

Abstract
Targeted drug delivery aims to increase the therapeutic index by making more drug molecules available at the diseased sites while reducing systemic drug exposure. In this update, we provide an overview of polymer-drug conjugates that have advanced into the clinical trials. These systems use synthetic water-soluble polymers as the drug carriers. The preclinical pharmacology and recent data in clinical trials with poly(L-glutamic acid)-paclitaxel (PG-TXL) are discussed first. This is followed by a summary of conjugates of a variety of polymeric conjugates with chemotherapeutic agents. Results from early clinical trials of these polymer-drug conjugates have demonstrated several advantages over the corresponding parent drugs, including fewer side effects, enhanced therapeutic efficacy, ease of drug administration, and improved patient compliance. Collectively, these data warrant further clinical development of polymer-drug conjugates as a new class of anticancer agents.
Keywords: Polymer-Drug Conjugates, Anticancer Drugs, Nanocarreirs, Paclitaxel, PG-TXL
1. Introduction
Most chemotherapeutic drugs used clinically are limited by a relatively low therapeutic index, owing to toxic side effects. In the field of anticancer therapy, two strategies for improving the therapeutic efficacy of anticancer agents have emerged over the past several decades. The first approach is the design and development of agents that modulate the molecular processes and pathways specifically associated with tumor progression. The success of this approach is shown by the successful introduction of a new breed of molecularly targeted anticancer agents such as imatinib mesylate (Gleevec), gefitinib (Iressa), trastuzumab (Herceptin), and cetuximab (C225, Erbitux). Alternatively, existing anticancer agents can be made more effective by using nanocarriers that bring more drug molecules to the tumor site, compared with the conventional formulation, while reducing exposure of normal tissues to the drug.
The idea of covalently attaching chemotherapeutic agents to a water-soluble polymer was first proposed by Ringsdorf [1] in the mid-1970s. In this model, it was envisioned that not only could the pharmacokinetics of the drug attached to the polymeric carrier be modulated but also that active targeting could be achieved if a homing moiety was introduced to the same polymeric carrier (Fig. 1). Since then, polymer-drug conjugates have become a fast-growing field, with nearly a dozen polymeric conjugates advancing to the clinical trial stage (Table 1). Results from early clinical trials of these polymer-drug conjugates have demonstrated several advantages over the corresponding parent drugs, including fewer side effects, enhanced therapeutic efficacy, ease of drug administration, and improved patient compliance. Increased therapeutic efficacy is achieved primarily through an enhanced permeability and retention (EPR) effect of long-circulating polymers [2]. Among synthetic polymer-drug conjugates, poly(L-glutamic acid) (PG)-paclitaxel (PG-TXL) (CT-2103, Xyotax®) has advanced to Phase III clinical trials and is positioned to be the first of its class to reach the market. We previously reviewed the chemistry and applications of PG conjugates with various chemotherapeutic agents [3]. In this article, we will update the current clinical development of polymer-drug conjugates as novel anticancer agents, with particular emphasis on PG-TXL. Many excellent reviews with different focuses have been published, including those based on polymer-protein conjugates [4–7], most of which use polyethylene glycol (PEG) as the drug carrier [5–9]. These materials will not be covered here.
Fig. 1.
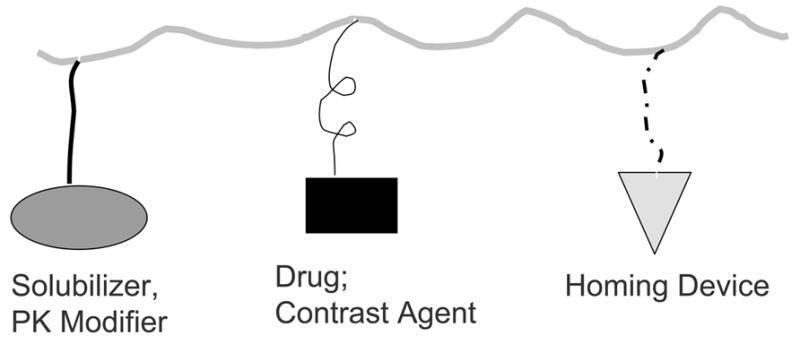
Model for targetable polymer-drug conjugates according to Ringsdof [1], in which a solubility enhancer, a pharmacokinetic modifier, a homing device, and specific drugs or imaging probes could be attached to the same polymeric chain.
