

Abstract
Factor VIII circulates as a heterodimer composed of heavy (A1A2B domains) and light (A3C1C2 domains) chains, whereas the contiguous A1A2 domains are separate subunits in the active cofactor, factor VIIIa. Whereas the A1 subunit maintains a stable interaction with the A3C1C2 subunit, the A2 subunit is weakly associated in factor VIIIa and its dissociation accounts for the labile activity of the cofactor. In examining the ceruloplasmin-based factor VIII A domain model, potential hydrogen bonding based upon spatial separations of <2.8Å were found between side chains of 14 A2 domain residues and 7 and 9 residues in the A1 and A3 domains, respectively. These residues were individually replaced with Ala, except Tyr residues were replaced with Phe, and proteins stably expressed to examine the contribution of each residue to protein stability. Factor VIII stability at 55 °C and factor VIIIa activity were monitored using factor Xa generation assays. Fourteen of 30 factor VIII mutants showed >2-fold increases in either or both decay rates compared with wild type; whereas, 7 mutants showed >2-fold increased rates in factor VIIIa decay compared with factor VIII decay. These results suggested that multiple residues at the A1-A2 and A2-A3 domain interfaces contribute to stabilizing the protein. Furthermore, these data discriminate residues that stabilize interactions in the procofactor from those in the cofactor, where hydrogen bonding in the latter appears to contribute more significantly to stability. This observation is consistent with an altered conformation involving new inter-subunit interactions involving A2 domain following procofactor activation.
Factor VIII, a plasma protein that participates in the blood coagulation cascade, is decreased or defective in individuals with hemophilia A. Factor VIII circulates as a non-covalent, metal ion-dependent heterodimer consisting of a heavy chain comprised of A1(a1)A2(a2)B2 domains and a light chain comprised of (a3)A3C1C2 domains (see Ref. 1 for review). The factor VIII procofactor is activated by limited proteolysis catalyzed by thrombin or factor Xa to yield the cofactor, factor VIIIa, following cleavages at the a1A2, a2B, and a3A3 junctions. Thus, factor VIIIa is a heterotrimer of subunits designated A1, A2, and A3C1C2. Factor VIIIa functions as a cofactor for the serine protease factor IXa in the membrane-dependent conversion of zymogen factor X to the serine protease, factor Xa (see Ref. 1 for review). The role of the cofactor in the intrinsic factor Xase complex is to increase the catalytic efficiency of this reaction by several orders of magnitude.
Factor Xase is self-dampening as a result of instability in the factor VIIIa cofactor due to weak electrostatic interactions between the A2 subunit and the A1/A3C1C2 dimer (2, 3). Limited information is available regarding the association of the A2 subunit in factor VIIIa. Fluorescence energy transfer results (4) and activity assays following swapping the human A1 domain for the porcine homolog (5) suggest that interactions of the A2 subunit with the A1 subunit of the A1/A3C1C2 dimer account for the bulk of the binding energy. However, contributions to inter-A2 subunit affinity are also derived from the A3C1C2 subunit. Several factor VIII point mutations have been shown to facilitate A2 dissociation compared with WT3 factor VIIIa, and include R531H, A284E, A284P, and S289L at the modeled A1-A2 domain interface (6, 7) and N694I, R698L, and R698W at the modeled A2-A3 domain interface (8). These molecules demonstrate a characteristic one-stage/two-stage assay discrepancy (9, 10), reflecting as much as 50% reduced activity in the latter assay as a result of increased A2 dissociation.
Examination of the ceruloplasmin-based homology model for the A domains of factor VIII (11) suggests a relatively large area contributes to the A2-interactive interface with A1 and A3 domains. Consistent with this observation, we identified 30 charged or polar residues including Glu1829 (12) buried at the A2-interactive surface that may potentially form inter-domain hydrogen bonds, based upon spatial separations of <2.8 Å, contributing to the overall binding energy (13, 14). To determine a role for these residues in the stability of factor VIIIa, as well as the factor VIII procofactor, we employed a site-directed mutagenesis approach where each of these residues was individually replaced by Ala (Phe for Tyr residues) and the resulting factor VIII variants were stably expressed as B-domainless factor VIII in baby hamster kidney cells. The purified proteins were evaluated for thermostability of the factor VIII procofactor following incubation at 55 °C and the intrinsic stability of factor VIIIa following activation of the procofactor by thrombin. Results from this study identify residues that contribute to protein stability, likely through participation in hydrogen bonding at the inter-subunit interface. Furthermore, a subset of these residues makes differential contributions to stability of the two protein forms consistent with a change in bonding pattern upon procofactor activation.
MATERIALS AND METHODS
Reagents—Recombinant factor VIII (Kogenate™) was a gift from Dr. Lisa Regan of Bayer Corporation (Berkeley, CA). Phospholipid vesicles containing 20% PC, 40% PE, and 40% PS were prepared using octyl glucoside as described previously (15). The reagents α-thrombin, factor IXaβ, factor X, and factor Xa (Enzyme Research Laboratories, South Bend, IN), hirudin and phospholipids (DiaPharma, West Chester, OH), the chromogenic Xa substrate, Pefachrome Xa (Pefa-5523, CH3OCO-d-CHA-Gly-Arg-pNA·AcOH; Centerchem Inc. Norwalk CT), and Phe-Pro-Arg-chloromethyl ketone (PPACK, Calibiochem, La Jolla, CA) were purchased from the indicated vendors.
