

Abstract
Therapeutic application of recombinant adeno-associated virus (AAV) has been limited by its small carrying capacity. To overcome this limitation trans-splicing vectors were developed recently. However, the transduction efficiency of trans-splicing vectors is considerably lower than that of a single intact vector in skeletal muscle. To improve trans-splicing vectors for skeletal muscle gene therapy, we examined whether coinfection efficiency is a rate-limiting factor in the mdx mouse, a model for Duchenne muscular dystrophy. Two different AAV viruses were delivered to the mdx muscle. Similar to previous reports in normal muscle, coinfection efficiency reached approximately 90% in the diseased muscle. This result suggests that coinfection is not a hurdle in dystrophic muscle. Another critical step in the trans-splicing method is the transcription and splicing across the inverted terminal repeat (ITR) junction in the reconstituted genome. To test whether this represented a significant obstacle, we systematically evaluated the transcription, pre-mRNA stability and splicing, and translation in a synthetic lacZ construct that mimicked the reconstituted genome. Although inserting an intron in the lacZ gene had no effect on its expression, inclusion of the ITR junction in the intron reduced expression by 50%. In construct containing the ITR junction, the mRNA transcript level was significantly reduced. This mRNA level reduction was associated with decreased pre-mRNA stability. These data suggest that the accumulation of mRNA is a rate-limiting factor in trans-splicing vector-mediated gene therapy.
OVERVIEW SUMMARY
Dual-viral trans-splicing vectors have been developed to overcome the size limitation of recombinant adeno-associated virus (AAV). Despite the success in doubling the packaging capacity, transduction efficiency of this novel approach is substantially lower than that of a single intact AAV vector. Several factors could influence the trans-splicing vector-mediated gene therapy. These include dual-vector coinfection, viral genome recombination, transcription and splicing across the inverted terminal repeat (ITR) junction, and pre-mRNA stability. A coinfection study with two different AAV viruses excluded coinfection efficiency as a rate-limiting factor in dystrophic muscle. To examine whether mRNA production represented a barrier, we quantified RNA transcripts from a reconstituted genome by RNase protection assay. Our results suggest that decreased mRNA accumulation may account for a 50% reduction of the transduction efficiency in the trans-splicing vectors. Furthermore, the attenuated mRNA steady-state level correlated with a rapid degradation of pre-mRNA containing double-D ITR.
INTRODUCTION
Adeno-associated virus (AAV) is a single-strand DNA virus. Recombinant AAV has become a powerful gene therapy vehicle in the last decade. A long list of therapeutic proteins has been successfully expressed via AAV vectors in many different animal models of human diseases. Recent developments in clinical trials brings the hope of eventually curing some of these diseases at the gene level in the near future (Duan et al., 2002). Despite this promising progress, the therapeutic application of AAV vectors has been restricted mainly to diseases that involve small cDNAs. The size limitation is inherent to the physical property of the viral particle itself. The maximal genome length that can be encapsidated into the 20-nm AAV virion is approximately 5 kb (Dong et al., 1996). This capacity hurdle has excluded the direct packaging of larger therapeutic genes, such as the 6-kb mini-dystrophin gene for Duchenne's muscular dystrophy (DMD) therapy (Chamberlain, 2002).
To overcome the size limitation, researchers have recently developed several mechanistically different dual-vector approaches to expand AAV packaging capacity (Duan et al., 2000a; Nakai et al., 2000; Sun et al., 2000; Yan et al., 2000; Duan et al., 2001b; Chao et al., 2002; Halbert et al., 2002; Reich et al., 2003). The common theme is to split the target gene into two parts and then to package them into two AAV particles. Transgene expression is reconstituted in infected cells either through viral genome recombination and subsequent splicing, or through homologous recombination of the overlapping regions. Among these, the trans-splicing strategy has shown a potential for muscle-based gene therapy (Duan et al., 2001b). Therapeutic level of secreted proteins, such as erythropoietin, has been produced from trans-splicing vectors in muscle (Yan et al., 2000). However, the level of expression from the trans-splicing vector seems to be insufficient to treat inherited muscle diseases (such as DMD) that involve structure proteins (Duan et al., 2001b). In this regard, novel strategies are needed to boost expression from trans-splicing vectors before they can be used for treating dystrophic muscle diseases.
The trans-splicing-mediated AAV gene therapy involves at least four steps: (1) coinfection of two independent AAV vectors into the same cell; (2) head-to-tail recombination between incoming vector genomes; (3) transcription elongation through the inverted terminal repeat (ITR) junction and the stability of pre-mRNA; and (4) splicing across the ITR junction. Defect in any one of above steps will compromise the overall therapeutic efficacy of this unique gene delivery system. As a first step toward improving the trans-splicing approach, we explored some potential early rate-limiting steps. We first examined whether coinfection efficiency would be a problem in the diseased muscle. Because the formation of the irregular double-D ITR structure is a unique feature of AAV intermolecular recombination (Xiao et al., 1997; Duan et al., 1999), we also evaluated whether mRNA production from the reconstituted AAV genome represents a significant hurdle for the trans-splicing method in general. Our results suggest that dystrophic skeletal muscle can be efficiently cotransduced by two different AAV vectors simultaneously. However, the double-D ITR junction prevents the accumulation of spliced mRNA and subsequent protein expression by 50%. Our results suggest that increasing mRNA production may improve the overall efficiency of the trans-splicing vector-mediated gene therapy.