Table 1.
Summary of clinical studies of PG-TXL (CT-2103)
| Effect Studied | Status | Results | References |
|---|---|---|---|
| Safety and Pharmacokinetics | Phase Ia (n = 19 on 3- weekly schedule); Phase Ib (n = 11 on 2-weekly schedule) | Phase Ia: MTD, 233 mg/m2; DLT, neutropenia. Phase 1b: MTD, 177 mg/m2; DLT, neuropathy. Observed prolonged plasma half-life (>100 h) and low steady-state volume of distribution, indicating a distribution of PG-TXL restricted to plasma and extracellular fluids | [29] |
| Safety | Phase I (n = 22) PG-TXL used in combination with carboplatin; solid tumors | MTD, 250 mg/m2; DLTs, neutropenia and thrombocytopenia. 3 patients with ovarian cancer had partial responses | [30] |
| Safety | Phase I (n = 21) PG-TXL plus concurrent radiation; esophageal and gastric cancer | MTD, 70 mg/m2/week; DLTs, gastritis, esophagitis, eutropenia. 4 of 12 patients with loco-regional disease had a complete response; 7 patients had a partial response | [31] |
| Safety and Efficacy | Phase II (n = 99) patients with heavily pretreated ovarian cancer | Efficacy: Response rate in platinum-sensitive patients, 28%; in platinum-resistant patients, 10%. Toxicity: grade 3 neutropenia (15%); grade 4 neutropenia (9%); grade 2 neuropathy(15%); grade 3 neuropathy (15%) | [32] |
| Safety and Efficacy | Phase II (n = 18) patients with metastatic breast cancer | Efficacy: objective response 22%. Toxicity: grade 3/4 hypersensitivity reaction (22%); grade3/4 neuropathy (22%). The study was terminated due to unexpected high incident of hypersensitivity reaction | [33] |
| Safety and Efficacy | Phase II (n = 21) patients with androgen independent prostate cancer | Efficacy: 2 previously treated and 1 untreated pts responded based on a 50% PSA reduction. Toxicity: hypersensitivity reaction (1 pts), grade2/3 neuropathy (7 pts), grade 3/4 neutropenia (8 pts) and grade 3 thrombocytopenia (1 pts) | [34] |
| Efficacy | Phase III (n = 400) CT-2103 plus carboplatin compared to Taxol® plus carboplatin; STELLAR 3 trial in PS2 pts with NSCLC | Efficacy: no difference in the duration of overall survival. Women with estrogen >30 pg/dl had a significant survival benefit when receiving CT-2103 compared to women in control arm (10.2 vs. 5.5 mo; p = 0.039). | [18, 35, 36] |
| Efficacy | Phase III (n = 477) CT-2103 as a single agent compared to gemcitabine or vinorelbine; STELLAR4 trial in PS2 patients with NSCLC | Efficacy: no difference in the duration of overall survival. Combined analysis of STELLAR 3&4 shows median survival advantage for CT-2103 treated women <55 years old (10.0 vs. 5.2 mo, p = 0.038) compared with women in the control arm | [18, 35, 36] |
| Efficacy | Phase III (n = 850) CT-2103 as a single agent compared to docetaxel; STELLAR 2 trial in PS0-2 patients with NSCLC | Efficacy: no difference in the duration of overall survival as compared to the control arm | [18, 35] |
2. PG-TXL (CT-2103)
2.1. Antitumor Activity
Paclitaxel (Taxol®, TXL) is a mitotic spindle poison that promotes microtubular aggregation and interferes with essential cellular functions such as mitosis, cell transport, and cell motility [10]. Taxol® has shown a remarkable anti-neoplastic effect in a wide range of human cancers. Initially approved for the treatment of refractory ovarian cancer in 1992, it is now the first-line therapy for metastatic breast cancer and advanced ovarian cancer. In women with metastatic breast cancer, Taxol® produced an objective response rate of 32–62% [11]. It’s effectiveness has also been demonstrated against non-small cell lung cancer (NSCLC), head and neck cancer, melanoma, and Kaposi’s sarcoma. Because of its poor aqueous solubility, use of Taxol® requires a mixture of polyethoxylated castor oil (Cremophor) and ethanol as a vehicle, and it must be infused over 3 to 24 h.
