fig. 4. Values are given as the means ± S.E. of triplicate determinations. †, p > 0.05; ***, p < 0.001 as compared with control (mock), ANOVA with Dunnett's post test. Nuclei were stained with Hoechst 33342 (blue). Scale bar, 5 μm.
FIGURE 9.

Trafficking of progressively truncated C-terminal domains of SgII fused to GFP or EAP in PC12 cells. PC12 cells expressing the indicated photoproteins were processed for deconvolution microscopy (A). A transition from a punctate/vesicular distribution (e.g. SIG-SgII-GFP) to a juxtanuclear localization (e.g. SIG-GFP) is seen as the N-terminal domain of SgII is reduced from +348 to +333 residues. B, secretagogue-evoked release of SgII domain-EAP chimeras. PC12 cells expressing the indicated EAP fusion proteins were exposed for 15 min to secretion medium alone (mock) or to 2 mm Ba2+. The enzymatic activity of EAP chimeras was assayed in the culture supernatant and cell lysate, and relative secretion was determined as described in the legend of Fig. 4. Values are given as the means ± S.E. of triplicate determinations. †, p > 0.05; *, p < 0.05; ***, p < 0.001 as compared with control (mock), ANOVA with Dunnett's post test. Scale bar, 5 μm.
FIGURE 10.

The domain SgII-(333-348) functions as a transferable DCG-sorting determinant in PC12 cells. SIG-SgII-(334-348)-GFP colocalizes with secretory granule marker CgB in PC12 cells (A). Cells transfected with pCMV-SgII-(1181-1225)-GFP were processed for immunocytochemistry using a polyclonal anti-CgB primary and an Alexa Fluor 594-conjugated secondary antibodies, followed by imaging by deconvolution microscopy. Yellow regions indicate an overlap in the distribution of SIG-SgII-(334-348)-GFP (green) and endogenous secretory granules marker CgB (red), and the extent was determined using ImageMaster 5.0 (Table 1). B, accumulation of SIG-SgII-(334-348)-GFP puncta along and at the tip of neurite processes. NGF-differentiated PC12 cells were transiently transfected with pCMV-SgII-(1181-1225)-GFP and labeled with CellTracker Orange CMRA dye (red) prior to fixation to visualize the cell body and neurite processes. Deconvolution microscopy imaging reveals a substantial accumulation of SIG-SgII-(334-348)-GFP chimera at the tip of neurites, as shown in the enlarged inset. C, colocalization between SIG-SgII-(334-348)-EAP and secretory granule marker CgB. Cells expressing SIG-SgII-(334-348)-EAP were processed for immunocytochemistry using polyclonal anti-CgB and anti-human placental alkaline phosphatase primary and Alexa Fluor 594-conjugated and 488-conjugated secondary antibodies. Quantification of fluorescence overlap is reported in Table 1. D, regulated secretion of SIG-SgII-(334-348)-EAP. PC12 cells expressing SIG-EAP, SIG-SgII-EAP, or SIG-SgII-(334-348)-EAP were incubated for 15 min in secretion medium alone (mock) or 2 mm Ba2+. The enzymatic activity of EAP chimeras was assayed in the culture supernatant and cell lysate, and relative secretion was determined as described in the legend of Fig. 4. Values are given as the means ± S.E. of triplicate determinations. †, p > 0.05; ***, p < 0.001 as compared with control (mock), ANOVA with Dunnett's post test. Nuclei are stained with Hoechst 33342 (blue). Scale bar, 5 μm.
RESULTS
A Human SgII-GFP Chimeric Photoprotein Localizes to Dense-core Secretory Granules in Sympathoadrenal Cells
Intracellular Distribution in Sympathoadrenal PC12 Cells and Primary Chromaffin Cells—We designed expression plasmids encoding the predicted 30-amino acid signal peptide of human SgII (-30MAEAKTHWLGAALSLIPLIFLISGAEAASF-1, or SIG; pCMV-SgII181-EGFP, see Fig. 1), as defined in Ref. 24, or full-length human SgII (including SIG; pCMV-SgII-EGFP, see Fig. 1) fused in-frame at their C termini to the N-terminal of enhanced GFP (EGFP). The subcellular distribution of the resulting chimeric photoproteins was first examined by three-dimensional deconvolution fluorescence microscopy after transient expression in PC12 cells and primary bovine chromaffin cells (Fig. 2A). SIG-GFP accumulated in the perinuclear region of the cells, consistent with the localization of the chimera in the Golgi apparatus (Fig. 2A). Indeed, quantification in three dimensions of the colocalization between SIG-GFP and the trans-Golgi/TGN resident glycoprotein β1,4-galactosyltransferase fused to cyan fluorescent protein (GalT-CFP), in sets of 200-nm xy image stacks, revealed high overlap in their distribution (Fig. 5A), with a Pearson's correlation coefficient Rp of 0.73 ± 0.07 and an overlap coefficient Ro of 0.74 ± 0.07 between the two molecules (n = 27; Table 1). In addition, the subcellular distribution of SIG-GFP in PC12 and primary chromaffin cells (Fig. 2A) was remarkably similar to that of the constitutive secretory pathway markers SgP-GFP (where SgP is the predicted 18-amino acid signal peptide of human CgA) (13) and SEAP (21, 25) in the sympathoadrenal cell lineage, which further indicates that SIG-GFP is likely routed to the constitutive branch of the secretory pathway.
FIGURE 1.

Schematic representation of SgII domain GFP, EAP, or CFP chimeric proteins designed to study SgII trafficking to DCGs in sympathoadrenal cells. Sequences corresponding to full-length human SgII or indicated fragment were fused in-frame to GFP, EAP, or GalT-CFP reporters, under the control of the cytomegalovirus promoter. Plasmid numbering refers to the position of the SgII fragment subcloned, within the original human SgII cDNA (NCBI NM_003469). For instance, in pCMV-SgII1084-EGFP (or EAP), the 3′ end of the subcloned fragment (at amino acid 301) is located at 1084 bp in the original cDNA. Numbers along the diagram of the chimeric proteins indicate amino acid residues positions. Proteins are drawn proportional to scale.
In sharp contrast, the distribution of full-length SgII fused to GFP (SIG-SgII-GFP) in PC12 cells and primary chromaffin cells was highly punctate (Fig. 2A), suggesting storage of the chimera in secretory granules. Examination of individual 200-nm xy optical sections of SIG-SgII-GFP-expressing PC12 cells revealed a ring of subplasmalemmal vesicular fluorescence (Fig. 2A) characteristic of the DCG distribution previously reported for other regulated secretory GFP fusion proteins after transient expression in PC12 cells (i.e. CgA-GFP, atrial natriuretic factor-GFP, or neuropeptide Y-GFP (13, 30, 31)). On the other hand, intracellular distribution of SIG-SgII-GFP in primary chromaffin cells appeared more uniform, with punctate fluorescence present throughout the cell body, likely produced by secretory granules (Fig. 2A).