Construction, Expression, and Purification of WT and Variant Factor VIII—Mutants and WT factor VIII forms were constructed as a B-domainless factor VIII, stably expressed in baby hamster kidney cells, and purified as described previously (16). Protein yields for the variants ranged from >10 to ∼100 μg from two 750-cm2 culture flasks, with purity from ∼85 to >95% as judged by SDS-PAGE. Factor VIII concentration was measured by enzyme-linked immunosorbent assay and factor VIII activity was determined by a one-stage clotting assay and a two-stage chromogenic factor Xa generation assay (see below).
Enzyme-linked Immunosorbent Assay—A sandwich enzyme-linked immunosorbent assay was performed to measure the concentration of factor VIII proteins as previously described (17) using purified commercial recombinant factor VIII (Kogenate, Bayer Corporation) as a standard.
One-stage Clotting Assay—One-stage clotting assays were performed using substrate plasma chemically depleted of factor VIII (18) and assayed using a Diagnostica Stago clotting instrument. Plasma was incubated with activated partial thromboplastin time reagent (General Diagnostics) for 6 min at 37 °C at which time a dilution of factor VIII was added to the cuvette. After 1 min the mixture was recalcified and the time to clot formation determined and compared with a pooled normal plasma standard.
Two-stage Chromogenic Factor Xa Generation Assay—The rate of conversion of factor X to factor Xa was monitored in a purified system (19) according to methods previously described (20, 21). Briefly, factor VIII (1 nm) in buffer containing 20 mm HEPES, pH 7.2, 0.1 m NaCl, 0.01% Tween 20, 0.01% bovine serum albumin, 5 mm CaCl2, and 10 μm PS/PC/PE vesicles (Buffer A) was activated with 20 nm α-thrombin for 1 min. The reaction was stopped by adding 10 units/ml hirudin and the resultant factor VIIIa was reacted with factor IXa (40 nm) for 1 min. Factor X (300 nm) was added to initiate the reaction and after 1 min, the reaction was quenched by addition of 50 mm EDTA. Factor Xa generated was determined following reaction with the chromogenic substrate Pefachrome Xa (0.46 mm final concentration). All reactions were run at 23 °C.
Factor VIII Activity Decay at Elevated Temperature—Wild type and factor VIII variants (4 nm) in buffer A were incubated at 55 °C. Aliquots were removed at the indicated times and residual activity was determined using a two-stage chromogenic factor Xa generation assay as described above.
Factor VIIIa Activity Decay—For assays performed in the presence of 40 nm factor IXa and 10 μm PS/PC/PE vesicles, WT and factor VIII variants (4 nm) in buffer A were activated by 20 nm thrombin for 1 min. Thrombin was quenched with 10 units/ml hirudin and reactions incubated at 23 °C. Aliquots were removed at the indicated times and residual factor VIIIa activity was determined using the factor Xa generation assay following initiation of the reactions with factor X. For factor VIIIa decay experiments performed in the absence of factor IXa, the time for thrombin activation was shortened to 10 s to minimize subsequent factor VIIIa decay. Factor IXa (40 nm) was added to the reaction time course aliquots just prior to initiation of the reactions with factor X.
Data Analysis—Factor VIIIa activity values as a function of time were fitted to a single exponential decay curve by nonlinear least square regression using the equation,
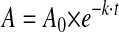 |
(Eq.1) |
where A is residual cofactor activity (nanomolar/min/nm factor VIII), A0 is the initial activity, k is the apparent rate constant, and t is the time in minutes of reaction of factor VIII at elevated temperatures (for factor VIII decay experiments) or following thrombin activation (for factor VIIIa decay experiments). Nonlinear least-squares regression analysis was performed by Kaleidagraph (Synergy, Reading, PA). Comparison of average values was performed by the Student's t test. The factor VIII A domain modeled structure was analyzed by the Swiss PDB Viewer to detect hydrogen bond at a threshold of 2.8 Å between hydrogen donor and acceptor atoms (22).
RESULTS
Activity Values for Factor VIII Mutants Targeting Hydrogen Bonding Interactions—Bonding interactions involving the A2 domain in factor VIII remain poorly understood yet represent a primary mechanism for the regulation of cofactor activity. The factor VIII homology model (11) identifies the potential for many hydrogen bonds linking residues in the A2 domain with those in the A1 or A3 domains. Using a criterion for a spatial separation of <2.8 Å between hydrogen donor and acceptor atoms (23) we identified 30 residues in which a side chain atom may be involved in hydrogen bonding with an atom from a complementary A domain (Table 1). In approximately half of the residues identified, side chain atoms were juxtaposed with either backbone carbonyl oxygen or amide hydrogen atoms, whereas the remainder represented possible interactions between neighboring side chains. Target residues in the factor VIII A domains were individually mutated to Ala, with the exception that Tyr residues were replaced with Phe, and the point mutations were stably expressed as B-domainless factor VIII.
TABLE 1.