MATERIALS AND METHODS
Plasmids for evaluating the effect of the double-D ITR junction on transcription and splicing
Three plasmid constructs were used in this study including, pCMVβ (Clontech, Palo Alto, CA; catalog #6177-1), pZX18 and pZX19.
pCMVβ contains the full-length prokaryotic β-galactosidase gene. It was used as a template for introducing intron and the double-D ITR junction. This plasmid was also used as a reference for the maximal achievable levels of transcription and translation.
pZX18 was generated by inserting a chimeric intron from pCI (Promega, Madison, WI; catalog #E1731) at a putative splicing site (AAG/G, between nt 2406 and nt 2407) in pCMVβ(the nucleotide numbering accords with the β-galactosidase gene sequence in pCMVβ). This was accomplished by a three-stage cloning procedure. First, we modified pCMVβ by inactivating several restriction sites that are located outside β-galactosidase coding region, including a NarI site (nt 7006), two XbaI sites (nt 625, nt 4507), two SphI sites (nt 18, nt 4529) and a SacI site (nt 535) (a second SacI site located within the β-galactosidase gene was not inactivated). The restriction site inactivation was achieved either by blunting or by making small deletions. The resultant plasmid was named pZX7. In the second stage, a polymerase chain reaction (PCR)-mediated approach was used to create a new XbaI site and a new NarI site at 19 bp upstream and 7 bp downstream of the putative splicing site, respectively, in pZX7. The sequence changes represent silent mutations (TCCCGC to TCTAGA for XbaI site, GGAGGC to GGCGCC for NarI site). The PCR primers were DL191 (first forward primer), CGGCGGTGAAATTATCGATG; DL192 (first reverse primer), GGCGCCCGCGCGTCTAGAAGGATCGACAGATTTGATCCAGCG (the XbaI site is underlined); DL193 (second forward primer), TCTAGACGCGCGGGCGCCGACACCACGGCCACCGATATTATT (the NarI site is underlined); and DL175 (second reverse primer), CAGATGGCGATGGCTGGTTTCCATC. Briefly, a 600-bp PCR fragment from the first PCR primer set and a 485-bp PCR fragment from the second PCR primer set were sequentially cloned into the ClaI-XbaI-SacI sites in pBluescript II SK + (Stratagene, La Jolla, CA; catalog #212205) to produce an intermediate plasmid pZX6. A 1091-bp ClaI/SacI fragment from pZX6 was subsequently swapped into pZX7 to generate pZX10. At the last stage, we used a PCR-mediated approach again to insert the chimeric intron into the putative splicing site. The PCR primers included DL197 (forward primer), GCGCTCTAGACCGGTGCAGTATGAAGGTAAGTATCAAGGTTACAAGACAGG (the intron sequence is underlined); and DL198 (reverse primer) GCGCGGCGCCGCCGCCTGTGGAGAGAAAGGCAAAGTGGATG (the intron sequence is underlined). To facilitate subsequent insertion of the double-D ITR junction, we also introduced a SphI site between the splicing donor signal and the splicing branch point in the chimeric intron. Briefly, a 173-bp PCR product from DL197 and DL198 primer set was used as the template and amplified again with DL197 (forward primer) and DL199 (reverse primer, TCGCGAGCATGCGGTACCTCAGAAACGCAAGAGTCTTCT, SphI site is underlined), and DL200 (forward primer, GGTACCGCATGCTCGCGATAGGCACCTATTGGTCTTACTAG, SphI site is underlined) and DL198 (reverse primer), respectively. An equal molar ratio mixture of the above two PCR products was taken as the template and amplified by DL197 and DL198. The final 218-bp PCR product was inserted into the XbaI/NarI sites in pZX10 to create pZX18 (Fig. 2).
FIG. 2.
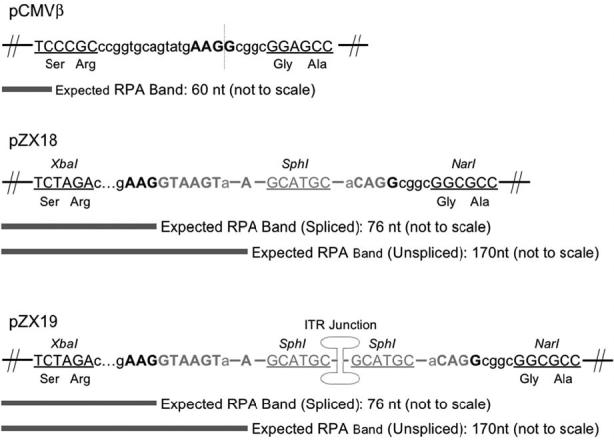
Schematic diagram of the constructs used in RNase protection assay (RPA). pCMVβ is used as a template to introduce intron and the double-D ITR junction. The dotted vertical line indicates the gene splitting site. pZX18 contains an engineered intron. Silent mutations are introduced in pZX18 to create an XbaI site upstream of intron and a NarI site downstream of intron, respectively. pZX19 is generated by inserting the double-D ITR junction in the SphI site in pZX18. The fragments that are detected by antisense RNA probe in RPA are marked at the corresponding regions (not drawn to scale). Bold: the conserved splicing signal sequences. Underlined: restriction sites. Black: nucleotides in the lacZ gene. Gray: nucleotides in intron.
pZX19 was constructed by introducing the double-D ITR junction into the chimeric intron of pZX18. Briefly, a 259-bp SphI fragment containing the head-to-tail AAV ITR junction was released from pDD212, a circular AAV genome retrieved from AAV infected muscle. pDD212 shared the exactly same double-D ITR junction structure as a previously described circular AAV transduction intermediate, p81 (Duan et al., 1999). The 259-bp SphI fragment was then inserted into the SphI site in pZX18 to create pZX19 (Fig. 2). To monitor potential PCR-introduced mutations, we have sequenced the PCR fragments and the cloning junctions in the intermediates and the final cloning products. Furthermore, β-galactosidase expression was examined at each cloning step.
Recombinant AAV production and skeletal muscle infection
The cis plasmid, pCisRSV.AP, for rAAV expressing the heat-resistant alkaline phosphatase (AP) has been described previously (Yue et al., 2003). The cis plasmid, pCisCMV.ntLacZ, for rAAV expressing the nuclear-localized lacZ was generated by cloning a 3.2-kb XhoI/NotI fragment from pAdCMV.ntLacZ into the plasmid pDD2 (Yue and Duan, 2002). The pseudotyped rAAV-5 was generated according to a previously described protocol with modification (Duan et al., 2001a). Briefly, 60% confluent 293 cells were cotransfected with cis-plasmid, AAV-2 Rep plasmid, AAV-5 helper plasmid, and adenoviral helper plasmid at a ratio of 1:1:1:3. Crude viral lysate was harvested at 60 hr post-transfection. To facilitate viral particle release, crude lysate was sequentially treated by three rounds of freeze-thaw, 10-min sonication at the power output of 10 (Sonifier Cell Disruptor, Heat Systems Inc., Farmingdale, NY), DNaseI digestion (33,000 Kunitz units per 20 × 150 mm plate cell lysate), 1% sodium deoxycholate treatment and 0.05% tissue-culture grade trypsin (final concentration) digestion. Finally, the crude viral lysate was purified through three rounds of CsCl isopycnic ultracentrifugation. The viral titer determination and quality control were carried out as described previously (Duan et al., 2001a).