Colocalization with a Marker of DCGs—To further define the subcellular localization of SIG-SgII-GFP in sympathoadrenal cells, we assessed its colocalization with CgB, a classical marker of neuroendocrine and neuronal secretory granules (17). Detection of endogenous CgB was achieved by immunocytochemistry in PC12 cells expressing SIG-SgII-GFP or SIG-GFP. As anticipated, immunostaining of CgB and fluorescence of SIG-SgII-GFP displayed highly punctate patterns (Fig. 2B), and the extent of colocalization between the two molecules was substantial (Rp = 0.39 ± 0.05 and Ro = 0.41 ± 0.05; n = 121; Table 1), although incomplete, as compared with that of the maximum possible colocalization of endogenous SgII and CgB assessed by immunofluorescence (Rp = 0.76 ± 0.02 and Ro = 0.75 ± 0.02; n = 254; Table 1; visual output not shown). On the contrary, colocalization of SIG-GFP with endogenous CgB was poor (Fig. 2B) with Rp = 0.13 ± 0.05 and Ro = 0.17 ± 0.04 (n = 72; Table 1), as predicted for a fusion protein primarily destined to the constitutive secretory pathway (21, 25). Partial rather than absolute overlap in the distribution of SIG-SgII-GFP with endogenous CgB can best be explained by the extended half-life of DCGs, which gives rise to pools of old granules (pre-transfection; lacking SIG-SgII-GFP) and young post-transfection granules (i.e. containing SIG-SgII-GFP). Lack of CgB immunostaining of granules positive for SIG-SgII-GFP may also be the result of incomplete penetration of the antibodies into the secretory granule core. Nonetheless, such partial colocalization is consistent with values reported in studies localizing other GFP fusion proteins, such as CgA-GFP (13) or pro-atrial natriuretic factor-GFP (13, 32), with endogenous markers of DCGs.
NGF Differentiation to Neurites—Redistribution of DCGs away from the peripheral/subplasmalemmal region of the cell body into the tips of neurite-type processes has been documented in PC12 cells after neuronal differentiation by NGF (13, 31). PC12 cells transiently expressing SIG-GFP or SIG-SgII-GFP were exposed to NGF (100 ng/ml, 96 h), and neurite extensions were visualized with the cytosolic fluorescent dye Cell-Tracker Orange CMRA. Consistent with routing of SIG-SgII-GFP to DCGs, substantial accumulation of chimeric granin fluorescence was seen at the termini of growth cones (Fig. 3), whereas NGF treatment did not affect the juxtanuclear/Golgi apparatus distribution of SIG-GFP (Fig. 3). Taken together, these findings provide evidence that human SgII flanked by a signal peptide sequence and fused in-frame to GFP is correctly targeted into DCGs of sympathoadrenal cells.
A Human SgII-EAP Fusion Protein Is Sorted to DCGs and Released by Exocytosis in Response to Secretagogue
We previously reported that CgA fused to an engineered form of secreted EAP is trafficked to DCGs in PC12 cells and release by exocytosis (25). We reasoned that an SgII/EAP fusion protein might also become a soluble component of DCGs when expressed in PC12 cells. We designed plasmids encoding EAP fused at the C termini of full-length SgII (SIG-SgII-EAP; Fig. 1) or SgII's signal sequence SIG (SIG-EAP; Fig. 1). We first examined the subcellular distribution of SIG-EAP and SIG-SgII-EAP by immunofluorescence microscopy after transient expression in PC12 cells (Fig. 4A). Both SIG-EAP and SIG-SgII-EAP chimeras showed an intracellular distribution similar if not identical to their GFP-labeled counterparts. Thus, SIG-EAP exhibited a perinuclear distribution highly indicative of a Golgi apparatus localization, although the punctate pattern of SIG-SgII-EAP was likely because of storage of the recombinant protein into DCGs. Consistent with visual inspection, the extent of colocalization between endogenous CgB and SIG-EAP was poor (Rp = 0.19 ± 0.02 and Ro = 0.25 ± 0.02; n = 85; Table 1), whereas SIG-SgII-EAP showed substantial overlap with CgB (Rp = 0.49 ± 0.05 and Ro = 0.52 ± 0.05; n = 95; Table 1; visual output not shown).
To further ascertain the localization of SIG-SgII-EAP into the lumen of DCGs, we measured secretion of SIG-SgII-EAP and SIG-EAP from transfected PC12 cells stimulated with Ba2+. Secretion of SIG-SgII-EAP was low (2.8 ± 0.2%) under basal, nonstimulating condition, consistent with efficient storage of the fusion protein within DCGs (Fig. 4B and Table 1). Ba2+ (2 mm, 15 min) triggered 39.5 ± 1.8% secretion of SIG-SgII-EAP into the extracellular medium, which represented a highly significant ∼14.3-fold increase over basal (p < 0.0001; Fig. 4B), clearly demonstrating regulated trafficking of the chimera. In contrast, the release of the constitutive secretory protein SEAP (11, 25) and that of SIG-EAP was elevated (8.6-8.9%), even in the absence of secretagogue (Fig. 4B), and was slightly augmented to 15.2-16.8% in response to secretagogue. Although such a secretory profile might suggest some degree of nonselective entry of SEAP and SIG-EAP from the constitutive into a regulated pathway of secretion, as suggested previously for SEAP in PC12 cells (26), the marginal increase in relative stimulated release (∼1.7-1.8-fold) of the chimeras was not significant as compared with control (mock) treatment (p > 0.05; Fig. 4B), suggesting that SEAP and SIG-EAP may primarily traffic through the constitutive pathway of secretion.
Thus, these results indicate that a SgII/EAP chimera may be expressed in PC12 cells where it is targeted to DCGs and released by exocytosis in response to secretagogue. We show that fluorescent and EAP-mediated chemiluminescence SgII chimeras may be used as tracers of the regulated secretory pathway in living cells, providing both qualitative (GFP) and quantitative (EAP) means to study the mechanisms by which SgII is sorted into DCGs and to determine whether such a process requires discrete regions within the primary structure of the molecule.
Dominant Inhibitory Effect of a trans-Golgi/TGN Resident Form of SgII on the Targeting of Soluble SgII to DCGs
On the premise that sorting of SgII into the regulated pathway may depend on aggregative signals and/or recognition signals, we questioned whether a mutant form of SgII designed to be selectively retained within the distal regions of the Golgi apparatus might compromise targeting of a soluble cargo of SgII to DCGs. We fused the C-terminal end of the trans-Golgi/TGN retention signal of β1,4-galactosyltransferase (GalT) to the N terminus of full-length SgII (excluding its signal sequence SIG) to create GalT-CFP-SgII (Fig. 1). Analysis of the subcellular distribution of GalT-CFP-SgII revealed a cluster of CFP fluorescence in a perinuclear region of PC12 cells characteristic of the Golgi apparatus (Fig. 5). The distribution of GalT-CFP-SgII was virtually identical to that of SIG-GFP, as shown by fluorescence microscopy image overlays of volume views and 0.2-μm xy optical sections (Fig. 5A). The extent of the overlap between GalT-CFP-SgII and SIG-GFP was high, with colocalization parameters Rp = 0.64 ± 0.03 and Ro = 0.65 ± 0.03 comparable with the high overlap in the distribution of SIG-GFP with the trans-Golgi/TGN marker GalT-CFP (Rp = 0.73 ± 0.07 and Ro = 0.74 ± 0.07; see Fig. 5A and Table 1). These results provide evidence that SgII fused to the membrane-anchoring region of GalT is efficiently retained/retrieved within the lumen of the trans-Golgi/TGN.
