

Abstract
Alpha 1 (XI) collagen (Col11a1) is essential for normal skeletal development. Mutations in Col11a1 cause Marshall and Stickler syndromes, both of which are characterized by craniofacial abnormalities, nearsightedness and hearing deficiencies. Despite its link to human diseases, few studies have described factors that control Col11a1 transcription. We previously identified Col11a1 as a differentially expressed gene in Lef1-suppressed MC3T3 preosteoblasts. Here we report that Lef1 activates the Col11a1 promoter. This activation is dependent upon the DNA binding domain of Lef1, but does not require the ²-catenin interaction domain, suggesting that it is not responsive to Wnt signals. Targeted suppression of Col11a1 with an antisense morpholino accelerated osteoblastic differentiation and mineralization in C2C12 cells, similar to what was observed in Lef1-suppressed MC3T3 cells. Moreover incubation with a purified Col11a1 N-terminal fragment, V1B, prevented alkaline phosphatase expression in MC3T3 and C2C12 cells. These results suggest that Lef1 is an activator of the Col11a1 promoter and that Col11a1 suppresses terminal osteoblast differentiation.
Keywords: Lef1, Wnt, beta-catenin, collagen 11a1, VO, V1b, osteoblasts
1. Introduction
Extracellular matrix proteins are essential for the normal formation and function of all tissues (Comper, 1996). Skeletal tissue is composed of mineral and organic components in combination with various cell types (Gokhale et al., 2001). Collagens comprise a large percentage of the organic extracellular matrix in bone. Type I collagen is the major scaffolding protein for mineralized bone and constitutes 90% of the organic matrix (Robey and Boskey, 2003). Other collagens, however, are also important for proper bone formation. Minor collagens, such as types III, V, VI and XI collagen, control fibril growth, diameter and assembly of major collagens (Gelse et al., 2003; Robey and Boskey, 2003).
Type XI collagen is a minor fibril-forming collagen. It is a heterotrimer of alpha 1 (XI) collagen (Col11a1), alpha 2 (XI) collagen (Col11a2) and alpha 1 (II) collagen (Col2a1), except in the vitreous of the eye where Col11a2 is replaced with alpha 1 (V) collagen (Col5a1) (Fichard et al., 1995). Type XI collagen assists in proper type II collagen fibril formation (Gelse et al., 2003). Col11a1 is mainly expressed in articular cartilage and the vitreous fluid of the eye (Lui et al., 1995; Yoshioka et al., 1995; Iyama et al., 2001; So et al., 2001); however, Col11a1 expression is detectable in many other human fetal tissues including the bone (Lui et al., 1995). In mice lacking Col11a1 (Cho mice), chondrocytes fail to fully differentiate causing a chondrodystrophic phenotype. Cho mice also have enhanced long bone mineralization, thicker trabeculae, and increased bone mass at the diaphysis (Seegmiller et al., 1971). Observations from the Cho mice suggest that Col11a1 is essential for skeletal morphogenesis because it controls type II collagen fibrillogenesis, chondrocyte maturation and bone mineralization (Seegmiller et al., 1971; Li et al., 1995).
In humans, mutations in Col11a1 cause Marshall and type II Stickler syndromes (Griffith et al., 1998; Annunen et al., 1999). Marshall syndrome is an autosomal dominant inherited disease characterized by short stature, nearsightedness, hearing loss, flattened facial features, thickened calvarium and intracranial ossifications. Type II Stickler syndrome patients have similar symptoms but are of near normal height and exhibit no bony overgrowths (Snead and Yates, 1999). Both syndromes occasionally present with cleft palate and patients frequently develop early osteoarthritis (Olsen, 1995; Snead and Yates, 1999; Rodriguez et al., 2004; Jakkula et al., 2005). Whether Marshall and Stickler syndromes are variants of the same disease or separate syndromes is controversial, but it is apparent that they are very similar and many patients display symptoms of both diseases (Winter et al., 1983; Ayme and Preus, 1984; Stratton et al., 1991; Annunen et al., 1999).
Lef1 is one four nuclear high mobility group (HMG) proteins that mediate gene transcription in response to canonical Wnt signaling, which is an important regulator of skeletal development and bone homeostasis (reviewed in (Westendorf et al., 2004;Baron et al., 2006; Glass and Karsenty, 2007). Lef1 is essential for mesenchymal/epithelial interactions that occur during the development of tissues such as mammary glands, teeth and whiskers (van Genderen et al., 1994). Lef1 null mice are smaller than wild type mice and die shortly after birth (van Genderen et al., 1994). A mouse model expressing a mutant form of Lef1 lacking the HMG domain (Lef1-βgal) displays several skeletal abnormalities, mostly associated with skeletal patterning (Galceran et al., 2004). Recently, we recently discovered that Lef1 binds to Runx2 through the Lef1 HMG domain and represses Runx2-induced transcription of the osteocalcin promoter. This repression is further enhanced by mimicking canonical Wnt signaling with stabilized β-catenin (Kahler and Westendorf, 2003). We previously showed that osteoblasts with reduced Lef1 expression mineralize at an accelerated rate while osteoblasts over expressing Lef1 do not mineralize (Kahler et al., 2006). These data suggest that Lef1 may have an important role in normal osteoblast maturation.
While we reported a role for Lef1 in osteoblast maturation, the exact mechanism by which Lef1 affects osteoblast differentiation is unknown. In this report, we describe a role for Lef1 in the expression of Col11a1. We demonstrate that Lef1-suppressed preosteoblasts express Col11a1 mRNA at reduced levels compared to control cells. An antisense morpholino that inhibits Col11a1 translation enhances differentiation of C2C12 cells along the osteoblast lineage in a manner similar to Lef1-suppression (Kahler et al., 2006). Moreover, incubation with an N-terminal fragment of Col11a1 slows alkaline phosphatase production by pre-osteoblasts. These data suggest that Lef1 is important for Col11a1 expression and Col11a1, in turn, plays a role in controlling osteoblast differentiation and mineralization.
2. Results
2.1. Lef1-suppressed cells have reduced Col11a1 expression
To identify genes that were differentially regulated as a result of Lef1 suppression, we subjected RNA samples from control (Ffl cells) and Lef1-suppressed (L2) MC3T3 cells to gene chip analysis (Kahler et al., 2006). One group of genes that was consistently and significantly downregulated in Lef1-suppressed osteoblasts was the collagen family. The gene chip analyses indicated that collagens 3a1, 5a1, 6a2 and 11a1 were all downregulated 1.5-fold or more at a significance of p ≤ 0.005 (Fig. 1A and (Kahler et al., 2006)). Quantitative real time PCR confirmed that each of these genes were downregulated in Lef1-suppressed cells compared to control cells (Fig. 1A). Col11a1 expression was reduced between 3.5- and 7-fold (Fig. 1A and (Kahler et al., 2006)).
Figure 1. Col11a1 expression is reduced in Lef1-deficient cells.
