

Abstract
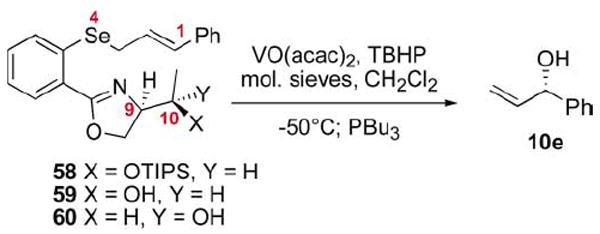
A vanadium-catalyzed method for the oxidation of prochiral aryl, allylic selenides with tandem [2,3] sigmatropic rearrangement has been developed. This protocol has been screened on a series of substrates to test for its generality and effectiveness. The applicability of this methodology to the synthesis of enantiomerically enriched allylic alcohols has been studied on a series of chiral oxazole-containing systems with a diastereomeric ratio (d.r.) of up to 85 : 15. The chiral transfer observed in the allyl alcohol products is the result of a net 1,9- and/or 1,10-induction. Finally, the first example of a selenium–oxygen nonbonding interaction in oxazole-containing selenide appears to have been observed via X-ray crystal analysis.
Introduction
Allylic alcohols are vital cogs in organic synthesis. Because of the importance of allylic alcohols as synthons, many chemists have explored a variety of pathways for the construction of this intricate subunit.1 Chiral and achiral allylic alcohols have also been exploited to create other useful precursors including epoxides, cyclopropanes, and unsaturated diols.2 In addition, allylic alcohols are present in numerous complex natural products.3
One potentially attractive method for accessing the allylic alcohol functionality is by selenide oxidation with in situ [2,3] sigmatropic rearrangement (SOS reaction, Scheme 1). Several laboratories have exploited this strategy and the area has been recently reviewed.4 A sulfur-based variant for the SOS reaction (the Mislow–Braverman–Evans rearrangement) is also well-known and has been extensively studied.5,6 Interestingly, the equilibrium of the sulfur-based [2,3] sigmatropic rearrangement often favors the sulfoxide, thereby requiring an in situ “trapping agent” such as P(OMe)3 to force the equilibrium in the desired direction. In contrast, the selenoxide intermediate 2 undergoes rapid sigmatropic rearrangement, even at low temperatures, with the equilibrium strongly favoring the selenenates 5 and 6. Finally, most protocols for the key selenide oxidation step rely upon stoichiometric oxidants, such as H2O2 and m-CPBA. In fact, prior to our work,7 no general method8 had been reported for a metal-catalyzed system for accomplishing this dual reaction sequence.
Scheme 1.
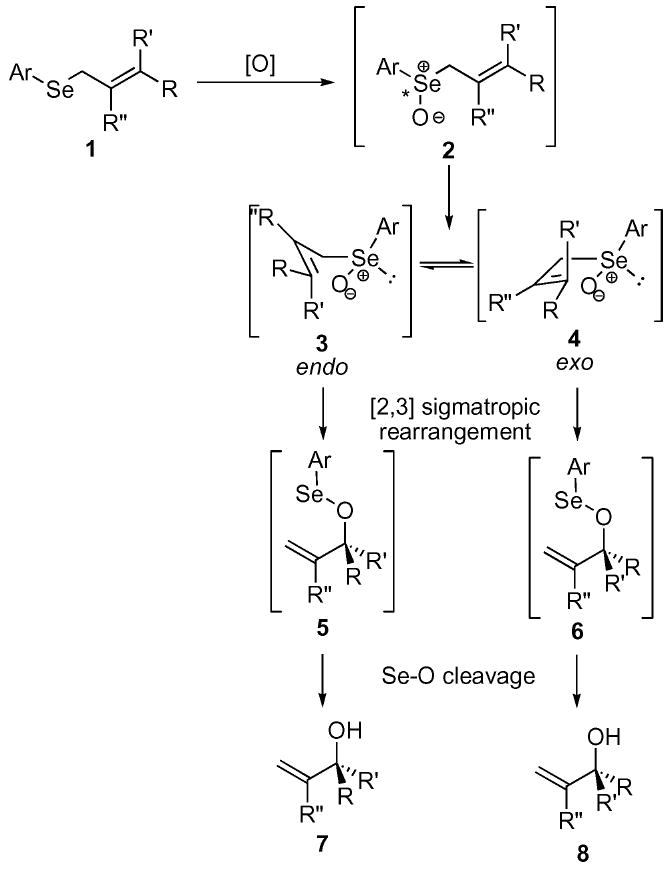
Selenide oxidation with in situ [2,3] sigmatropic rearrangement (SOS reaction).
Several important stereochemical issues are also present in the SOS reaction. Oxidation of the prochiral allylic selenide yields a chiral selenoxide 2 which can transfer its chiral information via a [2,3] sigmatropic rearrangement to the resultant selenenates 5 and 6. Cleavage of the Se–O bond provides the allylic alcohols 7 and 8. Two potential pathways exist for the sigmatropic rearrangement involving either an endocyclic or exocyclic transition state. Reich and co-workers calculated the endo pathway 3 to be approximately 2 kcal mol−1 more stable than its exo counterpart 4.9 Interestingly, Davis and co-workers studied the oxidation of prochiral selenides not capable of undergoing sigmatropic rearrangement using a chiral oxaziradine. They were able to achieve up to 97 : 3 enantiomeric ratio (e.r.) on sterically demanding aryl selenide systems. Application of their chiral oxaziradines to selenide systems capable of undergoing [2,3] sigmatropic rearrangement yielded enantiomeric excesses ranging from 40% e.e. for E-alkenes to 60% e.e. for Z-alkenes.10 These results would indicate that a considerable amount of chiral information is lost in the sigmatropic rearrangement. Despite these apparent limitations, several laboratories have been able to achieve levels of selectivity that are larger than should be possible, based on the oxaziradine experiments.11-15 It would appear that the exact nature of the chirality transfer in the sigmatropic rearrangement continues to not be fully understood.
The potential applications of an asymmetric selenide oxidation with in situ [2,3] sigmatropic rearrangement (ASOS reaction) would be significant. The considerable efforts by several laboratories in this area have primarily been focused on the utilization of stoichiometric oxidants in both the SOS and the ASOS reactions.9-14 Due to the prevalent interest in the development of metal-catalyzed processes, our laboratory sought to explore a metal-catalyzed method for accomplishing this dual reaction sequence.
Results and discussion
Vanadium-catalyzed SOS reaction
Our preliminary investigations involved the use of a Ti(OR)4/t-butyl hydroperoxide (TBHP) system, which has proven an ideal oxidation combination in numerous asymmetric transformations.16 We were disappointed to observe that both the racemic and the asymmetric variants 15 of this system performed poorly in our hands with sluggish reaction rates and stoichiometric levels of the metal catalyst to proceed to completion. These results are consistent with the work done independently by the Tingoli 17 and Kamigata 18 laboratories using a titanium catalyst to effect selenide oxidation.
Vanadium-catalyzed oxidations 19 have proven effective in the oxidation of sulfides to the corresponding sulfoxide.20 Unlike titanium, these sulfide-based reactions proceed readily and usually require only a small amount of the metal catalyst. We were gratified to find that 10 mol% vanadyl acetylacetonate [VO(acac)2], with TBHP or cumene hydroperoxide (CHP) as the co-oxidant, effciently and rapidly induced the SOS reaction. The VO(acac)2 was key to this tandem reaction sequence; the control experiment without VO(acac)2 led to <5% conversion after 24 h. This catalyst system was screened on a diverse series of allylic selenides, possessing a variety of substituents on the alkene (Table 1). The selenides 9a–h were constructed in one step from their corresponding alcohols using the commercially available o-NO2C6H4SeCN and PBu3 in high yield.21 The vanadium-catalyzed SOS reaction proved amenable to all substrates that were screened with yields ranging from 65–89%. Cleavage of the Se–O bond of the selenenate species was best accomplished using PBu3. Traditional cleavage methods (such as pyridine/H2O and PPh3) proved slow and inconsistent. It is important to note, however, that Bu3P must be added to the reaction at −10 °C. If the Se–O cleavage was performed at room temperature, a considerable amount of the 2° allylic selenide was recovered, resulting from a Mitsunobu-type SN2 inversion.
Table 1.
Selected examples of a vanadium-catalyzed SOS reaction a
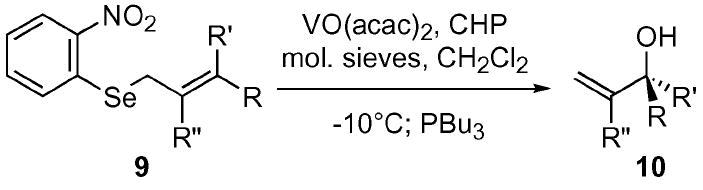
| Entry | R | R′ | R″ | Yield b |
|---|---|---|---|---|
| a c | −C6H13 | H | H | 70% |
| b |
|
H | H | 75% |
| c |
|
H | H | 84% |
| dd |
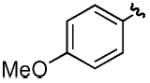
|
H | H | 66% |
| e |
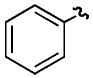
|
H | H | 65% |
| f |
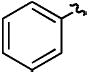
|
H | CH3 | 70% |
| g | H | −C6H13 | H | 89% |
| hd | H |
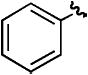
|
H | 86% |
All reactions unless otherwise noted were performed at 0.3 M concentration of substrate using 1.2 eq. tributylphosphine to quench the selenenate intermediate.
All yields are isolated yields after chromatography over silica gel.
This reaction was quenched with triphenyl-phosphine.
t-Butyl hydroperoxide was substituted for cumene hydroperoxide to simplify purification.
One additional area that we felt it was prudent to study was the effect of a proximal stereogenic center on the diastereomeric ratio of the resultant allylic alcohol (Scheme 2). The required chiral allylic selenides 12 and 14 were synthesized from the known Evans alkylation derivative 11 22 via standard methods. We were intrigued to discover that the neighboring stereogenic center had little effect on the stereochemical outcome of the SOS reaction. This result is consistent with a hypothesis that the stereochemistry of the selenoxide is the major determining factor in the configuration of the resultant allylic alcohol.10
Scheme 2.
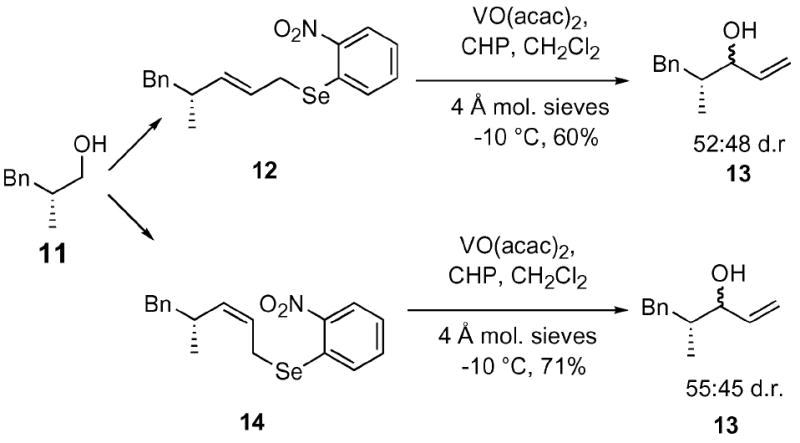
Effect of proximal stereogenic centers on the vanadium-catalyzed SOS reaction.
Vanadium-catalyzed, ligand-based ASOS reaction
With the utility of the vanadium-catalyzed SOS reaction firmly established, we shifted our focus to the development of the ASOS reaction. We initially hoped to employ a chiral tri- or tetradentate Schiff base ligand system to facilitate asymmetric induction to the ultimate allylic alcohol product. Several laboratories have exploited this strategy in the asymmetric oxidation of sulfides to sulfoxides with good levels of enantio-selectivity.20 Based on the work by other laboratories, which showed increased levels of selectivity using the Z-alkene,10,11 the selenide 9g was chosen as our initial substrate (Table 2). We were disappointed to observe essentially no meaningful levels of selectivity. It was thought that a more sterically demanding substituent on the alkene might help to increase the level of chiral induction from the Schiff base ligands to the selenoxide by increasing a disruptive steric interaction in the exo transition state of the [2,3] sigmatropic rearrangement.10,11,14,23 To this effect, the cyclohexyl selenide 15 was constructed via standard methods. This substrate proved only marginally more effective with up to 55 : 45 e.r. observed using the tetradentate ligand 17. Ligands such as the t-leucinol derived ligand 20, which provided good levels of e.r. on the sulfide to sulfoxide system, proved ineffective. It became apparent that extension of the vanadium-catalyzed methodology to a ligand-based ASOS reaction was premature.
Table 2.
Selected examples of a ligand-based, vanadium-catalyzed ASOS reaction
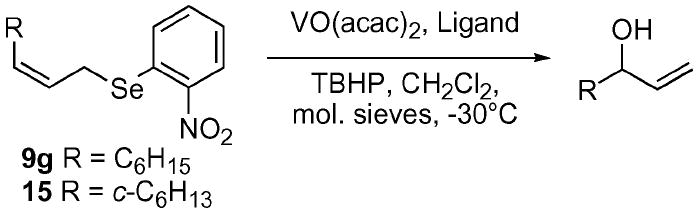
| Entry | Selenide | Ligand | e.r. |
|---|---|---|---|
| a | 9g | 16 | <51 : 49 |
| b | 9g | 17 | <51 : 49 |
| c | 9g | 20 | <51 : 49 |
| d | 15 | 16 | <51 : 49 |
| e | 15 | 17 | 55 : 45 |
| f | 15 | 18 | <51 : 49 |
| g | 15 | 19 | <51 : 49 |
| h | 15 | 20 | 53 : 47 |
| i | 15 | 21 | <51 : 49 |
| j | 15 | 22 | <51 : 49 |
| k | 15 | 23 | 53 : 47 |
| l | 15 | 24 | 52 : 48 |
Vanadium-catalyzed, oxazole-based ASOS reaction
While the promising results obtained in the catalytic, asymmetric oxidation of prochiral sulfides by other laboratories 20 did not correlate well with ASOS reaction using a ligand-based strategy, several other approaches have been employed for the synthesis of enantioenriched sulfoxides. Williams and co-workers published an elegant solution to the oxidation of prochiral sulfides using an oxazole-based strategy (Scheme 3).24 They were able to achieve a diastereomeric ratio (d.r.) of up to 97 : 3 using a metal-catalyst [VO(acac)2 or Ti(Oi–Pr)4 with the latter giving superior results]. The Williams laboratory proposed that a precoordinated complex between the metal and the oxazole facilitated the observed level of selectivity. This system would appear ideally suited for extension to the ASOS reaction.
Scheme 3.
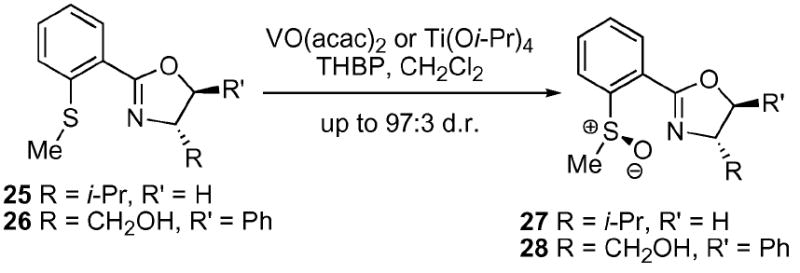
Williams and co-workers oxazole-based sulfide oxidation system.
Construction of the required selenide precursors was available in 3 straightforward steps (Scheme 4). The commercially available amino alcohols were condensed with 2-bromobenzoyl chloride (31) to give benzamides 32 and 33. The crude benzamides were converted to the oxazole using SOCl2. It should be noted that oxazoles 34 and 35 have been previously prepared by Zhou and Pfaltz.25 Halogen–metal exchange using t-BuLi followed by the addition of selenium powder yielded the lithiated selenolate intermediates 36 and 37. Subsequent alkylation with cinnamyl bromide provided the required selenides 38 and 39.
Scheme 4.