We assessed the influence of GalT-CFP-SgII on the trafficking of SIG-SgII-GFP in PC12 cells. First, we examined the distribution of SIG-SgII-GFP in cells coexpressing GalT-CFP. As expected, SIG-SgII-GFP showed a typical DCG localization, whereas GalT-CFP accumulated in the trans-Golgi/TGN region of the cells (Fig. 5B). Consequently, the extent of colocalization between SIG-SgII-GFP and GalT-CFP was poor, as judged by visual inspection of the image overlays (Fig. 5B) and determination of Pearson's and overlap coefficients (Rp = 0.15 ± 0.06 and Ro = 0.17 ± 0.06; see Table 1). In sharp contrast, expression of SIG-SgII-GFP together with GalT-CFP-SgII caused substantial redistribution of SIG-SgII-GFP to the perinuclear region of the cells (Fig. 5B). In such conditions, SIG-SgII-GFP showed high colocalization with GalT-CFP-SgII (Rp and Ro = 0.77 ± 0.03; see Fig. 5B and Table 1), indeed suggesting retention of SIG-SgII-GFP into the trans-Golgi/TGN in the presence of GalT-CFP-SgII. To further probe the dominant inhibitory effect of the trans-Golgi/TGN-resident form of SgII, we also quantified the size and abundance of SIG-SgII-GFP puncta in three dimensions. As shown in Fig. 5C, the size of fluorescent SIG-SgII-GFP puncta was similar in either GalT-CFP-SgII- or GalT-CFP-expressing PC12 cells, with an average diameter of ∼308 nm. Considering that the resolution of fluorescence puncta containing the photoprotein is diffraction-limited, this value is consistent with our earlier analyses by electron microscopy, reporting a DCG diameter of ∼100-130 nm in PC12 cells (21, 25). The number of SIG-SgII-GFP puncta per xy optical section was significantly reduced in cells coexpressing GalT-CFP-SgII (22 ± 4 puncta/xy plan, n = 43; p < 0.05; Fig. 5C), as compared with PC12 cells expressing SIG-SgII-GFP and GalT-CFP (66 ± 14 puncta/xy plan; n = 43; Fig. 5C). Thus, these results clearly suggest that transient expression of GalT-CFP-SgII may reduce targeting of coexpressed SIG-SgII-GFP to DCGs, while promoting its retention within the trans region of the Golgi apparatus.
We also analyzed the secretory profile of SIG-EAP and SIG-SgII-EAP in the presence of GalT-CFP or GalT-CFP-SgII (Fig. 5D). The release of SIG-EAP was unchanged in the presence of either photoproteins and was not significantly stimulated by 2 mm Ba2+ (p > 0.05; Fig. 5D), consistent with trafficking of SIG-EAP into the constitutive branch of the secretory pathway (Fig. 4B). In contrast, expression of SIG-SgII-EAP together with GalT-CFP-SgII lowered Ba2+-evoked secretion of SIG-SgII-EAP by ∼48% (p < 0.001; Fig. 5D), as compared with cells coexpressing GalT-CFP. It is noteworthy that expression of GalT-CFP (lacking SgII) did not reduce the vesicular localization of coexpressed SIG-SgII-GFP (Fig. 5B) nor Ba2+-stimulated secretion of SIG-SgII-EAP (Figs. 4B and 5D). This clearly establishes that impaired regulated trafficking of soluble SgII chimeras specifically results from the expression of a trans-Golgi cisternae membrane-anchored form of SgII and is not merely a consequence of protein overexpression that could, for instance, exacerbate GFP and CFP susceptibility to self-associate/cluster (33).
Altogether, these findings provide evidence in cella that a chimeric form of SgII designed to be selectively retained/recycled to the Golgi apparatus may act as dominant negative, impairing the trafficking of coexpressed, secreted forms of SgII (i.e. SIG-SgII-GFP or SIG-SgII-EAP) from the lumen of the trans-Golgi/TGN into the regulated secretory pathway.
DCG Targeting of N-terminal and C-terminal Half-domains of SgII
Earlier studies in PC12 cells have identified sorting determinants for the regulated secretory pathway within the N-terminal region of CgA and CgB (8, 11, 13, 25, 34). We questioned whether a signal for the vesicular targeting of SgII might also locate within a discrete region of the primary structure of granin.
We designed expression plasmids encoding the N- and C-terminal half-region of SgII with GFP or EAP reporters fused at the C-terminal end (SIG-SgII-(1-301)-GFP/EAP and SIG-SgII-(302-587)-GFP/EAP; Fig. 1). Fluorescence microscopy analysis of the distribution of SIG-SgII-(1-301)-GFP and SIG-SgII-(302-587)-GFP after transient expression in PC12 showed a highly punctate distribution of the chimera similar to that of SIG-SgII-GFP, suggesting secretory granule storage of both SgII-(1-301) and SgII-(302-587) photoproteins (Fig. 6A). Ba2+ (2 mm, 15 min) triggered the release of SIG-SgII-(1-301)-EAP and SIG-SgII-(302-587)-EAP from PC12 cells by ∼7.7- and ∼12-fold over basal (mock medium), respectively (p < 0.01; Fig. 6B), and the extent of the regulated release of both half-regions of SgII was comparable with that of full-length SIG-SgII-EAP (∼14.5-fold over basal; Fig. 6B). Taken together, these results demonstrate that both the N-terminal and C-terminal half-regions of SgII are able to steer GFP and EAP reporters into DCGs for exocytosis, suggesting that SgII contains independent sorting signals for the regulated secretory pathway.