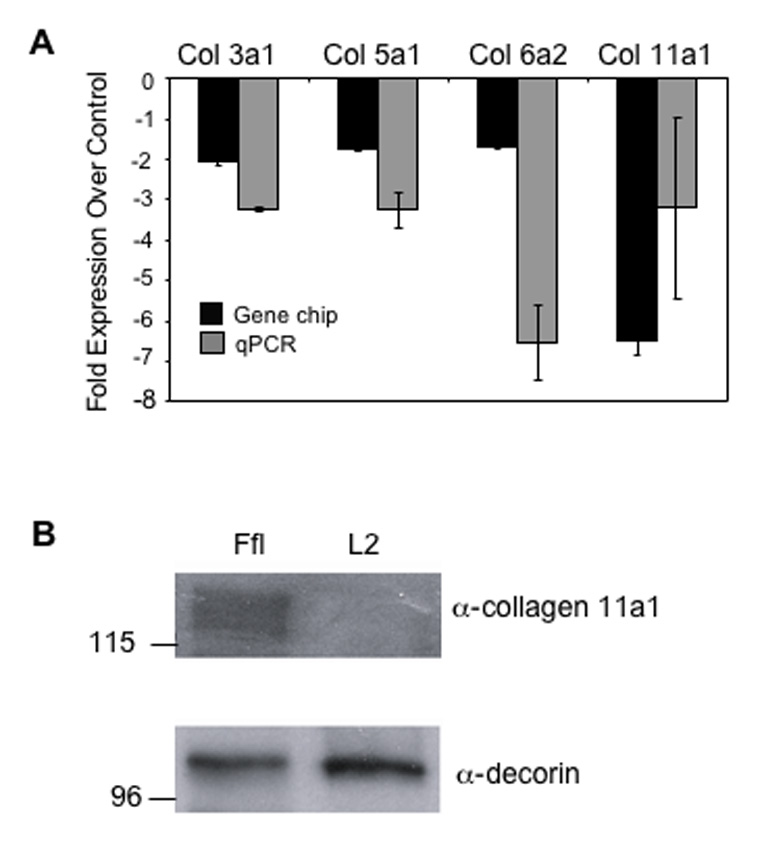
A. The relative change in expression of collagen 11a1 in Lef1-suppressed MC3T3 cells (L2) as compared to control cells expressing a short hairpin RNA against firefly luciferase (Ffl). Black bars depict fold reduction of expression ± SD as measured by gene chip analysis with a p-value ≤ 0.05. Grey bars depict fold reduction of expression ± SD as measured by quantitative RT-PCR. B. Immunoblot analysis of protein precipitated from L2 or Ffl cell-conditioned media. The upper panel is blotted with α-collagen 11a1 antibody. The lower panel is blotted with α-decorin antibody as a protein loading control.
To verify that Col11a1 protein levels were concomitantly decreased in Lef1-suppressed cells, we treated control (Ffl) or Lef1-suppressed (L2) MC3T3 cells with osteogenic medium for 14 days. Protein extracts from both the conditioned media and extracellular matrices were subjected to immunoblot analysis. Collagen 11a1 was detectable in the conditioned medium of the control cells but not the Lef1-suppressed cells or in the extracellular matrices of either cell line (Fig. 1B and data not shown). Blotting with anti-decorin antibody shows that both samples contained similar amounts of protein (Fig. 1B). Similar results were obtained with MC3T3 clone 14 cell lines (data not shown). These data show that Lef1 suppression leads to reduced expression of Col11a1 mRNA and protein. In these conditions, it also appears that Col11a1 is mostly secreted into the medium and not integrated into the extracellular matrix. Decorin was readily detectable in the extracellular matrix (data not shown).
2.2. Lef1 activates the Col11a1 promoter
Because Col11a1 was so strongly downregulated in Lef1-suppressed cells, we asked if Lef1 could activate the collagen 11a1 promoter. We used a collagen 11a1 promoter reporter, pCol11a1(−1454)-luc, containing 1454 nucleotides upstream of the transcriptional start site and the first exon of human Col11a1 (Fig. 2A) (Yoshioka et al., 1995). When transiently transfected into C2C12 cells, Lef1 activated the reporters 4–8-fold and in a concentration-dependent manner (Fig. 2B & C), but had no effect on the empty luciferase reporter plasmid, pGL3. Lef1 also activated the Col11a1 promoter in other cell lines, including MC3T3, NIH3T3 and ROS cells, albeit at lower levels (1.5 to 6 fold) (data not shown).
Figure 2. Lef1 activates the Col11a1 promoter.
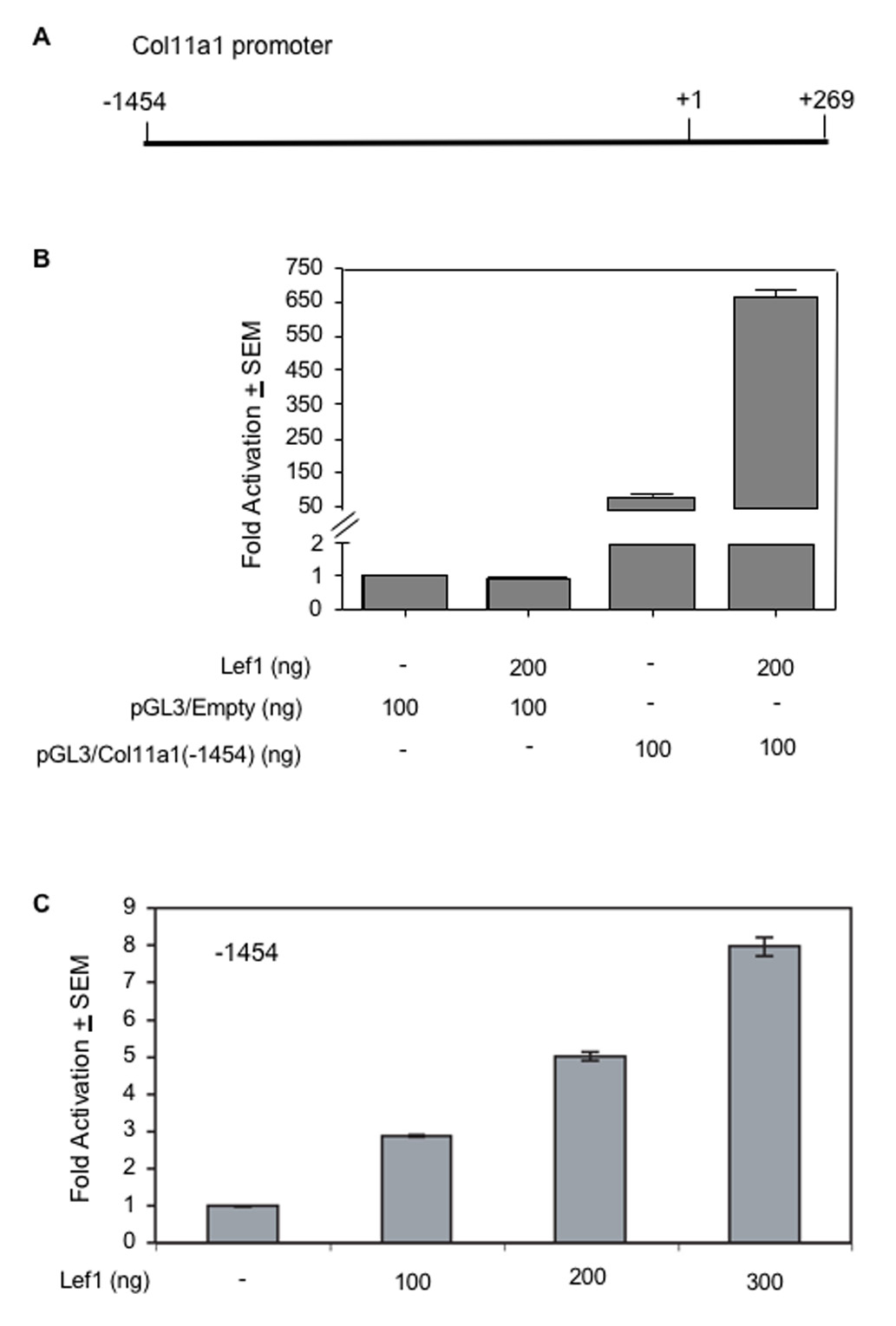
A. This schematic depicts the Col11a1 promoter used in this study. pCol11a1(−1454)-luc encompasses bases −1454 to +269. B. C2C12 cells were transfected with pGL3 or pGL3-Col11a1(−1454)-luc, pRL-null and 200 ng of either pcDNA (−) or pCMV5B-Lef1-HA. Transcription activation is shown as fold luciferase activity over control ± SEM and corrected for transfection efficiency relative to renilla expression. C. C2C12 cells were transfected with pGL3-Col11a1(−1454)-luc, pRL-null and the indicated concentrations of pCMV5B-Lef1-HA. Data were analyzed as in B.
2.3. The Lef1 HMG domain is necessary for activation of the Col11a1 promoter
We next sought to determine whether Lef1 DNA binding activity is necessary for activation of the Col11a1 promoter. To test this, we used a Lef1 construct that lacks the HMG domain (Lef1ΔHMG), which is necessary for DNA binding (Fig. 3A). Lef1ΔHMG was unable to activate the Col11a1 reporter (Fig. 3B). These data suggest that Lef1 requires the HMG domain to activate the Col11a1 promoter, either through its DNA binding domain or through interaction with other proteins.