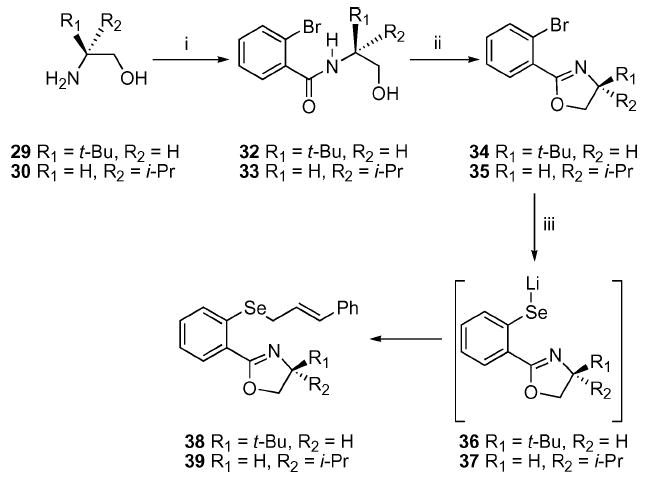
Synthesis of the iso-propyl and tert-butyl oxazoles. i. 2-Br–C6H4COCl (31), Et3N, CH2Cl2; ii. SOCl2, CH2Cl2, 50–74%; iii. t-BuLi, THF, −78 °C; Se powder, −78 to 0 °C then E-PhCH=CHCH2Br, 61–73%.
With the oxazoles 38–40 in hand, the ASOS reaction was explored under the standard VO(acac)2 conditions (Table 3). We were pleased to find a direct correlation between the amount of steric bulk on the oxazole and the level of diastereomeric excess. In addition, these oxazole substrates 38–40 proceeded at −50 °C, unlike the o-NO2 selenides 5g and 15 in the ligand-based series which typically required temperatures of −30 °C to proceed at a reasonable rate. The (S)-tert-leucinol-based oxazole 38 gave the highest level (70 : 30 d.r.) of selectivity, which is the result of net-1,9 asymmetric induction. It is important to note that treatment of the t-butyl oxazole 38 with m-CPBA, the standard oxidant for most previous examples of the ASOS reaction reported in the literature,12-14 yielded little diastereoselectivity (52 : 48 d.r.). It was clear that the vanadium system was able to provide a significant improvement over previous oxidation systems. Based on these encouraging levels of induction, the construction of more sterically demanding oxazole systems was warranted.
Table 3.
Selected examples of amino alcohol derived oxazoles in the vanadium-catalyzed ASOS reaction
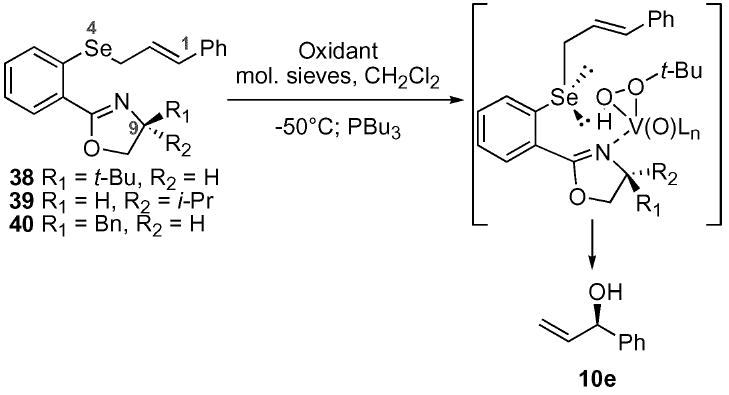
| Entry | Selenide | Oxidant | Yield | d.r.b |
|---|---|---|---|---|
| a | 40a | VO(acac)2/TBHP | 78% | 55 : 45 (R) |
| b | 39 | VO(acac)2/TBHP | 56% | 60 : 40 (S) |
| c | 38 | VO(acac)2/TBHP | 52% | 70:30 (R) |
| d | 38 | m-CPBA | 41% | 52 : 48 |
Selenide 40 was prepared via a two step protocol starting from the commercially available amino alcohol and 2-bromobenzonitrile in the presence of zinc chloride in chlorobenzene at 140 °C followed by the standard halogen–metal exchange protocol described for selenides 38 and 39.
The d.r. was determined by Mosher ester analysis using 1H and/or 19F NMR.
The corresponding camphor-based oxazoles were constructed from the known exo and endo amino alcohols 41 and 42 26 (Scheme 5). Formation of the amide linkage was accomplished in an analogous fashion to the previous series. Subsequent cyclization to the required oxazole, however, required an alternative approach. Fortunately, treatment of the amides 43 and 44 with TFA in benzene at elevated temperatures cleanly provided the cyclized products 45 and 46. Subsequent halogen–metal exchange followed by incorporation of the allylic selenides 47 and 48 proceeded in 17–27% unoptimized yield.
Scheme 5.
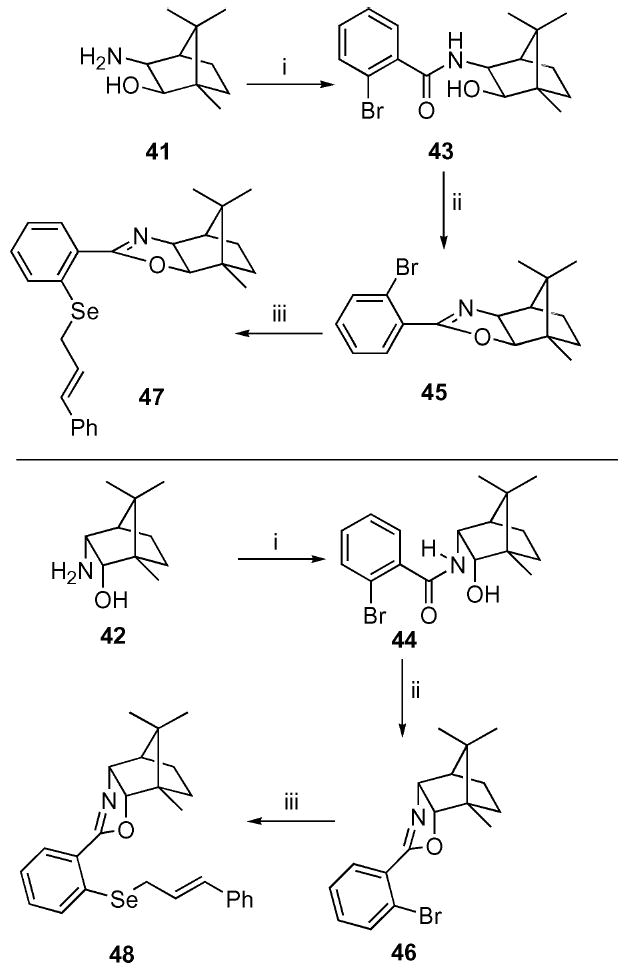
Synthesis of the camphor-based oxazoles. i. 2-Br–C6H4COCl (31), Et3N, CH2Cl2, 60%; ii. TFA, PhH, 140 °C, 50–81%; iii. t-BuLi, THF, −78 °C; Se powder, −78 to 0 °C then E-PhCH=CHCH2Br, 17–27%.
Submission of the camphor-based selenides 47 and 48 to the standard vanadium-catalyzed ASOS reaction conditions yielded informative results (Table 4). We were surprised to discover that both the exo and the endo camphor-based oxazoles performed poorly in the vanadium-catalyzed ASOS reaction. The diastereomeric ratios for selenides 47 and 48 were significantly below the levels previously observed with the t-butyl oxazole 38 (70 : 30 d.r.). One possible explanation can be found in the fact that these selenides did not readily undergo oxidation at −50 °C. Only after warming the reaction an additional 10 to 15 °C was a sluggish rate observed. This apparent decrease in rate could potentially be explained by the inability of the vanadium/oxidant complex to preorganize with the oxazole prior to selenide oxidation. One option to encourage this preorganization of the vanadium/oxidant complex to the oxazole would be the incorporation of an additional Lewis base(s) on the oxazole ring system. This multidentate system should more tightly associate with the metal, thereby providing a better opportunity for transfer of the chiral information.
Table 4.
Synthesis of the camphor-based oxazoles
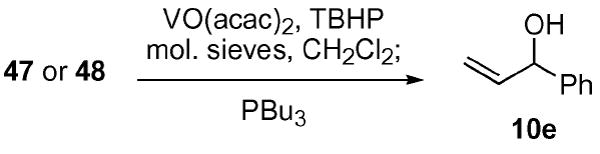
| Entry | Selenide | Temperature | Yield | d.r.(R : S)a |
|---|---|---|---|---|
| a | 47 | −35 °C | 50% | 53 : 47 |
| B | 48 | −40 °C | 57% | 43 : 57 |
The d.r. was determined by Mosher ester analysis using 1H and/or 19F NMR.
A serine-based oxazole would appear ideally suited to provide an additional Lewis-base for coordination (Scheme 6). The required oxazole was readily available from d- or l-serine methyl ester. The amide linkage was constructed as described previously27 to provide 50. Silylation of the free hydroxyl function followed by reduction yielded the alcohols 51 and 52. Oxazole formation followed by halogen–metal exchange with allylic selenide incorporation yielded the silyl protected selenides 53 and 54. Removal of the silyl ethers was best accomplished under buffered TBAF conditions to yield the free alcohol 55. Unbuffered TBAF conditions led to a considerable decrease in yield. The free alcohol 55 was also methylated to provide an additional selenide substrate for screening.
Scheme 6.
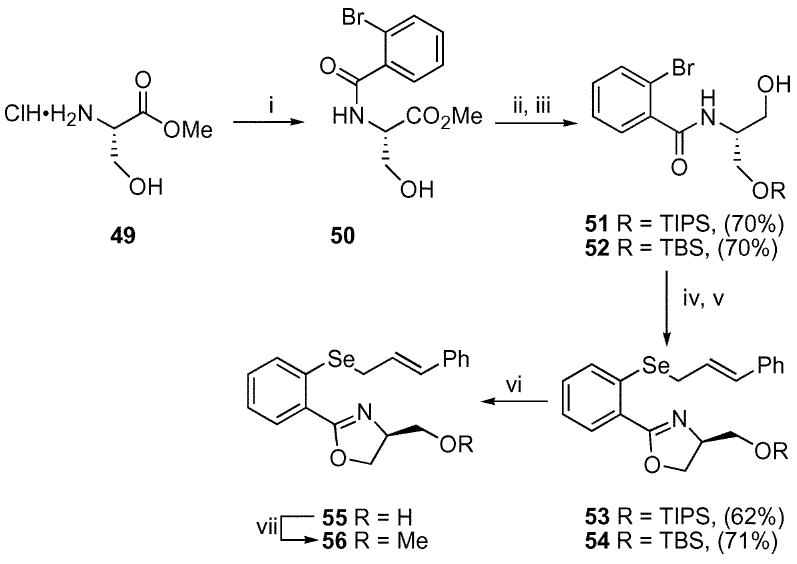
Synthesis of the serine-based oxazoles. i. 2-Br–C6H4COCl (31), Et3N, CH2Cl2, 72%; ii. TBSCl or TIPSOTf, Et3N, DMAP, CH2Cl2, 70%; iii. LiAlH4, THF, 0 °C, 62–71%; iv. TsCl, DMAP, Et3N, CH2Cl2, 70–72%; v. t-BuLi, THF, −78 °C; Se powder, −78 to 0 °C then E–PhCH=CHCH2Br, 62–71%; vi. TBAF, HOAc, THF, 76–94%; vii. MeI, NaH, THF, 82%.
The structure of selenide 55 was conclusively established by X-ray crystal analysis (Fig. 1).¶ It is interesting to note that the oxygen, not the nitrogen, appears to interact with the selenium atom (2.709 Å Se–O distance). To the best of our knowledge, this compound represents the first example of a selenium–oxygen nonbonding interaction in oxazole-containing selenides.28 The free hydroxyl participates in intermolecular hydrogen bonding with the oxazole nitrogen of another molecule, thereby blocking any nonbonding interaction between the nitrogen and selenium. Interestingly, 77Se NMR of compounds 55 and 54 show a small difference between each other is (349.3 ppm for 55, 354.4 ppm for 54 in CDCl3 versus 335.8 ppm for PhSeAllyl and 387.2 ppm for 9d) which indicates there may be a change in the nature of the interaction between the oxazole and the selenide. In comparison, 77Se NMR chemical shifts of other oxazole-containing selenides tend to fall between 300 and 425 ppm.28b
Fig. 1.
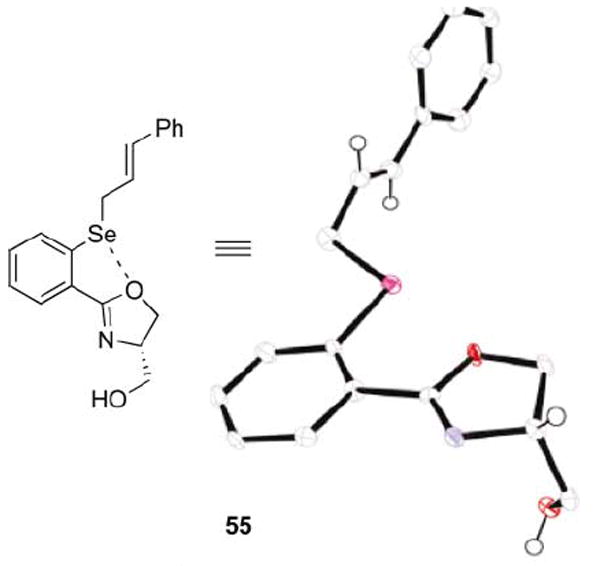
ORTEP representation of the X-ray data for the serine-derived selenide 55.
The selenides 53, 55 and 56 were screened for utility in the vanadium-catalyzed ASOS reaction (Table 5). Silyl ether 53 provided low levels of induction, which is consistent with the TIPS ether’s inability to chelate.29 We were gratified to discover that the hydroxyl containing selenide 55 yielded an increase in diastereoselectivity (80 : 20 d.r.). Further support for the chelation model can be found in a reversal in the absolute configuration of the resultant alcohol 10e versus the selenide 38. These results support a model involving chelation of both the oxazole nitrogen and the free hydroxyl to the metal/oxidant complex, thereby situating the oxidant complex in the opposite face relative to the selenide 38. Williams and co-workers put forth a similar model to explain their stereochemical outcome in the sulfide oxidation series (Scheme 3). It is also interesting to note that 80 : 20 d.r. is considerably higher than the level of selectivity observed by Davis and co-workers using the chiral oxaziradine oxidants on their E-allylic selenide (70 : 30 e.r.).10 Interestingly, submission of the methyl ether 56 to the standard vanadium-catalyzed system yielded essentially no diastereoselectivity. It would appear that the free hydroxyl function is required for selectivity.
Table 5.
Synthesis of the camphor-based oxazoles
| Entry | Selenide | Yield | d.r.(R : S) a |
|---|---|---|---|
| a | 53 | 52% | < 45 : 55 |
| b | 55 | 63% | 80:20(S) |
| c | 56 | 50% | < 45 : 55 |
The d.r. was determined by Mosher ester analysis using 1H and/or 19F NMR.
In order to ensure that the vanadium-catalyzed system continued to be the optimum protocol for accomplishing the ASOS reaction, a series of alternative oxidant systems were screened (Table 6). As shown in Table 6, all other systems screened led to diminished levels of selectivity versus the vanadium-catalyzed protocol. It is particularly interesting to note that the titanium-based catalyst performed poorly in the ASOS reaction. The result is in stark contrast to the analogous sulfide systems by Williams in which the titanium-based system provided superior results.24 In addition, m-CPBA, the oxidant of choice for the majority of previously reported auxiliary-based ASOS systems, yielded comparable levels of diastereoselectivity to the titanium system.
Table 6.
Variation of oxidant system for ASOS reaction
| Entry | Oxidant system | Yield | d.r. (S : R) b |
|---|---|---|---|
| a a | Ti(Oi–Pr)4 (10 mol%) | 47% | 65 : 35 |
| b | Ti(Oi–Pr)4 (100 mol%) | 60% | 65 : 35 |
| c | MoO2(acac)2 (10 mol%) | 53% | 62 : 38 |
| d | Zr(acac)2 (10 mol%) | 35% | 67 : 33 |
| e | Mn(acac)2 (10 mol%) | 19% | 67 : 34 |
| f | m-CPBA (10 mol%) | 63% | 63 : 37 |
| g | VO(acac)2 (10 mol%) | 63% | 80 : 20 |
This reaction was performed at −50 °C to −25 °C as little to no reaction was observed at lower temperatures.
The d.r. was determined by Mosher ester analysis using 1H and/or 19F NMR.
Due to the increased level of diastereoselectivity observed with the serine-based series, we sought to study the effect of an additional stereogenic center located on the hydroxyl function. Using an analogous approach as described previously, the threonine-derived oxazole was prepared (Scheme 7). Silyl deprotection of 58 using buffered TBAF conditions yielded the desired alcohol 59. The epimeric hydroxyl function 60 was readily accessible via Mitsunobu inversion with p-NO2 benzoic acid 30 followed by transesterification.
Scheme 7.