Amino acid residues capable of hydrogen bonding
| Residue (atom) | Domain | Paired residue (atom) | Domain | Distance |
|---|---|---|---|---|
| Å | ||||
| Asp27 (Oδ) | A1 | Asn538 (Hδ) | A2 | 2.16 |
| His281 (Nδ) | A1 | Ser524 (Hγ) | A2 | 2.12 |
| Arg282 (Hη) | A1 | Gly520 (CO)a | A2 | 2.02 |
| Glu287 (Hε) | A1 | Pro672 (CO) | A2 | 1.79 |
| Asp302 (Hδ) | A1 | Asp482 (CO) | A2 | 1.98 |
| Ser313 (Hγ) | A1 | Gly643 (CO) | A2 | 1.87 |
| His317 (Nδ) | A1 | Glu540 (Hε) | A2 | 2.78 |
| Tyr476 (Hη) | A2 | Glu272 (CO) | A1 | 1.62 |
| Thr522 (Oγ) | A2 | Arg282 (NH)b | A1 | 2.39 |
| Ser524 (Hγ) | A2 | His281 (Nδ) | A1 | 2.12 |
| Arg531 (Hη) | A2 | Arg282 (CO) | A1 | 2.33 |
| Asn538 (Hδ) | A2 | Asp27 (Oδ) | A1 | 2.16 |
| Glu540 (Hε) | A2 | His317 (Nδ) | A1 | 2.78 |
| Ser650 (Hγ) | A2 | Pro1980 (CO) | A3 | 1.54 |
| Ser654 (Hγ) | A2 | Tyr1786 (Oη) | A3 | 1.65 |
| Tyr664 (Hη) | A2 | His1822 (CO) | A3 | 1.94 |
| Asp666 (Oδ) | A2 | Leu1789 (NH) | A3 | 1.93 |
| Glu683 (Oε, Hε) | A2 | Gln1820 (Hε, Oε) | A3 | 2.58, 1.72 |
| Asn684 (Oε) | A2 | Ser1791 (Hγ) | A3 | 1.76 |
| Ser695 (Hγ) | A2 | Leu1843 (CO) | A3 | 2.03 |
| Asp696 (Hδ) | A2 | Ser1949 (Oγ), Asn1950 (NH) | A3 | 1.99, 2.21 |
| Tyr1786 (Oη) | A3 | Ser654 (Hγ) | A2 | 1.65 |
| Ser1791 (Hγ) | A3 | Asn684 (Oε) | A2 | 1.76 |
| Tyr1792 (Hη) | A3 | Ser654 (CO) | A2 | 2.27 |
| Asp1795 (Oδ) | A3 | Leu687 (NH) | A2 | 1.99 |
| Gln1820 (Oε, Hε) | A3 | Glu683 (Hε, Oε) | A2 | 1.72, 2.58 |
| Glu1829 (Oε, Hε) | A3 | Tyr664 (NH, CO) | A2 | 2.15, 1.95 |
| Ser1949 (Oγ) | A3 | Asp696 (Hδ) | A2 | 1.99 |
| Asn1950 (Hδ) | A3 | Thr646 (CO) | A2 | 2.39 |
| Arg1966 (Hη-1, Hη-2) | A3 | Lys661 (CO) | A2 | 2.79, 2.01 |
Backbone carbonyl oxygen atom.
Backbone amide hydrogen atom.
Factor VIII activity was measured for the purified proteins using a one-stage clotting assay and a (two-stage) factor Xa generation assay (Fig. 1). Results from the one-stage assay indicated that 9 of the 30 point mutants showed <50% activity relative to WT factor VIII. Five of these variants demonstrated a one-stage/two-stage assay discrepancy (>1.5-fold difference), with three mutants (S524A, H281A, and E287A) showing marked reduction in only the two-stage assay. The reduced activity values for mutation in several targeted residues were consistent with a contribution of those side chains to the structural stability of factor VIII and/or factor VIIIa.
FIGURE 1.

Activity of factor VIII mutants relative to WT factor VIII as measured by a one-stage clotting assay (solid bar) and two-stage chromogenic factor Xa generation assay (hatched bar). Activity for WT and mutant factor VIII forms were measured as described under “Materials and Methods.” Error bars show the values for standard deviation averaged from three separate determinations.
Thermostability of Factor VIII Variants—To assess the heat stability of the WT procofactor and variants, a temperature at 55 °C was employed based upon factor VIII inactivation results described in an earlier study (24). For these reactions, factor VIII was incubated for the indicated times at the elevated temperature, after which the reaction mixture was immediately cooled to room temperature, and factor VIII reacted with thrombin and assayed for cofactor activity using a factor Xa generation assay. Rates of loss a factor VIII activity to the heat treatment, as judged residual cofactor function, was determined as described under “Materials and Methods.” Fig. 2A shows results for variants showing the greatest and the least sensitivities to heat treatment compared with WT. Results for the remaining variants are presented as supplemental data.
FIGURE 2.