Figure 3. The HMG domain of Lef1 is required for activation of Col11a1 promoter.
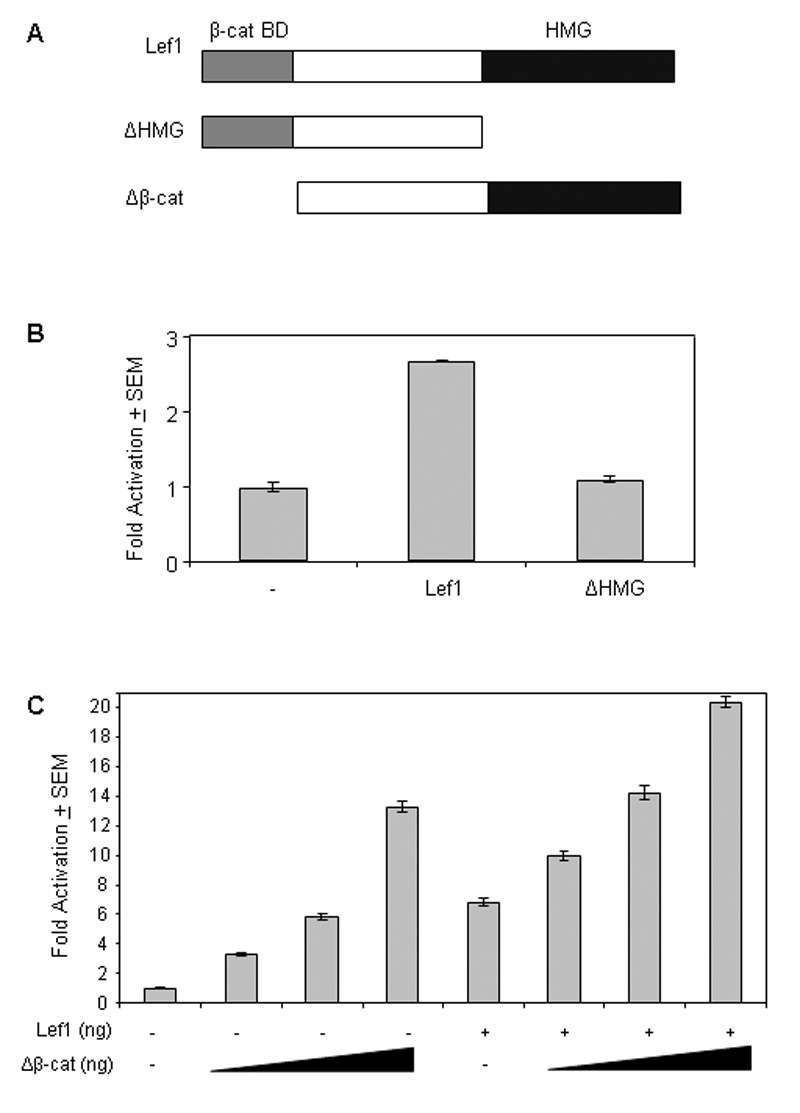
A. This schematic depicts the Lef1, Lef1ΔHMG, and Lef1Δβ-cat expression constructs used in these experiments. β-cat BD, β-catenin binding domain; TLE BD, TLE binding domain; HMG,,high mobility group domain. B. C2C12 cells were transfected with pCol11a1(−1454)-luc, pRL-null and 200 ng of either pcDNA3 (−), pCMV5-Lef1-HA (Lef1), or pCMV5-Lef1ΔHMG-HA (ΔHMG) as indicated. Transcription is measured as fold luciferase activity over control ± SEM. C. C2C12 cells were transfected with pCol11(−1454)-luc, pRL-null, 200 ng pCMV5B-Lef1-HA and either 200 ng pcDNA3 (−) or the indicated amount of Lef1Δβ-cat. Transcription is measured as fold luciferase activity over control ± SEM.
2.4. Lef1’s β-catenin binding domain is not required for activation of the Col11a1 promoter
In many systems, Lef1 interaction with β-catenin is necessary for Lef1-mediated activation of target genes (reviewed in (Behrens et al., 1996; Clevers and van de Wetering, 1997). Lef1 lacking the β-catenin binding domain is still able to bind DNA and thus could compete with β-catenin-responsive Lef1 isoforms for DNA binding sites (reviewed in (Waterman, 2004)). To determine whether Lef1-mediated activation of the Col11a1 promoter was dependent upon interaction with β-catenin, we utilized an N-terminally truncated Lef1 mutant protein lacking the high affinity β-catenin binding domain (Fig. 3A). This construct, Lef1Δβ-cat, activated the Col11a1 promoter in a dose dependent manner (Fig. 3C) and enhanced activation by full length Lef1 in an additive manner (Fig. 3C). These data indicate that Lef1 does not require interaction with β-catenin to activate the Col11a1 promoter, suggesting that expression of Col11a1 by Lef1 may be Wnt-independent.
2.5. Reduced Col11a1 expression enhances osteoblastic differentiation of C2C12 cells
When induced to differentiate, Lef1-suppressed MC3T3 preosteoblasts express osteoblast markers and mineralize earlier than control cells (Kahler et al., 2006). To determine the role of Col11a1 in osteoblast maturation, C2C12 cells were cultured with BMP-2 in the presence or absence of an antisense morpholino specifically designed to downregulate the protein expression of Col11a1 by sterically blocking the translation of the Col11a1 mRNA. Control C2C12 cells that were either untransfected or transfected with a non-specific morpholino expressed Col11a1 protein at the time of induction and Col11a1 levels increased until 24 hours after the addition of BMP-2 (Fig. 4A,). By 48 hours following the addition of BMP-2, Col11a1 expression was nearly undetectable. C2C12 cells transfected with the Col11a1 antisense morpholino expressed considerably less Col11a1 protein at any time point, illustrating the effectiveness of the antisense morpholino. These data suggest that Col11a1 is expressed early in osteoblastic differentiation but that its expression is quickly downregulated as osteoblasts mature.
Figure 4. Col11a1 antisense morpholinos induce increased expression of osteoblast markers.
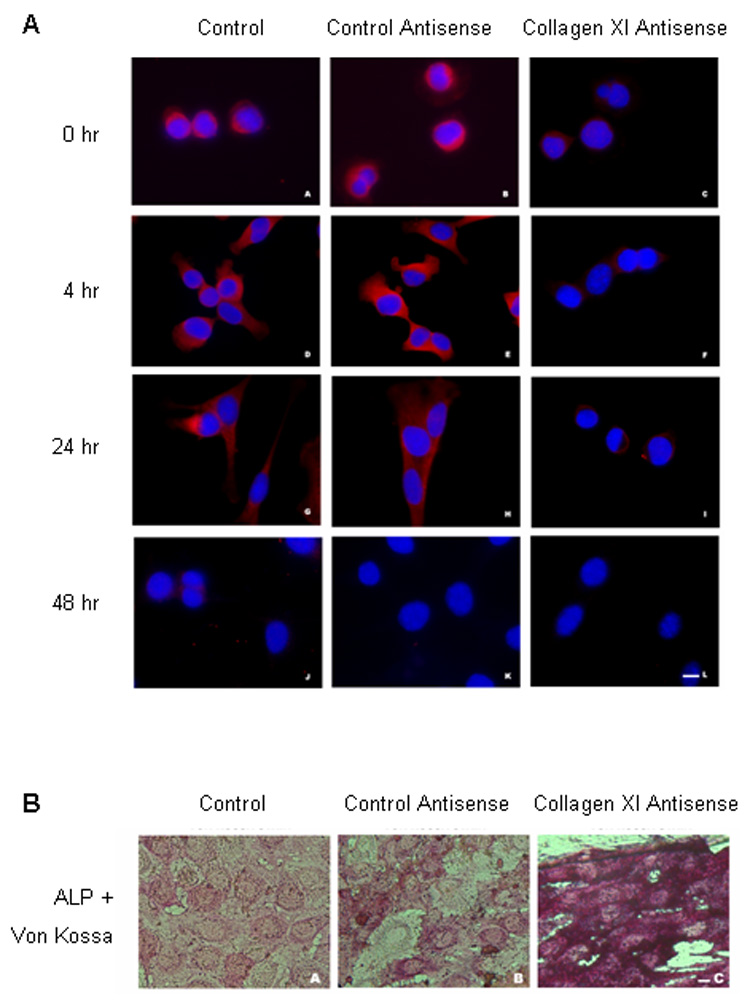
A. C2C12 cells were treated with either no (left column), control (center column) or Col11a1 antisense morpholinos (right column) and immunofluorescently stained with an anti-Col11a1 primary antibody and a TRITC-labeled secondary antibody. C2C12 cells were induced to differentiate into the osteoblastic lineage by treatment with BMP-2 for the indicated length of time. B. C2C12 cells were treated either with no (A), control (B) or Col11a1 antisense morpholinos (C) and stained for alkaline phosphatase activity (red) and mineralization (von Kossa staining, black) after 14 days treatment with BMP-2.