PG-TXL was initially developed to overcome the poor aqueous solubility of Taxol® and hypersensitivity reaction associated with the use of Taxol® formulated with Cremophor/alcohol. In PG-TXL, paclitaxel is conjugated to a synthetic polyamino acid, poly(L-glutamic acid) (PG) through its 2′-hydroxyl group via an ester linkage (Fig. 2). The resulting conjugate is highly water soluble (>20 mg/kg) and has demonstrated significantly enhanced antitumor efficacy and improved safety compared with paclitaxel in preclinical studies [12]. In a well-established syngeneic murine ovarian OCa-1 carcinoma model, a single dose of PG-TXL in saline (10 mg equiv. paclitaxel/ml) at a dose of 80 mg equiv. paclitaxel/kg caused significant tumor growth delay compared with the same dose of Taxol® in Cremophor/alcohol. Furthermore, a complete tumor cure was observed in all mice injected (n = 26) with PG-TXL at its maximal tolerated dose (MTD) of 160 mg equivalent paclitaxel/kg, which is two-fold higher than that of free paclitaxel. Similarly, treatment with PG-TXL caused complete tumor eradication at the MTD of PG-TXL (60 mg equiv. paclitaxel/kg) and at a lower dose of 40 mg/kg in Fischer rats with well-established rat breast 13762F adenocarcinoma. In contrast, paclitaxel at the dose of 40 mg/kg (its MTD) caused only a transient growth delay of approximately 15 days (Fig. 3).
Fig. 2.
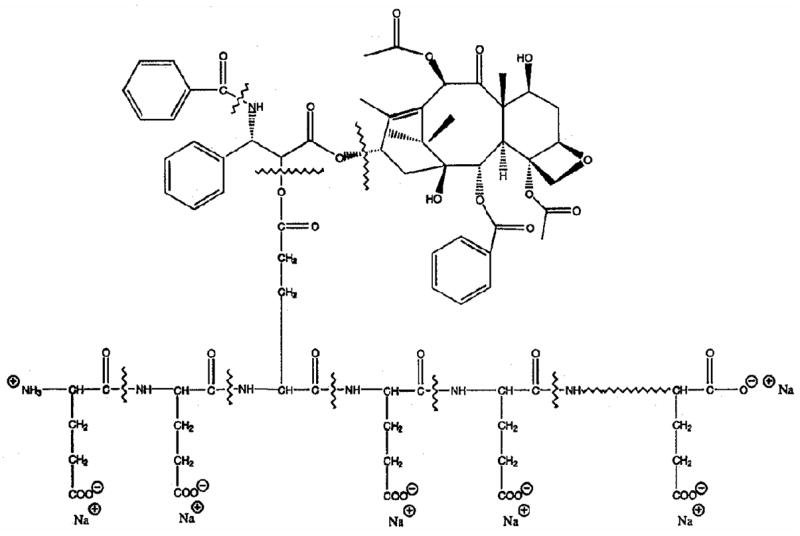
Structure of PG-TXL.
Fig. 3.