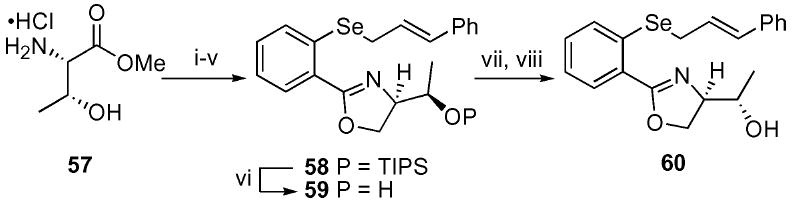
Synthesis of the serine-based oxazoles. i. 2-Br–C6H4COCl (31), Et3N, CH2Cl2, 58%; ii. TIPSOTf, Et3N, CH2Cl2, 62%; iii. LiAlH4, THF, 0 °C, 71%; iv. TsCl, DMAP, Et3N, CH2Cl2, 63%; v. t-BuLi, THF, −78 °C; Se powder, −78 to 0 °C then E–PhCH=CHCH2Br, 54%; vi. TBAF, HOAc, THF, 78%; vii. DEAD, p-NO2-C6H4CO2H, PPh3, THF, 80%; viii. K2CO3, MeOH, 50%.
The threonine-based oxazoles 58–60 were screened in accord with the established vanadium-catalyzed protocol (Table 7). Once again, the silyl protected system 58 yielded low levels of diastereoselectivity. Interestingly, the free alcohol 59, possessing the natural configurations of the amino acid, resulted in a decrease in selectivity versus the serine series. We were gratified to find, however, that the epimeric alcohol 60 lead to a further modest increase in diastereoselectivity to 83 : 17 d.r. It became apparent that the second stereogenic center, located in a net-1,10 relationship, may be able to facilitate the transfer of additional levels of chiral information.
Table 7.
Threonine-based oxazole in the ASOS reaction
| Entry | Selenide | Yield | d.r.a |
|---|---|---|---|
| a | 58 | 69% | 58 : 42 (R) |
| b | 59 | 74% | 75 : 25 (S) |
| c | 60 | 74% | 83 : 17 (S) |
The d.r. was determined by Mosher ester analysis using 1H and/or 19F NMR.
While the amino acid l-threonine did provide access to the oxazole possessing additional stereochemical information on the hydroxyl function, the route for the construction of the selenide oxazoles such as 59 and 60 had become lengthy. An alternate approach to the construction of oxazoles possessing this type of functionality needed to be developed in order to study the variation of the nature of the substitution on the hydroxyl function.
The previously described serine-derived alcohol 50 would appear to be the ideal starting point for a second generation synthetic strategy (Scheme 8). One important advantage to this starting point is the commercial availability of both enantiomers (d- or l- serine methyl ester) for a reasonable price. Cyclization using DAST31 readily provided the desired oxazole in high yield. While the reduction of the similar oxazole ester functionality has been reported in the literature,32 the resultant aldehyde is notoriously unstable. An alternative approach would involve selective monoaddition of an appropriately chosen nucleophile to the ester. While esters do not normally undergo the desired process, it was hoped that the neighboring oxazole nitrogen might help to accomplish this goal.33 Subsequent reduction of the resultant ketone would provide access to a wide range of oxazole possessing the desired substitution pattern. We were gratified to find that addition of i-PrMgCl to the ester provided the desired mono addition product 62 in 74% yield. Subsequent reduction using NaBH4 in MeOH provided both epimeric alcohols in a nearly equal (58 : 42) ratio (63 : 64). These alcohols were readily separable and both epimers were carried through the remaining sequence individually to provide selenides 65 and 66. The stereochemistry of 63 and 64 was conclusively established by X-ray crystal analysis of 66.¶
Scheme 8.
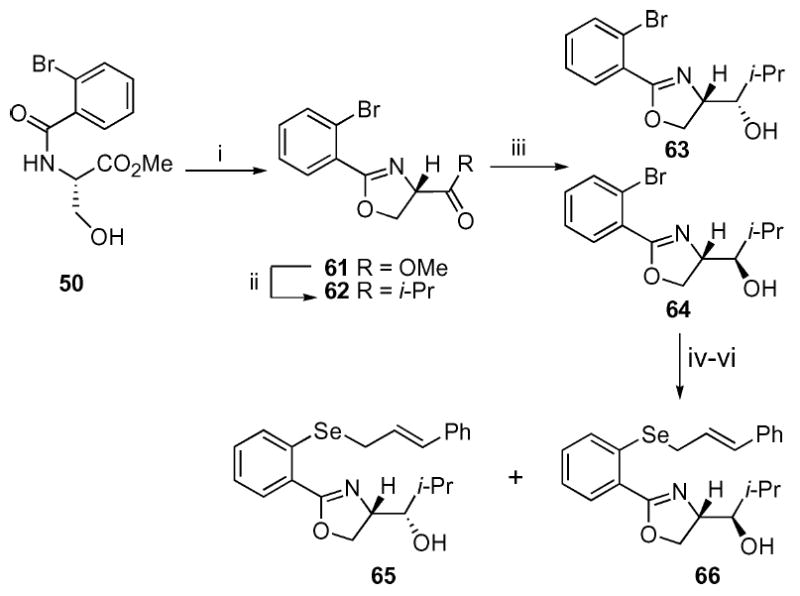
Synthesis of the second generation serine-based oxazoles. i. DAST, CH2Cl2; K2CO3, −78 °C, 91%; ii. i-PrMgCl, THF, −78 °C, 74%; iii. NaBH4, MeOH, 0 °C, 58% (63), 42% (64); iv. TBSOTf, 2,6-lutidine, CH2Cl2, 0 °C, 94% (from 63), 62% (from 64); v. t-BuLi, THF, −78 °C; Se powder then E–PhCH=CHCH2Br, 71% (from 63), 74% (from 64); vi. TBAF, AcOH, CH2Cl2, rt, 53% (65), 60% (66)
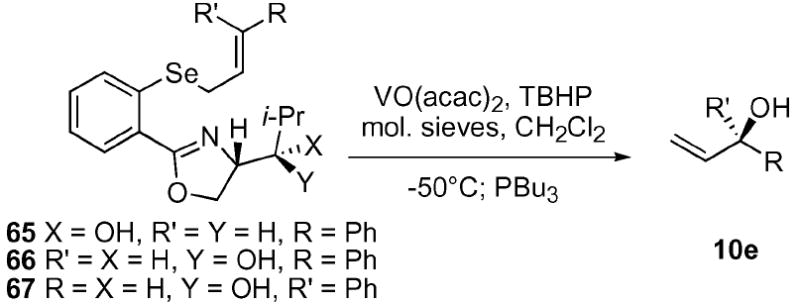
We were gratified to discover that the added substitution on the oxazole did provide a further increase in diastereoselectivity (Table 8). The anti-alcohol selenide 66 yielded an 85 : 15 d.r. while the syn-alcohol selenide 65 provided a still significant (81 : 19 d.r.) level of chiral induction. These results are consistent with the threonine-derived series and point to the modest importance of the stereogenic center on the 2° alcohol despite its remote distance (ten atoms away) from the resultant allyl alcohol. Finally, we constructed the corresponding Z-alkene substrate 6734 and submitted the substrate to our standard reaction conditions. Interestingly, this compound provided inferior results, with only a modest 71 : 29 d.r. observed. This result differs from previous work by several laboratories in which the Z-alkene provided superior results.9-11
Table 8.
Second generation serine-based oxazoles in the ASOS reaction
| Entry | Selenide | Yield | d.r.a |
|---|---|---|---|
| a | 65 | 61% | 81 : 19 (R) |
| b | 66 | 71% | 85 : 15 (R) |
| c | 67 | 58% | 71 : 29 (S) |
The d.r. was determined by Mosher ester analysis using 1H and/or 19F NMR.
A possible mechanistic picture is proposed in Scheme 9 to explain the observed selectivity. A bidentate complex 68/69 is formed which directs oxidation of the pro-S lone pair on selenium to provide the selenoxide 70/71. Given Davies and co-workers’ finding that the maximum level of induction possible with a 1,2-disubstituted E-alkene from a chiral selenoxide to the resultant chiral alcohol 10e is 70 : 30 e.r. (40% chiral transfer),10 it is reasonable to assume that the vanadium/oxazole complex interacts with the intermediate selenoxide in a complementary fashion to increase the observed level of chirality transfer. The exocyclic transition states 70/71-exo should be disfavored by the unfavorable interaction with the bicyclic vanadyl moiety. A similar argument could be put forth to explain why a decrease in selectivity is observed with the Z-cinnamyl selenide 67 (via72-endo). It should be noted that the effect of the relative stereochemistry at C9,10 is complicated by the fact that two chirality transfer steps are occurring in the same reaction. It is possible that the alternate relative stereochemistry at these positions (e.g.59 and 65) may not work at the same synergistic level as the optimum substrates 60 and 66. In fact, the oxidation step may proceed with greater (or reduced) levels of chirality transfer.
Scheme 9.
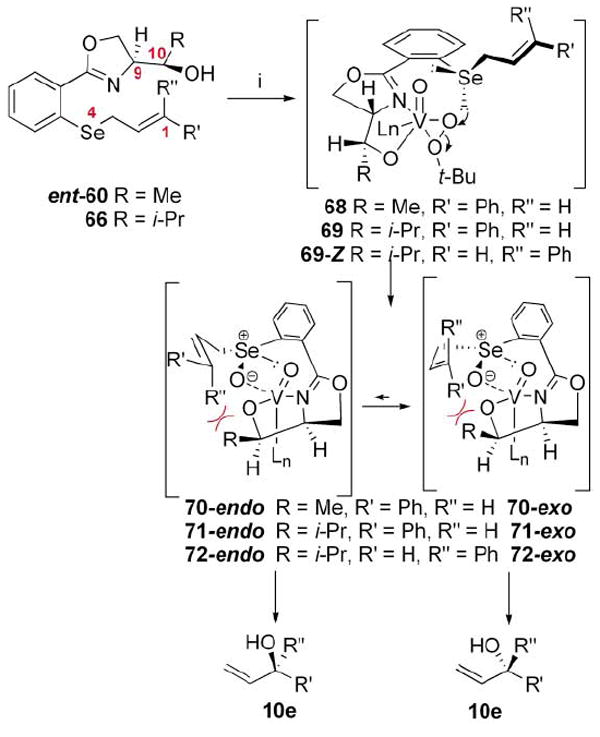
Possible mechanistic cartoon for vanadium-catalyzed ASOS reaction. i. VO(acac)2, TBHP, CH2Cl2, mol. sieves, −50 °C; PBu3.
Conclusion
A catalytic, metal-based SOS reaction has been developed using only 10 mol% of VO(acac)2 with either CHP or TBHP as a co-oxidant. These reactions proceed rapidly and cleanly to provide the resultant allylic alcohol in good yield. A broad spectrum of aryl, allylic selenides have been screened to demonstrate the generality of this methodology.
Considerable progress has been made toward the development of an effcient, vanadium-catalyzed ASOS reaction using a chiral oxazole-based approach. The optimum observed level of diastereomeric excess (85 : 15 d.r. for selenide 66) is among the highest values for comparable auxiliary-based, trans-cinnamyl systems.12 The chirality transfer observed in the product allyl alcohol is the result of a net 1,9- and/or 1,10-induction. A possible mechanistic cartoon is proposed to explain the observed selectivity.
It should be noted that the recovery of the chiral auxiliary from substrates such as selenide 66 should be possible. Treatment of the diselenide by-product produced in the tandem sequence with a reducing agent, such as NaBH4,should regenerate the necessary nucleophile for alkylation with the corresponding allyl halide. While this avenue was not explored during the course of this study, other laboratories have demonstrated its feasibility.11 Finally, this auxiliary-based approach should lay the foundation for the development of a ligand-based, vanadium-catalyzed ASOS reaction.
Experimental
Melting points were taken on a Mel-Temp apparatus and are uncorrected. Infrared spectra were recorded on a Perkin-Elmer paragon 500 FT-IR spectrophotometer neat unless otherwise indicated. 1H NMR spectra were recorded on a Bruker 300 spectrometer at 300 MHz or a Bruker 500 spectrometer at 500 MHz in the indicated solvent and are reported in ppm relative to trimethylsilane and referenced internally to the residually protonated solvent. 13C NMR spectra were recorded on a Bruker 300 spectrometer at 75 MHz or a Bruker 500 spectrometer at 125 MHz in the solvent indicated and are reported in ppm relative to trimethylsilane and referenced internally to the residually protonated solvent. 77Se NMR spectra were recorded on a Bruker 400 spectrometer at 76 MHz in CDCl3 and are reported in ppm relative to dimethylselenide and referenced internally to the added dimethylselenide in the solution. Optical rotations were determined on a Rudolph Research Analytical Autopol III automatic polarimeter using a sodium lamp at 589 nm in CHCl3.
Routine monitoring of reactions was performed using EM Science DC-Alufolien silica gel, aluminium back TLC plates. Flash chromatography was performed with the indicated eluents on Silicycle 230–400 mesh silica gel.
Air and/or moisture sensitive reactions were performed under usual inert atmosphere conditions. Reactions requiring anhydrous conditions were performed under a blanket of argon, in glassware dried in an oven at 120 °C or by a bunsen flame, then cooled under argon. Solvents and commercial reagents were purified in accord with Perrin and Armarego 35 or used without further purification.
(4S)-tert-Butyl-2-[2-(3-phenyl-allylselanyl)-phenyl]-4,5-dihydro-oxazole (38)
To a stirred solution of oxazole 34 (122 mg, 0.43 mmol) in THF (6.5 mL) at −78 °C was added t-BuLi (0.4 mL, 0.65 mmol, 1.7 M in pentane) dropwise over 20 min resulting in an orange color. After 1 h, Se powder (34.2 mg 0.43 mmol) was added in 1 portion under an inverse argon funnel and warmed to 0 °C. After 1 h or until the slurry was homogeneous, cinnamyl bromide (85 mg, 53 μL, 0.43 mmol) was added. After an additional 1 h, the reaction was quenched with sat. aq. NH4Cl (5 mL) and the product was extracted with Et2O (3 × 50 mL). The dried (MgSO4) organic layer was concentrated in vacuo and the residue was purified by chromatography on silica gel eluting with 2–20% EtOAc/hexane. The final product was recrystallized in hexane to yield 38 (105 mg, 61%) as a white solid: mp 138–141 °C; [α]D 23 −77.9 (c 1.00, CHCl3); IR (neat) 2959, 1644, 1353, 957 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.82 (d, J = 7.7 Hz, 1H), 7.48 (d, J = 8.0 Hz, 1H), 7.17–7.37 (m, 7H), 6.83 (d, J = 15.7 Hz, 1H), 6.42 (dt, J = 7.4, 15.7 Hz, 1H), 4.28–4.34 (m, 1H), 4.14–4.23 (m, 2H), 3.71 (d, J = 7.2 Hz, 2H), 0.99 (s, 9H); 13C NMR (75 MHz, CDCl3) δ 162.7, 137.2, 136.8, 133.1, 130.9, 130.3, 128.7, 128.1, 127.6, 126.7, 126.5, 125.5, 124.7, 76.8, 68.4, 34.2, 28.8, 26.2; HRMS (FAB+) calcd. for C22H26NO80Se (M + H+) 400.1180. Found 400.1172.
(4R)-Isopropyl-2-[2-(3-phenyl-allylselanyl)-phenyl]-4,5-dihydro-oxazole (39)
To a stirred solution of t-BuLi (4.12 mL, 7.0 mmol, 1.7 M in pentane) in THF (21 mL) at −78 °C was added the bromide 35 (0.94 g, 3.50 mmol) in THF (11 mL) dropwise via syringe pump over 30 min resulting in an orange solution. The benzamide conical vial was further rinsed with an additional amount of THF (2 mL). After 1 h, Se powder (290 mg, 3.7 mmol) was added under an inverse argon funnel and the reaction was then warmed to 0 °C. After 1 h or until the solution was homogeneous, cinnamyl bromide (723 mg, 0.53 mL, 3.70 mmol) was added. After 1 h, the reaction was quenched with sat. aq. NH4Cl (5 mL) and extracted with Et2O (3 × 75 mL). The dried (MgSO4) organic layer was concentrated in vacuo and the residue was purified by chromatography on silica gel, eluting with 2–20% EtOAc/hexane. The final product was recrystallized in hexane to give 39 (981 mg, 73%) as a white solid: mp 122–125 °C; [α]D 23 +67.6 (c 1.00, CHCl3); IR (neat) 2955, 1645, 1363, 1251 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.82 (dd, J = 1.6, 7.8 Hz, 1H), 7.48 (d, J = 8.1 Hz, 1H), 7.18–7.37 (m, 7H), 6.61 (d, J = 15.7 Hz, 1H), 6.40 (dt, J = 7.4, 15.7 Hz, 1H), 4.39 (dd, J = 7.8, 8.9 Hz, 1H), 4.20 (dd, J = 7.8, 14.7 Hz, 1H), 4.10 (dd, J = 7.6, 15.3 Hz, 1H), 3.71 (d, J = 7.5 Hz, 2H), 1.80–1.84 (m, 1H), 1.08 (d, J = 6.7 Hz, 3H), 0.97 (d, J = 6.7 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 161.4, 137.1, 136.5, 133.1, 130.9, 130.2, 128.7, 128.1, 127.6, 126.4, 125.4, 124.7, 77.2, 73.6, 70.2, 33.4, 28.8, 19.3, 18.8; HRMS (FAB+) calcd. for C21H24NO80Se (M + H+) 386.1023. Found 386.1024.