To determine the effect of the Col11a1 morpholino on osteoblastic differentiation, we cultured C2C12 cells in osteogenic medium containing BMP-2 in the presence or absence of the Col11a1 antisense or the control morpholino and then stained the cells for alkaline phosphatase activity and mineralization. C2C12 cells that were transfected with the Col11a1 antisense morpholino exhibited increased alkaline phosphatase activity and von Kossa staining as compared to C2C12 cells that were untreated or transfected with a control morpholino (Fig. 4B). These data suggest that Col11a1 I a negative regulator of osteoblast differentiation.
2.6. An N-terminal fragment of Col11a1, V1B, blocks alkaline phosphatase production by pre-osteoblasts
We next sought to confirm the role of Col11a1 as a negative regulator of osteoblast maturation by determining the consequence of increased Col11a1 protein during osteoblast differentiation. To test this, we cultured C2C12 cells in osteogenic medium containing BMP-2 in the presence of Col11a1 N-terminal fragments V0 or V1B. V0 and V1B represent variations of the domain of Col11a1 that remains associated with the surface of collagen fibril for an extended period of time. V0 and V1B are the two most prevalent isoforms implicated in mineralization of the bone collar in long bone development (Davies et al., 1998; Morris et al., 2000). The N-terminal fragments are proteolytically released from the surface of collagen fibrils slowly but at isoform-specific rates of processing (Thom and Morris, 1991; Medeck et al., 2003). As compared to control cells, C2C12 cells treated with V1B had a 60% decrease in alkaline phosphatase production after two days and 28% reduction after five days (Fig. 5A), but only the former level reached statistical significance (p<0.5). Similarly, MC3T3 cells treated with Col11a1 V1B fragment exhibited reduced expression of alkaline phosphatase compared to control MC3T3 cells at days three and nine of differentiation in osteogenic medium (Fig. 5B), but only the 73% reduction at day three was statistically significant (p<0.001). Fragment V0 also suppressed alkaline phosphates production in MC3T3 cells albeit to a lesser (39%) but significant level (p<0.01) at day three. These data support the role of Col11a1 as a negative regulator of osteoblast maturation in an isoform-specific manner.
Figure 5. Alkaline phosphatase expression is inhibited during osteoblast differentiation by an N-terminal Col11a1 fragment, V1B.
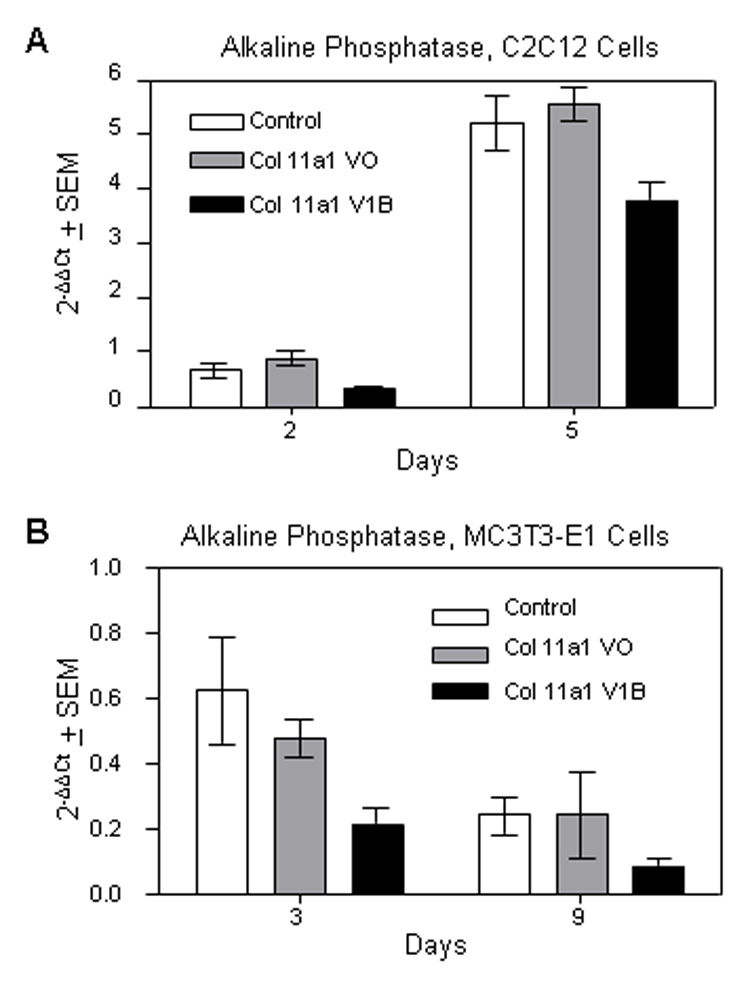
C2C12 cells were differentiated in osteogenic medium supplemented with BMP2 for five days (A) and MC3T3-E1 cells were differentiated in osteogenic medium for nine days (B) in the presence of vehicle alone, Col11a1 V0 fragment, or Col11a1 V1B fragment. RNA was collected at the indicated time points and quantitative PCR was performed using alkaline phosphatase and actin specific primers. Alkaline phosphatase values were normalized to actin.
3. Discussion
Collagen 11a1 is mutated in Marshall and Stickler syndromes (Snead et al., 1996; Griffith et al., 1998; Meisler et al., 1998; Annunen et al., 1999; Martin et al., 1999; Snead and Yates, 1999). Patients with Marshall syndrome exhibit short stature and bony overgrowths on the skull (Stratton et al., 1991; Meisler et al., 1998), suggesting a role for Col11a1 in controlling bone mineralization. Further evidence for this conclusion is apparent in the Cho mice, which exhibit excess mineralization in the long bones (Seegmiller et al., 1971; Li et al., 1995). Sequence variations in Col11a1 are also associated with osteoarthritis, early-onset osteoarthritis, degenerative lumbar spinal stenosis and cleft palate (Melkoniemi et al., 2003; Noponen-Hietala et al., 2003; Rodriguez et al., 2004; Jakkula et al., 2005). Its association with a variety of common diseases makes Col11a1 an important target for musculoskeletal disease research.
This report highlights a role for Lef1 in regulating Col11a1 expression. Reduced Lef1 expression coincides with decreased expression of Col11a1 mRNA and protein (Fig. 1 and (Kahler et al., 2006)). Transient transfection assays indicated that Lef1 activates the Col11a1 promoter in a concentration-dependent manner (Fig. 2). These data are consistent with a study which found a correlation between Col11a1 expression in colorectal carcinoma and an activated β-catenin-Lef1/Tcf pathway (Fischer et al., 2001). Analysis of the Col11a1 promoter identified at least six potential Lef1 binding sites, but only two were found to bind Lef1 in vitro (data not shown). Mutating these sites individually or all together did not eliminate the activation of the reporter by Lef1. These data suggest that Lef1 activates the Col11a1 promoter indirectly, perhaps through the HMG domain, which is essential for Lef-1 mediated activation (Figure 3).