A, activity decay of WT and mutant factor VIII. Factor VIII (4 nm) was incubated at 55 °C and at the indicated times and aliquots were removed and assayed for activity by factor Xa generation assays as described under “Materials and Methods.” Results are shown for WT (dashed line, open circles), R282A (open triangles), S524A (open squares), N684A (open diamonds), R531A (closed circles), S650A (closed triangles), E287A (closed squares), and D302A (closed diamonds). Results for other variants are presented as supplemental data. B, activity decay of WT and mutant factor VIIIa in the presence of factor IXa. Thrombin-activated factor VIIIa (4 nm) in the presence of 40 nm factor IXa was incubated at 23 °C, aliquots were taken at the indicated time points and activity was measured by the factor Xa generation assay as described under “Materials and Methods.” Results are shown for WT (dashed line, open circles), R282A (open triangles), S524A (open squares), Y1792F (open diamonds), N684A (closed circles), Y1786F (closed triangles), R531A (closed squares), E287A (closed diamonds), and D302A (hatched circles). Results for other variants are presented as supplemental data. Results for selected fast decay variants are shown in an expanded scale in the inset. C, activity decay of WT and mutant factor VIIIa in the absence of factor IXa. Thrombin-activated factor VIIIa (4 nm) was incubated at 23 °C, aliquots were taken at the indicated time points and activity was measured by factor Xa generation assay as described under “Materials and Methods.” Results are shown for WT (dashed line, open circles), N684A (open triangles), Y1786F (open squares), Y1792F (open diamonds), R282A (closed circles), and S524A (closed triangles). The inset shows an expanded scale for the early time points. Data were fitted by non-linear least squares regression and each point represents the value averaged from three separate determinations.
Table 2 and Fig. 3 summarize the results obtained from factor VIII thermostability assays for the 30 variants. Overall, these activity data fit well to a single exponential decay function with correlation coefficients in most cases >0.98. Whereas a number of mutations were benign with respect to the amino acid replacement (21 showing <2-fold differences in rates of decay), several residues including Arg282 (A1 domain), and A2 domain residues Ser524, Asn684, and Ser650 showed ∼5-20-fold increased rates in factor VIII decay suggesting an important role for these residues in maintaining factor VIII stability. Furthermore, the R282A and N684A variants showed significantly reduced specific activity values suggesting both activity and stability parameters were affected by the single point mutations. On the other hand, replacement of Glu287 and Asp302 with Ala yielded reduced rates for factor VIII decay at the elevated temperature. This apparent increase in protein stability following mutation is consistent with these acidic side chains destabilizing inter-domain interactions.
TABLE 2.
Factor VIII and VIIIa decay rates and activity values
Mutant factor VIII forms are ordered based on decreasing rates of factor VIII decay. Standard deviations for rate decay values are estimated based on least squares curve-fitting and are within ~10% of mean values.
|
Decay rates in min−1
|
Specific activity
|
||||
|---|---|---|---|---|---|
|
Factor VIII
|
Factor VIIIa
|
One-stage assay
|
Two-stage assay
|
||
| FIXa (+)a | FIXa (−)b | ||||
| WT | 0.0473 (1.00)c | 0.0113 (1.00) | 0.0631 (1.00) | 4.77d (1.00) | 44.5e (1.00) |
| R282A | 0.9646 (20.4) | 0.4708 (41.7) | 0.6738 (10.7) | 0.95 (0.20) | 1.77 (0.04) |
| S524A | 0.4332 (9.16) | 0.4554 (40.4) | 0.4416 (7.00) | 4.20 (0.88) | 1.02 (0.02) |
| N684A | 0.4002 (8.46) | 0.4096 (36.3) | 1.1837 (18.8) | 0.41 (0.09) | 2.15 (0.05) |
| R531A | 0.2448 (5.18) | 0.0758 (6.72) | 2.62 (0.55) | 24.0 (0.54) | |
| S650A | 0.1395 (2.95) | 0.0317 (2.81) | 4.41 (0.93) | 45.5 (1.02) | |
| Y664F | 0.1173 (2.48) | 0.0148 (1.31) | 5.25 (1.10) | 47.4 (1.07) | |
| H281A | 0.1170 (2.47) | 0.0450 (3.99) | 3.70 (0.78) | 21.1 (0.47) | |
| Y1786F | 0.1138 (2.41) | 0.2361 (20.9) | 1.0740 (17.0) | 1.43 (0.30) | 6.21 (0.14) |
| D696A | 0.0889 (1.88) | 0.0118 (1.05) | 4.82 (1.01) | 45.0 (1.01) | |
| S313A | 0.0770 (1.63) | 0.0210 (1.86) | 4.34 (0.91) | 36.5 (0.82) | |
| E683A | 0.0743 (1.57) | 0.0263 (2.33) | 1.00 (0.21) | 15.8 (0.36) | |
| D1795A | 0.0697 (1.47) | 0.0238 (2.11) | 3.82 (0.80) | 32.5 (0.73) | |
| E540A | 0.0691 (1.46) | 0.0091 (0.81) | 4.40 (0.92) | 37.9 (0.85) | |
| R1966A | 0.0682 (1.44) | 0.0163 (1.44) | 3.74 (0.78) | 36.6 (0.82) | |
| D666A | 0.0646 (1.37) | 0.0545 (4.83) | 2.47 (0.52) | 17.5 (0.39) | |
| N538A | 0.0630 (1.33) | 0.0144 (1.28) | 4.00 (0.84) | 35.7 (0.80) | |
| H317A | 0.0629 (1.33) | 0.0145 (1.28) | 3.83 (0.80) | 30.8 (0.69) | |
| N1950A | 0.0618 (1.31) | 0.0195 (1.73) | 3.46 (0.72) | 25.7 (0.58) | |
| S654A | 0.0599 (1.27) | 0.0145 (1.28) | 5.02 (1.05) | 45.2 (1.02) | |
| T522A | 0.0596 (1.26) | 0.0270 (2.39) | 0.83 (0.18) | 24.5 (0.55) | |