The N-terminal Domain SgII-(25-41) Contains Sufficient Information for Sorting into the Regulated Secretory Pathway
To further refine the presence of a domain within the N-terminal region of SgII that may function in cis as a sorting signal for the regulated pathway, we used a strategy based on C-terminal truncations of the 1-301 domain of SgII. We generated expression plasmids encoding a series of SgII fragment-GFP fusion proteins spanning the region +1 to +301 of the mature SgII (SIG-SgII-(1-145)-GFP, SIG-SgII-(1-76)-GFP, SIG-SgII-(1-41)-GFP, and SIG-SgII-(1-24)-GFP; Fig. 1). The subcellular distribution of each photoprotein was first analyzed by deconvolution fluorescence microscopy after transient expression in PC12 cells. SIG-SgII-(1-145)-GFP, SIG-SgII-(1-76)-GFP, and SIG-SgII-(1-41)-GFP localized to vesicular structures with a distribution similar to that of SIG-SgII-GFP (Fig. 7A), suggesting storage of the chimeric proteins into secretory granules. On the other hand, the subcellular localization of the shorter domain of SgII (SIG-SgII-(1-24)-GFP) appeared identical to that of SIG-GFP and showed accumulation of GFP fluorescence in a perinuclear region containing the Golgi apparatus (Fig. 7A). Such marked difference in the distribution of SIG-SgII-(1-41)-GFP and SIG-SgII-(1-24)-GFP prompted us to examine the trafficking behavior of these chimeras during extension and remodeling of neurites in PC12 cells. Neuronal differentiation of SIG-SgII-(1-41)-GFP-expressing PC12 cells by NGF (2.5 S, 100 ng/ml, 96 h) induced the accumulation of SIG-SgII-(1-41)-GFP puncta along and at the tip of neurite processes (Fig. 3), which is characteristic of the spatial distribution of DCGs after phenotype conversion (13, 31). No accumulation of fluorescence was seen at the termini of processes in NGF-differentiated cells expressing SIG-SgII-(1-24)-GFP (Fig. 3), suggesting that a targeting signal may be contained in a region of SgII spanning from amino acid +25 to +41. To further test this proposition, we studied the release of a series of SgII fragment-EAP chimeras (SIG-SgII-(1-145)-EAP, SIG-SgII-(1-76)-EAP, SIG-SgII-(1-41)-EAP, and SIG-SgII-(1-24)-EAP) from transiently transfected PC12 cells, in response to a secretagogue. Exposure of such transfected cells to Ba2+ (2 mm, 15 min) triggered the release of SIG-SgII-(1-145)-EAP, SIG-SgII-(1-76)-EAP, SIG-SgII-(1-41)-EAP, and SIG-SgII-EAP (p < 0.01, p < 0.001; Fig. 7C) but not that of SIG-EAP and SIG-SgII-(1-24)-EAP (p > 0.05; Fig. 7C).
Thus, our results show that the domain SgII-(1-41) appears sufficient to target GFP and EAP to DCGs for exocytosis and identify the amino acid 25-41 sequence (25MIRALEYIENLRQQAHK41) as a necessary region within residues 1-41 for DCG targeting of GFP or EAP fusion proteins. Next, we questioned whether residues 25-41 would also contain sufficient (transferable) information for granule sorting activity. As shown earlier, GFP or EAP flanked by the signal sequence of SgII (SIG) primarily traffics through the constitutive pathway of secretion (Figs. 2, 3, 4, 5, 6 and 7). The domain SgII-(25-41) was inserted in-frame into SIG-GFP to create SIG-SgII-(25-41)-GFP (Fig. 1), and its subcellular distribution was assessed after transient expression in PC12 cells (Fig. 8A). SIG-SgII-(25-41)-GFP exhibited a scattered, somewhat diffuse distribution throughout the cell body (Fig. 8A), which did not colocalize with endogenous CgB (Rp = 0.10 ± 0.05 and Ro = 0.17 ± 0.04; Table 1). Also, the distribution of SIG-SgII-(25-41)-GFP appeared quite different from the Golgi localization of SIG-GFP and SIG-SgII-(1-24)-GFP (Figs. 2, 3, 4, 5 and 7) and is perhaps a consequence of aberrant structure of SgII-(25-41)-GFP, and failure to pass the early secretory pathway quality control machinery leads to mistrafficking of the chimera. For instance, we did find substantial colocalization of SIG-SgII-(25-41)-GFP with the ER marker calreticulin and lysosome marker LGP120 (data not shown). In contrast, transfer of residues 25-41 to EAP (SIG-SgII-(25-41)-EAP; Fig. 1) resulted in a punctate distribution of the chimera that overlapped that of endogenous CgB (Rp = 0.52 ± 0.07 and Ro = 0.60 ± 0.06; Table 1 and Fig. 8B). Furthermore, the release of SIG-SgII-(25-41)-EAP was stimulated in response to Ba2+, further suggesting a DCG localization of the fusion protein (Fig. 8C). Hence, the domain SgII-(25-41) not only appears necessary in the context of targeting residues 1-41 to DCG (Fig. 7) but may also function as a sufficient sorting signal for the regulated secretory pathway.
Localization of a DCG-sorting Signal within the C-terminal Half-region of SgII
Our results also indicate the presence of a signal for sorting within the C-terminal region of SgII (Fig. 6) that may function independently from the granule sorting activity of the N-terminal SgII-(25-41) domain. This finding prompted us to further narrow down the location of such determinant, perhaps to a discrete sequence of SgII. Applying our progressive deletion paradigm to the C-terminal half-region of SgII, we generated expression plasmids encoding a series of SgII domain-GFP chimeras, spanning the region +301 to +587 of SgII (SIG-SgII-(302-505)-GFP, SIG-SgII-(302-469)-GFP, SIG-SgII-(302-371)-GFP, SIG-SgII-(302-348)-GFP, and SIG-SgII-(302-333)-GFP; Fig. 1). Overall, the intracellular distribution of SIG-SgII-(302-505)-GFP, SIG-SgII-(302-469)-GFP, SIG-SgII-(302-402)-GFP, SIG-SgII-(302-371)-GFP, and SIG-SgII-(302-348)-GFP fluorescence was punctate and virtually indistinguishable from that of full-length SIG-SgII-GFP (Fig. 9A). However, we noted that ∼10% of cells transfected with the shorter chimeras SIG-SgII-(302-371)-GFP or SIG-SgII-(302-348)-GFP showed a substantial accumulation of fluorescence in the Golgi apparatus (data not shown), suggesting some degree of inefficiency in the sorting of the newly synthesized chimeras into DCGs. On the other hand, ∼78% of cells expressing SIG-SgII-(302-333)-GFP displayed a characteristic Golgi apparatus accumulation of fluorescence similar to that of SIG-GFP (Fig. 9A), whereas only ∼22% of PC12 cells exhibited a punctate distribution SIG-SgII-(302-333)-GFP (data not shown). Such a marked difference in the intracellular distribution of SIG-SgII-(302-348)-GFP versus SIG-SgII-(302-333)-GFP suggests that the domain of SgII spanning amino acids 334 and 348 acts as a trafficking determinant for sorting of SgII into DCGs. Indeed, neuronal differentiation of SIG-SgII-(302-348)-GFP-expressing PC12 cells by NGF showed a clear redistribution of the chimera along and at the tip of neurite processes (Fig. 3), whereas the fluorescence of SIG-SgII-(302-333)-GFP remained localized in perinuclear clusters within the cell body, with no detectable GFP fluorescence at the neurites (Fig. 3). To further explore the secretory granule sorting activity of SgII-(302-348) region, we studied secretagogue-evoked release of the C-terminal fragments of SgII fused to EAP in PC12 cells (Fig. 9B). Ba2+ triggered the release of SIG-SgII-(302-505)-EAP, SIG-SgII-(302-469)-EAP, SIG-SgII-(302-402)-EAP, SIG-SgII-(302-371)-EAP, and SIG-SgII-(302-348)-EAP into the extracellular medium, with relative secretion values ranging from ∼4.5- to ∼14.6-fold over the basal release (mock medium) (p < 0.05; Fig. 9B). In contrast, the release of SIG-SgII-(302-333)-EAP was not significantly augmented in the presence of Ba2+ (∼2.5-fold, p > 0.05; Fig. 9B) and was of the same magnitude as the constitutively secreted SIG-EAP chimera (∼1.8-fold, p > 0.05; Fig. 9B). These results demonstrate that the SgII-(302-348) domain is targeted to DCGs for exocytosis, whereas the SgII-(302-333) domain may primarily traffic to the constitutive secretory pathway, thereby indicating that the region of SgII spanning amino acids 334 and 348 may be required (necessary) to steer GFP or EAP reporters to the regulated pathway.