Despite the seriousness of Marshall and Stickler syndromes and the implications of Col11a1 expression in osteoarthritis and other common disorders, there are only a handful of reports on the transcriptional control of the Col11a1 gene. The initial studies defining the Col11a1 promoter identified the minimal promoter containing cell type specificity as the fragment from −199 to +269 (Yoshioka et al., 1995). However, the most active promoter fragment in this study contained nucleotides −541 to +269. We found that this fragment remains responsive to Lef1 (data not shown). Yoshioka, et al. identified nine DNase I protected footprints in the region of the promoter containing bases −541 to + 269, most of which were identified as probable binding sites for ubiquitously expressed proteins NF-kB, CF1, AP2, AP3 and Sp1. (Yoshioka et al., 1995). One footprint was determined to be a binding site for a zinc-finger protein that was termed FP9C (Kinoshita et al., 1997). The only reported zinc-finger protein to interact with Tcf/Lef1 is Kaiso, which competes with β-catenin for binding to Tcf-3 resulting in transcriptional repression of the Samois gene in X. laevis (Park et al., 2005). This is unlikely to be the factor through which Lef1 activates the Col11a1 promoter for several reasons. First, the Kaiso binding sequence does not match that of the unidentified zinc-finger protein FP9C. Second, Kaiso competes with β-catenin binding to Tcf/Lef1 and the β-catenin binding domain is not necessary for activation of the Col11a1 promoter (Fig. 2 and 3). Finally, Kaiso is reported to be a repressor of transcription so interaction between Lef1 and Kaiso is unlikely to result in transcriptional activation (Kim et al., 2004; Park et al., 2005; Spring et al., 2005).
A second transcription factor reported to interact with the Col11a1 promoter is CBF/NF-Y (Matsuo et al., 2003). This transcription factor binds to the Col11a1 promoter within the −147 to −121 region the and activates transcription of promoter (Matsuo et al., 2003). At this time, there is no evidence to suggest that Lef1 could be activating the Col11a1 promoter indirectly through CBF/NF-Y. We found that Lef1 did not cooperate with NF-Y (data not shown). We also did not detect any changes in CBF/NF-Y expression in the gene chip experiment performed with Lef1-suppressed cells, suggesting that the activity of Lef1 on the Col11a1 promoter is not through changes in CBF/NF-Y expression (data not shown). Additional studies are needed to determine the nature of Lef1’s interaction with the Col11a1 promoter.
Though the importance of Col11a1 has been established through the detrimental effects of the loss of normal Col11a1 expression in Marshall and Stickler syndromes, the exact role Col11a1 plays in bone formation has not yet been identified. This report shows a link between Lef1 and Col11a1 both in the ability of Lef1 to activate the Col11a1 promoter and the osteoblastic phenotype resulting from reduced expression of either Lef1 or Col11a1. We previously reported that using RNAi to reduce Lef1 expression leads to accelerated osteoblast maturation characterized by early expression of osteoblast markers and enhanced matrix mineralization (Kahler et al., 2006). Lef1-suppressed cells have reduced Col11a1 expression (Fig. 1). When we used an antisense morpholinos to mimic the reduced Col11a1 expression seen in Lef1-suppressed cells, we observed that alkaline phosphatase expression and mineralization were enhanced (Fig. 6). These data suggest that one of the main mechanisms for the enhanced differentiation we observed as a result of reducing Lef1 levels is the reduction of Col11a1. They also suggest that Col11a1 is important for controlling the rate of osteoblast mineralization.
In accordance with shRNA experiments, exposure to a Col11a1 N-terminal fragment delayed early osteoblast maturation as measured by alkaline phosphatase expression. The V0 and V1B containing isoforms of Col11a1 constitute 70% to 80% of the total Col11a1 transcripts in growth plate cartilage, including the region undergoing mineralization to form the bone collar between the cartilage and the perichondrium. V1B Col11a1 shows a restricted distribution in the cartilage of the developing long bones. Before primary ossification, V1B is expressed only in the diaphysis, primarily adjacent to the perichondrium, a location consistent with a role in the inhibition of osteoblast differentiation and mineralization. This unique localization of V1B may indicate that it is involved in the signaling between the cells of the perichondrium, associated chondrocytes and differentiating osteoblasts.
The main function of type XI collagen is to control fibrillogenesis of type II collagen (Gelse et al., 2003). However, this function requires the integration of type XI collagen into matrix containing type II collagen (Gelse et al., 2003). Furthermore, the expression of each of the individual type XI collagen genes varies from tissue to tissue, suggesting that they do not form heterotrimers in all tissues and hence the function of each type XI collagen molecule might be tissue specific (Lui et al., 1995). When we examined the expression of Col11a1 upon induction of osteoblastic differentiation, we observed very early expression of Col11a1 with nearly a complete loss of expression by 48 hours (Fig. 4). It is possible that Col11a1 is incorporated into the matrix but cannot be detected by immunofluorescence. However, we were unable to detect Col11a1 protein in the matrix of differentiating osteoblasts by immunoblotting, a method that easily detected Col11a1 in the supernatant (Fig. 1). The absence of Col11a1 in the matrix suggests that the role of Col11a1 in osteoblast differentiation is separate from its role in controlling fibrillogenesis in other tissues. The mechanism by which the reduction of Col11a1 expression induces enhanced mineralization and alkaline phosphatase remains to be determined. However, its presence outside of the matrix might suggest a role in regulating the availability of growth factors or as a signaling molecule itself (reviewed in (Tran et al., 2004; Bi et al., 2005; Bix and Iozzo, 2005; Iozzo, 2005; Tran et al., 2005)).
4. Experimental Procedures
4.1. Plasmids
The luciferase reporter construct (pCol11a1(−1454)-luc was kindly provided by Dr. Francesco Ramirez (Kinoshita et al., 1997; Matsuo et al., 2003). Dr. Liliana Attisano provided pCMV5B-Lef1-HA and pCMV5B-Lef1ΔHMG-HA. Dr. Randall Moon provided pCMV5-Lef1Δβ-catenin.
4.2. Cell Culture and Differentiation
MC3T3 Ffl and MC3T3 L2 cells were described previously (Kahler et al., 2006). MC3T3 cells were maintained in minimal essential medium (MEM) supplemented with 10% fetal bovine serum (FBS), 1% non-essential amino acids, 200 mM L-glutamine, 50 units/ml penicillin and 50 µg/ml streptomycin. To induce differentiation, confluent MC3T3 cells were transferred to differentiation medium (α-MEM supplemented with 10% FBS, 50 units/ml penicillin, 50 µg/ml streptomycin, 50 µg/ml ascorbic acid and 10 mM β-glycerolphosphate), which was replaced every three days.
C2C12 cells were maintained at 1.5 × 105 cells per 75 cm2 in Dulbecco’s modified Eagle’s medium (DMEM) (Gibco) containing 10% FBS (Gibco), 100U/ml penicillin and 100 µg/ml streptomycin, at 37 °C in a humidified atmosphere, 5% CO2. To induce differentiation along the osteoblastic lineage, C2C12 cells were plated at 2 × 104 cells/cm2 and the medium was replaced with DMEM containing 5% FBS, 100U/ml penicillin, 100 µg/ml streptomycin and 300 ng/ml of BMP-2 for the indicated lengths of time.
Twelve micromolar of either Col11a1 antisense (5’-ACCTGGACCAGGGCTCCATCACCAA -3’) or standard control morpholino, (5’-CCTCTTACCTCAGTTACAATTTATA-3’) (Gene Tools) and 6 µM of Endo-Porter (Gene Tools) were added to C2C12 cells at 2 × 104 cells/cm2. Cells were incubated for 24 hours in medium containing 5% FBS, 100 U/ml penicillin and 100 µg/ml streptomycin prior to inducing osteoblastic differentiation as described above.
One microgram of either Col11a1 N-terminal fragment V0 or V1B purified protein was added to C2C12 cells and MC3T3 cells cultured in osteogenic medium as described above at a density of 1.5 × 105 cells per well of a 12-well plate. Col11a1 V0 or V1B protein was added to the cells every 3 days over the course of the differentiation when the medium was replaced.