All the animal experiments were carried out in accordance with National Institutes of Health (NIH) and institutional guidelines of the University of Missouri. Four-month-old male mdx mice were purchased from Jackson Laboratory (Bar Harbor, ME). Recombinant AAV was delivered to the anterior tibialis (TA) muscle according to a previously described protocol (Duan et al., 1998). In single vector infection group, 8 × 1010 viral particles (either AV.CMVntLacZ or AV.RSVAP) were injected to one TA muscle in a final volume of 40 μl. In coinfection group, 8 × 1010 AAV particles of each virus were thoroughly mixed and then injected at a final volume of 40 μl per muscle. At 1 month postinfection, muscle was harvested to determine the level of transgene expression.
Reporter gene expression assay in cultured cells
Using Lipofectamine and Plus reagent (Gibco BRL, Gaithersburg, MD), 60–70% confluent 293 cells (Graham et al., 1977) and the undifferentiated C2C12 muscle cells (Yaffe and Saxel, 1977) were cotransfected with the indicated experimental plasmids (2 μg per well in 6-well plates) and a luciferase control plasmid pDD12 (0.2 μg per well in 6-well plates), respectively. In pDD12, the luciferase expression is under the transcription control of the SV40 promoter and the SV40 polyA. The level of β-galactosidase expression in transfected 293 cells was evaluated at 24 hr posttransfection by cytochemical X-gal (5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside) staining and direct quantification of β-galactosidase enzymatic activity in cell lysate according to a published protocol (Duan et al., 2001b). The X-gal staining was carried out at 37°C for 2 hr. The β-galactosidase activity assay was measured with a Galacto-Light Plus system (Applied Biosystems, Bedford, MA; catalog BL100P) in a luminometer (TD-20/20; Turner Designs Instrument, Sunnyvale, CA). The sensitivity was set at the 60%. The transfection efficiency was normalized by luciferase activity. To measure luciferase activity, 10 μg of cell lysate was mixed with the luciferase assay buffer containing 25 mM glycylglycine (pH 7.8), 15 mM potassium phosphate (pH 7.8), 15 mM MgSO4, 4 mM EGTA, 2 mM adenosine triphosphate (ATP), and 1 mM dithiothreitol (DTT). After addition of 200 μM of luciferin, the luciferase activity was immediately determined in a TD-20/20 luminometer at a sensitivity of 90%.
Reporter gene expression assay in murine skeletal muscle
Histochemical staining for heat-resistant AP and nuclear-localized lacZ was performed at 37°C for 10 min and 2 hr, respectively, in 8 μm cryosections according to previously published protocols (Duan et al., 2000b, 2001b). To facilitate quantification of coinfection efficiency, cryosections from coinfected muscles were examined by a double-staining protocol. At first, tissue sections were stained for lacZ for 2 hr. Endogenous AP was then inactivated by incubating slides at 60°C for 45 min. Finally, viral-mediated heat-resistant AP expression was determined by a 10-min staining at 37°C. Morphometric quantification of AAV-transduced cells was performed in at least 15 cross-sections for each muscle sample. To minimize sampling error, the entire TA muscle was first divided into five approximately 2-mm thick blocks before embedding. Staining positive cells were manually counted with an electronic colony counter (Fisher Scientific, catalog # 07-910-15) from digitized images of the entire muscle cross-sections.
RNA quantification by RNase protection assay
RNase protection assay (RPA) was performed essentially as described before (Qiu et al., 2002). Briefly, 70–80% confluent 293 cells and the undifferentiated C2C12 cells were cotransfected with experimental plasmids (3.5 μg per 60-mm plate; including pCMVβ, pZX18, and pZX19, respectively) and the luciferase control plasmid pDD12 (0.2 μg per 60-mm plate), respectively, using Lipofectamine and Plus reagent (Gibco BRL). At 48 hr posttransfection, cells were trypsinized from culture plates. Protein lysate was prepared from one fifth of the cells for luciferase activity assay. Total RNA was extracted from remaining cells according to a previously published protocol (Qiu et al., 2002). Briefly, cells were washed in ice-cold phosphate-buffered saline (PBS) twice and then lysed in 4 M guanidine isothiocyanate. RNA pellet was subsequently isolated by ultracentrifugation in 5.7 M CsCl.
To generate RPA probe, a 170-bp fragment in pZX18 was PCR-amplified with a forward primer DL245, gcgcgaattcGGCCACGGCGCTAATCACGA (underlined sequence represents an EcoRI site; capital letters are nucleotides in pZX18) and a reverse primer DL246, gcgcggattcTCAGAAACGCAAGAGTCTTC (underlined sequence represents a BamHI site; capital letters are nucleotides in pZX18). The PCR product was subsequently cloned into the EcoRI/BamHI sites in pGEM-3Z (Promega). After linearization at the EcoRI site, a 220-nt anti-sense RNA probe for β-galactosidase transcripts was synthesized with SP6 RNA polymerase by in vitro transcription with [α-32P]-rUTP substrate. This probe also contained a 6-nt tail corresponding to the EcoRI cloning site and an additional 44-nt tail corresponding to the cloning junction region in pGEM-3Z. This probe could detect a 170-nt band representing unspliced transcripts from pZX18 and pZX19, a 76-nt band representing spliced transcripts from pZX18 and pZX19, and a 60-nt band representing transcripts from pCMVβ (Fig. 2). RNA ladder was generated with T7 RNA polymerase as we described before (Qiu et al., 2002).