The Domain SgII-(334-348) Is a Sufficient Sorting Signal for Trafficking into the Regulated Secretory Pathway
To test whether the SgII-(333-348) domain might be also be a sufficient DCG-sorting determinant, we generated GFP- and EAP-tagged chimeric proteins consisting of SgII's signal peptide sequence SIG fused to residues 334-348 (SIG-SgII-(334-348)-GFP and SIG-SgII-(334-348)-EAP; Fig. 1). Expression of SIG-SgII-(334-348)-GFP or SIG-SgII-(334-348)-EAP in PC12 cells resulted in a highly punctate distribution of the chimeras throughout the cell body, which showed substantial overlap with endogenous CgB (Fig. 10, A and C). Quantitative analysis of such colocalization yielded an Rp of 0.39 ± 0.04 and an Ro of 0.44 ± 0.01 for SIG-SgII-(334-348)-GFP versus CgB, and an Rp of 0.67 ± 0.08 and an Ro of 0.72 ± 0.06 for SIG-SgII-(334-348)-EAP versus CgB (Fig. 10, A and C; Table 1). Note that these values were comparable and even exceeded the colocalization coefficients of full-length SgII-GFP (SIG-SgII-GFP) versus endogenous CgB (Rp = 0.39 ± 0.05 and Ro = 0.41 ± 0.05; Fig. 2B and Table 1). Accumulation of SIG-SgII-(334-348)-GFP fluorescence at the termini of neurite processes was obvious in cells differentiated by NGF (Fig. 10B), consistent with DCG storage of the chimera. Finally, we found that SIG-SgII-(334-348)-EAP was secreted in response to secretagogue. As shown in Fig. 10C, Ba2+ triggered the release of SIG-SgII-(334-348)-EAP from transfected PC12 cells by ∼4-fold over basal (p < 0.001). The stimulated secretion of SIG-SgII-(334-348)-EAP was lower than that of full-length SgII-EAP chimera (SIG-SgII-EAP; ∼14.3-fold over basal, p < 0.001) but significantly higher than the release of SIG-EAP, which showed no significant response to Ba2+ (p > 0.05; Fig. 10C). Taken together, the results provide evidence that SgII-(334-348) (334DLIEMLKTGEKPNGS348) functions as a necessary determinant, in the context of sorting of the larger SgII-(302-371) domain into the regulated pathway, and contains a sufficient (transferable) granule-targeting signal, regardless of the reporter tag (GFP or EAP).
Potential Structures Involved in the Sorting of SgII Results from Secondary Structure Predictions
Our deletion scheme (Fig. 1) takes its cue from in silico data predicting α-helical domains across full-length human SgII (minus its signal sequence) by neural network methods, using NNPREDICT (27) and NPS@ suite of 11 algorithms for consensus secondary structure prediction (28). The predicted secondary structure across SgII was on average 41.7 ± 1.7% α-helix, 7.0 ± 2.5% strand/β-sheet, and 46.8 ± 3.8% coil. We find that SgII-(1-41) and SgII-(334-348) may function as sorting signals to target SgII into the regulated secretory pathway of PC12 cells. Consensus secondary structure prediction show highly conserved α-helices of moderate amphipathicity at residues 24-41 (24EMIRALEYIENLRQQAHK41; ∼4 turns) and residues 333-340 (333EDLIEMLK340; ∼2 turns) (Fig. 11). Helical wheel presentation of the sequence 24-41 reveals a hydrophobic patch, consisting of Met25, Ala28, Leu29, Tyr31, Ile32, Leu35, and Ala39, lying on one face of the helix (Fig. 11A), whereas the other side consists mostly of hydrophilic residues. An α-helical domain is also predicted to exist at the N-terminal side of SgII-(334-348), but with a much shorter span. Modeling residues 333-340 to an α-helix shows a characteristic amphipathic structure with hydrophobic residues (Leu335, Ile336, Met338, and Leu339) forming the putative hydrophobic face of the helix (Fig. 11B).
FIGURE 11.

Sorting domains within N- and C-terminal region of SgII; conservation among vertebrates and secondary structure prediction. Multiple sequence alignment of full-length SgII proteins was performed using ClustalW and the amino acid sequences of Homo sapiens (NCBI NP_003460), Pan troglodytes (NCBI XP_001166617), Sus scrofa (NCBI NP_001012299), Canis lupus (NCBI XP_545669), Bos taurus (NCBI NP_776601), Rattus norvegicus (NCBI NP_073160), Mus musculus (NCBI NP_033155), Xenopus laevis (NCBI NP_001081590), and Rana ridibunda (NCBI AAB17470). The boxed SgII-(25-41) (A) and SgII-(334-348) (B) domains and the surrounding region are shown. Identical amino acids are highlighted by gray shading. Secondary structure of human SgII was predicted with NNPREDICT and further analyzed with the NPS@ consensus secondary structure prediction program that includes the algorithms DPM, DSC, GOR1, GOR3, GOR4, HNNC, PHD, Predator, SIMPA96, SOPM, and SOPMA. Consensus secondary structure prediction is indicated below the multiple sequence alignment of each region. Also shown is the helical wheel projection (PEPWHEEL) of predicted α-helices within SgII-(25-41) and SgII-(334-348). The hydrophobic face of the α-helices is highlighted with light gray shading, and hydrophobic amino acids residues are indicated in black boxes.
DISCUSSION
Understanding the molecular mechanisms by which secretory proteins are sorted into DCGs has been the subject of intensive research for many years, and it underscores the remarkable heterogeneity of this process (6, 7). This study defines sequence features of the secretory prohormone SgII that are involved in the targeting of the molecule to DCGs in sympathoadrenal cells. We find that information necessary for such a process is dependent upon a saturable sorting machinery within the lumen of the trans-Golgi/TGN, and recruitment of SgII-(25-41) and SgII-(334-348) regions, which are predicted to contain α-helical structural motifs that may function as sufficient, independent sorting domains for the regulated pathway.