4.3. Collagen Fragment Synthesis
V0 and V1B sequences were amplified, cloned and expressed as described previously (Gregory et al., 2000). Recombinant proteins were purified, refolded and characterized as previously described (Warner et al., 2007).
4.4. Alkaline Phosphatase and von Kossa Staining
C2C12 cells were plated at 2 × 104 cells/cm2 on tissue culture-treated glass slides (Nunc) in the presence or absence of the collagen 11a1 antisense or control morpholino for 14 days with 300 ng/ml of BMP-2 (R&D Systems). C2C12 cells were then fixed for 5 minutes in cold methanol and incubated for 45 minutes in 0.1 mg/ml of naphthol AS-MX phosphate, 0.4% N, N-Dimethylformamide, 0.2M Tris-HCl pH 8.3 and 0.6mg/ml of Red Violet LB salt. The slides were then washed in distilled water four times and stained with 2.5% silver nitrate for 30 minutes, followed by 4 washes in distilled water.
4.5. Cell Transfection and Luciferase Assays
Prior to transfection, C2C12 cells were seeded at a density of 4 × 104 cells/well into 12-well plates and allowed to adhere overnight. C2C12 cells were transiently transfected with pGL3 or pGL3-Col11a1-luc (200 ng/well), pRL-null (50 ng/well), and pCMV5B-Lef1-HA, pCMV5B-Lef1ΔHMG-HA or pCMV5-Lef1Δβ-catenin as indicated using LipofectAMINE (Invitrogen) as directed by the manufacturer. Transcription of luciferase reporter constructs was measured using the Dual-Luciferase Reporter Assay System kit (Promega) according to the manufacturer’s instructions 24 hours after transfection.
4.6. Reverse Transcriptase Polymerase Chain Reaction
RT-PCR was performed by reverse transcribing 1 µg RNA using the Invitrogen Superscript First-Strand Synthesis System for RT-PCR. Targets were amplified from the cDNA (1 µl of a 1:10 dilution) using the sequence-specific primers: Lef1 forward 5’-TCACTGTCAGGCGACACTTC-3’, Lef1 reverse 5’-TGAGGCTTCACGTGCATTAG-3’, Col3a1 forward 5’-GTCCACGAGGTGACAAAGGT-3’, Col3a1 reverse 5’-CATCTTTTCCAGGAGGTCCA-3’, Col5a1 forward 5’-TGCCCTCTGACTGCCTCTAT-3’, Col5a1 reverse 5’-CACATTGCAGCCTGAAAGAA-3’, Col6a2 forward 5’-CTTCCCCTACCCCAAGTCTC-3’, Col6a2 reverse 5’-TGATATGGGGCTCTCAGGTC-3’, Col11a1 forward 5’-CTGGTCATCCTGGGAAAGAA-3’, Col11a1 reverse 5’-TTGAATCCTGGAAAGCCATC-3’, Actin forward 5’-AAGGAAGGCTGGAAAAGAGC-3’, Actin reverse 5’-GCTACAGCTTCACCACCACA-3’, Alkaline phosphatase forward 5’-TGTTGACAAGGCAGACAAGC-3’, Alkaline phosphatase reverse 5’-CAGGACCGTTGCCGTATAGT-3’. Standard RT-PCRs were performed in a Bio Rad iCycler. All quantitative PCRs were done in a Roche Light Cycler. Amplification of targets was performed on cDNA using the SYBR Green Taq ReadyMix, Capillary Formulation (Sigma).
4.7. Extracellular Matrix Preparation
Media were collected from 14-day differentiation cultures and the remaining cells were washed once with 0.5 M acetic acid. The washes were pooled with the conditioned media before equal volumes of 4 M NaCl in 0.5 M acetic acid were added. Proteins were precipitated by incubating this mixture for 48 hours at 4° C. To collect the extracellular matrix proteins, the washed cells were treated overnight with 1 mg/ml pepsin in 0.5 M acetic acid at 4° C. The supernatants were collected and proteins precipitated as described above. All protein precipitates were collected by centrifugation for 1 hour at 26,000 × g, resuspended in 0.5 M acetic acid and dialyzed against 0.5 M acetic acid to remove the salts. The samples were then lyophilized and resuspended in 0.5 M acetic acid.
4.8. Immunoblotting
Protein was quantitated using the Bio-Rad DC Protein Assay kit. Protein aliquots were mixed with non-reducing protein sample dye and resolved by SDS 7% PAGE. Proteins were transferred to PVDF membranes for immunoblotting with a rabbit polyclonal antibody recognizing the C-telopeptide domain of collagen 11a1 (20545) (Li et al., 1995; Davies et al., 1998) or mouse monoclonal anti-decorin antibody (Scott et al., 1993). Col11a1 and decorin antibodies were diluted 1:1000 and 1:40, respectively, in TBST (TBS, 0.4% tween-20) with 3% BSA. Dr. Paul G. Scott graciously provided the anti-decorin antibody.
4.9. Immunofluorescence and Microscopy
C2C12 cells were plated as described above on glass slides in the presence or absence of Col11a1 antisense or control morpholinos. All treatment groups received 300 ng/ml of BMP-2 following 24 hours incubation with the indicated morpholino. The cells were fixed for 5 minutes in cold methanol at the indicated time points ranging from 0 hours to 48 hours following treatment with BMP-2. The slides were then rinsed in TBS and permeablized in TBS-0.5% Triton X-100 (TX) for 10 minutes. The slides were washed 3 × 5 minutes in TBS-0.1% TX, followed by blocking with 2% BSA in TBS-0.1% TX for 10 minutes. Anti-collagen 11a1 antibody was added to the slide at a dilution of 1:100 in 2% BSA, TBS-0.1% TX for 1.5 hours in a humidified chamber at 37°C. The cells were subsequently washed 5× 5minutes in TBS-0.1% TX. Following the last wash, the secondary antibody, a Rhodamine (TRITC)-conjugated AffiniPure Donkey Anti-Rabbit IgG (Jackson ImmunoResearch Laboratories) diluted 1:100 in TBS-0.1% TX was added and incubated for 1 hour in a humidified chamber at 37°C. The slides were then washed 3× 5 minutes in TBS-0.1% TX and incubated in 3.0 µg/ml of DAPI for 10 minutes (Molecular Probes). The slides were again washed 3× 5 minutes in TBS-0.1% TX, rinsed once in TBS, drained, mounted with Vectashield according to the manufacturer’s protocol (Vector Laboratories) and sealed.
For microscopic observation and photomicrography of the fluorescently labeled cells, an Olympus BX60 fluorescence microscope equipped with a PM-10AD system for photomicrography was used. The fluorescent molecules were excited with a 100 W mercury lamp. TRITC-labeled molecules were detected with a filter set having a 510–560 nm wavelength bandpass, a 565 nm dichroic beamsplitter and a 575 to 645 nm emission filter (scale 10 nm).
For microscopic observation and photomicrography of the general morphology and of alkaline phosphatase and Von Kossa staining of the cells, a Nikon Inverted Microscope Eclipse TS100-F equipped with a Spot Insight Color digital camera and software was used (Nikon Instruments Inc.). The cells were detected with a 40× objective using Hoffman Modulation Contrast (HMC) microscopy (scale 10nm).