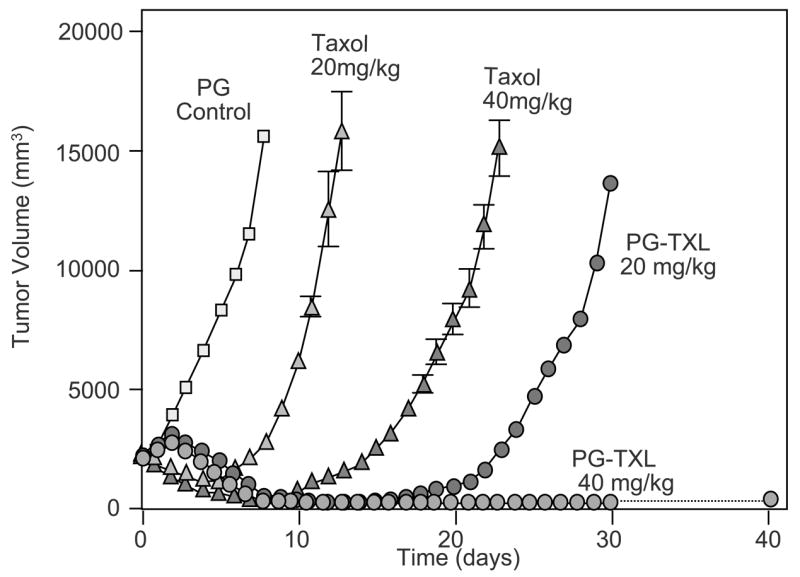
Antitumor activity of PG-TXL in rats bearing rat breast tumor 13762F. Each drug was injected i.v. in a single dose at the indicated equivalent paclitaxel concentration. Data are presented as means ± SD of tumor volume (adapted from reference [12]).
The antitumor activity of PG-TXL was further evaluated in a panel of syngeneic murine tumors (MCa-4 and MCa-35 breast cancer, HCa-1 hepatocellularcarcinoma, and FSa-II sarcoma; intramuscular inoculation), a human SKOV3ip1 ovarian tumor injected intraperitoneally, and an orthotopic human MDA-MB-435Lung2 breast tumor grown in the mammary fat pad of nude mice [13, 14]. All treatments were given intravenously as a single bolus injection. All murine tumors, whether sensitive or resistant to paclitaxel treatment, showed significant growth delay with PG-TXL at its maximum tolerated dose (160 mg equivalent paclitaxel/kg) or even at lower doses. In SKOV3ip1 tumor, PG-TXL significantly extended the survival of the mice. The median survival time for PG-TXL at 120 mg equivalent paclitaxel /kg was 75 days. In contrast, no difference in median survival time was found between paclitaxel -treated mice (56 days) and Cremophor vehicle-treated mice (59 days). In an orthotopic, metastatic MDA-MB-435Lung2 tumor model, PG-TXL at 120 mg equivalent paclitaxel/kg produced regression of the tumor in 50% of the cases. In the remaining mice with progressively growing tumors, micrometastases in the lung were found in only 25% of the animals. In comparison, treatment with paclitaxel at 60 mg/kg did not result in tumor regression, and metastases were found in 42% of the mice. In nude mice bearing i.p. implanted NMP-1 and HEY ovarian tumors, which were paclitaxel-resistant, significant improvement in survival and even some cures were observed after a single i.p. treatment with PG-TXL [14]. These studies demonstrated that PG-TXL has significant therapeutic activity against a broad range of solid tumors and metastatic disease.