(4S)-Benzyl-2-[2-(3-phenyl-allylselanyl)-phenyl]-4,5-dihydro-oxazole (40)
To a stirred solution of t-BuLi (1.18 mL, 2.00 mmol, 1.7 M in pentane) in THF (6 mL) at −78 °C was added a solution of the known oxazole 34a25 (311 mg, 1.00 mmol) in THF (3 mL), dropwise via syringe pump over 20 min resulting in an orange solution. After 1 h, Se powder (79 mg, 1.00 mmol) was added under an inverse argon funnel and the reaction was warmed to 0 °C. After 1 h or until the mixture was homogeneous, cinnamyl bromide (196.7 mg, 0.15 mL, 1 mmol) was added. After an additional 1 h, the reaction was quenched with sat. aq. NH4Cl (10 mL) and extracted with Et2O (3 × 75 mL). The dried (MgSO4) organic layer was concentrated in vacuo and the residue was purified by chromatography on silica gel, eluting with 2–20% EtOAc/hexane to yield 40 (46.3 mg, 10%) as a pale yellow oil: IR (neat) 2959, 1643; 1589 cm−1; 1H NMR (CDCl3, 300 MHz) δ 7.80 (d, J = 7.8 Hz, 1H), 7.48 (d, J = 7.9 Hz, 1H), 7.18–7.37 (m, 12H), 6.62 (d, J = 15.6 Hz, 1H), 6.42 (dt, J = 7.4, 15.2 Hz, 1H), 4.68 (m, 1H), 4.31 (dd, J = 8.7, 8.7 Hz, 1H), 4.08–4.15 (m, 1H), 3.73 (d, J = 7.3 Hz, 2H), 3.29 (dd, J = 5.4, 13.7 Hz, 1H), 2.78 (dd, J = 8.6, 13.6 Hz, 1H).
2-Bromo-N-{(3R)-hydroxy-4R,7,7-trimethyl-bicyclo[2.2.1]hept-(2S)-yl}-benzamide (43)
To a stirred solution of amino alcohol 4126 (808.0 mg, 4.78 mmol) in CH2Cl2 (9 mL) at 0 °C were sequentially added DMAP (146.0 mg, 1.20 mmol) and Et3N (627.1 mg, 0.86 mL). After 10 min, a solution of 2-bromobenzoyl chloride 31 (1.05 g, 0.63 mL, 4.00 mmol) in CH2Cl2 (4.8 mL) was added dropwise via syringe pump over 20 min. After an additional 30 min, the reaction was warmed to rt. After 2 h, the reaction was quenched with sat. aq. NH4Cl (25 mL) and extracted with Et2O (3 × 75 mL). The organic layer was sequentially washed with aq. HCl (5%, 50 mL) and sat. aq. NaHCO3 (100 mL). The dried (MgSO4) organic layer was concentrated in vacuo and the residue was purified by chromatography on silica gel, eluting with 2–40% EtOAc/hexane to give 43 (842 mg, 60%) as a light pink solid: mp 169–170 °C; [α]D 23 −3.1 (c 0.72, CHCl3); IR (neat) 3381; 1651, 1506 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.58 (d, J = 7.9 Hz, 1H), 7.52 (dd, J = 1.5, 7.7 Hz, 1H), 7.23–7.37 (m, 2H), 6.67 (br s, 1H), 4.00 (dd, J = 6.8, 13.4 Hz, 1H), 3.91 (dd, J = 7.6, 4.14 Hz, 1H), 2.17 (br s, 1H), 1.99 (d, J = 4.4 Hz, 1H), 1.27–1.57 (m, 4H), 1.23 (s, 3H), 1.21 (s, 3H), 1.15 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 167.7, 138.3, 133.6, 131.3, 129.6, 127.6, 119.6, 80.1, 58.6, 50.4, 49.4, 47.1, 33.4, 26.4, 21.7, 21.4, 11.5; HRMS (FAB+) calcd. for C17H23NO279Br (M + H+) 352.0912. Found 352.0922.
4-(2-Bromo-phenyl)-1,10,10-trimethyl-(3R)-oxa-(5S)-aza-tricyclo[5.2.1.0]dec-4-ene (45)
To a stirred solution of alcohol 43 (137.9 mg, 0.393 mmol) in benzene (1.1 mL) was added TFA (9.1 mg, 6.0 μL, 0.08 mmol) and powdered 4 Å mol. sieves (50 mg). After 10 min, the reaction was heated to 140 °C in a sealed tube. After 8 h, the reaction was cooled to rt and quenched with sat. aq. NaHCO3 (10 mL) and extracted with Et2O (3 × 50 mL). The dried (MgSO4) organic layer was concentrated in vacuo and the residue was purified by chromatography on silica gel, eluting with 2–25% EtOAc/hexane to give 45 (65 mg, 50%) as a white solid: mp 142 °C; [α]D 23 +10.9 (c 1.3, CHCl3); IR (neat) 2950; 1651, 1506 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.58–7.80 (m, 2H), 7.26–7.38 (m, 2H), 4.43 (d, J = 8.6 Hz, 1H), 4.24 (d, J = 8.6 Hz, 1H), 2.20 (d, J = 4.5 Hz, 1H), 1.65–1.85 (m, 1H), 1.45–1.60 (m, 1H), 0.95–1.10 (m, 2H), 1.07 (s, 3H), 1.01 (s, 3H), 0.87 (s, 3H);13C NMR (75 MHz, CDCl3) δ 164.1, 134.4, 131.7, 131.5, 129.9, 127.2, 122.0, 91.7, 77.0, 49.0, 48.9, 47.3, 32.3, 26.2, 23.6, 19.0, 11.6; HRMS (FAB+) calcd. for C17H21NO79Br (M + H+) 334.0807. Found 334.0813.
1,10,10-Trimethyl-4-[2-(3-phenyl-allylselanyl)-phenyl]-(3R)-oxa-(5S)-aza-tricyclo[5.2.1.0]dec-4-ene (47)
To a stirred solution of t-BuLi (1.18 mL, 2.00 mmol, 1.7 M in pentane) in THF (6.3 mL) at −78 °C was added a solution of benzamide 45 (328 mg, 0.99 mmol) in THF (3 mL) dropwise via syringe pump over 10 min. The benzamide conical vial was further rinsed with an additional amount of THF (1.5 mL). After 1 h, Se powder (79 mg, 1.00 mmol) was added under an inverse argon funnel and the reaction was warmed to 0 °C. After 1 h or until the solution was homogeneous, cinnamyl bromide (200 mg, 0.15 mL, 1.00 mmol) was added to the reaction. After 1 h, the reaction was quenched with sat. aq. NH4Cl (15 mL) and extracted with Et2O (3 × 75 mL). The dried (MgSO4) organic layer was concentrated in vacuo and purified by chromatography on silica gel, eluting with 2–20% EtOAc/hexane. The final product was recrystallized in hexane to give 47 (122 mg, 27%) as a pale yellow solid: mp 155–160 °C; [α]D 23 +5.9 (c 2.76, CHCl3); IR (neat) 2955, 1641 cm −1; 1H NMR (300 MHz, CDCl3) δ 7.82 (dd, J = 1.1, 7.7 Hz, 1H), 7.45 (d, J = 8.0 Hz, 1H), 7.17–7.36 (m, 7H), 6.61 (d, J = 15.7 Hz, 1H), 6.41 (dt, J = 7.5, 15.7 Hz, 1H), 4.37 (d, J = 8.4 Hz, 1H), 4.31 (d, J = 8.4 Hz, 1H), 3.71 (d, J = 7.4 Hz, 2H), 2.20 (d, J = 4.5 Hz, 1H), 1.70–1.90 (m, 1H), 1.50–1.65 (m, 1H), 0.95–1.10 (m, 2H), 1.11 (s, 3H), 1.09 (s, 3H), 0.89 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 164.6, 137.1, 136.6, 133.2, 131.0, 130.5, 128.7, 128.0, 127.6, 127.0, 126.4, 125.1, 124.7, 91.0, 77.0, 49.2, 48.8, 47.2, 32.3, 28.7, 26.2, 23.7, 18.9, 11.7; HRMS (FAB+) calcd. for C26H30NO80Se(M + H+) 452.1493. Found 452.1486.
2-Bromo-N-[(3S)-hydroxy-4,7,7-trimethyl-bicyclo[2.2.1]hept-(2R)-yl)]-benzamide (44)
To a stirred solution the amino alcohol 42 26 (890.1 mg,5.23 mmol) in CH2Cl2 (10 mL) at 0 °C was added Et3N (700 mg, 0.951 mL, 6.85 mmol) and DMAP (160 mg, 1.30 mmol). After 10 min, a solution of 2-bromobenzoyl chloride 31 (1.25 g, 0.69 mL, 5.30 mmol) in CH2Cl2 (5 mL) was added dropwise via syringe pump over a period of 20 min and warmed to rt. After 2 h, the reaction was quenched with sat. aq. NH4Cl (10 mL) and extracted with Et2O (3 × 75 mL). The organic layer was sequentially washed with aq. HCl (50 mL, 5%) and sat. aq. NaHCO3 (100 mL). The dried (MgSO4) organic layer was concentrated in vacuo and the residue was purified by chromatography on silica gel, eluting with 3–40% EtOAc/hexane to give 44 (1.53 g, 81%) as a white solid: mp 92–95 °C; [α]D 23 −10.4 (c 1.29, CHCl3); IR (neat) 3381; 1651, 1506 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.23–7.59 (m, 4H), 6.79 (br s, 1H), 4.00 (dd, J = 6.8, 13.4 Hz, 1H), 3.91 (dd, J = 4.14, 7.6 Hz, 1H), 2.17 (br s, 1H), 1.27–1.57 (m, 5H), 1.23 (s, 3H), 1.21 (s, 3H), 1.15 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 167.7, 143.0, 133.6, 131.3, 129.6, 127.6, 119.6, 80.1, 58.6, 50.4, 49.4, 47.1, 33.4, 26.4, 21.7, 21.4, 11.5; HRMS (FAB+) calcd. for C17H23NO279Br (M + H+) 352.0912. Found 352.0911.
4-(2-Bromo-phenyl)-1,10,10-trimethyl-(3S)-oxa-(5R)-aza-tricyclo[5.2.1.0]dec-4-ene (46)
To a stirred solution of alcohol 44 (1.50 g, 4.32 mmol) in benzene (12 mL) at rt was added TFA (90 mg, 66 μL, 0.85 mmol) and powdered 4 Å mol. sieves (200 mg). After 10 min, the reaction was heated to 140 °C in a sealed tube. After 24 h, the reaction was cooled to rt and quenched with sat. aq NaHCO3 (40 mL) and extracted with Et2O (3 × 100 mL). The dried (MgSO4) organic layer was concentrated in vacuo and the residue was purified by chromatography on silica gel, eluting with 2–25% EtOAc/hexane to give 46 (587 mg, 41%) as a white solid: mp 110 °C; [α]D 23 −6.9 (c 0.85, CHCl3); IR (neat) 2950; 1651, 1506 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.79 (dd, J = 1.2, 7.7 Hz, 1H), 7.66 (dd, J = 1.2, 7.9 Hz, 1H), 7.26–7.38 (m, 2H), 4.77 (dd, J = 4.8, 9.8 Hz, 1H), 4.68 (dd, J = 1.5, 9.9 Hz, 1H), 2.19 (t, J = 4.2 Hz, 1H), 1.51–1.69 (m, 2H), 1.25–1.29 (m, 2H), 1.00 (s, 3H), 0.99 (s, 3H), 0.98 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 163.0, 134.2, 131.7, 131.6, 130.3, 127.2, 121.9, 89.4, 72.3, 49.7, 49.5, 49.4, 27.4, 20.9, 20.0, 18.6, 15.2; HRMS (FAB+) calcd. for C17H21NO79Br (M + H+) 334.0807. Found 334.0818.
1,10,10-Trimethyl-4-[2-(3-phenyl-allylselanyl)-phenyl]-(3S)-oxa-(5R)-aza-tricyclo[5.2.1.0]dec-4-ene (48)
To a stirred solution of t-BuLi (3.33 mL, 5.64 mmol, 1.7 M in pentane) in THF (18 mL) at −78 °C, was added a solution of benzamide 46 (938 mg, 2.82 mmol) in THF (8.5 mL) dropwise over 20 min resulting in an orange solution. The benzamide conical vial was further rinsed with an additional amount of THF (2 mL). After 1 h, Se powder (223 mg, 2.82 mmol) was added under an inverse argon funnel and the reaction was then warmed to 0 °C. After 1 h or until the solution was homogeneous, cinnamyl bromide (800 mg, 0.43 mL, 2.9 mmol) was added. After 1 h, the reaction was quenched with sat. aq. NH4Cl (20 mL) and extracted with Et2O (3 × 75 mL). The dried (MgSO4) organic layer was concentrated in vacuo and the residue was purified by chromatography on silica gel, eluting with 2–20% EtOAc/hexane. The final product was recrystallized in hexane to give 48 (215 mg, 17%) as a white solid: mp 154–155 °C; [α]D 23 −12.4 (c 1.19, CHCl3); IR (neat) 2949, 1635 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.92 (dd, J = 1.4, 7.7 Hz, 1H), 7.47 (d, J = 7.7 Hz, 1H), 7.20–7.38 (m, 7H), 6.61 (d, J = 15.7 Hz, 1H), 6.41 (dt, J = 7.5, 15.2 Hz, 1H), 4.81 (dd, J = 4.7, 9.7 Hz, 1H), 4.64 (dd, J = 1.6, 9.8 Hz, 1H), 3.71 (d, J = 7.4 Hz, 2H), 2.19 (t, J = 4.3 Hz, 1H), 1.40–1.60 (m, 3H), 1.10–1.35 (m, 1H), 1.00 (s, 6H), 0.96 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 163.2, 137.1, 136.5, 133.2, 130.9, 130.4, 128.7, 128.1, 127.6, 127.2, 126.4, 125.2, 124.7, 88.5, 72.4, 49.7, 49.5, 49.3, 28.8, 27.3, 21.0, 20.1, 18.6, 15.2; HRMS (FAB+) calcd. for C26H30NO80Se (M + H+) 452.1493. Found 452.1492.
(2S)-(2-Bromo-benzoylamino)-3-hydroxy-propionic acid methyl ester (50)
To a stirred solution of l-serine methyl ester hydrochloride (49) (2.11 g, 13.56 mmol) in CH2Cl2 (26 mL) at 0 °C, was added Et3N (3.2 g, 4.34 mL, 31.2 mmol) and DMAP (0.331 g, 2.71 mmol). After 10 min, a solution of 2-bromobenzoyl chloride (31) (3.30 g, 1.85 mL, 14.9 mmol) in CH2Cl2 (15 mL) was added dropwise to the amino alcohol solution via syringe pump over a period of 20 min. Next, the solution was warmed to rt. After 2 h, the reaction mixture was quenched with sat. aq. NH4Cl (50 mL) and extracted with Et2O (3 × 75 mL). The organic layer was sequentially washed with aq. HCl (50 mL, 5%) and sat. aq. NaHCO3 (100 mL). The dried (MgSO4) organic layer was concentrated in vacuo and purified by chromatography on silica gel, eluting with 2–100% EtOAc/hexane to give 50 (2.94 g, 72%) as a clear oil: [α]D 23 +21.2 (c 2.67, CHCl3); IR (neat) 3335, 1734, 1652 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.54–7.61 (m, 2H), 7.25–7.38 (m, 2H), 7.00 (d, J = 6.8 Hz, 1H), 4.80–4.90 (m, 1H), 4.06–4.14 (m, 2H), 3.83 (s, 3H), 1.24 (t, J = 7.0 Hz, 1H); 13C NMR (75 MHz, CDCl3) δ 170.7, 168.3, 137.0, 133.4, 131.6, 129.4, 127.5, 119.5, 62.4, 55.2, 52.7.