Acknowledgments
This work was supported by NIH grants AR050074 and AR048147 to JJW, T32-AR050938, AR47985, AR48672, RR16454, Idaho SBOE Musculoskeletal Research Center, Lori and Duane Stueckle Professorship, and funding from the M. J. Murdock Foundation to JTO.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Annunen S, Korkko J, Czarny M, Warman ML, Brunner HG, Kaariainen H, Mulliken JB, Tranebjaerg L, Brooks DG, Cox GF, Cruysberg JR, Curtis MA, Davenport SL, Friedrich CA, Kaitila I, Krawczynski MR, Latos-Bielenska A, Mukai S, Olsen BR, Shinno N, Somer M, Vikkula M, Zlotogora J, Prockop DJ, Ala-Kokko L. Splicing mutations of 54-bp exons in the COL11A1 gene cause Marshall syndrome, but other mutations cause overlapping Marshall/Stickler phenotypes. Am. J. Hum. Genet. 1999;65:974–983. doi: 10.1086/302585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayme S, Preus M. The Marshall and Stickler syndromes: objective rejection of lumping. J. Med. Genet. 1984;21:34–38. doi: 10.1136/jmg.21.1.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baron R, Rawadi G, Roman-Roman S. Wnt signaling: a key regulator of bone mass. Curr. Top. Dev. Biol. 2006;76:103–127. doi: 10.1016/S0070-2153(06)76004-5. [DOI] [PubMed] [Google Scholar]
- Behrens J, von Kries JP, Kuhl M, Bruhn L, Wedlich D, Grosschedl R, Birchmeier W. Functional interaction of beta-catenin with the transcription factor LEF-1. Nature. 1996;382:638–642. doi: 10.1038/382638a0. [DOI] [PubMed] [Google Scholar]
- Bi Y, Stuelten CH, Kilts T, Wadhwa S, Iozzo RV, Robey PG, Chen XD, Young MF. Extracellular matrix proteoglycans control the fate of bone marrow stromal cells. J. Biol. Chem. 2005;280:30481–30489. doi: 10.1074/jbc.M500573200. [DOI] [PubMed] [Google Scholar]
- Bix G, Iozzo RV. Matrix revolutions: "tails" of basement-membrane components with angiostatic functions. Trends Cell Biol. 2005;15:52–60. doi: 10.1016/j.tcb.2004.11.008. [DOI] [PubMed] [Google Scholar]
- Clevers H, van de Wetering M. TCF/LEF factor earn their wings. Trends Genet. 1997;13:485–489. doi: 10.1016/s0168-9525(97)01305-x. [DOI] [PubMed] [Google Scholar]
- Comper WD. Extracellular Matrix. Amsterdam, The Netherlands: Harwood Academic Publishers; 1996. [Google Scholar]
- Davies GB, Oxford JT, Hausafus LC, Smoody BF, Morris NP. Temporal and spatial expression of alternative splice-forms of the alpha1(XI) collagen gene in fetal rat cartilage. Dev. Dyn. 1998;213:12–26. doi: 10.1002/(SICI)1097-0177(199809)213:1<12::AID-AJA2>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- Fichard A, Kleman JP, Ruggiero F. Another look at collagen V and XI molecules. Matrix Biol. 1995;14:515–531. doi: 10.1016/s0945-053x(05)80001-0. [DOI] [PubMed] [Google Scholar]
- Fischer H, Salahshor S, Stenling R, Bjork J, Lindmark G, Iselius L, Rubio C, Lindblom A. COL11A1 in FAP polyps and in sporadic colorectal tumors. BMC Cancer. 2001;1:17. doi: 10.1186/1471-2407-1-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galceran J, Sustmann C, Hsu SC, Folberth S, Grosschedl R. LEF1-mediated regulation of Delta-like1 links Wnt and Notch signaling in somitogenesis. Genes Dev. 2004;18:2718–2723. doi: 10.1101/gad.1249504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelse K, Poschl E, Aigner T. Collagens--structure, function, and biosynthesis. Adv Drug Deliv Rev. 2003;55:1531–1546. doi: 10.1016/j.addr.2003.08.002. [DOI] [PubMed] [Google Scholar]
- Glass DA, 2nd, Karsenty G. In vivo analysis of Wnt signaling in bone. Endocrinology. 2007;148:2630–2634. doi: 10.1210/en.2006-1372. [DOI] [PubMed] [Google Scholar]
- Gokhale JA, Robey PG, Boskey AL. The Biochemistry of Bone. Osteoporosis. San Diego, CA: Academic Press; 2001. [Google Scholar]
- Gregory KE, Oxford JT, Chen Y, Gambee JE, Gygi SP, Aebersold R, Neame PJ, Mechling DE, Bachinger HP, Morris NP. Structural organization of distinct domains within the non-collagenous N-terminal region of collagen type XI. J. Biol. Chem. 2000;275:11498–11506. doi: 10.1074/jbc.275.15.11498. [DOI] [PubMed] [Google Scholar]
- Griffith AJ, Sprunger LK, Sirko-Osadsa DA, Tiller GE, Meisler MH, Warman ML. Marshall syndrome associated with a splicing defect at the COL11A1 locus. Am. J. Hum. Genet. 1998;62:816–823. doi: 10.1086/301789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iozzo RV. Basement membrane proteoglycans: from cellar to ceiling. Nat Rev Mol Cell Biol. 2005;6:646–656. doi: 10.1038/nrm1702. [DOI] [PubMed] [Google Scholar]
- Iyama K, Sumiyoshi H, Khaleduzzaman M, Matsuo N, Ninomiya Y, Yoshioka H. Differential expression of two exons of the alpha1(XI) collagen gene (Col11a1) in the mouse embryo. Matrix Biol. 2001;20:53–61. doi: 10.1016/s0945-053x(00)00130-x. [DOI] [PubMed] [Google Scholar]
- Jakkula E, Melkoniemi M, Kiviranta I, Lohiniva J, Raina SS, Perala M, Warman ML, Ahonen K, Kroger H, Goring HH, Ala-Kokko L. The role of sequence variations within the genes encoding collagen II, IX and XI in non-syndromic, early-onset osteoarthritis. Osteoarthritis Cartilage. 2005;13:497–507. doi: 10.1016/j.joca.2005.02.005. [DOI] [PubMed] [Google Scholar]
- Kahler RA, Galindo M, Lian J, Stein GS, van Wijnen AJ, Westendorf JJ. Lymphocyte enhancer-binding factor 1 (Lef1) inhibits terminal differentiation of osteoblasts. J. Cell. Biochem. 2006;97:969–983. doi: 10.1002/jcb.20702. [DOI] [PubMed] [Google Scholar]
- Kahler RA, Westendorf JJ. Lymphoid enhancer factor-1 and beta-catenin inhibit Runx2-dependent transcriptional activation of the osteocalcin promoter. J. Biol. Chem. 2003;278:11937–11944. doi: 10.1074/jbc.M211443200. [DOI] [PubMed] [Google Scholar]
- Kim SW, Park JI, Spring CM, Sater AK, Ji H, Otchere AA, Daniel JM, McCrea PD. Non-canonical Wnt signals are modulated by the Kaiso transcriptional repressor and p120-catenin. Nat Cell Biol. 2004;6:1212–1220. doi: 10.1038/ncb1191. [DOI] [PubMed] [Google Scholar]
- Kinoshita A, Greenwel P, Tanaka S, Di Liberto M, Yoshioka H, Ramirez F. A transcription activator with restricted tissue distribution regulates cell-specific expression of alpha1(XI) collagen. J. Biol. Chem. 1997;272:31777–31784. doi: 10.1074/jbc.272.50.31777. [DOI] [PubMed] [Google Scholar]
- Li Y, Lacerda DA, Warman ML, Beier DR, Yoshioka H, Ninomiya Y, Oxford JT, Morris NP, Andrikopoulos K, Ramirez F, et al. A fibrillar collagen gene, Col11a1, is essential for skeletal morphogenesis. Cell. 1995;80:423–430. doi: 10.1016/0092-8674(95)90492-1. [DOI] [PubMed] [Google Scholar]
- Lui VC, Kong RY, Nicholls J, Cheung AN, Cheah KS. The mRNAs for the three chains of human collagen type XI are widely distributed but not necessarily co-expressed: implications for homotrimeric, heterotrimeric and heterotypic collagen molecules. Biochem. J. 1995;311(Pt 2):511–516. doi: 10.1042/bj3110511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin S, Richards AJ, Yates JR, Scott JD, Pope M, Snead MP. Stickler syndrome: further mutations in COL11A1 and evidence for additional locus heterogeneity. Eur. J. Hum. Genet. 1999;7:807–814. doi: 10.1038/sj.ejhg.5200377. [DOI] [PubMed] [Google Scholar]