2.2. Preclinical Pharmacology and Mechanism of Action
The observation that PG-TXL had antitumor activity superior to that of free paclitaxel and that PG-TXL was active against paclitaxel-resistant tumors in preclinical studies suggests that PG-TXL might possess favorable pharmacokinetic properties and/or have a mechanism of action different from that of paclitaxel. In vitro, both paclitaxel and PG-TXL induced extensive telomere erosion and telomeric associations, which are the early manifestations of programmed cell death [15]. Morphological analysis and biochemical characterizations in a panel of breast cancer cell lines confirmed that both PG-TXL and paclitaxel had similar abilities to induce apoptosis, independent of p53 status. Flow cytometric analysis further revealed that both PG-TXL and the free drug induced a characteristic G2/M arrest in the cell cycle [16]. However, compared with paclitaxel, PG-TXL appears to have reduced potency in vitro. For example, PG-TXL is >20 times less potent in supporting the growth of a paclitaxel-dependent CHO mutant cell line [12]. These data are consistent with the disturbance of microtubule polymerization as the major mechanism of action for PG-TXL and suggest that the release of paclitaxel or active species from PG-TXL is required for PG-TXL to exert its action.
In vivo, PG-TXL showed a biodistribution pattern different from that of free paclitaxel [17]. On the basis of area under the tissue concentration-time curve (AUC) values, tumor exposure to paclitaxel was five times greater when administered as PG-TXL than as paclitaxel formulated in Cremophor EL/alcohol vehicle. PG-TXL was retained much longer than free paclitaxel in the tumor due to a slower elimination of the drug conjugate. Furthermore, concentrations of free paclitaxel released from PG-TXL remained relatively constant in tumor tissue over a period of 144 h, whereas the concentration of free paclitaxel in the tumor of mice injected with paclitaxel in Cremophor EL/alcohol was reduced more than 6-fold by 144 h after injection. Singer et al. [18] showed that PG-TXL had a prolonged blood half-life and negligible release of the activity agent, paclitaxel (<1% of the total taxane). Thus, enhanced permeability and retention effect of PG-TXL are likely important contributing factors responsible for the superior therapeutic index of PG-TXL observed in preclinical studies.
Results comparing cellular uptake of tritium-labeled [3H]PG-TXL ([3H] labeled on PG polymer) and PG-[3H]TXL ([3H] labeled on paclitaxel) in tumor cells are intriguing. Whereas an insignificant amount of radioactivity was found taken up by MDA-MB-453 breast cancer cells when [3H]PG-TXL was used, there was a time-dependent increase of cell-associated radioactivity when PG-[3H]TXL was incubated with MDA-MB-453 cells. Moreover, cellular uptake of PG-[3H]TXL was more than 100 times higher than that of [3H]PG-TXL [16]. These results suggest that it was the free paclitaxel released into the culture media that had been taken up by the cells. Intact PG-TXL was probably taken up by cancer cells through less efficient pinocytosis. PG-TXL, however, can be taken up efficiently by phagocytosis. Using HPLC/mass spectral analysis, Shaffer et al. [19] recently compared the profiles of PG-TXL metabolites in murine RAW 264.7 cells with monocyte/macrophage-like activity and in cancer cells (NCI-H460 lung cancer cells and HT-29 colon cancer cells). They showed that the major intracellular metabolites of PG-TXL resulting from the degradation of polymer backbone were monoglutamyl-2′-paclitaxel (Glu-2′-TXL) and diglutamyl-2′-paclitaxel (H2N-Glu-Glu-2′-TXL). Significantly, the intracellular concentrations of Glu-2′-TXL and H2N-Glu-Glu-2′-TXL were 100–1000 times higher in RAW 264.7 cells than in the cancer cells [19].