The probe for endogenous human β-actin and mouse β-actin was produced by in vitro transcription with SP6 RNA polymerase on the pTRI-β-actin-human antisense (Ambion, Austin, TX; catalog # 7424) and pTRI-β-actin-mouse antisense (Ambion; catalog # 7423) control templates, respectively. RNA hybridization and protection were performed with 10 μg (for 293 cells) or 20 μg (for C2C12 cells) total RNA and excessive amounts of radiolabeled β-galactosidase and β-actin probes. In 293 cell RPA, the unprotected RNA was digested by RNaseT1 and RNaseA at the final concentration of 0.923 μg/ml and 18.46 μg/ml, respectively. In C2C12 cell RPA, the unprotected RNA was digested by RNaseT1 and RNaseA at the final concentration of 0.615 μg/ml and 12.27 μg/ml, respectively. RPA reaction products were resolved in 8% denaturing polyacrylamide urea gel. The protected RNA signals were quantified with the Molecular Imager FX and Quantity One (version 4.2.2 image software; Bio-Rad, Hercules, CA). Relative signal intensity was normalized for transfection efficiency (by luciferase activity), RNA extraction, RPA assay, and loading (by the internal human or mouse β-actin control).
Measurement of pre-mRNA stability by RPA
As described above, 70–80% confluent 293 cells were cotransfected with experimental plasmids (pZX18, pZX19) and the luciferase control plasmid pDD12. At 48 hr after transfection, actinomycin D (Sigma, St. Louis, MO; catalog #A1410) was added to the medium at the final concentration of 5 μg/ml. The actinomycin D concentration was decided according to previous publications and a pilot study evaluating the toxicity of different concentrations of actinomycin D (Yang et al., 1996). The 5 μg/ml concentration was the maximal concentration that permitted cell viability in 293 cells. Cell pellets were harvested at 0, 0.5, 1, and 3 hr later for protein and RNA extraction as described above. RPA was performed exactly as described above and the unspliced pre-mRNA signals at each time point were quantified.
Statistical analysis
Data are expressed as mean ± standard error of the mean (SEM) and analyzed by one-way analysis of variance (ANOVA). In the case of significant difference in ANOVA (p < 0.05), post hoc analyses were performed. Statistical significance level was set at p < 0.05.
RESULTS
Trans-splicing AAV vector-mediated expression is not limited by the coinfection efficiency in dystrophic muscle
Simultaneous uptake of both viral vectors by the same cell has to occur prior to intermolecular viral genome recombination. To compensate for the loss of a muscle structure protein by the trans-splicing approach, a fairly large population of myofibers has to be cotransduced by both vectors. Previous study demonstrated a greater than 90% coinfection efficiency in normal skeletal muscle (Yang et al., 1999). To test whether this is the case in dystrophic muscle, we coinfected mdx TA muscle with two separate AAV vectors, AV.RSVAP and AV.CMVntLacZ.
We have previously shown that respiratory syncytial virus (RSV) promoter and cytomegalovirus (CMV) promoter were equally efficient in driving transgene expression in the mdx muscle (Yue and Duan, 2002). Therefore, promoter strength will not skew data interpretation. To facilitate concomitant quantification of both AP and ntLacZ expression in the same tissue section, we developed a double staining protocol. As shown in Figure 1A, transgene-positive cells were detected at a similar sensitivity by either single- or double-staining protocol. Interestingly, ntLacZ-positive cells (22.49 ± 2.06%) were consistently lower than those of AP positive cells (55.18 ± 2.89%) despite the fact that an equal amount of viral particles and an equal volume were used in injection for both AV.RSVAP and/or AV.CMVntLacZ. This is unlikely to be caused by the saturation of one or both vectors because neither of them resulted in 100% transduction in muscle cells. Additional plasmid transfection study suggests that the discrepancy may relate to the difference in assay sensitivity in lacZ and AP staining protocol (data not shown).
FIG. 1.
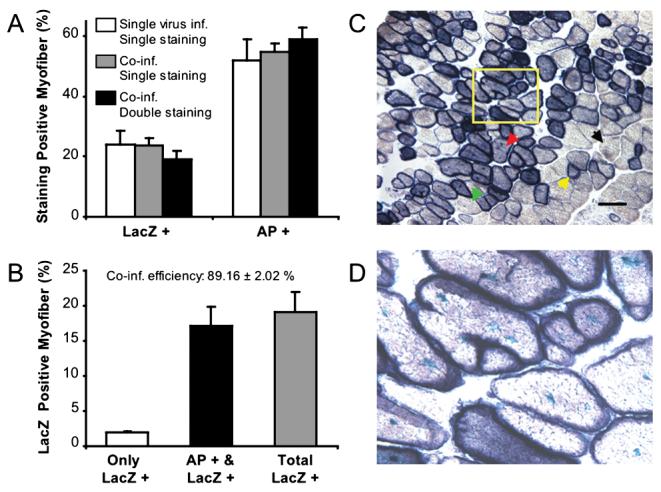
Morphometric analysis of dual-vector coinfection efficiency in mdx mouse skeletal muscle. A: Transgene expressing myofibers can be detected by either single or double staining; n = 8 for each group. B: Quantification of LacZ-positive myofibers in coinfected samples; n = 8 for each group. Coinfection efficiency was calculated as the percentage of LacZ/alkaline phosphatase (AP) double-positive myofibers in total lacZ-positive cells. C: A representative photomicrograph from a coinfected muscle sample. Double-staining protocol was used to reveal lacZ and/or AP-positive myofibers in the same section. The centrally localized nucleus is the characteristic feature of the dystrophic mdx muscle. Yellow arrow, a muscle cell expressing AP only; green arrow, a muscle cell expressing lacZ only; red arrow, a muscle cell that expresses both lacZ and AP; black arrow, a nontransduced muscle cell. D: A higher magnification photomicrograph of the boxed region in (C). Scale bar in (C) represents 200 μm.
AP is usually expressed in cytosol. Interestingly, we found a predominant AP expression at sarcolemma in mdx muscle. This is in sharp contrast to the nuclei-restricted ntLacZ expression (Figure 1C and D). The distinct physical locations of the two proteins greatly facilitated morphometric quantification. To determine co-infection efficiency, we counted the cells that were only positive for ntLacZ and cells that expressed both AP and ntLacZ. As shown in Figure 1B, only 1.93 ± 0.31% muscle cells were infected by AV.CMVntLacZ alone. However, approximately 89.16 ± 2.02% of the ntLacZ-positive cells were also positive for AP. Taken together, our results indicate that the dystrophic pathology does not influence the coinfection efficiency. Post-entry events are the rate limiting factors in dual vector-mediated gene therapy of muscular dystrophy.