- Matsuo N, Yu-Hua W, Sumiyoshi H, Sakata-Takatani K, Nagato H, Sakai K, Sakurai M, Yoshioka H. The transcription factor CCAAT-binding factor CBF/NF-Y regulates the proximal promoter activity in the human alpha 1(XI) collagen gene (COL11A1) J. Biol. Chem. 2003;278:32763–32770. doi: 10.1074/jbc.M305599200. [DOI] [PubMed] [Google Scholar]
- Medeck RJ, Sosa S, Morris N, Oxford JT. BMP-1-mediated proteolytic processing of alternatively spliced isoforms of collagen type XI. Biochem. J. 2003;376:361–368. doi: 10.1042/BJ20030894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meisler MH, Griffith AJ, Warman M, Tiller G, Sprunger LK. Gene symbol: COL11A1. Disease: Marshall syndrome. Hum. Genet. 1998;102:498. doi: 10.1007/s004390050731. [DOI] [PubMed] [Google Scholar]
- Melkoniemi M, Koillinen H, Mannikko M, Warman ML, Pihlajamaa T, Kaariainen H, Rautio J, Hukki J, Stofko JA, Cisneros GJ, Krakow D, Cohn DH, Kere J, Ala-Kokko L. Collagen XI sequence variations in nonsyndromic cleft palate, Robin sequence and micrognathia. Eur. J. Hum. Genet. 2003;11:265–270. doi: 10.1038/sj.ejhg.5200950. [DOI] [PubMed] [Google Scholar]
- Morris NP, Oxford JT, Davies GB, Smoody BF, Keene DR. Developmentally regulated alternative splicing of the alpha1(XI) collagen chain: spatial and temporal segregation of isoforms in the cartilage of fetal rat long bones. J. Histochem. Cytochem. 2000;48:725–741. doi: 10.1177/002215540004800601. [DOI] [PubMed] [Google Scholar]
- Noponen-Hietala N, Kyllonen E, Mannikko M, Ilkko E, Karppinen J, Ott J, Ala-Kokko L. Sequence variations in the collagen IX and XI genes are associated with degenerative lumbar spinal stenosis. Ann. Rheum. Dis. 2003;62:1208–1214. doi: 10.1136/ard.2003.008334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen BR. Mutations in collagen genes resulting in metaphyseal and epiphyseal dysplasias. Bone. 1995;17:45S–49S. doi: 10.1016/8756-3282(95)00208-u. [DOI] [PubMed] [Google Scholar]
- Park JI, Kim SW, Lyons JP, Ji H, Nguyen TT, Cho K, Barton MC, Deroo T, Vleminckx K, Moon RT, McCrea PD. Kaiso/p120-catenin and TCF/beta-catenin complexes coordinately regulate canonical Wnt gene targets. Dev Cell. 2005;8:843–854. doi: 10.1016/j.devcel.2005.04.010. [DOI] [PubMed] [Google Scholar]
- Robey PG, Boskey AL. Extracellular Matrix and Biomineralization of Bone. Primer on the Metabolic Bone Diseases Disorders and Mineral Metabolism. Washington, D.C.: American Society for Bone and Mineral Research; 2003. [Google Scholar]
- Rodriguez RR, Seegmiller RE, Stark MR, Bridgewater LC. A type XI collagen mutation leads to increased degradation of type II collagen in articular cartilage. Osteoarthritis Cartilage. 2004;12:314–320. doi: 10.1016/j.joca.2003.12.002. [DOI] [PubMed] [Google Scholar]
- Scott PG, Dodd CM, Pringle GA. Mapping the locations of the epitopes of five monoclonal antibodies to the core protein of dermatan sulfate proteoglycan II (decorin) J. Biol. Chem. 1993;268:11558–11564. [PubMed] [Google Scholar]
- Seegmiller R, Fraser FC, Sheldon H. A new chondrodystrophic mutant in mice. Electron microscopy of normal and abnormal chondrogenesis. J. Cell Biol. 1971;48:580–593. doi: 10.1083/jcb.48.3.580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snead MP, Yates JR. Clinical and Molecular genetics of Stickler syndrome. J. Med. Genet. 1999;36:353–359. [PMC free article] [PubMed] [Google Scholar]
- Snead MP, Yates JR, Williams R, Payne SJ, Pope FM, Scott JD. Stickler syndrome type 2 and linkage to the COL11A1 gene. Ann. N. Y. Acad. Sci. 1996;785:331–332. doi: 10.1111/j.1749-6632.1996.tb56300.x. [DOI] [PubMed] [Google Scholar]
- So CL, Kaluarachchi K, Tam PP, Cheah KS. Impact of mutations of cartilage matrix genes on matrix structure, gene activity and chondrogenesis. Osteoarthritis Cartilage. 2001;9 Suppl A:S160–S173. [PubMed] [Google Scholar]
- Spring CM, Kelly KF, O'Kelly I, Graham M, Crawford HC, Daniel JM. The catenin p120ctn inhibits Kaiso-mediated transcriptional repression of the beta-catenin/TCF target gene matrilysin. Exp. Cell Res. 2005;305:253–265. doi: 10.1016/j.yexcr.2005.01.007. [DOI] [PubMed] [Google Scholar]
- Stratton RF, Lee B, Ramirez F. Marshall syndrome. Am. J. Med. Genet. 1991;41:35–38. doi: 10.1002/ajmg.1320410111. [DOI] [PubMed] [Google Scholar]
- Thom JR, Morris NP. Biosynthesis and proteolytic processing of type XI collagen in embryonic chick sterna. J. Biol. Chem. 1991;266:7262–7269. [PubMed] [Google Scholar]
- Tran KT, Griffith L, Wells A. Extracellular matrix signaling through growth factor receptors during wound healing. Wound Repair Regen. 2004;12:262–268. doi: 10.1111/j.1067-1927.2004.012302.x. [DOI] [PubMed] [Google Scholar]
- Tran KT, Lamb P, Deng JS. Matrikines and matricryptins: Implications for cutaneous cancers and skin repair. J. Dermatol. Sci. 2005;40:11–20. doi: 10.1016/j.jdermsci.2005.05.001. [DOI] [PubMed] [Google Scholar]
- van Genderen C, Okamura RM, Farinas I, Quo RG, Parslow TG, Bruhn L, Grosschedl R. Development of several organs that require inductive epithelial-mesenchymal interactions is impaired in LEF-1-deficient mice. Genes Dev. 1994;8:2691–2703. doi: 10.1101/gad.8.22.2691. [DOI] [PubMed] [Google Scholar]
- Warner LR, Blasick CM, Brown RJ, Oxford JT. Expression, purification, and refolding of recombinant collagen alpha1(XI) amino terminal domain splice variants. Protein Expr. Purif. 2007;52:403–409. doi: 10.1016/j.pep.2006.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterman ML. Lymphoid enhancer factor/T cell factor expression in colorectal cancer. Cancer Metastasis Rev. 2004;23:41–52. doi: 10.1023/a:1025858928620. [DOI] [PubMed] [Google Scholar]
- Westendorf JJ, Kahler RA, Schroeder TM. Wnt signaling in osteoblasts and bone diseases. Gene. 2004;341:19–39. doi: 10.1016/j.gene.2004.06.044. [DOI] [PubMed] [Google Scholar]
- Winter RM, Baraitser M, Laurence KM, Donnai D, Hall CM. The Weissenbacher-Zweymuller, Stickler, and Marshall syndromes: further evidence for their identity. Am. J. Med. Genet. 1983;16:189–199. doi: 10.1002/ajmg.1320160209. [DOI] [PubMed] [Google Scholar]
- Yoshioka H, Greenwel P, Inoguchi K, Truter S, Inagaki Y, Ninomiya Y, Ramirez F. Structural and functional analysis of the promoter of the human alpha 1(XI) collagen gene. J. Biol. Chem. 1995;270:418–424. doi: 10.1074/jbc.270.1.418. [DOI] [PubMed] [Google Scholar]
- Yoshioka H, Iyama K, Inoguchi K, Khaleduzzaman M, Ninomiya Y, Ramirez F. Developmental pattern of expression of the mouse alpha 1 (XI) collagen gene (Col11a1) Dev. Dyn. 1995;204:41–47. doi: 10.1002/aja.1002040106. [DOI] [PubMed] [Google Scholar]