(2S)-(2-Bromo-benzoylamino)-3-triisopropylsilanyloxy-propionic acid methyl ester (73)
To a stirred solution of amino alcohol methyl ester 50 (2.68 g, 8.90 mmol) in CH2Cl2 (25 mL) at 0 °C, was added Et3N (2.07 g, 2.85 mL, 20.5 mmol). After 10 min, TIPSOTf (3.25 g, 2.9 mL, 10.6 mmol) was added. After 2 h, the reaction was quenched with sat. aq. NH4Cl (50 mL) and extracted with Et2O (3 × 100 mL). The dried (MgSO4) organic layer was concentrated in vacuo and the residue was purified by chromatography on silica gel, eluting with 2–20% EtOAc/hexane to give 73 (3.11 g, 70%) as a yellow oil: [α]D 23 +50.8 (c 0.86, CHCl3); IR (neat) 2942, 1748, 1652 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.58–7.63 (m, 2H), 7.21–7.34 (m, 2H), 6.97 (d, J = 8.0 Hz, 1H), 4.91 (dt, J = 2.8, 8.1 Hz, 1H), 4.28 (dd, J = 2.5, 9.9 Hz, 1H), 4.12 (dd, J = 2.9, 9.8 Hz, 1H), 3.79 (s, 3H), 1.00–1.10 (m, 21H); 13C NMR (75 MHz, CDCl3) δ 170.7, 167.2, 137.2, 133.7, 131.7, 129.9, 127.7, 119.6, 64.1, 55.1, 52.6, 18.0, 12.0; HRMS (FAB+) calcd. for C20H32Si79BrNO4 (M + H+) 458.1362. Found 458.1370.
2-Bromo-N-[(2R)-hydroxy-1-(triisopropyl-silanyloxymethyl)-ethyl-benzamide (51)
To a stirred solution of LiAlH4 (3.5 mL, 3.5 mmol, 1 M in Et2O) in THF (26 mL) at 0 °C was added a solution of benzamide 73 (1.50 g, 3.19 mmol) in THF (17.4 mL) dropwise via syringe pump over 30 min. The benzamide flask was rinsed with an additional amount of THF (2 mL). After 2 h, the reaction was quenched with a 1 : 1 mixture of EtOAc/H2O (20 mL) and extracted with EtOAc (3 × 75 mL). The dried (MgSO4) organic layer was concentrated in vacuo and the residue was purified by chromatography on silica gel, eluting with 2–50% EtOAc/hexane to give 51 (1.01 g, 71%) as a light pink oil: [α]D 23 +10.0 (c 4.48, CHCl3); IR (neat) 3419, 2942, 2865, 1651, 1635, 1539 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.55–7.61 (m, 2H), 7.26–7.39 (m, 2H), 6.78 (d, J = 7.3 Hz, 1H), 4.15–4.30 (m, 1H), 4.00 (d, J = 3.6 Hz, 2H), 3.98–4.00 (m, 1H), 3.84–3.90 (m, 1H), 2.9 (br s, 1H), 1.00–1.10 (m, 21H); 13C NMR (75 MHz, CDCl3) δ 167.9, 137.6, 133.6, 131.5, 129.8, 127.7, 119.4, 64.5, 63.9, 52.6, 18.1, 11.9; HRMS (FAB+) calcd. for C19H3279BrNO3Si (M + H+) 430.1413. Found 430.1421.
(2S)-(2-Bromo-benzoylamino)-3-(tert-butyl-dimethyl-silanyloxy)-propionic acid methyl ester (74)
To a stirred solution of amino alcohol methyl ester 50 (1.60 g, 5.32 mmol) in CH2Cl2 (11 mL) at 0 °C was added Et3N (1.13 g, 1.55 mL, 20.5 mmol) and DMAP (0.129 g, 1.06 mmol). After 10 min, TBSCl (1.20 g, 8.02 mmol) was added and warmed to rt. After 2 h, the reaction was quenched with sat. aq. NH4Cl (50 mL) and extracted with Et2O (3 × 100 mL). The dried (MgSO4) organic layer was concentrated in vacuo and the residue was purified by chromatography on silica gel, eluting with 1–20% EtOAc/hexane to give 74 (1.55 g, 70%) as a yellow oil: [α]D 23 +26.3 (c 0.50, CHCl3); IR (neat) 2360, 1748, 1652, 1506 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.58–7.63 (m, 2H), 7.21–7.34 (m, 2H), 6.97 (d, J = 7.8 Hz, 1H), 4.91 (dt, J = 2.8, 8.1 Hz, 1H), 4.18 (dd, J = 2.8, 10.2 Hz, 1H), 4.00 (dd, J = 2.7, 10.3 Hz, 1H), 3.79 (s, 3H), 0.87 (s, 9H), 0.04 (s, 6H); 13C NMR (75 MHz, CDCl3) δ 170.7, 167.2, 137.2, 133.7, 131.7, 130.0, 127.7, 119.6, 63.6, 55.0, 52.6, 25.8, 18.3, −5.2, −5.4; HRMS (FAB+) calcd. for C17H26BrSiNO4 (M + H+) 416.0893. Found 416.0903.
2-Bromo-N-[1-(tert-butyl-dimethyl-silanyloxymethyl)-(2R)-hydroxy-ethyl]-benzamide (52)
To a stirred solution of LiAlH4 (19.8 mL, 19.8 mmol, 1 M in Et2O) in THF (150 mL) at 0 °C was added a solution of benzamide 74 (7.45 g, 18.04 mmol) in THF (100 mL) dropwise via syringe pump over 30 min. The benzamide flask was rinsed with an additional amount of THF (5 mL). After 2 h, the reaction was quenched with a 1 : 1 mixture of EtOAc/H2O (100 mL) and extracted with EtOAc (3 × 75 mL). The dried (MgSO4) organic layer was concentrated in vacuo and the residue was purified by chromatography on silica gel, eluting with 2–80% EtOAc/hexane to give 52 (4.62 g, 62%) as a pink oil: [α]D 23 +15.7 (c 0.50, CHCl3); IR (neat) 3419, 2953, 1652, 1635, 1540 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.55–7.61 (m, 2H), 7.26–7.39 (m, 2H), 6.78 (br d, J = 7.3 Hz, 1H), 4.15–4.25 (m, 1H), 3.90–4.00 (m, 1H), 3.95 (d, J = 6.6 Hz, 2H), 3.80–3.90 (m, 1H), 2.85–2.95 (m, 1H), 0.89 (s, 9H), 0.10 (s, 3H), 0.08 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 167.9, 137.6, 133.6, 131.6, 129.9, 127.7, 119.4, 64.1, 63.9, 52.4, 26.0, 18.3, −5.2, −5.3.
2-(2-Bromo-phenyl)-(4R)-(triisopropyl-silanyloxymethyl)-4,5-dihydro-oxazole (75)
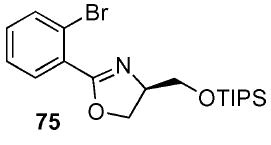
To a stirred solution of benzamide 51 (1.29 g, 3.01 mmol) in CH2Cl2 (10.3 mL) at 0 °C, was sequentially added DMAP (74 mg, 0.6 mmol) and Et3N (0.46 g, 0.63 mL, 4.58 mmol). After 10 min, TsCl (68 mg, 3.6 mmol) was added and the reaction was warmed to rt. After 12 h, the reaction was quenched with sat. aq. NH4Cl (50 mL) and extracted with Et2O (3 × 75 mL). The dried (MgSO4) organic layer was concentrated in vacuo and the residue was purified by chromatography on silica gel, eluting with 2–20% EtOAc/hexane to give 75 (997 mg, 70%) as a light pink oil: [α]D 23 −26.5 (c 1.87, CHCl3); IR (neat) 2941, 1648, 1465 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.70 (dd, J = 1.9, 7.6 Hz, 1H), 7.65 (dd, J = 1.5, 7.5 Hz, 1H), 7.25–7.36 (m, 2H), 4.45–4.55 (m, 3H), 4.00–4.10 (m, 1H), 3.75–3.85 (m, 1H), 1.00–1.10 (m, 21H); 13C NMR (75 MHz, CDCl3) δ 164.4, 134.0, 131.7, 131.5, 130.1, 127.2, 121.9, 70.7, 68.8, 65.3, 18.1, 12.1; HRMS (FAB+) calcd. for C19H31NO279BrSi (M + H+) 412.1307. Found 412.1314.
2-[2-(3-Phenyl-allylselanyl)-phenyl]-(4R)-(triisopropyl-silanyloxymethyl)-4,5-dihydro-oxazole (53)
To a stirred solution of t-BuLi (1.23 mL, 2.10 mmol, 1.7 M in pentane) in THF (6.6 mL) at −78 °C, was added a solution of benzamide 75 (342 mg, 0.84 mmol) in THF (2.7 mL) dropwise via syringe pump over 20 min. The benzamide conical vial was further rinsed with an additional amount of THF (0.5 mL). After 1 h, Se powder (66 mg, 0.84 mmol) was added under an inverse argon funnel and the reaction was warmed to 0 °C. After 1 h or until the solution was homogeneous, cinnamyl bromide (197 mg, 0.26 mL, 1.0 mmol) was added. After 1 h, the reaction was quenched with sat. aq. NH4Cl (20 mL) and extracted with Et2O (3 × 50 mL). The dried (MgSO4) organic layer was concentrated in vacuo and purified by chromatography on silica gel, eluting with 2–20% EtOAc/hexane. The final product was recrystallized in hexane to give 53 (266 mg, 62%) as a white solid: mp 73 °C; [α]D 23 −4.2 (c 3.41, CHCl3); IR (neat) 2941, 1644, 1469 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.85 (d, J = 7.6 Hz, 1H), 7.46 (d, J = 8.0 Hz, 1H), 7.18–7.37 (m, 7H), 6.63 (d, J = 15.7 Hz, 1H), 6.42 (dt, J = 7.3, 15.3 Hz, 1H), 4.50–4.55 (m, 1H), 4.41–4.44 (m, 2H), 4.07 (dd, J = 3.9, 9.7 Hz, 1H), 3.65–3.75 (m, 3H), 1.00–1.10 (m, 21H); 13C NMR (75 MHz, CDCl3) δ 164.3, 137.0, 136.3, 133.2, 131.0, 130.4, 128.7, 128.3, 127.6, 127.0, 126.4, 125.1, 124.8, 70.3, 69.1, 65.8, 28.8, 18.1, 12.1; HRMS (FAB+) calcd. for C28H10NO280SeSi (M + H+) 530.1994. Found 530.1986.
2-(2-Bromo-phenyl)-(4R)-(tert-butyl-dimethyl-silanyloxy-methyl)-4,5-dihydro-oxazole (76)
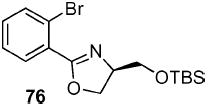
To a stirred solution of benzamide 52 (4.49 g, 11.6 mmol) in CH2Cl2 (22 mL) at 0 °C, was added DMAP (283 mg, 2.3 mmol) and Et3N (2.47 g, 3.4 mL, 24.4 mmol). After 10 min, TsCl (2.41 g, 12.8 mmol) was added and the reaction was warmed to rt. After 12 h, the reaction was quenched with aqueous NH4Cl (50 mL) and extracted with Et2O (3 × 75 mL). The dried (MgSO4) organic layer was concentrated in vacuo and purified by chromatography on silica gel, eluting with 2–20% EtOAc/hexane to give 76 (3.42 g, 72%) as a light pink oil: [α]D 23 −27.7 (c 1.00, CHCl3); IR (neat) 2929, 1652 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.70 (dd, J = 1.9, 7.6 Hz, 1H), 7.65 (dd, J = 1.5, 7.5 Hz, 1H), 7.26–7.33 (m, 2H), 4.40–4.50 (m, 3H), 3.90 (dd, J = 3.4, 9.8 Hz, 1H), 3.70 (dt, J = 3.1, 5.9 Hz, 1H), 0.89 (s, 9H), 0.08 (s, 3H), 0.07 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 164.4, 134.0, 131.8, 131.5, 130.0, 127.2, 121.9, 70.5, 68.7, 64.9, 26.0, 18.4, −5.0, −5.1; HRMS (FAB+) calcd. for C16H25NO279BrSi (M + H+) 370.0838. Found 370.0842.
(4R)-(tert-Butyl-dimethyl-silanyloxymethyl)-2-[2-(3-phenyl-allylselanyl)-phenyl]-4,5-dihydro-oxazole (54)
To a stirred solution of oxazole 76 (1.08 g, 2.93 mmol) in THF (27 mL) at −78 °C, was added t-BuLi (3.45 mL, 5.86 mmol 1.7 M in pentane) dropwise via syringe pump over 20 min. After 1 h, Se powder (234 mg, 2.92 mmol) was added under an inverted argon funnel and the reaction was warmed to 0 °C. After 1 h or until the solution was homogeneous, cinnamyl bromide (583 mg, 0.44 mL, 2.96 mmol) was added. After 1 h, the reaction was quenched with sat. aq. NH4Cl (50 mL) and extracted with Et2O (3 × 50 mL). The dried (MgSO4) organic layer was concentrated in vacuo and purified by chromatography on silica gel, eluting with 0–10% Et2O/hexane to give 54 (1.01 g, 71%) as a white solid: mp 104–106 °C; [α]D 23 −10.2 (c 1.20, CHCl3); IR (neat) 2926, 1639, 1469 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.84 (dd, J = 1.6, 7.8 Hz, 1H), 7.46 (d, J = 7.8 Hz, 1H), 7.18–7.37 (m, 7H), 6.63 (d, J = 15.7 Hz, 1H), 6.42 (dt, J = 7.6, 15.2 Hz, 1H), 4.46–4.53 (m, 1H), 4.40–4.42 (m, 2H), 3.96 (dd, J = 4.0, 10.0 Hz, 1H), 3.73 (d, J = 7.4 Hz, 2H), 3.61 (dd, J = 7.2, 9.8 Hz, 1H), 0.86 (s, 9H), 0.06 (s, 3H), 0.04 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 164.3, 137.0, 136.2, 133.2, 131.0, 130.4, 128.7, 128.3, 127.6, 126.9, 126.4, 125.1, 124.8, 70.1, 68.9, 65.3, 28.8, 26.0, 18.0, −5.0; 77Se NMR (CDCl3) 354.4; HRMS (FAB+) calcd. for C25H34NO280SeSi (M + H+) 488.1524. Found 488.1526.
{2-[2-(3-Phenyl-allylselanyl)-phenyl]-4,5-dihydro-oxazol-(4S)-yl}-methanol (55)
To a stirred solution of selenide 53 (97 mg, 0.18 mmol) in THF (1.8 mL) at rt were sequentially added acetic acid (33 mg, 32 μL, 0.55 mmol) and TBAF (0.74 mL, 0.74 mmol, 1 M in THF). After 1 h, the reaction was quenched with sat. aq. NH4Cl (10 mL) and extracted with EtOAc (3 × 25 mL). The dried (MgSO4) organic layer was concentrated in vacuo and the residue was purified by chromatography on silica gel, eluting with 2–60% EtOAc/hexane to give 53 (52 mg, 76%) as a white solid; mp 113–115 °C; [α]D 23 −40.2 (c 1.08, CHCl3); IR (neat) 3200, 1652 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.81 (dd, J = 1.5, 7.8 Hz, 1H), 7.52 (d, J = 7.9 Hz, 1H), 7.20–7.40 (m, 7H), 6.61 (d, J = 15.6 Hz, 1H), 6.40 (dt, J = 7.5, 15.3 Hz, 1H), 4.53–4.61 (m, 1H), 4.49 (dd, J = 7.9, 9.6 Hz, 1H), 4.34 (dd, J = 7.5, 7.5 Hz, 1H), 4.02 (dd, J = 5.8, 9.3 Hz, 1H), 3.74 (d, J = 7.6 Hz, 2H), 3.64 (dd, J = 3.7, 11.4 Hz, 1H), 2.90 (t, J = 7.5 Hz, 1H); 13C NMR (75 MHz, CDCl3) δ 164.3, 136.9, 136.0, 133.4, 131.3, 130.3, 129.0, 128.7, 127.7, 127.3, 126.5, 125.2, 124.9, 69.2, 68.7, 64.4, 29.4; 77Se NMR (CDCl3) 349.3; HRMS (FAB+) calcd. For C19H19NO280Se (M + H+) 374.0659. Found 374.0666.