Constructs for evaluating the effect of the double-D ITR junction on gene expression
Efficient RNA transcription elongation and splicing over the ITR junction are critical steps for successful gene expression from trans-splicing vectors. Head-to-tail recombination between AAV viral genomes results in an unusual double-D ITR structure (Xiao et al., 1997; Duan et al., 1999). This structure resembles the cruciform Holiday junction. To test whether the presence of this irregular DNA structure affects mRNA production, we inserted an artificial intron in the middle of the lacZ gene (Fig. 2, pZX18). This intron has been previously used to test the trans-splicing strategy in muscle (Duan et al., 2001b). A second construct was generated by inserting a previously isolated double-D ITR fragment in this intron (Fig. 2, pZX19). This construct mimics the structure of a reconstituted viral genome in the trans-splicing method. As a control for intron size change, we generated an additional control construct (pYL26). In pYL26, a nonspecific stuffer DNA from alkaline phosphatase gene (exactly the same size as the double D-ITR fragment) was inserted in pZX18. A side-by-side comparison of lacZ expression in these constructs will help us to determine whether the double-D ITR junction is a significant hurdle in the trans-splicing vector-mediated gene therapy.
The ITR junction reduces expression at the protein level
We first examined whether inserting an intron in the lacZ gene affected gene expression. At 24 hr posttransfection, similar levels of lacZ expression were observed in pCMVβ-, pZX18-, and pYL26-transfected cells (data not shown). We then examined the effect of the double-D ITR junction on gene expression. As shown in Figure 3, lacZ expression in pZX19 transfected cells reached only half of what was seen in pCMVβ and pZX18 groups (p < 0.01). Similar results were also observed at 48 hr posttransfection in 293 cells and at 24 hr and 48 hr posttransfection in C2C12 muscle cell line (data not shown). As we expected, the inclusion of a synthetic intron in lacZ gene did not diminish lacZ expression. However, our findings suggest that the presence of the double-D ITR junction within the intron reduced transgene expression.
FIG. 3.
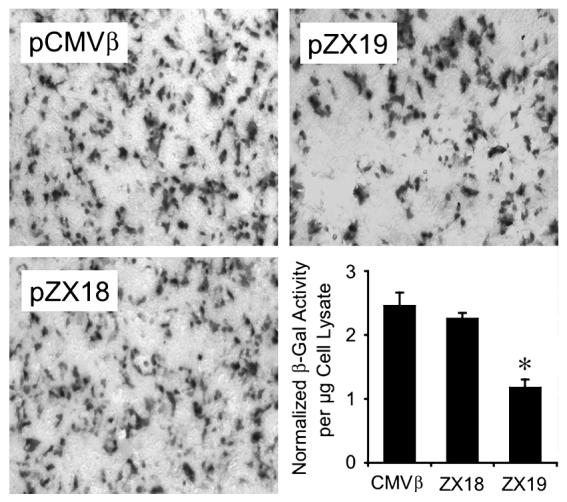
Effect of the double-D ITR junction on protein expression in 293 cells. LacZ expression from pCMVβ, pZX18, and pZX19 was examined at 24 hr posttransfection by in situ cytochemical X-gal staining and enzymatic measurement of β-galactosidase activity in cell lysates. The photomicrographs were taken under 10× objective lens in an Olympus CK40 inverted microscope (Olympus, Orangeburg, NY). These images are representative photomicrographs from four independent experiments. Data in bar graph are normalized for transfection efficiency and represent five independent experiments. *lacZ expression from pZX19 is significantly lower than that from pCMVβ and pZX18.
The double-D ITR junction prevents the accumulation of mRNA
To clarify the molecular mechanism(s) underlying the reduced protein expression further, we directly measured the transcript levels by RPA. A representative RPA result (in 293 cells) is shown in Figure 4. pCMVβ was directly transcribed into mRNA. pZX18 and pZX19 were transcribed into the unspliced pre-mRNA forms that were subsequently spliced into mRNA. Consistent with the protein expression data described above, the level of the spliced mRNA was reduced in pZX19. Furthermore, the reduction in spliced mRNA was accompanied with a reduction in unspliced RNA transcripts generated by pZX19 (Fig. 4).
FIG. 4.
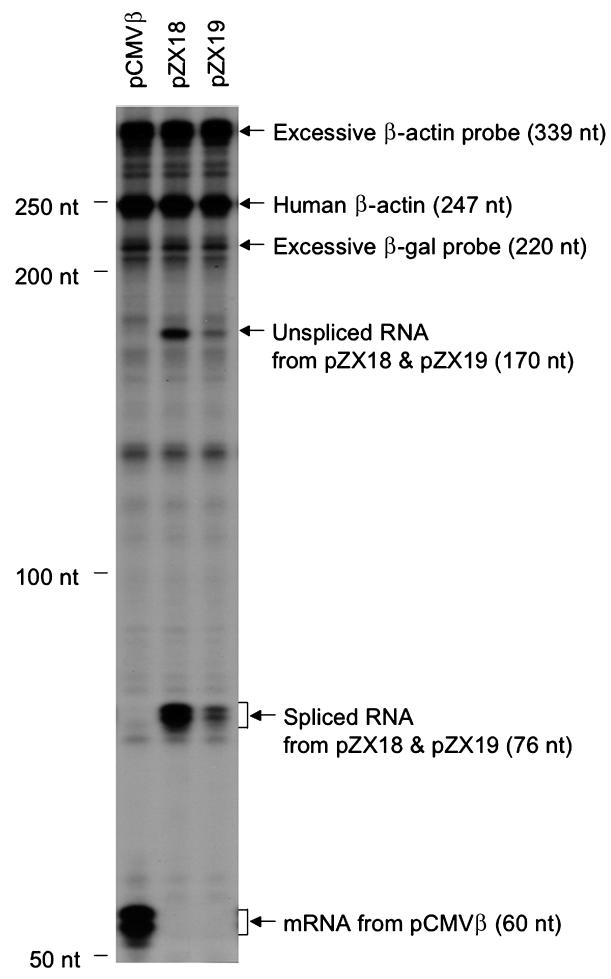
Effect of the double-D ITR junction on transcription and splicing in 293 cells. The levels of unspliced and spliced RNA transcripts were evaluated by RNase protection assay (RPA) at 48 hr posttransfection. Antisense probes for both β-galactosidase gene and human β-actin gene were included in RPA assay to simultaneously detect the RNA transcripts from the transfected plasmids (60 nt, 76 nt, and 170 nt) and the endogenous human β-actin gene (247 nt). Remaining excessive antisense probes for β-galactosidase (220 nt) and β-actin (339 nt) are marked. This is a representative photomicrograph from three independent experiments. β-gal, β-galactosidase.