| S1791A | 0.0595 (1.26) | 0.0208 (1.85) | 3.73 (0.78) | 28.9 (0.65) | |
| Y1792F | 0.0577 (1.22) | 0.4335 (38.4) | 0.7237 (11.5) | 1.41 (0.30) | 3.42 (0.08) |
| Y476F | 0.0579 (1.22) | 0.0139 (1.23) | 4.57 (0.96) | 41.8 (0.94) | |
| S1949A | 0.0573 (1.21) | 0.0129 (1.14) | 3.17 (0.66) | 28.6 (0.64) | |
| S695A | 0.0524 (1.11) | 0.0085 (0.75) | 5.15 (1.08) | 45.4 (1.02) | |
| D27A | 0.0489 (1.03) | 0.0089 (0.79) | 4.53 (0.95) | 40.1 (0.90) | |
| Q1820A | 0.0480 (1.01) | 0.0114 (1.01) | 4.91 (1.03) | 44.0 (0.99) | |
| E287A | 0.0367 (0.78) | 0.0088 (0.78) | 2.86 (0.60) | 16.4 (0.37) | |
| D302A | 0.0369 (0.78) | 0.0049 (0.43) | 5.38 (1.03) | 49.0 (1.10) | |
Decay experiments performed in the presence of factor IXa.
Decay experiments performed in the absence of factor IXa.
Value in parentheses are relative to wild type.
Unit/μg.
Nanomolar factor Xa generated/min/nm factor VIII.
FIGURE 3.

Summary of factor VIII/VIIIa activity decay rates. Factor VIII (hatched bar) and factor VIIIa (solid bar) activity decay rates relative to wild type factor VIII are based on the results shown in Fig. 2. Data are ordered beginning with mutants showing the highest rates for factor VIII activity decay.
Factor VIIIa Decay Rates—Factor VIIIa activity is labile due to A2 subunit dissociation (2, 3). Results from earlier studies showed that inclusion of factor IXa and phospholipid vesicles with factor VIIIa to form the Xase complex reduced the lability of the cofactor (25, 26) by partially stabilizing the A2 subunit within factor Xase (27). We recently used this approach to examine the decay rate for an E1829A factor VIIIa mutant (12) because the activity decay of this variant factor VIIIa in the absence of factor IXa and membrane was too rapid to accurately measure. This approach was similarly employed to assess rates for factor VIIIa decay for the panel of variants described in the present study. Factor VIII (4 nm) was incubated with a molar excess of factor IXa (40 nm) and phospholipid vesicles, rapidly activated with thrombin, and subsequent factor Xase activity was measured over a time course at 23 °C. Rates of decay of factor Xase activity was attributed to A2 subunit dissociation and data were fitted using a single exponential decay. Given the high Kd value for the affinity of A2 subunit within factor VIIIa (144 nm) (12) and the low factor VIIIa concentration (4 nm) used in the reactions, the effect of reassociation of dissociated A2 subunit is negligible, supporting use of a simple single exponential applied for this regression analysis.
Results are presented in Fig. 2B that shows data for the most severely affected variants as well as those variants showing a positive response to the mutation. Data for the remaining variants showing intermediate parameter values are presented as supplemental data. We observed 7 variants possessing significant (>5-fold) increases in rates of factor VIIIa decay compared with WT (Table 2 and Fig. 3). These mutations included R282A, S524A, N684A, E1829A,4 Y1786F, D666A, and Y1792F. Factor VIII activity values for these variants as measured by a two-stage assay were significantly lower than those determined by the one-stage assay (Fig. 1), consistent with the mutations leading to appreciable rates of A2 subunit dissociation. Furthermore, several of these mutations including R282A, N684A, and Y1792F showed overall low specific activity in the one-stage assay. As is the case for factor VIII mutants possessing this assay discrepancy, activity determined from the one-stage assay was also reduced (6-8), possibly reflecting direct effects of A2 dissociation rates on determining factor VIII activity. Conversely, variants E287A and D302A that possessed greater thermostabilities than WT factor VIII also yielded enhanced stability of factor VIIIa as judged by reductions in the rates of cofactor decay following activation by thrombin. Results with the D302A variant were more pronounced and showed an ∼2-fold reduced rate of cofactor decay relative to WT factor VIIIa, retaining ∼90% of its original activity after 40 min.
Although the concentration of factor IXa used in the above reactions (40 nm) saturated 4 nm factor VIII (data not shown), those mutations showing unusually fast rates for decay (>10-fold) relative to wild type were also evaluated in the absence of added factor IXa to eliminate concerns that the fast decay resulted from a defect in interactions of the cofactors with factor IXa (Fig. 2C and Table 2). Comparison of the WT factor VIII forms showed an ∼5-fold increase in the factor VIIIa decay rate observed in the absence of factor IXa compared with its presence, and this result was consistent with our earlier observations showing partial stabilization of the cofactor under these conditions (27). The “fast decay” variants assayed, R282A, S524A, N684A, Y1786F, and Y1792F, all demonstrated significantly faster decay rates (>5-20-fold) relative to the WT value indicating that the rapid decay did not result from a defect in factor IXa interaction.