{2-[2-(3-Phenyl-allylselanyl)-phenyl]-4,5-dihydro-oxazol-(4S)-yl}-methanol (55)
To a stirred solution of selenide 54 (970 mg, 2.0 mmol) in THF (5 mL) at rt was sequentially added acetic acid (314 mg, 0.3 mL, 5.2 mmol) and TBAF (7.6 mL, 7.6 mmol, 1 M in THF). After 1 h, the reaction was quenched with sat. aq. NaHCO3 (20 mL), and extracted with Et2O (3 × 50 mL). The dried (MgSO4) organic layer was concentrated in vacuo and the residue was purified by chromatography on silica gel, eluting with 5–75% EtOAc/hexane to give 55 (700 mg, 94%) as a white solid.
(4S)-Methoxymethyl-2-[2-(3-phenyl-allylselanyl)-phenyl]-4,5-dihydro-oxazole (56)
To a stirred solution of NaH (6 mg, 0.148 mmol 60% in mineral oil) and MeI (21 mg, 9.2 μL, 0.15 mmol) in THF (0.4 mL) at 0 °C, was added a solution of 55 (50 mg, 0.14 mmol) in THF (0.4 mL) dropwise over 5 min. After 1 h, the reaction was quenched with sat. aq. NH4Cl (5 mL) and extracted with Et2O (3 × 25 mL). The dried (MgSO4) organic layer was concentrated in vacuo and the residue was purified by chromatography on silica gel eluting with 2–20% EtOAc/hexane to yield 56 (41 mg, 82%) as a white solid: mp 103–105 °C; [α]D 23 −1.6 (c 1.15, CHCl3); IR (neat) 2924, 1642, 1471, 1029 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.85 (dd, J = 1.4, 7.8 Hz, 1H), 7.48 (d, J = 7.9 Hz, 1H), 7.18–7.37 (m, 7H), 6.63 (d, J = 15.7 Hz, 1H), 6.36 (dt, J = 7.8, 15.7 Hz, 1H), 4.55–4.64 (m, 1H), 4.44 (dd, J = 8.3, 8.3 Hz, 1H), 4.32 (dd, J = 7.5, 7.5 Hz, 1H), 3.71–3.75 (m, 3H), 3.47 (dd, J = 1.9, 7.6 Hz, 1H), 3.41 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 164.4, 137.0, 136.3, 133.2, 131.2, 130.5, 128.7, 128.4, 127.6, 126.8, 126.4, 125.1, 124.9, 75.0, 70.4, 67.0, 59.5, 28.9; HRMS (FAB+) calcd. for C20H22NO280Se (M + H+) 388.0816. Found 388.0813.
(2S)-(2-Bromo-benzoylamino)-(3R)-hydroxy-butyric acid methyl ester (77)
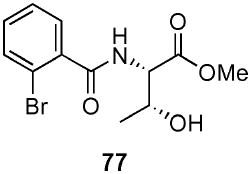
To a stirred solution of l-threonine methyl ester hydrochloride (57) (1.65 g, 9.7 mmol) in CH2Cl2 (19 mL) at 0 °C was added Et3N (2.92 g, 4.05 mL, 29.1 mmol) and DMAP (0.297 g, 2.4 mmol). After 10 min, a solution of 2-bromobenzoyl chloride (31) (2.34 g, 1.4 mL, 14.9 mmol) in CH2Cl2 (10 mL) was added dropwise via syringe pump over a period of 20 min. Next, the solution was warmed to rt. After 2 h, the reaction mixture was quenched with sat. aq. NH4Cl (25 mL) and extracted with Et2O (3 × 75 mL). The organic layer was sequentially washed with aq. HCl (5%, 50 mL) and sat. aq. NaHCO3 (100 mL). The dried (MgSO4) organic layer was concentrated in vacuo and purified by chromatography over silica gel, eluting with 5–100% EtOAc/hexane to give 77 (1.74 g, 58%) as a white oil: [α]D 23 +1.5 (c 1.91, CHCl3); IR (neat) 3332, 1744, 1644 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.62 (dd, J = 1.1, 7.9 Hz, 1H), 7.55 (dd, J = 1.8, 7.6 Hz, 1H), 7.26–7.40 (m, 2H), 6.80 (br d, J = 8.1 Hz, 1H), 4.79 (dd, J = 2.2, 8.9 Hz, 1H), 4.44–4.49 (m, 1H), 3.81 (s, 3H), 2.28 (br s, 1H), 1.36 (d, J = 6.4 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 171.3, 168.1, 137.3, 133.6, 131.7, 129.8, 127.7, 119.5, 68.3, 57.8, 52.9, 20.3; HRMS (FAB+) calcd. for C12H15NO479Br (M + H+) 316.0184. Found 316.0181.
(2S)-(2-Bromo-benzoylamino)-(3R)-triisopropylsilanyloxy-butyric acid methyl ester (78)
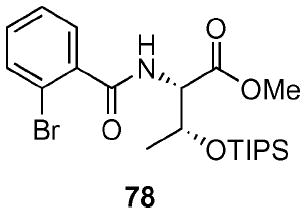
To a stirred solution of amino alcohol methyl ester 77 (1.74 g, 5.52 mmol) in CH2Cl2 (11 mL) at 0 °C was added Et3N (1.11 g, 1.5 mL, 11 mmol). After 10 min, TIPSOTf (4.9 g, 6 mL, 16 mmol) was added. After 1 h, the reaction was warmed to rt. After 2 h, the reaction was quenched with sat. aq. NH4Cl (50 mL) and extracted with Et2O (3 × 100 mL). The dried filtrate (MgSO4) was concentrated in vacuo and purified by chromatography over silica gel, eluting with 2–15% EtOAc/hexane to give 78 (1.60 g, 62%) as a yellow oil: [α]D 23 −6.8 (c 1.14, CHCl3); IR (neat) 1744, 1644 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.62 (dd, J = 7.8, 7.9 Hz, 1H), 7.55 (dd, J = 7.8, 7.6 Hz, 1H), 7.26–7.40 (m, 2H), 6.80 (br d, J = 8.1 Hz, 1H), 4.73–4.81 (m, 2H), 3.81 (s, 3H), 1.36 (d, J = 6.4 Hz, 3H), 1.00–1.10 (m, 21H); 13C NMR (75 MHz, CDCl3) δ 170.9, 168.0, 139.0, 133.7, 132.0, 130.1, 128.4, 119.5, 69.5, 59.1, 52.5, 21.2, 18.6, 12.9; HRMS (FAB+) calcd. for C21H35NO4BrSi (M + H+) 472.1519. Found 472.1519.
2-Bromo-N-[(1R)-hydroxymethyl-(2R)-(triisopropyl-silanyloxyl)-propyl]-benzamide (79)
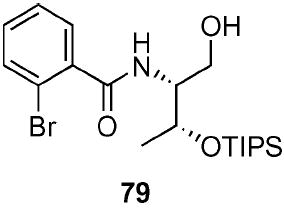
To a stirred solution of LiAlH4 (3.51 mL, 3.51 mmol, 1.0 M in Et2O) in THF (26 mL) at 0 °C, was added a solution of benzamide 78 (1.50 g, 3.20 mmol) in THF (17.4 mL) dropwise via syringe pump over 30 min. The benzamide syringe was rinsed with an additional amount of THF (2 mL). After 30 min the reaction was warmed to rt. After 2 h, the reaction was quenched with EtOAc/H2O (100 mL, 1 : 1) and extracted with ethyl acetate (3 × 75 mL). The dried (MgSO4) organic layer was concentrated in vacuo and the residue was purified by chromatography on silica gel, eluting with 2–50% EtOAc/hexane to give 79 (1.03 g, 71%) as a light pink oil: [α]D 23 −6.3 (c 1.61, CHCl3); IR (neat) 2942, 1651, 1506 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.62 (dd, J = 1.7, 4.0 Hz, 1H), 7.60 (dd, J = 1.8, 4.4 Hz, 1H), 7.26–7.40 (m, 2H), 6.80 (br d, J = 9.0 Hz, 1H), 4.43 (dd, J = 4.9, 6.5 Hz, 1H), 4.08–4.10–4.20 (m, 1H), 3.77–3.88 (m, 2H), 2.66 (br s, 1H), 1.36 (d, J = 6.5 Hz, 3H), 1.00–1.10 (m, 21H); 13C NMR (75 MHz, CDCl3) δ 168.0, 137.5, 133.7, 131.6, 130.2, 127.8, 119.2, 68.1, 64.0, 57.5, 21.7, 18.3, 12.8; HRMS (FAB+) calcd. for C20H35NO379BrSi (M + H+) 444.1570. Found 444.1574.
2-(2-Bromo-phenyl)-(4R)-(triisopropyl-R-silanyloxymethyl)-4,5-dihydro-oxazole (80)
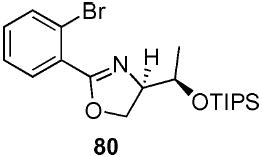
To a stirred solution of benzamide 79 (750 mg, 1.69 mmol) in CH2Cl2 (5 mL) at 0 °C, were added DMAP (41 mg, 0.34 mmol) and Et3N (340 mg, 0.5 mL, 3.38 mmol). After 10 min, TsCl (600 mg, 3.04 mmol) was added and the reaction was warmed to rt. After 12 h, the reaction was quenched with sat. aq. NH4Cl (50 mL) and extracted with Et2O (3 × 75 mL). The dried (MgSO4) organic layer was concentrated in vacuo and purified by chromatography on silica gel, eluting with 2–20% EtOAc/hexane to give 80 (400 mg, 63%) as a light pink oil: [α]D 23 −24.0 (c 1.66, CHCl3); IR (neat) 2942, 1651, 1463 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.62–7.67 (m, 2H), 7.26–7.37 (m, 2H), 4.54–4.61 (m, 2H), 4.35–4.43 (m, 2H), 1.18 (d, J = 5.5 Hz, 3H), 1.00–1.10 (m, 21H); 13C NMR (75 MHz, CDCl3) δ 164.2, 133.9, 131.7, 131.4, 130.2, 127.2, 121.8, 72.0, 68.7, 68.4, 17.6, 17.3, 13.0; HRMS (FAB+) calcd. for C20H33NO279BrSi (M + H+) 426.1464. Found 426.1477.
2-[2-(3-Phenyl-allylselanyl)-phenyl]-(4R)-(triisopropyl-R-silanyloxymethyl)-4,5-dihydro-oxazole (58)
To a stirred solution of t-BuLi (1.3 mL, 2.21 mmol, 1.7 M in pentane) in THF (6.7 mL) at −78 °C was added a solution of the bromide 80 (434 mg, 1.02 mmol) in THF (3.4 mL) dropwise via syringe pump over 20 min resulting in an orange solution. The benzamide conical vial was further rinsed with an additional amount of THF (0.5 mL). After 1 h, Se powder (81 mg, 1.02 mmol) was added under an inverse argon funnel and the reaction was warmed to 0 °C. After 1 h or until the solution was homogeneous, cinnamyl bromide (220 mg, 0.17 mL, 1.12 mmol) was added. After 1 h, the reaction was quenched with sat. aq. NH4Cl (20 mL) and extracted with Et2O (3 × 75 mL). The dried (MgSO4) organic layer was concentrated in vacuo and the residue was purified by chromatography on silica gel, eluting with 2–20% EtOAc/hexane. The final product was recrystallized in hexane to give 58 (300 mg, 54%) as a yellow solid: mp 59 –63 °C; [α]D 23 −8.0 (c 6.50, CHCl3). IR (neat) 2941, 1644, 1464 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.82 (dd, J = 1.6, 7.5 Hz, 1H), 7.49 (d, J = 7.9 Hz, 1H), 7.18–7.38 (m, 7H), 6.64 (d, J = 15.6 Hz, 1H), 6.40 (dt, J = 7.5, 15.0 Hz, 1H), 4.67 (dt, J = 4.5, 4.7 Hz, 1H), 4.55 (dd, J = 6.9, 8.6 Hz, 1H), 4.44 (dd, J = 4.5, 6.2 Hz, 1H), 4.35 (dd, J = 8.7, 9.9 Hz, 1H), 3.73 (d, J = 7.5, 2H), 1.18 (d, J = 6.2 Hz, 3H), 1.00–1.10 (m, 21H); 13C NMR (75 MHz, CDCl3) δ 164.0, 137.1, 136.3, 133.2, 131.0, 130.4, 128.7, 128.3, 127.6, 127.0, 126.4, 125.2, 124.8, 72.3, 68.9, 67.7, 28.8, 18.3, 17.7, 12.5; HRMS (FAB+) calcd. for C29H42NO280SeSi (M + H+) 544.2150. Found 544.2147.
1R-{2-[2-(3-Phenyl-allylselanyl)-phenyl]-4,5-dihydro-oxazol-(4R)-yl}-ethanol (59)
To a stirred solution of selenide 58 (41 mg, 0.10 mmol) in THF (0.8 mL) at rt was added acetic acid (15 mg, 13 μL, 0.2 mmol) and TBAF (1.9 mL, 1.9 mmol, 1 M in THF). After 1 h, the reaction was quenched with sat. aq. NH4Cl (10 mL) and extracted with EtOAc (3 × 25 mL). The dried (MgSO4) organic layer was concentrated in vacuo and the residue was purified by chromatography over silica gel, eluting with 2–60% EtOAc/hexane to yield 59 (52 mg, 78%) as a white solid: mp 105 °C; [a]D23 −11.7 (c 0.50, CHCl3); IR (neat) 3445, 1644, 1472 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.84 (dd, J = 1.5, 7.7 Hz, 1H), 7.52 (d, J = 7.8 Hz, 1H), 7.20–7.40 (m, 7H), 6.64 (d, J = 15.7 Hz, 1H), 6.43 (dt, J = 7.4, 15.1 Hz, 1H), 4.48 (dd, J = 7.4, 9.3 Hz, 1H), 4.30–4.37 (m, 1H), 4.23 (dd, J = 7.6 Hz, 2H), 3.70 (dd, J = 7.0, 7.9 Hz, 2H), 2.50 (br s, 1H), 1.36 (d, J = 6.5 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 164.6, 136.9, 136.3, 133.3, 131.3, 130.4, 128.7, 128.7, 127.7, 126.7, 126.5, 125.1, 125.0, 73.4, 70.3, 69.6, 29.3, 20.3; HRMS (FAB+) calcd. for C20H22NO280Se (M + H+) 388.0816. Found 388.0824.
4-Nitro-benzoic acid (1S)-{2-[2-(3-phenyl-(E)-allylselanyl)-phenyl]-4,5-dihydro-oxazol-(4R)-yl}-ethyl ester (81)
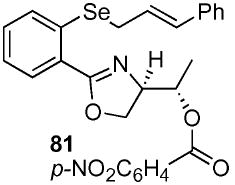
To a stirred solution of selenide 59 (35 mg, 0.087 mmol) in THF (0.72 mL) at −78 °C were added PPh3 (47 mg, 0.18 mmol) and p-NO2C6H4CO2H (30 mg, 0.18 mmol). After 5 min, DEAD (31 mg, 28 μL, 0.18 mmol) was added dropwise to the reaction. Over 2 h, the reaction was warmed slowly from −78 °C to rt. After an additional 5 h at rt, the reaction was quenched with sat. aq. NH4Cl (5 mL) and extracted with Et2O (3 × 50 mL). The dried (MgSO4) organic layer was concentrated in vacuo and the residue was purified by chromatography on silica gel, eluting with 0.5–10% EtOAc/hexane to yield 81 (39 mg, 80%) as a white solid: mp 97–99 °C; [α]D 23 +18.4 (c 0.50, CHCl3); IR (neat) 1723, 1645, 1525, 1272 cm−1; 1H NMR (300 MHz, CDCl3) δ 8.11–8.19 (m, 4H), 7.80–7.83 (dd, J = 1.1, 7.9 Hz, 1H), 7.44 (d, J = 8.0 Hz, 1H), 7.20–7.37 (m, 7H), 6.58 (d, J = 15.5 Hz, 1H), 6.31 (dt, J = 7.8, 15.5 Hz, 1H), 5.30–5.37 (m, 1H), 4.52–4.62 (m, 1H), 4.40–4.50 (dt, J = 8.8, 17.9 Hz, 2H), 3.67 (d, J = 7.4 Hz, 2H), 1.54 (d, J = 6.3 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 164.7, 164.2, 150.6, 136.9, 136.6, 135.9, 133.2, 131.4, 130.9, 130.4, 128.7, 128.4, 127.7, 126.4, 126.3, 124.9, 123.6, 74.1, 71.0, 68.8, 28.8, 17.2, 14.4; HRMS (FAB+) calcd. for C27H25N2O580Se (M + H+) 537.0929. Found 537.0942.