To measure transcription and splicing more accurately, we normalized the RPA results for transfection efficiency, and various parameters of the RPA assay including RNA extraction and gel loading. Quantitative data from triplicate experiments are shown in Table 1. As expected, the inclusion of an intron in the lacZ gene (pZX18) had no apparent effect on RNA transcription. The total amount of RNA transcripts was approximately the same for pCMVβ and pZX18, and the majority of the pre-mRNAs generated from pZX18 were efficiently spliced. However, the accumulation of total RNA transcripts (spliced plus unspliced) from pZX19 was dramatically reduced. They only reached approximately 60% of those generated from pCMVβ and pZX18. Furthermore, the level of spliced RNA transcripts in pZX19 was also significantly reduced. Interestingly, however, the ratio of spliced to unspliced products generated from the ITR-containing construct pZX19 was not reduced. Taken together, the RPA results matched closely with the protein expression data in Figure 3 and suggest that a reduction in the accumulated levels of spliced mRNA is responsible for the reduced protein expression from the ITR containing construct pZX19.
Table 1.
Quantitative Analysis of 293 Cell RPA Results
| Total RNA | Unspliced RNA | Spliced RNA | Spliced/Unspliced | |
|---|---|---|---|---|
| pCMVβ | 109.45 ± 7.04 | 109.45 ± 7.04 | ||
| pZX18 | 103.39 ± 12.67 | 7.71 ± 1.40 | 95.68 ± 12.88 | 12.55 ± 2.10 |
| pZX19 | 62.78 ± 9.47a | 2.07 ± 0.34a | 60.71 ± 9.32a | 30.06 ± 4.69a |
Values are significantly different from pCMVβ or pZX18 groups.
RPA, RNase protection assay.
To further confirm our finding, we also repeated the RPA studies in C2C12 muscle cell line. Consistent with the 293 cell results, the reduced lacZ expression in pZX19 was also associated with the reduced mRNA accumulation in C2C12 cells (data not shown).
The double-D ITR containing RNA is less stable
Apparently, the lower levels of accumulated mRNA were responsible for the lower levels of protein expression in the ITR-containing construct (pZX19) transfected cells. Many factors can lead to a lower mRNA level. These include poor transcription, diminished splicing, and/or a reduced stability of either pre-mRNA or mRNA. Because the exact same mRNA was generated from pZX18 and pZX19, and the relative ratios of spliced versus unspliced transcripts were not reduced in pZX19, the most likely explanations in our case would be an inefficient transcription elongation and/or less stable pre-mRNA. To further distinguish between these two mechanisms, we compared the stability of pre-mRNA produced from pZX18 and pZX19. After blocking de novo RNA synthesis by actinomycin D (Yang et al., 1996), we measured the relative levels of pre-mRNA to that of β-actin RNA over a 3-hr time period. As expected, the pre-mRNA levels were gradually reduced after actinomycin D treatment. However, pre-mRNA from the ITR containing construct pZX19 was reduced at a much faster rate (Fig. 5). By 3 hr after actinomycin D administration, the pre-mRNA level was decreased to approximately 60% of the starting level in pZX19 while it only decreased to approximately 75% during the same period in pZX18 (Fig. 5). This result suggests that the relatively rapid degradation of pre-mRNA may have contributed to the diminution of the accumulated mRNA in the presence of the double-D ITR in pZX19.
FIG. 5.

Effect of the double-D ITR junction on pre-mRNA stability in 293 cells. The relative levels of pre-mRNA signals were quantified at different time points post actinomycin D treatment. The intensity of signal was normalized by transfection, RNA extraction, RNase protection assay (RPA), and RNA loading. The percentages of the signal intensity reduction from time zero were presented at 0.5 hr, 1 hr, and 3 hr after actinomycin D treatment (n = 6 for each time point). Filled triangle, pZX18 transfection. Solid circle, pZX19 transfection. The relative rates of pre-mRNA degradation for pZX18 and pZX19 are represented as dotted and solid trend-lines, respectively. Inserted table depicts the p values for statistical comparisons of pre-mRNA signal drops from time zero to later time points.
DISCUSSION
Recombinant AAV has been repeatedly demonstrated as one of the most efficient gene delivery vehicle. It encompasses several unique features of an ideal vector, such as low immune response, long-term transgene expression, and broad host range. However, because AAV is one of the smallest DNA viruses, the therapeutic cassettes that can fit into AAV vector are also limited. Consequently, the application of AAV vector to larger disease genes, such as the minidystrophin gene, has traditionally been prohibited. To take advantage of the superior features of AAV vectors, intensive attempts have been made to truncate the larger therapeutic gene to its smallest core unit. An extraordinary case is the molecular engineering of the dystrophin gene. Recently, a C-terminal deleted microdystrophin gene was developed for DMD gene therapy (Wang et al., 2000; Harper et al., 2002; Yue et al., 2003). This microgene encodes only approximately 30% of the sequence of the full-length dystrophin cDNA. The deletion of genetic material is likely going to be accompanied with a cost on the gene function. In fact, it has been shown that the microdystrophin gene is less functional than the minidystrophin gene in the diaphragm (Harper et al., 2002).