The effects of mutation on factor VIIIa stability appeared to represent a more common outcome than that observed for procofactor stability because 7 mutants showed >2-fold greater rates of factor VIIIa decay compared with factor VIII decay. On the other hand, only two variants (Y664F and D696A) showed significantly increased factor VIII decay rates compared with factor VIIIa decay rates. This latter observation was consistent with the mutations primarily altering conformation at the inter-domain interface in the procofactor. Taken together, these results identify contributions of multiple residues to inter-A2 (domain) subunit interactions in the procofactor and cofactor forms of factor VIII with selected residues making disparate contributions to protein stability.
DISCUSSION
Prior observations that the association of A2 subunit in factor VIIIa is mediated primarily by electrostatic rather than hydrophobic interactions (28) formed the basis for our evaluation of hydrogen bonding interactions at the A2 domain interface. Examination of the ceruloplasmin-based homology model of the A domains of factor VIII identified 30 residues that could contribute hydrogen bonding to stabilize interactions of the A2 domain in the pro- and cofactor forms of the protein. Point mutations to eliminate charge or reduce polarity resulted in approximately half of the variants showing increases in rates of inactivation following reaction of the procofactor to elevated temperatures and/or resulting from spontaneous decay of factor VIIIa. Loss of factor VIIIa activity dominated over loss of factor VIII stability with 7 mutations severely affecting the former parameter, many of which showed a characteristic one-stage/two-stage assay discrepancy consistent with accelerated dissociation of the A2 subunit. It is also noteworthy that mutations identified at 9 residues of the 30 residues targeted (R282H (or with Cys or Leu), T522S, R531C (or with Gly or His), Y664C, N684D (or with Ser), S1791P, E1829A (or with Gly), S1949R, and R1966Q) are listed in the Hemophilia A data base (29) as yielding varying severity of the disease. Our results show that mutation at 7 of these residues yielded significant increases in procofactor and/or cofactor decay rates consistent with destabilization of the protein structure.
Mapping the target residues to the modeled factor VIII A domains suggested discrete regions contributing to interaction of the A2 domain with the A1 and A3 domains. Fig. 4 shows the modeled factor VIII structure with a transparent A2 domain overlaying the A1 and A3 domains. Residue locations are indicated and are grouped into four regions. A gradient of color is used to indicate the relative effect of mutation at a given residue on either factor VIII (white to blue) or factor VIIIa (white to yellow) decay rates. Regions designated 3 and 4 that contain 8 and 5 target residues, respectively, showed little if any effect of mutation on stability parameters, suggesting a minimal role for side chains of these residues in forming an interacting surface. The lone exception is Ser650 in region 4, which showed <5-fold increases in both factor VIII and factor VIIIa decay rates. On the other hand, mutation of 4 of the 5 residues in Region 1, which represents an A2 interface with the A1 domain, yielded increases in the rates of factor VIII/VIIIa decay. Three of these residues, Arg282, Ser524, and Arg531, showed >5-fold increases in factor VIII decay and ∼8- to >40-fold increases in factor VIIIa decay, suggesting a significant contribution to inter-domain (-subunit) affinity is derived from this interface between A1 and A2. Furthermore, this observation is consistent with earlier studies demonstrating a primary role for the A1 subunit in A2 subunit retention following procofactor activation (4, 5).
FIGURE 4.

Visual representation of factor VIII and factor VIIIa activity decay rates mapped onto the factor VIII structure model. Factor VIII surface model of individual domains are drawn by Swiss PDB viewer as colored by yellow (A1), transparent blue (A2), red (A3), green (C1), and C2 (gray). Boxes indicate the location of side chain atoms of the indicated residues potentially involved in the hydrogen bonding. White boxes indicate that a mutation at a given residue resulted in essentially no effect on the rate of activity decay. Color gradients represent increasing rates of activity decay for factor VIII (white to blue) or factor VIIIa (white to yellow) for the mutated residue.
Region 2, which describes interaction of the A2 and A3 domains, contains 12 residues, many of which demonstrated little if any effect on protein stability following mutagenesis. However, several residues of interest were observed. Asn684 appeared critical for stability of both procofactor and cofactor forms, as judged by ∼8- and ∼35-fold increases in respective decay rates following replacement with Ala. Interestingly, a set of residues in this region appeared to make a greater contribution to cofactor stability compared with that of the procofactor. Residues where mutations yielded the greatest effect on factor VIII stability produced a rank order of Arg282 > Ser524 > Asn684 > Arg531 > Ser650 > Glu1829 > Tyr664 > His281 (see Table 2 and Fig. 3). However, ranking those residues having the greatest effect on factor VIIIa stability resulted in the order Arg282 > Ser524 > Tyr1792 > Asn684 > Glu1829 > Tyr1786 > Arg531 > D666A. Clearly, residues Arg282, Ser524, and Arg531 (Region 1) and Asn684 show intrinsic importance to the interaction of the A2 domain within factor VIII as well as retention of the A2 subunit following cleavage at the A1-A2 junction by thrombin. However, residues Glu1829, Tyr1792, Tyr1786, and Asp666 each make apparent significant contributions to A2 subunit retention in factor VIIIa while making marginal contributions to the stability of factor VIII. The disparate effects of this set of mutations suggest a change in the overall distribution of bonding interactions upon cleavage of the contiguous A1-A2 domains during activation of the procofactor.