(1S)-{2-[2-(3-Phenyl-(E)-allylselanyl)-phenyl]-4,5-dihydro-oxazol-4R-yl}-ethanol (60)
To a stirred solution of seleno-ester 81 (37 mg, 0.070 mmol) in MeOH (0.3 mL) at rt was added K2CO3 (2.0 mg, 0.010 mmol). After 30 min, the reaction was quenched with sat. aq. NH4Cl (3 mL) and extracted with EtOAc (3 × 20 mL), The dried (MgSO4) organic layer was concentrated in vacuo and the residue purified by chromatography on silica gel, eluting with 2–40% EtOAc/hexane to give 60 (13 mg, 50%) as a white solid: mp 100 °C; [α]D 23 −5.7 (c 1.20, CHCl3); IR (neat) 3445, 1644, 1472 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.84 (dd, J = 1.5, 7.7 Hz, 1H), 7.52 (d, J = 7.8 Hz, 1H), 7.20–7.40 (m, 7H), 6.61 (d, J = 15.7 Hz, 1H), 6.41 (dt, J = 7.4, 15.1 Hz, 1H), 4.38–4.42 (m, 3H), 4.11–4.19 (m, 1H), 3.73 (d, J = 7.5 Hz, 2H), 2.05 (br s, 1H), 1.24 (d, J = 6.5 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 164.7, 136.9, 135.9, 133.3, 131.2, 130.2, 129.0, 128.7, 127.7, 126.5, 125.2, 124.9, 73.0, 68.1, 67.5, 29.4, 18.8, 14.4; HRMS (FAB+) calcd. for C20H21NO278Se (M + H+) 386.0824. Found 386.0827.
2-(2-Bromo-phenyl)-4,5-dihydro-oxazole-(4S)-carboxylic acid methyl ester (61)
To a stirred solution of 50 (1.24 g, 4.10 mmol) in CH2Cl2 (35 mL) at −78 °C was added DAST (726 mg, 0.6 mL, 4.50 mmol) dropwise over 15 min. After 1 h, the slurry was quenched with K2CO3 (851 mg, 6.20 mmol) and warmed to rt. After 20 min, sat. aq. NaHCO3 (20 mL) was added and extracted with Et2O (3 × 75 mL). The dried (MgSO4) organic layer was concentrated in vacuo and the residue was purified by chromatography on silica gel to give 61 (1.06 g, 91%) as a light pink oil: [α]D 23 +33.2 (c 1.31, CHCl3); IR (neat) 2952, 1731, 1651 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.70 (dd, J = 1.9, 6.9 Hz, 1H), 7.62 (dd, J = 1.2, 7.3 Hz, 1H), 7.23–7.34 (m, 2H), 4.99 (dd, J = 8.0, 10.8 Hz, 1H), 4.71 (dd, J = 8.2, 16.9 Hz, 1H), 4.62 (dd, J = 8.6, 10.6 Hz, 1H), 3.78 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 171.4, 166.0, 134.0, 132.3, 131.8, 128.3, 127.3, 122.0, 69.9, 68.8, 52.9; HRMS (FAB+) calcd. for C11H10NO3Br (M + H+) 282.9844. Found 283.9922.
1-[2-(2-Bromo-phenyl)-4,5-dihydro-oxazol-(4S)-yl]-2-methyl-propan-1-one (62)
To a stirred solution 61 (2.22 g, 7.84 mmol) in THF (41 mL) at − 78 °C was added i-PrMgCl (9.4 mL, 9.4 mmol, 1 M in Et2O) dropwise via syringe pump over 60 min. After 1.5 h, the slurry was quenched with NH4Cl and extracted with Et2O (3 × 250 mL). The dried (MgSO4) organic layer was concentrated in vacuo and the residue was purified by chromatography on silica gel to give 62 (1.72 mg, 74%) as a light pink oil: [α]D23 +28.4 (c 1.19, CHCl3). IR (neat) 2970, 1715, 1651 cm−1. 1H NMR (300 MHz, CDCl3) δ 7.70 (dd, J = 1.9, 7.3 Hz, 1H), 7.66 (dd, J = 1.4, 7.4 Hz, 1H), 7.26–7.37 (m, 2H), 5.06 (dd, J = 7.4, 10.5 Hz, 1H), 4.82 (dd, J = 7.4, 8.5 Hz, 1H), 4.52 (dd, J = 8.9, 10.9 Hz, 1H), 3.25–3.35 (m, 1H), 1.27 (d, J = 7.2 Hz, 3H), 1.15 (d, J = 6.7 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 211.8, 164.6, 134.2, 132.1, 131.5, 129.2, 127.3, 122.1, 73.8, 68.5, 38.4, 18.8, 17.7; HRMS (FAB+) calcd. for C13H15NO279Br (M + H+) 296.0286. Found 296.0299.
1-[2-(2-Bromo-phenyl)-4,5-dihydro-oxazol-(4S)-yl]-2-methyl-propan-(1R/S)-ol (63 and 64)
To a stirred solution of 62 (250 mg, 0.9 mmol) in MeOH (3 mL) at rt was added NaBH4 (38 mg, 1 mmol) in portions. After 30 min, the slurry was quenched with sat. aq. NH4Cl (10 mL) and extracted with Et2O (3 × 50 mL). The dried (MgSO4) organic layer was concentrated in vacuo and the residue was purified by chromatography on silica gel, eluting with 2–30% EtOAc/hexane to give sequentially (S)-(S) 63 (72 mg, 58%) and (S)-(R) 64 (53 mg, 42%) as white solids. 63: [α]D 23 +22.5 (c 0.98, CHCl3); IR (neat) 3367, 2959 1715, 1654 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.63–7.68 (m, 2H), 7.26–7.37 (m, 2H), 4.50–4.64 (m, 2H), 4.20 (dt, J = 1.0, 10.9 Hz, 1H), 3.25 (dt, J = 4.5, 6.7 Hz, 1H), 2.28 (d, J = 7.1 Hz, 1H), 1.85–1.88 (m, 1H), 1.05 (d, J = 6.8 Hz, 6H); 13C NMR (75 MHz, CDCl3) δ 164.6, 133.9, 132.0, 131.3, 129.7, 127.4, 122.1, 79.1, 70.5, 69.6, 31.8, 19.7, 18.2; HRMS (FAB+) calcd. for C13H17NO279Br (M + H+) 298.0443. Found 298.0448. 64: [α]D 23 +5.9 (c 1.02, CHCl3); IR (neat) 3367, 2959 1715, 1654 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.62–7.67 (m, 2H), 7.26–7.37 (m, 2H), 4.39–4.56 (m, 2H), 3.72 (dd, J = 2.8, 7.7 Hz, 2H), 1.85 (br s, 1H), 1.71–1.77 (m, 1H), 1.07 (d, J = 6.5 Hz, 3H), 0.95 (d, J = 6.8 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 164.8, 133.9, 131.9, 131.2, 129.9, 127.4, 122.0, 79.0, 69.8, 68.0, 30.6, 19.2, 19.0; HRMS (FAB+) calcd. for C13H17NO279Br (M + H+) 298.0443. Found 298.0451.
2-(2-Bromo-phenyl)-(4S)-[(1S)-(tert-butyl-dimethyl-silanyloxy)-2-methyl-propyl]-4,5-dihydro-oxazole (82)
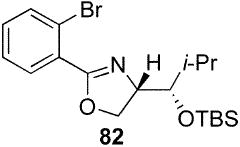
To a stirred solution of 63 (70 mg, 0.24 mmol) in CH2Cl2 (0.5 mL) at 0 °C were sequentially added 2,6-lutidine (39 mg, 42 μL, 0.36 mmol) and TBSOTf (75 mg, 65 μL, 0.28 mmol). After 1 h, the reaction was quenched with sat. aq. NH4Cl (5 mL) and extracted with Et2O (3 × 20 mL). The dried (MgSO4) organic layer was concentrated in vacuo and the residue was purified by chromatography on silica gel, eluting with 1–10% EtOAc/hexane to give 82 (90 mg, 94%) as a pink oil: [α]D 23 +8.1 (c 0.70, CHCl3); IR (neat) 2956 1653, 1471 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.71 (dd, J = 2.0, 7.5 Hz, 1H), 7.61 (dd, J = 1.6, 7.8 Hz, 1H), 7.24–7.36 (m, 2H), 4.55–4.60 (m, 1H), 4.36–4.40 (m, 2H), 3.72 (dd, J = 4.0, 5.4 Hz, 1H), 1.85–1.95 (m, 1H), 1.03 (d, J = 6.7 Hz, 3H), 0.97 (d, J = 6.8 Hz, 3H), 0.91 (s, 9H), 0.12 (s, 3H), 0.10 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 163.6, 134.0, 131.7, 130.3, 127.2, 121.8, 77.9, 70.8, 69.6, 31.0, 26.1, 21.0, 18.5, 17.4, −3.8, −4.2; HRMS (FAB+) calcd. for C19H31NO2Si79Br (M + H+) 412.1307. Found 412.1314.
(4S)-[(1S)-(tert-Butyl-dimethyl-silanyloxy)-2-methyl-propyl]-2-[2-(3-phenyl-allylselanyl)-phenyl]-4,5-dihydro-oxazole (83)
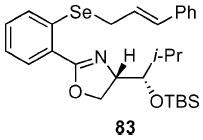
To a stirred solution of 82 (340 mg, 0.82 mmol) in THF (7.2 mL) at −78 °C was added t-BuLi (1.0 mL, 1.7 mmol, 1.7 M in pentane) dropwise via syringe pump over 20 min resulting in an orange solution. After 1 h, Se powder (65 mg, 0.82 mmol) was added to the reaction under an inverse argon funnel and warmed to 0 °C. After 2 h, cinnamyl bromide (162 mg, 0.12 mL, 0.82 mmol) was added. After 1 h, the reaction was quenched with sat. aq. NH4Cl (10 mL) and extracted with Et2O (3 × 50 mL). The dried (MgSO4) extract was concentrated in vacuo and the residue was purified by chromatography on silica gel, eluting with 0–10% EtOAc/hexane to give 83 (300 mg, 71%) as a white solid: [α]D 23 −10.9 (c 1.72, CHCl3); IR (neat) 2954, 1644, 1470 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.84 (dd, J = 1.5, 7.8 Hz, 1H), 7.47 (d, J = 8.1 Hz, 1H), 7.16–7.36 (m, 7H), 6.66 (d, J = 15.7 Hz, 1H), 6.45 (dt, J = 7.7, 15.7 Hz, 1H), 4.60–4.64 (m, 1H), 4.29–4.42 (m, 2H), 3.78–3.82 (m, 1H), 3.72 (d, J = 7.4 Hz, 2H), 1.96–2.01 (m, 1H), 1.00 (s, 9H), 1.06 (d, J = 6.8 Hz, 3H), 0.98 (d, J = 6.0 Hz, 3H), 0.25 (s, 3H), 0.16 (s, 3H); HRMS (FAB+) calcd. for C28H40NO2Si80Se (M + H+) 530.1994. Found 530.2008.
2-Methyl-1-{2-[(3-phenyl-allylselanyl)-phenyl]-4,5-dihydro-oxazol-(4S)-yl}-propan-(1S)-ol (65)
To a stirred solution of selenide 83 (320 mg, 0.6 mmol) in CH2Cl2 (1.23 mL) at rt were sequentially added acetic acid (78 mg, 70 μL, 1.35 mmol) and TBAF (7.5 mL, 7.5 mmol, 1 M in THF). After 1 h the reaction was quenched with sat. aq. NH4Cl (20 mL), and extracted with EtOAc (3 × 15 mL). The dried (MgSO4) filtrate was concentrated in vacuo and the residue was purified by chromatography on silica gel, eluting with 2–30% EtOAc/hexane to yield 65 (134 mg, 53%) as a white solid: [α]D 23 −22.4 (c 2.0, CHCl3); IR (neat) 3209, 1651, 1449, 1253 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.81 (d, J = 7.8 Hz, 1H), 7.51 (d, J = 8.0 Hz, 1H), 7.20–7.39 (m, 7H), 6.59 (d, J = 15.7 Hz, 1H), 6.37 (dt, J = 7.3, 15.4 Hz, 1H), 4.54–4.58 (m, 1H), 4.47 (dd, J = 7.8, 7.8 Hz, 1H), 4.23 (dd, J = 7.7, 7.7 Hz, 1H), 3.72 (d, J = 7.5 Hz, 2H), 3.23 (dd, J = 7.1, 11.8 Hz, 1H), 2.29 (d, J = 7.7 Hz, 1H), 1.90–1.97 (m, 1H), 1.07 (d, J = 6.7 Hz, 3H), 1.06 (d, J = 6.7 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 164.8, 137.2, 136.4, 133.5, 131.5, 130.5, 129.0, 128.9, 127.9, 127.2, 126.7, 125.3, 79.2, 70.4, 69.9, 32.4, 29.6, 20.0, 18.6; HRMS (FAB+) calcd. for C22H26NO280Se (M + H+) 416.1129. Found 416.1134.
2-(2-Bromo-phenyl)-(4S)-[(1R)-(tert-butyl-dimethyl-silanyloxy)-2-methyl-propyl]-4,5-dihydro-oxazole (84)
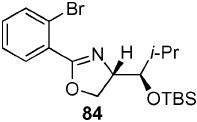
To a stirred solution of 64 (57 mg, 0.19 mmol) in CH2Cl2 (0.4 mL) at 0 °C were sequentially added 2,6-lutidine (32 mg, 27 μL, 0.29 mmol) and TBSOTf (61 mg, 52 μL, 0.23 mmol). After 1 h, the reaction was quenched with sat. aq. NH4Cl (5 mL) and extracted with Et2O (3 × 20 mL). The dried (MgSO4) organic layer was concentrated in vacuo and the residue was purified by chromatography on silica gel, eluting with 1–10% EtOAc/hexane to give 84 (48 mg, 62%) as a light pink oil: [α]D 23 +1.7 (c 0.78, CHCl3); IR (neat) 2956, 1653, 1471 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.76 (dd, J = 2.0, 7.5 Hz, 1H), 7.64 (dd, J = 1.6, 7.8 Hz, 1H), 7.24–7.36 (m, 2H), 4.53 (dd, J = 5.7, 7.0 Hz, 1H), 4.33–4.36 (m, 2H), 3.99 (dd, J = 2.0, 4.2 Hz, 1H), 1.81–1.87 (m, 1H), 0.96 (d, J = 7.0 Hz, 3H), 0.92 (d, J = 4.3 Hz, 3H), 0.90 (s, 9H), 0.07 (s, 3H), −0.02 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 163.6, 134.0, 131.7, 130.3, 127.2, 121.8, 77.9, 70.8, 69.6, 31.0, 26.1, 21.0, 18.5, 17.4, −3.8, −4.2; HRMS (FAB+) calcd. for C19H31NO2Si79Br (M + H+) 412.1307. Found 412.1318.
(4S)-[(1R)-(tert-Butyl-dimethyl-silanyloxy)-2-methyl-propyl]-2-[2-(3-phenyl-(E)-allylselanyl)-phenyl]-4,5-dihydro-oxazole (85)
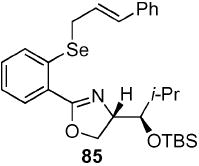
To a stirred solution of bromide 84 (176 mg, 0.43 mmol) in THF (3.8 mL) at −78 °C was added t-BuLi (0.5 mL, 0.85 mmol, 1.7 M in pentane) dropwise via syringe pump over 20 min resulting in an orange solution. After 1 h, Se powder (36 mg, 0.43 mmol) was added to the reaction under an inverse argon funnel and warmed to 0 °C. After 2 h, cinnamyl bromide (63 mg, 63 μL, 0.43 mmol) was added. After 1 h, the reaction was quenched with sat. aq. NH4Cl (10 mL) and extracted with Et2O (3 × 50 mL). The dried (MgSO4) extract was concentrated in vacuo and the residue was purified by chromatography on silica gel, eluting with 0–10% EtOAc/hexane to give 85 (169 mg, 74%) as a white solid: mp 122 °C; [α]D 23 −44.2 (c 1.23,CHCl3); IR (neat) 2954, 1644, 1470 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.83 (dd, J = 1.5, 7.8 Hz, 1H), 7.46 (d, J = 8.1 Hz, 1H), 7.16–7.36 (m, 7H), 6.62 (d, J = 15.7 Hz, 1H), 6.39 (dt, J = 7.7, 15.7 Hz, 1H), 4.43–4.50 (m, 2H), 4.27–4.33 (m, 1H), 3.92 (dd, J = 2.2, 4.2 Hz, 1H), 3.71 (d, J = 7.4 Hz, 2H), 1.82–1.87 (m, 1H), 0.96 (d, J = 7.0 Hz, 3H), 0.92 (d, J = 6.1 Hz, 3H), 0.88 (s, 9 H), 0.05 (s, 3H), −0.11 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 163.2, 137.1, 136.4, 133.1, 130.8, 130.4, 128.8, 128.7, 128.1, 127.6, 126.8, 126.5, 125.3, 124.7, 78.3, 70.0, 67.7, 33.5, 26.1, 19.0, 18.5, 18.4, −3.9, −4.0; HRMS (FAB+) calcd. for C28H10NO2Si80Se (M + H+) 530.1994. Found 530.1994.