To solve the AAV packaging problem, researchers have generated a dual-vector trans-splicing system to increase AAV loading capacity (Nakai et al., 2000; Sun et al., 2000; Yan et al., 2000; Duan et al., 2001b; Chao et al., 2002; Reich et al., 2003). This method is based on the ITR-mediated reconstitution of a small intron between two split parts of a cDNA. Despite the success in doubling AAV packaging capacity, the overall efficiency of the trans-splicing method remains to be improved to treat skeletal muscle diseases. In this study, we examined whether the coinfection efficiency and the mRNA production from the reconstituted genomes were the obstacles for a successful transduction by trans-splicing vectors.
Efficient cotransduction has been shown in normal skeletal muscle (Yang et al., 1999). However, dystrophic muscles are characterized by enormous myofiber degeneration and regeneration, inflammation, fibrosis, and necrosis. Surprisingly, this dramatic pathologic change in cellular microenvironment had no effect on AAV vectors' coinfection efficiency. We observed a nearly 90% cotransduction efficiency in the mdx muscle.
It is currently unclear whether the irregular DNA structure at the ITR junction affects the processing of the RNA polymerase II, assembly and function of the spliceosome, and stability of the double-D ITR containing RNA transcripts. The synthesis of RNA requires continuous unwinding and rewinding of the template DNA at the 3′ and the 5′ ends, respectively. It is possible that the complicated Holiday structure may slow down the movement of RNA polymerase II and affect transcription elongation (Saini et al., 1990; van Holde and Zlatanova, 1994). This hypothesis is supported by several studies (Bagga et al., 1990; Perros et al., 1994). However, other recent studies have suggested that the cruciform DNA structure may not block Escherichia coli. RNA polymerase (Lyu et al., 1999). To better understand this critical step, we compared the RNA profiles generated by the construct carrying the double-D ITR and the one without ITR. Our results suggest that the double-D ITR junction can block the accumulation of mRNA and reduce protein expression.
Several different mechanisms can account for mRNA reduction. First, the presence of the double-D ITR structure may block the RNA transcription. Second, the ITR containing RNA transcripts may be less stable and third, the double-D ITR structure may block the assembly and the function of the splicesome complex and reduce splicing efficiency. As an initial step to dissect the underlying mechanisms, we quantified the relative splicing efficiency and the pre-mRNA stability. Surprisingly, the irregular double-D ITR structure does not seem to effect splicing in the context of our construct. It remains to be tested whether this observation can be applied to other genes. Nevertheless, when we examined the pre-mRNA stability, transcripts containing ITR (pZX19) were degraded at a much faster rate. It is currently unclear how the presence of the double-D ITR induces pre-mRNA degradation. In this study, we did not evaluate the effect of the double-D ITR structure on transcription elongation. However, as suggested by other studies (Bagga et al., 1990; Perros et al., 1994), it remains quite possible that a blockage at this step may have also contributed to our observation.
Since its initial report in muscle, the trans-splicing approach has attracted a great attention for expanding AAV application in many other tissues such as the liver and the retina (Chao et al., 2002; Reich et al., 2003). Identifying and overcoming the barriers in trans-splicing vector-mediated gene therapy will eventually lead to clinical success of this novel gene delivery system.
ACKNOWLEDGMENTS
This work was supported by research grants from the Muscular Dystrophy Association (D.D.), the University of Missouri Research Board (D.D.) and the National Institutes of Health to D.D. (AR-49419), and D.J.P. (AI46458 and AI56310). We thank Bethany Kent for the assistance in morphometric quantification of the muscle histochemical staining. We thank Duan laboratory members for helpful discussion.
REFERENCES
- BAGGA R, RAMESH N, BRAHMACHARI SK. Supercoil-induced unusual DNA structures as transcriptional block. Nucleic Acids Res. 1990;18:3363–3369. doi: 10.1093/nar/18.11.3363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHAMBERLAIN JS. Gene therapy of muscular dystrophy. Hum. Mol. Genet. 2002;11:2355–2362. doi: 10.1093/hmg/11.20.2355. [DOI] [PubMed] [Google Scholar]
- CHAO H, SUN L, BRUCE A, XIAO X, WALSH CE. Expression of human factor VIII by splicing between dimerized AAV vectors. Mol. Ther. 2002;5:716–722. doi: 10.1006/mthe.2002.0607. [DOI] [PubMed] [Google Scholar]
- DONG JY, FAN PD, FRIZZELL RA. Quantitative analysis of the packaging capacity of recombinant adeno-associated virus. Hum. Gene Ther. 1996;7:2101–2112. doi: 10.1089/hum.1996.7.17-2101. [DOI] [PubMed] [Google Scholar]
- DUAN D, SHARMA P, YANG J, YUE Y, DUDUS L, ZHANG Y, FISHER KJ, ENGELHARDT JF. Circular intermediates of recombinant adeno-associated virus have defined structural characteristics responsible for long-term episomal persistence in muscle. J. Virol. 1998;72:8568–8577. doi: 10.1128/jvi.72.11.8568-8577.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DUAN D, YAN Z, YUE Y, ENGELHARDT JF. Structural analysis of adeno-associated virus transduction intermediates. Virology. 1999;261:8–14. doi: 10.1006/viro.1999.9821. [DOI] [PubMed] [Google Scholar]
- DUAN D, YUE Y, YAN Z, ENGELHARDT JF. A new dual-vector approach to enhance recombinant adeno-associated virus-mediated gene expression through intermolecular cis activation. Nat. Med. 2000a;6:595–598. doi: 10.1038/75080. [DOI] [PubMed] [Google Scholar]