If PG-TXL could be efficiently taken up by macrophages, what is the role of macrophages residing in the tumors in intratumoral dispersion of PG-TXL? An in vivo imaging study using PG-DTPA-Gd, a magnetic resonance imaging (MRI)-visible PG surrogate, showed that PG-DTPA-Gd was initially localized to the perivascular space after its intravenous injection but was distributed to the necrotic zone of the tumors at later times (2–4 days). Interestingly, immunohistochemical analysis revealed that biotinylated PG-DTPA-Gd co-localized with macrophages in the necrotic areas (Fig. 4) [20]. These results suggest that PG-DTPA-Gd, and possibly other PG-drug conjugates, may be captured by resident macrophages in the necrotic areas or by migrating macrophages that eventually infiltrate into the necrotic tissues. Consistent with this hypothesis, recent studies using a monoclonal antibody that specifically recognizes PG-TXL showed that PG-TXL was taken up by macrophages in vitro through endocytosis [21] and that PG-TXL preferentially localized to perinecrotic areas in tumors in vivo [22]. Further study showed that enhancement of the necrotic tissue of the tumors was observed only with polymeric contrast agents and not with low-molecular-weight contrast agents, including oligo(L-glutamic acid)-DTPA-Gd [20], suggesting that prolonged blood circulation is required for the polymer to interact with macrophages.
Fig. 4.
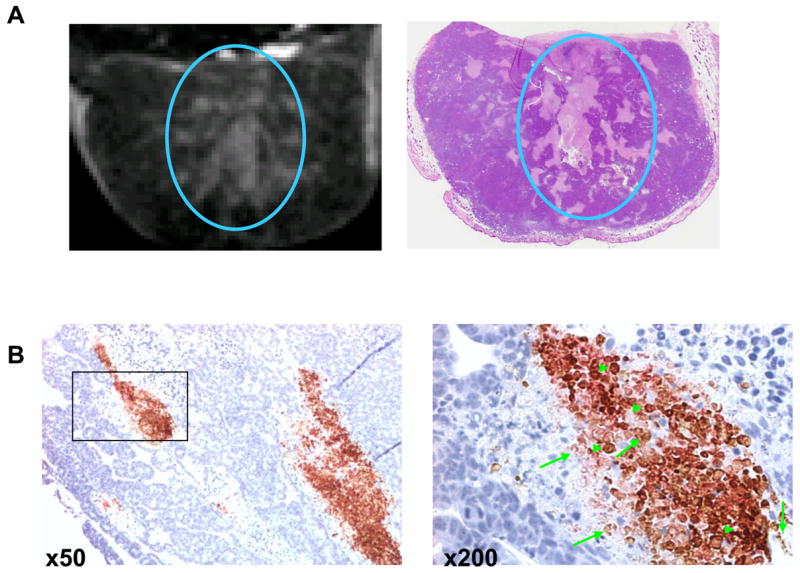
A: Representative T1-weighted MRI image from a mouse bearing a subcutaneously inoculated Colo-205 tumor (left) and a photograph of the corresponding H&E-stained section of the tumor (right). The mouse received intravenous injections of L-PG-DTPA-Gd (0.04 mmol Gd/kg). L-PG-DTPA-Gd was distributed within the necrotic area (circle). B: Co-localization of macrophages and biotinylated L-PG-DTPA-Gd in the necrotic area in OCA-1 tumors. Staining with antibody directed against macrophage antigen F4/80 (reddish-pink) and with streptavidin-horseradish peroxidase and DAB for biotinylated L-PG-DTPA-Gd showed widespread macrophage infiltration in necrotic areas as well as distribution of the polymer and its degradation products to the necrotic area (brown). The sections were counterstained with hematoxylin (blue) to label the nuclei of viable tumor cells. Examples of biotin-L-PG-DTPA-Gd staining and dual F4/80 and biotin staining are indicated with arrows and arrowheads, respectively (from reference [20].