2-Methyl-1-{2-[2-(3-phenyl-(E)-allylselanyl)-phenyl]-4,5-dihydro-oxazol-(4S)-yl}-propan-(1R)-ol (66)
To a stirred solution of selenide 85 (195 mg, 0.38 mmol) in CH2Cl2 (1 mL) at rt was sequentially added acetic acid (44 mg, 42 μL, 0.74 mmol) and TBAF (2.1 mL, 2.1 mmol, 1 M in THF). After 1 h, the reaction was quenched with sat. aq. NH4Cl (20 mL) and extracted with EtOAc (3 × 20 mL). The dried (MgSO4) organic layer was concentrated in vacuo and the residue was purified by chromatography on silica gel, eluting with 2–30% EtOAc/hexane to give 66 (75 mg, 60%) as a white solid: mp 125 °C; [α]D 23 −22.4 (c 2.0, CHCl3); IR (neat) 3209, 1651, 1449, 1253 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.70 (dd, J = 1.2, 7.60 Hz, 1H), 7.50 (d, J = 8.0 Hz, 1H), 7.20–7.39 (m, 7H), 6.58 (d, J = 15.7 Hz, 1H), 6.40 (dt, J = 7.3, 15.4 Hz, 1H), 4.56–4.64 (m, 1H), 4.37–4.41 (m, 2H), 3.71–3.76 (m, 3H), 2.12 (br s, 1H), 1.66–1.79 (m, 1H), 1.09 (d, J = 6.6 Hz, 3H), 0.95 (d, J = 6.8 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 164.6, 136.9, 135.9, 133.3, 131.2, 130.1, 129.0, 128.7, 127.7, 127.4, 126.5, 125.2, 125.0, 77.7, 70.0, 67.5, 30.6, 29.4, 19.3, 19.2; 77Se NMR (CDCl3) 349.0; HRMS (FAB+) calcd. for C22H26NO278Se (M + H+) 414.1137. Found 414.1133.
(4S)-[(1R)-(tert-Butyl-dimethyl-silanyloxy)-2-methyl-propyl]-2-[2-(3-phenyl-(Z)-allylselanyl)-phenyl]-4,5-dihydro-oxazole (86)
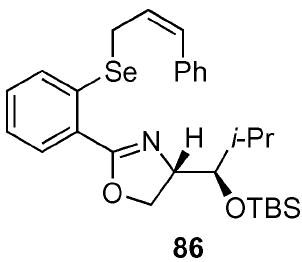
To a stirred solution of bromide 85 (150 mg, 0.37 mmol) in THF (1.2 mL) at −78 °C was added t-BuLi (0.45 mL, 0.77 mmol, 1.7 M in pentane) dropwise via syringe pump over 20 min resulting in an orange solution. After 1 h, Se powder (29 mg, 0.37 mmol) was added to the reaction under an inverse argon funnel and warmed to 0 °C. After 2 h, Z-cinnamyl chloride 34 (70 mg, 0.43 mmol) was added. After 1 h, the reaction was quenched with sat. aq. NH4Cl (10 mL) and extracted with Et2O (3 × 50 mL). The dried (MgSO4) extract was concentrated in vacuo and the residue was purified by chromatography on silica gel, eluting with 2–20% EtOAc/hexane to give impure 86 (76 mg) which was used in the next transformation without any additional purification.
2-Methyl-1-{2-[-(3-phenyl-(Z)-allylselanyl)-phenyl]-4,5-dihydro-oxazol-(4S)-yl}-propan-(1R)-ol (67)
To a stirred solution of selenide impure 86 (76 mg, 0.14 mmol) in CH2Cl2 (0.3 mL) at rt was sequentially added acetic acid (17 μL, 0.28 mmol) and TBAF (2.8 mL, 2.8 mmol, 1 M in THF). After 24 h, the reaction was quenched with sat. aq. NH4Cl (20 mL) and extracted with Et2O (3 × 20 mL). The dried (MgSO4) organic layer was concentrated in vacuo and the residue was purified by chromatography on silica gel, eluting with 2–30% EtOAc/hexane to give 67 (15 mg, 10% over 2 steps) as a white solid: The white solid was recrystallized twice in hexane to give a white solid: mp 58 °C; [α]D 23 +11.3 (c 0.4, CHCl3); 1H NMR (300 MHz, CDCl3) δ 7.78–7.82 (m, 1H), 7.16–7.38 (m, 8H), 6.56 (d, J = 11.3 Hz, 1H), 5.90 (dt, J = 8.5, 11.5 Hz, 1H), 4.44–4.66 (m, 2H), 4.23 (dd, J = 9.3, 17.1 Hz, 1H), 3.73–3.91 (m, 2H), 3.20 (dt, J = 4.9, 6.9 Hz, 1H), 2.30 (d, J = 7.4 Hz, 1H), 1.89–2.0 (m, 1H), 1.09 (d, J = 2.6 Hz, 3H), 1.07 (d, J = 2.6 Hz, 3H); 13C NMR (75 MHz, CDCl3) 164.6, 136.8, 136.4, 132.2, 131.3, 130.3, 129.0, 128.7, 128.6, 127.4, 127.0, 126.6, 124.9, 79.0, 70.2, 69.7, 32.2, 25.1, 19.8, 18.4; HRMS (FAB+) calcd. for C22H26NO278Se (M + H+) 414.1137. Found 414.1127.
General procedure for vanadium-catalyzed ASOS reaction with bidentate oxazoles [1-phenyl-prop-2-en-1-ol (10e)]
To a stirred solution of the selenide (1 mmol) in CH2Cl2 (4 mL) at rt was added VO(acac)2 (10 mol%) and powdered 4 Å mol. sieves (500 mg per mmol selenide). After 20 min, the reaction was cooled to −50 °C and TBHP (2 mmol, 5.5 M in decane) was added. After 12–20 h, the reaction was quenched with PBu3 (1.3 mmol). After 10 min, the slurry was diluted with sat. aq. NH4Cl (50 mL) and extracted with Et2O (3 × 200 mL). The dried (MgSO4) filtrate was concentrated in vacuo and the residue was purified by chromatography on silica gel, eluting with 2–20% EtOAc/hexane to yield 10e as a pale yellow oil. The diastereomeric excess was determined via conversion to the Mosher ester using 1H and/or 19F NMR in C6D6.
Acknowledgments
Financial support was provided by the National Institutes of Health (NIH) (GM63723), University of Mississippi and Oregon State University. This publication was also made possible in part by a grant from the NIH – National Institute of Environmental Health Sciences (P30 ES00210). The authors would like to thank Professor Max Dienzer (Mass Spectrometry Facility, Environmental Health Sciences Center, Oregon State University) and Dr Jeff Morré (Mass Spectrometry Facility, Environmental Health Sciences Center, Oregon State University) for mass spectra data, Roger Kohnert (Oregon State University) and Professor David E. Graves (University of Alabama-Birmingham) for NMR assistance and Dr Roger Hanselmann (Rib-X Pharmaceuticals) for his helpful discussions.
Footnotes
A portion of the this work was conducted in the Department of Chemistry and Biochemistry, University of Mississippi, Oxford, MS 38677.
Electronic Supplementary Information (ESI) available: Generalized procedures and spectral data for compounds 9–14, and X-ray crystallographic data for compounds 55 and 66. See http://www.rsc.org/suppdata/ob/b3/b316502g/
CCDC reference numbers 228854–228855. See http://www.rsc.org/suppdata/ob/b3/b316502g/ for crystallographic data in.cif or other electronic format.
References
- 1.(a) Corey EJ, Helal CJ. Angew Chem Int Ed. 1998;37:1986. doi: 10.1002/(SICI)1521-3773(19980817)37:15<1986::AID-ANIE1986>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]; (b) Ohkuma T, Koizumi M, Doucet H, Pham T, Kozawa M, Murata K, Katayama E, Yokozawa T, Ikariya T, Noyori R. J Am Chem Soc. 1998;120:13529. [Google Scholar]; (c) Gao Y, Hanson RM, Klunder JM, Ko SY, Masamune H, Sharpless KB. J Am Chem Soc. 1987;109:5765. [Google Scholar]; (d) Knochel P. In: Handbook of Reagents for Organic Synthesis: Reagents, Auxiliaries and Catalysts for C–C Bonds. Coates RM, Denmark SE, editors. John Wiley & Sons; New York: 1999. pp. 270–75. [Google Scholar]
- 2.Hoveyda AH, Evans DA, Fu GC. Chem Rev. 1993;93:1307. [Google Scholar]
- 3.Nicolaou KC, Sorenson EJ. Classics in Total Synthesis. VCH press; New York: 1996. [Google Scholar]
- 4.(a) Wirth T. Angew Chem Int Ed. 2000;39:3740. doi: 10.1002/1521-3773(20001103)39:21<3740::aid-anie3740>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]; (b) Nishibayashi Y, Uemura S. In: Organoslenium Chemistry-A Practical Approach. Back TG, editor. Oxford University Press; Oxford: 1999. pp. 207–221. [Google Scholar]; (c) Komatu N, Uemura S. In: Advances in Detailed Reaction Mechanisms. Coxon JM, editor. Volume 4. JAI Press; 1995. pp. 73–92. [Google Scholar]; (d) Reich HJ. In: Organoselenium Chemistry. Liotta D, editor. John Wiley & Sons; New York: 1987. pp. 365–93. [Google Scholar]; (e) Uemura S. Phosph Sulf and Sil. 2001;13:171–172. [Google Scholar]
- 5.It should be noted that a sulfur-based variant for the SOS reaction (the Mislow–Braverman–Evans rearrangement) is well-known and has been extensively reviewed. Jones-Hertzog DK, Jorgensen WL. J Org Chem. 1995;60:6682.Evans DA, Andrews GC. Acc Chem Res. 1974;7:147.
- 6.The Mislow–Braverman–Evans rearrangement has been employed in total synthesis. Mapp AK, Heathcock CH. J Org Chem. 1999;64:23. doi: 10.1021/jo9813742.Paquette LA, Meister PG, Friedrich D, Sauer DR. J Am Chem Soc. 1993;115:49.
- 7.Carter RG, Bourland TC. Chem Commun. 2000:2031. [Google Scholar]
- 8.Selected examples of enzymatic and nitrogen oxide-based processes for oxidation of selenides have been reported. Bosch E, Kochi JK. Nitrogen oxide catalysis. J Chem Soc Perkin Trans 1. 1996:2731.Chen G-P, Ziegler DM. Enzymatic catalysis. Arch Biochem Biophys. 1994;312:566. doi: 10.1006/abbi.1994.1346.Akerboom TPM, Sies H, Ziegler DM. Arch Biochem Biophys. 1995;316:220. doi: 10.1006/abbi.1995.1031.
- 9.Reich HJ, Yelm KE, Wollowitz S. J Am Chem Soc. 1983;105:2503. [Google Scholar]
- 10.Davis FA, Reddy RT. J Org Chem. 1992;57:2599. [Google Scholar]
- 11.Kurose N, Takahashi T, Koizumi T. Tetrahedron. 1997;53:12115. [Google Scholar]
- 12.Nishibayashi Y, Singh JD, Fukuzawa S, Uemura S. J Org Chem. 1995;60:4114. [Google Scholar]
- 13.Fujita K, Kanakubo M, Ushijima H, Oishi A, Ikeda Y, Taguchi Y. Synlett. 1998:987. [Google Scholar]
- 14.Reich HJ, Yelm KE. J Org Chem. 1991;56:5672. [Google Scholar]
- 15.Komatsu N, Nishibayashi Y, Uemura S. Tetrahedron Lett. 1993;34:2339. [Google Scholar]
- 16.(a) Gao Y, Hanson RM, Klunder JM, Ko SY, Masamune H, Sharpless KB. J Am Chem Soc. 1987;109:5765. [Google Scholar]; (b) Kagan HB, Pitchen P. Tetrahedron Lett. 1984;25:1049. [Google Scholar]
- 17.Tiecco M, Tingoli M, Testaferri L, Bartoli D. Tetrahedron Lett. 1987;28:3849. [Google Scholar]
- 18.Shimizu T, Kobayashi M, Kamigata N. Sulfur Lett. 1988;8:61. [Google Scholar]
- 19.For a general review of vanadium chemistry, see: Hirao T. Chem Rev. 1997;97:2707. doi: 10.1021/cr960014g.
- 20.(a) Cogan DA, Liu G, Kim K, Backes B, Ellman J. J Am Chem Soc. 1998;120:8011. [Google Scholar]; (b) Bolm C, Bienwald F. Angew Chem Int Ed Engl. 1995;34:2640. [Google Scholar]; (c) Nakijima K, Kojima M, Fujita J. Chem Lett. 1986:1483. [Google Scholar]
- 21.Comins DL, Dehgagani A. J Org Chem. 1995;60:794. [Google Scholar]
- 22.Evans DA, Ennis MD, Mathre DJ. J Am Chem Soc. 1982;104:1737. [Google Scholar]
- 23.This steric interaction between the aryl group on the selenide and the Z-substituent on the alkene has been put forth by other laboratories to explain the increased levels of selectivity observed in cis-alkene systems.
- 24.Bower JF, Martin CJ, Rawson DJ, Slawin MZA, Williams JMJ. J Chem Soc Perkin Trans 1. 1996:331. [Google Scholar]
- 25.Zhou Q, Pfaltz A. Tetrahedron. 1994;50:4467. [Google Scholar]
- 26.(a) M’Boungou-M’Passi A, Henin F, Muzart JP. Bull Soc Chim Fr. 1993;130:214. [Google Scholar]; (b) Gawley RE, Zhang P. J Org Chem. 1996;61:8103. doi: 10.1021/jo961164u. [DOI] [PubMed] [Google Scholar]; (c) Chittenden RA, Cooper GH. J Chem Soc C. 1970:49. [Google Scholar]
- 27.Pirrung MC, Tumey NL. J Comb Chem. 2000;2:675. doi: 10.1021/cc000047g. [DOI] [PubMed] [Google Scholar]
- 28.(a) Panda A, Mugesh G, Singh HB, Butcher RJ. Organometallics. 1999;18:1986. [Google Scholar]; (b) Mugesh G, Panda A, Singh HB, Butcher RJ. Chem Eur J. 1999;5:1411. [Google Scholar]; (c) Mugesh G, Singh HB, Butcher RJ. Tetrahedron: Asymmetry. 1999;10:237. [Google Scholar]
- 29.Keck GA, Castellino S. Tetrahedron Lett. 1987;28:281.Frye SV, Eliel E. Tetrahedron Lett. 1986;27:3223. It should be noted that there have been selected examples reported to the contrary; Willard PG, Hintze MJ. J Am Chem Soc. 1987;109:5539.Evans DA, Allison BD, Yang MG. Tetrahedron Lett. 1999;40:4457.Evans DA, Halstead DP, Allison BD. Tetrahedron Lett. 1999;40:4461.
- 30.Chen M-J, Narkunan K, Liu R-S. J Org Chem. 1999;64:8311. doi: 10.1021/jo991077c. [DOI] [PubMed] [Google Scholar]
- 31.Phillips AJ, Uto Y, Wipf P, Reno MJ, Williams DR. Org Lett. 2000;2:1165. doi: 10.1021/ol005777b. [DOI] [PubMed] [Google Scholar]
- 32.Mascavage LM, Lu Q, Vey J, Dalton DR, Carroll PJ. J Org Chem. 2001;66:3621. doi: 10.1021/jo001736h. and references cited within.
- 33.Selected examples have been reported that support this hypothesis. Onishi T, Otake Y, Hirose N, Nakano T, Torii T, Nakazawa M, Izawa K. Tetrahedron Lett. 2001;42:6337.Delair P, Einhorn C, Einhorn J, Luche JL. J Org Chem. 1999;59:4680.
- 34.The substrate 67 was made in an analogous fashion as described previously. The Z-alkene was incorporated by alkylation of the selenolate by Z-cinnamyl chloride. This chloride was prepared using the protocol outlined by Davis and co-workers.10 It should be noted that the corresponding Z-cinnamyl iodide proved problematic, as considerable isomerization was observed during its formation from the corresponding Z-allylic alcohol.
- 35.Perrin DD, Armarego WLF. Purification of Laboratory Chemicals. Third. Pergamon Press; New York: 1993. [Google Scholar]