- DUAN D, YUE Y, YAN Z, YANG J, ENGELHARDT JF. Endosomal processing limits gene transfer to polarized airway epithelia by adeno-associated virus. J. Clin. Invest. 2000b;105:1573–1587. doi: 10.1172/JCI8317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DUAN D, YAN Z, YUE Y, DING W, ENGELHARDT JF. Enhancement of muscle gene delivery with pseudotyped AAV-5 correlates with myoblast differentiation. J. Virol. 2001a;75:7662–7671. doi: 10.1128/JVI.75.16.7662-7671.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DUAN D, YUE Y, ENGELHARDT JF. Expanding AAV packaging capacity with trans-splicing or overlapping vectors: A quantitative comparison. Mol. Ther. 2001b;4:383–391. doi: 10.1006/mthe.2001.0456. [DOI] [PubMed] [Google Scholar]
- DUAN D, YUE Y, ENGELHARDT JF. Adeno-associated virus. In: Albelda SM, editor. Lung Biology in Health and Disease, Gene Therapy in Lung Disease. Marcel Dekker Inc.; New York, NY: 2002. pp. 51–92. [Google Scholar]
- GRAHAM FL, SMILEY J, RUSSELL WC, NAIRN R. Characteristics of a human cell line transformed by DNA from human adenovirus type 5. J. Gen. Virol. 1977;36:59–74. doi: 10.1099/0022-1317-36-1-59. [DOI] [PubMed] [Google Scholar]
- HALBERT CL, ALLEN JM, MILLER AD. Efficient mouse airway transduction following recombination between AAV vectors carrying parts of a larger gene. Nat. Biotechnol. 2002;20:697–701. doi: 10.1038/nbt0702-697. [DOI] [PubMed] [Google Scholar]
- HARPER SQ, HAUSER MA, DELLORUSSO C, DUAN D, CRAWFORD RW, PHELPS SF, HARPER HA, ROBINSON AS, ENGELHARDT JF, BROOKS SV, CHAMBERLAIN JS. Modular flexibility of dystrophin: Implications for gene therapy of Duchenne muscular dystrophy. Nat. Med. 2002;8:253–261. doi: 10.1038/nm0302-253. [DOI] [PubMed] [Google Scholar]
- LYU YL, LIN CT, LIU LF. Inversion/dimerization of plasmids mediated by inverted repeats. J. Mol. Biol. 1999;285:1485–1501. doi: 10.1006/jmbi.1998.2419. [DOI] [PubMed] [Google Scholar]
- NAKAI H, STORM TA, KAY MA. Increasing the size of rAAV-mediated expression cassettes in vivo by intermolecular joining of two complementary vectors [see comments] Nat. Biotechnol. 2000;18:527–532. doi: 10.1038/75390. [DOI] [PubMed] [Google Scholar]
- PERROS M, SPEGELAERE P, DUPONT F, VANACKER JM, ROMMELAERE J. Cruciform structure of a DNA motif of parvovirus minute virus of mice (prototype strain) involved in the attenuation of gene expression. J. Gen. Virol. 1994;75:2645–2653. doi: 10.1099/0022-1317-75-10-2645. [DOI] [PubMed] [Google Scholar]
- QIU J, NAYAK R, TULLIS GE, PINTEL DJ. Characterization of the transcription profile of adeno-associated virus type 5 reveals a number of unique features compared to previously characterized adeno-associated viruses. J. Virol. 2002;76:12435–12447. doi: 10.1128/JVI.76.24.12435-12447.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- REICH SJ, AURICCHIO A, HILDINGER M, GLOVER E, MAGUIRE AM, WILSON JM, BENNETT J. Efficient trans-splicing in the retina expands the utility of adeno-associated virus as a vector for gene therapy. Hum. Gene Ther. 2003;14:37–44. doi: 10.1089/10430340360464697. [DOI] [PubMed] [Google Scholar]
- SAINI KS, SUMMERHAYES IC, THOMAS P. Molecular events regulating messenger RNA stability in eukaryotes. Mol. Cell. Biochem. 1990;96:15–23. doi: 10.1007/BF00228449. [DOI] [PubMed] [Google Scholar]
- SUN L, LI J, XIAO X. Overcoming adeno-associated virus vector size limitation through viral DNA heterodimerization. Nat. Med. 2000;6:599–602. doi: 10.1038/75087. [DOI] [PubMed] [Google Scholar]
- VAN HOLDE K, ZLATANOVA J. Unusual DNA structures, chromatin and transcription. Bioessays. 1994;16:59–68. doi: 10.1002/bies.950160110. [DOI] [PubMed] [Google Scholar]
- WANG B, LI J, XIAO X. Adeno-associated virus vector carrying human minidystrophin genes effectively ameliorates muscular dystrophy in mdx mouse model. Proc. Natl. Acad. Sci. U.S.A. 2000;97:13714–13719. doi: 10.1073/pnas.240335297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- XIAO X, XIAO W, LI J, SAMULSKI RJ. A novel 165-base-pair terminal repeat sequence is the sole cis requirement for the adeno-associated virus life cycle. J. Virol. 1997;71:941–948. doi: 10.1128/jvi.71.2.941-948.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YAFFE D, SAXEL O. Serial passaging and differentiation of myogenic cells isolated from dystrophic mouse muscle. Nature. 1977;270:725–727. doi: 10.1038/270725a0. [DOI] [PubMed] [Google Scholar]
- YAN Z, ZHANG Y, DUAN D, ENGELHARDT JF. From the Cover: Trans-splicing vectors expand the utility of adeno-associated virus for gene therapy. Proc. Natl. Acad. Sci. U.S.A. 2000;97:6716–6721. doi: 10.1073/pnas.97.12.6716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YANG J, ZHOU W, ZHANG Y, ZIDON T, RITCHIE T, ENGELHARDT JF. Concatamerization of adeno-associated viral circular genomes occurs through intermolecular recombination. J. Virol. 1999;73:9468–9477. doi: 10.1128/jvi.73.11.9468-9477.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YANG UC, HUANG W, FLINT SJ. mRNA export correlates with activation of transcription in human subgroup C adenovirus-infected cells. J. Virol. 1996;70:4071–4080. doi: 10.1128/jvi.70.6.4071-4080.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YUE Y, DUAN D. Development of multiple cloning site cis-vectors for recombinant adeno-associated virus production. Bio-techniques. 2002;33:672, 674, 676–678. doi: 10.2144/02333dd03. [DOI] [PubMed] [Google Scholar]
- YUE Y, LI Z, HARPER SQ, DAVISSON RL, CHAMBERLAIN JS, DUAN D. Microdystrophin gene therapy of cardiomyopathy restores dystrophin-glycoprotein complex and improves sarcolemma integrity in the Mdx mouse heart. Circulation. 2003;108:1626–1632. doi: 10.1161/01.CIR.0000089371.11664.27. [DOI] [PMC free article] [PubMed] [Google Scholar]