Trafficking of SgII Fusion Proteins into the Regulated Secretory Pathway—Taking our cue from the experimental paradigm developed in our earlier studies on the vesicular trafficking of CgA in sympathoadrenal cells (13, 21, 25), we fused full-length human SgII to GFP or EAP, for fluorescent or enzymatic tracing of the regulated secretory pathway. This study establishes that SgII flanked by a signal sequence is able to convert GFP or EAP to soluble (releasable) components of DCGs, and it thus reveals that SgII contains a dominant sorting signal for the regulated secretory pathway, whereas its signal peptide alone does not (Figs. 2, 3, 4, 5, 6 and 7, 9, and 10). We find that the intracellular distribution and trafficking of SIG-SgII-GFP and SIG-SgII-EAP may reflect the behavior of endogenous regulated secretory proteins, providing unique tools to study qualitatively and quantitatively the intracellular fate of SgII (or a domain therein) and characterize the targeting mechanism of the protein in sympathoadrenal cells.
Two Independent, Minimum Sorting Domains within the N- and C-terminal Region of SgII—Peptide sequences acting as independent sorting motif for the regulated secretory pathway have been documented for several regulated proteins. A prime example is proinsulin, in which DCG-targeting determinants may include a binding domain for carboxypeptidase E (CPE), PCs cleavage sites, and aggregation domains (35, 36). Similarly, two distinct peptide sequences in the C-terminal region of PC1 may function as sufficient sorting signals to reroute a constitutively secreted IgG Fc fragment to DCGs in pituitary cells (15). Sorting of CgA into DCGs may rely on cell-specific mechanisms mediated by distinct sorting domains. Studies in PC12 cells have shown that information for sorting of CgA into the regulated pathway may be confined within the N-terminal region of the molecule that contains a hydrophobic disulfide-bonded loop and an α-helical motif (11, 13, 25), whereas only a C-terminal domain appears necessary for DCG targeting of CgA in pituitary cells (11). Our results point to the presence of multiple sorting signals, but with two discrete regions SgII-(25-41) and SgII-(334-348), functioning independently for sorting of SgII within the same cell type.
What might be the physiological significance of two domains acting independently to traffic SgII within the regulated pathway of PC12 cells? An intriguing parallel may be drawn from the vesicular targeting of the ELH prohormone in Aplysia californica neurons, in which peptides derived from proteolytic processing of ELH are sorted into separate DCGs, suggesting the existence of independent sorting domains within the prohormone (37). Thus, distinct structural motifs within a prohormone precursor may function independently in the segregation of processing intermediates, thereby determining their routing through distal compartments of the secretory pathway. SgII is also a precursor protein that can be proteolytically processed by PCs at dibasic amino acid cleavage sites into smaller peptides with important biological function. For instance, secretoneurin (SgII-(152-184)) acts as a chemotactic/angiogenic cytokine in vivo, with activity comparable in potency to vascular endothelial growth factor (38, 39). Whether full-length SgII and/or processing intermediates are uniformly distributed, or localize to distinct populations of secretory granules, remains an open question (40), and it is also unclear if proteolytic processing of SgII begins in the TGN prior to sorting or in later compartments of the regulated secretory pathway, for instance ISGs (41).
Conserved α-Helical Domains of SgII as Putative Sorting Signals for the Regulated Pathway—Our progressive truncation scheme (Fig. 1) takes its cue from in silico, secondary structure predictions across full-length human SgII (minus its signal sequence) by neural network methods (27, 28). We find that SgII's minimum sorting domains SgII-(25-41) and SgII-(334-348) contain putative α-helices of moderate amphipathicity (Fig. 11). Both sequences are preceded by a proline residue, which is frequently found at the beginning of an α-helix (42). Although a typical α-helix has an average span of 12 amino acids (∼3 turns), as low as five residues may be sufficient to form a helical hydrogen-bonded pattern. Both sequences 24-41 and 333-340, as well as residues in the immediate vicinity of these α-helical motifs, are highly conserved across vertebrates (Fig. 11), but the search for sequence similarity with other proteins by BLAST (Basic Local Alignment Search Tool) and PSI-BLAST (Position-specific Iterative-BLAST) did not reveal homology below the threshold of significance E = 0.01. α-Helical domains with amphipathic properties have been suggested to mediate targeting of several prohormones into the regulated pathway, for instance CgA (13), prosomatostatin (14), and PCs (15, 16), possibly by virtue of interaction with membrane components (5, 43). In support of this concept, we show that α-helix-containing domains SgII-(25-41) and SgII-(333-348) function as transferable DCG-sorting signals; the hydrophobic “faces” of these two amphipathic helical segments in cis (Fig. 11) readily rise to a model whereby such motifs might interact with similarly hydrophobic lipid membrane surfaces in trans within the Golgi or secretory granule compartments.
Integration of the Results and Putative Mechanisms—Considering a mechanism based on a sorting-for-entry model, we propose that particular regions of SgII, for instance SgII-(25-41) and SgII-(334-348), act as discrete sorting signals binding to particular lipid components of the membrane of the TGN or the nascent secretory granule. Consistent with this hypothesis, SgII may interact specifically with isolated vesicle membranes at a mildly acidic pH (44), a property shared by several secretory granule cargo proteins, including CgA and CgB (45, 46). A mechanism recruiting a specific sorting receptor at the membrane of the TGN may also be considered. For instance, CPE is thought to act as a common sorting receptor for proopiomela-nocortin, proinsulin, and proenkephalin (35, 47). Earlier studies have excluded a role of CPE in the sorting of CgA (35, 48), but new evidence suggests that a sorting complex consisting of CPE and SgIII mediates targeting of several prohormones in endocrine cells (49). However, interaction of SgII with SgIII has not been found (50), and whether CPE may contribute to the sorting process of SgII has yet to be determined. Another candidate sorting receptor is the inositol 1,4,5-trisphosphate receptor (IP3R). CgA and CgB have been documented to bind IP3R in vitro at the intravesicular pH of 5.5 and modulate the channel activity in living cells along several compartments of the secretory pathway, including DCGs (51, 52). Hence, the pH-dependent and functional interaction of IP3R with CgA and CgB raises the possibility of such a receptor serving as a membrane target for the vesicular sorting of other calcium storage proteins such as SgII.