The proteolytic activation of factor VIII to factor VIIIa is accompanied by changes in protein conformation. The A2 domain is a focal point for conformational change leading to altered function as this domain contacts the protease domain of factor IXa (30). For example, differences in fluorescence anisotropy of a fluorophor-labeled factor IXa active site were observed using an isolated A2 subunit in the presence and absence of the A1 subunit compared with factor VIII heavy chain (contiguous A1-A2 domains) (31). Based upon the present study, we speculate that conformational changes associated with activation are derived in large part from changes in bonding of the A2 subunit within factor VIIIa such that now residues including Tyr1792, Tyr1786, and Asp666 participate in this interaction.
An earlier report (12) showed that the low specific activity and relatively fast decay of factor VIIIa possessing the E1829A mutation resulted from an ∼4-fold reduction in the affinity of A2 subunit for the A1/A3C1C2 subunit harboring this mutation in the A3 domain. According to the ceruloplasmin-based homology model, the side chain carboxyl group of Glu1829 is predicted to form hydrogen bonds to both amide hydrogen and carbonyl oxygen of Tyr664 in the A2 subunit (see Table 1). Of the remaining 10 residues ranked above for showing the most marked increases to factor VIII (VIIIa) decay, side chains from 6 are proposed to bond backbone atoms in their partners. Two residues, His281 and Ser524 are proposed to form a bonding pair. The remaining two residues, Asn684 and Tyr1786 appear to bond with side chain atoms from Ser1791 and Ser654, respectively. Because mutations at these latter residues showed little effect on protein stability values, these discrepancies may suggest bonding interactions at alternate sites and/or differential mechanisms for reduced stability due to mutation at Asn684 and Tyr1786.
Whereas the observed effects of mutation at the target residues were for the most part either benign or detrimental, we did note that mutation at two A1 domain acidic residues, Asp302 and Glu287, yielded modest enhancement in stability in both pro- and active cofactor forms. The relative activity of Glu287 was somewhat reduced compared with WT, whereas the activity values for the Asp302 variant were indistinguishable from the WT protein, and suggest the latter represented a gain-of-function mutation. The reason(s) for the enhanced stabilities of the variants is not known but suggests some destabilization may result from burying the (negative) charge at the interface and/or an increase in stability when these residue side chains are hydrophobic.
Detailed structural information of factor VIII is limited. Our use of the ceruloplasmin-based homology model for factor VIII in this mutagenesis and functional study has proved a useful resource in that mutation at approximately half of the target residues predicted to participate in hydrogen bonding affected protein stability. During the preparation of this article, a 3.75-Å crystal structure of the B-domainless, human factor VIII procofactor was reported (32). Whereas this level of resolution does not allow assignment of hydrogen bonding interactions (<2.8 Å), the authors of the study indicated that the A domains of factor VIII could be superimposed onto those of ceruloplasmin with a high degree of accuracy. Availability of the structure coordinates (anticipated in September 2008) may yield additional insights into new target residues to test for functional contributions to the interactions involving A2 in factors VIII and VIIIa. However, the results obtained in the present study provide functional information to reconcile limitations in our knowledge of factor VIII structure. Furthermore, this approach applied to other bonding interactions should yield insights into additional structural changes that occur during conversion of the procofactor to cofactor and may identify other gain-of-function variants that could have application for therapeutic use.
Acknowledgments
We thank Lisa M. Regan for the gifts of recombinant human factor VIII and Qian Zhou for excellent technical assistance.
This work was supported by National Institutes of Health Grants HL38199 and HL76213. An account of this work was presented at the 49th Annual Meeting of the American Society of Hematology, December 8, 2007, in Atlanta, GA. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains a supplemental figure.
Footnotes
Lower case a represents short (∼30 residue) segments rich in acidic residues.
The abbreviations used are: WT, wild type; PC, phosphatidylcholine; PE, phosphatidylethanolamine; PS, phosphatidylserine.
Results obtained for the E1829A variant are from Ref. 12.