Although our findings provide evidence that SgII-(25-41) and SgII-(334-348) domains contain sorting information sufficient to redirect GFP and EAP into the regulated pathway, whether these two regions uniquely contribute to the sorting of SgII within the regulated pathway remains to be seen. Indeed, our results from secretagogue-stimulated release of SgII domain-EAP chimeras show a positive correlation between the sorting efficiency and the length of the SgII domain under test, i.e. the longer the domain, the better the sorting efficiency, with the highest efficiency achieved for full-length SgII (Figs. 6, 7, and 9). This secretory behavior does not prove but certainly points to a substantial contribution of other regions of the SgII to achieve optimal sorting efficiency, perhaps by promoting the ability of SgII to aggregate/multimerize within the TGN or ISGs and enhance segregation by a sorting-by-retention mechanism. One example of a synergistic action between sorting signals might be that of CgB. As outlined in the Introduction, an N-terminal disulfide-bonded loop domain acts as a transferable sorting determinant (8-10) but does not contribute to the aggregation/multimerization of the granin in PC12 cells (34). However, this sorting domain may not be functional in the absence of endogenous CgB, which underlies the contribution of aggregation in the sorting process of CgB (10).
Our results from the trans-Golgi resident GalT-CFP-SgII chimera are consistent with a contribution of aggregative signals to the sorting of SgII (Fig. 5). One possibility is that membrane-anchored SgII might compete with a multimerization/aggregation step common to both models for sorting, behaving as a bait for the nucleation and growth of an aggregate of preysoluble SgII, and recycle such aggregate within the trans-Golgi/TGN, thereby impairing further trafficking into the regulated secretory pathway. In the simplest scenario, membrane-anchored SgII would homomerize with endogenous and/or exogenous soluble SgII (GFP- or EAP-labeled), which is consistent with studies in vitro reporting pH- and Ca2+-regulated self-aggregation of SgII (24, 44). However, the situation could be more complex, with a role of heteromeric association with other cargo proteins to the overall multimerization/aggregation process. For instance, CgA and CgB form homo- as well as hetero-aggregates in vitro in a mildly acidic pH and millimolar Ca2+ environment (53). However, heteromeric interaction of SgII with other granins has yet to be reported in sympathoadrenal cells, although studies in pituitary cells showed no interaction of SgII with CgA or secretogranin III (SgIII) (50). An equally plausible explanation is that GalT-CFP-SgII may compete out binding of soluble SgII to a putative membrane receptor at the TGN, leading to the accumulation of soluble SgII within this compartment. Indeed, sorting of SgII via a sorting-for-entry mechanism is expected to be limited by the number of receptors and therefore saturable.
In conclusion, this work elucidates some of the molecular details that underlie sorting of SgII within the regulated secretory pathway of sympathoadrenal cells. We find that discrete domains within the primary structure of SgII act as sufficient sorting signals, perhaps by virtue of aggregation/multimerization properties and/or by interaction with a specific component of the membrane of the budding secretory granule. The ability of the TGN-resident SgII chimera to readily saturate the sorting machinery at the trans-Golgi/TGN may provide essential information for future attempts to identify protein-protein interaction and/or putative receptor that mediate efficient and optimal sorting of SgII into DCGs.
Acknowledgments
We appreciate the technical assistance of Katherine Harding, Carrie Rodemer, and Tom Toneff. We thank Dr. Daniel T. O'Connor (University of California, San Diego) for critical reading of the manuscript. We also thank Dr. Hans-Hermann Gerdes (University of Bergen, Norway) for the gift of pGEM4-hSgII. Digital imaging was performed at the University of California, San Diego Cancer Center Shared Resource Facilities (funded in part by NCI grants from the National Institutes of Health).
This work was supported by postdoctoral research fellowships from the Fondation Pour la Recherche Médicale and The National Kidney Foundation (to M. C.) and by National Institutes of Health Grants DK59628 and HL58120 (to L. T.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: DCG, dense-core secretory granule; CgA, chromogranin A; CgB, chromogranin B; CPE, carboxypeptidase E; EAP, embryonic alkaline phosphatase; GalT, β1,4-galactosyltranferase; GFP, green fluorescent protein; IP3R, receptor for inositol 1,4,5-trisphosphate; ISG, immature secretory granule; TGN, trans-Golgi network; SEAP, secreted form of embryonic alkaline phosphatase; SgII, secretogranin II; SgIII, secretogranin III; SIG, predicted 30 amino acid signal peptide of human SgII; ANOVA, analysis of variance; PBS, phosphate-buffered saline; NGF, nerve growth factor; CFP, cyan fluorescent protein; EGFP, enhanced GFP.
References
- 1.Kelly, R. B. (1985) Science 230 25-32 [DOI] [PubMed] [Google Scholar]
- 2.Blazquez, M., and Shennan, K. I. (2000) Biochem. Cell Biol. 78 181-191 [PubMed] [Google Scholar]
- 3.Arvan, P., and Castle, D. (1998) Biochem. J. 332 593-610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dannies, P. S. (1999) Endocr. Rev. 20 3-21 [DOI] [PubMed] [Google Scholar]
- 5.Dikeakos, J. D., and Reudelhuber, T. L. (2007) J. Cell Biol. 177 191-196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Glombik, M. M., and Gerdes, H. H. (2000) Biochimie (Paris) 82 315-326 [DOI] [PubMed] [Google Scholar]
- 7.Gorr, S. U., Jain, R. K., Kuehn, U., Joyce, P. B., and Cowley, D. J. (2001) Mol. Cell. Endocrinol. 172 1-6 [DOI] [PubMed] [Google Scholar]
- 8.Chanat, E., Weiss, U., Huttner, W. B., and Tooze, S. A. (1993) EMBO J. 12 2159-2168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cool, D. R., Fenger, M., Snell, C. R., and Loh, Y. P. (1995) J. Biol. Chem. 270 8723-8729 [DOI] [PubMed] [Google Scholar]
- 10.Kromer, A., Glombik, M. M., Huttner, W. B., and Gerdes, H. H. (1998) J. Cell Biol. 140 1331-1346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cowley, D. J., Moore, Y. R., Darling, D. S., Joyce, P. B., and Gorr, S. U. (2000) J. Biol. Chem. 275 7743-7748 [DOI] [PubMed] [Google Scholar]
- 12.Thiele, C., and Huttner, W. B. (1998) J. Biol. Chem. 273 1223-1231 [DOI] [PubMed] [Google Scholar]
- 13.Taupenot, L., Harper, K. L., Mahapatra, N. R., Parmer, R. J., Mahata, S. K., and O'Connor, D. T. (2002) J. Cell Sci. 115 4827-4841 [DOI] [PubMed] [Google Scholar]
- 14.Mouchantaf, R., Kumar, U., Sulea, T., and Patel, Y. C. (2001) J. Biol. Chem. 276 26308-26316 [DOI] [PubMed] [Google Scholar]
- 15.Jutras, I., Seidah, N. G., and Reudelhuber, T. L. (2000) J. Biol. Chem. 275 40337-40343 [DOI] [PubMed] [Google Scholar]
- 16.Lacombe, M. J., Mercure, C., Dikeakos, J. D., and Reudelhuber, T. L. (2005) J. Biol. Chem. 280 4803-4807 [DOI] [PubMed] [Google Scholar]
- 17.Taupenot, L., Harper, K. L., and O'Connor, D. T. (2003) N. Engl. J. Med. 348 1134-1149 [DOI] [PubMed] [Google Scholar]
- 18.Huh, Y. H., Jeon, S. H., and Yoo, S. H. (2003) J. Biol. Chem. 278 40581-40589 [DOI] [PubMed] [Google Scholar]
- 19.Mahapatra, N. R., O'Connor, D. T., Vaingankar, S. M., Hikim, A. P., Mahata, M., Ray, S., Staite, E., Wu, H., Gu, Y., Dalton, N., Kennedy, B. P., Ziegler, M. G., Ross, J., and Mahata, S. K. (2005) J. Clin. Investig. 115 1942-1952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim, T., and Loh, Y. P. (2006) Mol. Biol. Cell 17 789-798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Courel, M., Rodemer, C., Nguyen, S. T., Pance, A., Jackson, A. P., O'Connor D, T., and Taupenot, L. (2006) J. Biol. Chem. 281 38038-38051 [DOI] [PubMed] [Google Scholar]
- 22.Taupenot, L., Mahata, M., Mahata, S. K., and O'Connor, D. T. (1999) Hypertension 34 1152-1162 [DOI] [PubMed] [Google Scholar]
- 23.O'Connor, D. T., Mahata, S. K., Mahata, M., Jiang, Q., Hook, V. Y., and Taupenot, L. (2007) Nat. Protoc. 2 1248-1253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gerdes, H. H., Rosa, P., Phillips, E., Baeuerle, P. A., Frank, R., Argos, P., and Huttner, W. B. (1989) J. Biol. Chem. 264 12009-12015 [PubMed] [Google Scholar]
- 25.Taupenot, L., Harper, K. L., and O'Connor, D. T. (2005) J. Biol. Chem. 280 3885-3897 [DOI] [PubMed] [Google Scholar]
- 26.Eskeland, N. L., Zhou, A., Dinh, T. Q., Wu, H., Parmer, R. J., Mains, R. E., and O'Connor, D. T. (1996) J. Clin. Investig. 98 148-156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kneller, D. G., Cohen, F. E., and Langridge, R. (1990) J. Mol. Biol. 214 171-182 [DOI] [PubMed] [Google Scholar]
- 28.Combet, C., Blanchet, C., Geourjon, C., and Deleage, G. (2000) Trends Biochem. Sci. 25 147-150 [DOI] [PubMed] [Google Scholar]
- 29.Rice, P., Longden, I., and Bleasby, A. (2000) Trends Genet. 16 276-277 [DOI] [PubMed] [Google Scholar]
- 30.Lin, C. C., Huang, C. C., Lin, K. H., Cheng, K. H., Yang, D. M., Tsai, Y. S., Ong, R. Y., Huang, Y. N., and Kao, L. S. (2007) J. Cell. Physiol. 211 316-326 [DOI] [PubMed] [Google Scholar]
- 31.Ng, Y. K., Lu, X., Watkins, S. C., Ellis-Davies, G. C., and Levitan, E. S. (2002) J. Neurosci. 22 3890-3897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lang, T., Wacker, I., Steyer, J., Kaether, C., Wunderlich, I., Soldati, T., Gerdes, H. H., and Almers, W. (1997) Neuron 18 857-863 [DOI] [PubMed] [Google Scholar]
- 33.Zacharias, D. A., Violin, J. D., Newton, A. C., and Tsien, R. Y. (2002) Science 296 913-916 [DOI] [PubMed] [Google Scholar]
- 34.Glombik, M. M., Kromer, A., Salm, T., Huttner, W. B., and Gerdes, H. H. (1999) EMBO J. 18 1059-1070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Normant, E., and Loh, Y. P. (1998) Endocrinology 139 2137-2145 [DOI] [PubMed] [Google Scholar]
- 36.Kuliawat, R., Prabakaran, D., and Arvan, P. (2000) Mol. Biol. Cell 11 1959-1972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jung, L. J., and Scheller, R. H. (1991) Science 251 1330-1335 [DOI] [PubMed] [Google Scholar]
- 38.Fischer-Colbrie, R., Laslop, A., and Kirchmair, R. (1995) Prog. Neurobiol. 46 49-70 [DOI] [PubMed] [Google Scholar]
- 39.Kirchmair, R., Gander, R., Egger, M., Hanley, A., Silver, M., Ritsch, A., Murayama, T., Kaneider, N., Sturm, W., Kearny, M., Fischer-Colbrie, R., Kircher, B., Gaenzer, H., Wiedermann, C. J., Ropper, A. H., Losordo, D. W., Patsch, J. R., and Schratzberger, P. (2004) Circulation 109 777-783 [DOI] [PubMed] [Google Scholar]
- 40.Laslop, A., Weiss, C., Savaria, D., Eiter, C., Tooze, S. A., Seidah, N. G., and Winkler, H. (1998) J. Neurochem. 70 374-383 [DOI] [PubMed] [Google Scholar]
- 41.Urbe, S., Dittie, A. S., and Tooze, S. A. (1997) Biochem. J. 321 65-74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kim, M. K., and Kang, Y. K. (1999) Protein Sci. 8 1492-1499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gorr, S. U., and Darling, D. S. (1995) FEBS Lett. 361 8-12 [DOI] [PubMed] [Google Scholar]
- 44.Park, H. Y., So, S. H., Lee, W. B., You, S. H., and Yoo, S. H. (2002) Biochemistry 41 1259-1266 [DOI] [PubMed] [Google Scholar]
- 45.Yoo, S. H. (1993) Biochemistry 32 8213-8219 [DOI] [PubMed] [Google Scholar]
- 46.Yoo, S. H. (1995) J. Biol. Chem. 270 12578-12583 [DOI] [PubMed] [Google Scholar]
- 47.Cool, D. R., Normant, E., Shen, F., Chen, H. C., Pannell, L., Zhang, Y., and Loh, Y. P. (1997) Cell 88 73-83 [DOI] [PubMed] [Google Scholar]
- 48.Cool, D. R., and Loh, Y. P. (1998) Mol. Cell. Endocrinol. 139 7-13 [DOI] [PubMed] [Google Scholar]
- 49.Hosaka, M., Watanabe, T., Sakai, Y., Kato, T., and Takeuchi, T. (2005) J. Cell Sci. 118 4785-4795 [DOI] [PubMed] [Google Scholar]
- 50.Hosaka, M., Watanabe, T., Sakai, Y., Uchiyama, Y., and Takeuchi, T. (2002) Mol. Biol. Cell 13 3388-3399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yoo, S. H., So, S. H., Kweon, H. S., Lee, J. S., Kang, M. K., and Jeon, C. J. (2000) J. Biol. Chem. 275 12553-12559 [DOI] [PubMed] [Google Scholar]
- 52.Choe, C. U., Harrison, K. D., Grant, W., and Ehrlich, B. E. (2004) J. Biol. Chem. 279 35551-35556 [DOI] [PubMed] [Google Scholar]
- 53.Yoo, S. H. (1996) J. Biol. Chem. 271 1558-1565 [PubMed] [Google Scholar]