References
- 1.Fay, P. J. (2004) Blood Rev. 18 1-15 [DOI] [PubMed] [Google Scholar]
- 2.Fay, P. J., Haidaris, P. J., and Smudzin, T. M. (1991) J. Biol. Chem. 266 8957-8962 [PubMed] [Google Scholar]
- 3.Lollar, P., and Parker, C. G. (1990) J. Biol. Chem. 265 1688-1692 [PubMed] [Google Scholar]
- 4.Nogami, K., Wakabayashi, H., Schmidt, K., and Fay, P. J. (2003) J. Biol. Chem. 278 1634-1641 [DOI] [PubMed] [Google Scholar]
- 5.Parker, E. T., Doering, C. B., and Lollar, P. (2006) J. Biol. Chem. 281 13922-13930 [DOI] [PubMed] [Google Scholar]
- 6.Pipe, S. W., Eickhorst, A. N., McKinley, S. H., Saenko, E. L., and Kaufman, R. J. (1999) Blood 93 176-183 [PubMed] [Google Scholar]
- 7.Pipe, S. W., Saenko, E. L., Eickhorst, A. N., Kemball-Cook, G., and Kaufman, R. J. (2001) Blood 97 685-691 [DOI] [PubMed] [Google Scholar]
- 8.Hakeos, W. H., Miao, H., Sirachainan, N., Kemball-Cook, G., Saenko, E. L., Kaufman, R. J., and Pipe, S. W. (2002) Thromb. Haemostasis 88 781-787 [PubMed] [Google Scholar]
- 9.Duncan, E. M., Duncan, B. M., Tunbridge, L. J., and Lloyd, J. V. (1994) Br. J. Haematol. 87 846-848 [DOI] [PubMed] [Google Scholar]
- 10.Rudzki, Z., Duncan, E. M., Casey, G. J., Neumann, M., Favaloro, E. J., and Lloyd, J. V. (1996) Br. J. Haematol. 94 400-406 [DOI] [PubMed] [Google Scholar]
- 11.Pemberton, S., Lindley, P., Zaitsev, V., Card, G., Tuddenham, E. G., and Kemball-Cook, G. (1997) Blood 89 2413-2421 [PubMed] [Google Scholar]
- 12.Wakabayashi, H., Zhou, Q., Varfaj, F., and Fay, P. J. (2007) J. Thromb. Haemostasis 5 996-1001 [DOI] [PubMed] [Google Scholar]
- 13.Pace, C. N., Shirley, B. A., McNutt, M., and Gajiwala, K. (1996) FASEB J. 10 75-83 [DOI] [PubMed] [Google Scholar]
- 14.Thurlkill, R. L., Grimsley, G. R., Scholtz, J. M., and Pace, C. N. (2006) J. Mol. Biol. 362 594-604 [DOI] [PubMed] [Google Scholar]
- 15.Mimms, L. T., Zampighi, G., Nozaki, Y., Tanford, C., and Reynolds, J. A. (1981) Biochemistry 20 833-840 [DOI] [PubMed] [Google Scholar]
- 16.Wakabayashi, H., Freas, J., Zhou, Q., and Fay, P. J. (2004) J. Biol. Chem. 279 12677-12684 [DOI] [PubMed] [Google Scholar]
- 17.Wakabayashi, H., Su, Y. C., Ahmad, S. S., Walsh, P. N., and Fay, P. J. (2005) Biochemistry 44 10298-10304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Over, J. (1984) Scand. J. Haematol. Suppl. 41 13-24 [DOI] [PubMed] [Google Scholar]
- 19.Lollar, P., Fay, P. J., and Fass, D. N. (1993) Methods Enzymol. 222 128-143 [DOI] [PubMed] [Google Scholar]
- 20.Wakabayashi, H., Koszelak, M. E., Mastri, M., and Fay, P. J. (2001) Biochemistry 40 10293-10300 [DOI] [PubMed] [Google Scholar]
- 21.Wakabayashi, H., Schmidt, K. M., and Fay, P. J. (2002) Biochemistry 41 8485-8492 [DOI] [PubMed] [Google Scholar]
- 22.Guex, N., and Peitsch, M. C. (1997) Electrophoresis 18 2714-2723 [DOI] [PubMed] [Google Scholar]
- 23.Weiner, S. J., Kollman, P. A., Case, D. A., Singh, U. C., Ghio, C., Alagona, G., Profeta, S., and Weiner, P. K. (1984) J. Am. Chem. Soc. 106 765-784 [Google Scholar]
- 24.Ansong, C., and Fay, P. J. (2005) Biochemistry 44 8850-8857 [DOI] [PubMed] [Google Scholar]
- 25.Lollar, P., Knutson, G. J., and Fass, D. N. (1984) Blood 63 1303-1308 [PubMed] [Google Scholar]
- 26.Lamphear, B. J., and Fay, P. J. (1992) J. Biol. Chem. 267 3725-3730 [PubMed] [Google Scholar]
- 27.Fay, P. J., Beattie, T. L., Regan, L. M., O'Brien, L. M., and Kaufman, R. J. (1996) J. Biol. Chem. 271 6027-6032 [DOI] [PubMed] [Google Scholar]
- 28.Fay, P. J., and Smudzin, T. M. (1992) J. Biol. Chem. 267 13246-13250 [PubMed] [Google Scholar]
- 29.Kemball-Cook, G., Tuddenham, E. G., and Wacey, A. I. (1998) Nucleic Acids Res. 26 216-219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bajaj, S. P., Schmidt, A. E., Mathur, A., Padmanabhan, K., Zhong, D., Mastri, M., and Fay, P. J. (2001) J. Biol. Chem. 276 16302-16309 [DOI] [PubMed] [Google Scholar]
- 31.Fay, P. J., Mastri, M., Koszelak, M. E., and Wakabayashi, H. (2001) J. Biol. Chem. 276 12434-12439 [DOI] [PubMed] [Google Scholar]
- 32.Shen, B. W., Spiegel, P. C., Chang, C. H., Huh, J. W., Lee, J. S., Kim, J., Kim, Y. H., and Stoddard, B. L. (2008) Blood 111 1240-1247 [DOI] [PMC free article] [PubMed] [Google Scholar]