Consistent with the MRI findings obtained with PG-DTPA-Gd, autoradiographic studies in mice injected with PG-[3H]TXL found that radioactivity was primarily located in the periphery of the tumor initially and was heterogeneously distributed throughout the tumor at a later time (i.e., by day 6) (Fig. 5) [17]. Three possible routes may account for the dispersion of radioactive species towards the center of the tumor: 1) diffusion of intact PG-[3H]TXL; 2) diffusion of small-molecular-weight metabolites of PG-[3H]TXL such as free [3H]paclitaxel, Glu-2′-TXL, and H2N-Glu-Glu-2′-TXL; and 3) uptake of PG-[3H]TXL by macrophages and subsequent migration of macrophages within the tumor mass. Intact PG-TXL, as has been found with other macromolecules and nanoparticles, is more likely to localize in the perivascular space after extravasation from tumor microvessels with minimal penetration into the tumor matrix [23, 24]. Therefore, in addition to the possible role of macrophages in the dispersion of PG-TXL within the tumor interstitium, the role of PG-TXL degradation in perivascular fluid space on the dispersion of active species (i.e., radioactivity associated with PG-[3H]TXL and its metabolites) throughout the tumor mass should be further examined.
Fig. 5.
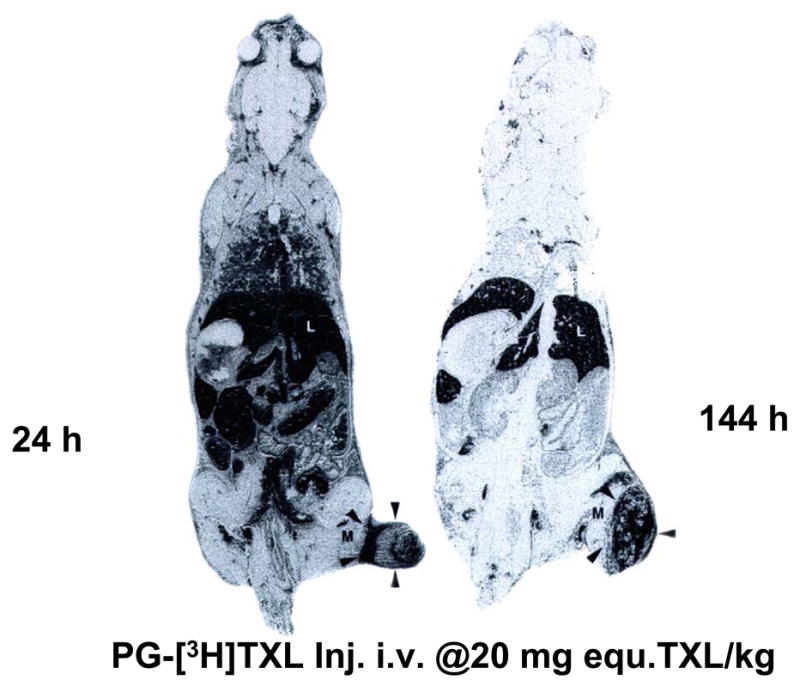
Whole-body autoradiographs of mice killed 1 day (A) and 6 days (B) after tail vein injection of PG-[3H]TXL. Most radioactivity was localized to tumor periphery at 1 day after injection, but by day 6, radioactivity had diffused into the center of the tumor. L: liver; M: muscle; Arrow head: tumor (from reference [17].
Recent studies suggest that selective proteolysis of the PG backbone through the action of cellular proteases in tumors, particularly cathepsin B, may be responsible for the increased site-specific delivery and enhanced antitumor activity of PG-TXL [19, 25]. Cathepsin B is a lysosomal cysteine protease in normal cells and tissues. In premalignant and malignant lesions, the expression of cathepsin B is highly upregulated, and the enzyme is secreted and becomes associated with the cell surface [26]. Cathepsin B has been implicated in the degradation of the extracellular matrix, a crucial step in tumor dissemination and angiogenesis. Using a noninvasive near-infrared fluorescence optical imaging technique, we recently demonstrated selective degradation of L-PG polymer but not D-PG polymer in an orthotopic U87 glioma tumor model (Fig. 6) [25]. Such a technique can be very useful in investigating the spatial and temporal distribution of the degradation products of PG-based polymeric drugs and the effect of polymer degradation on the antitumor activity of polymer-drug conjugates.
Fig. 6.
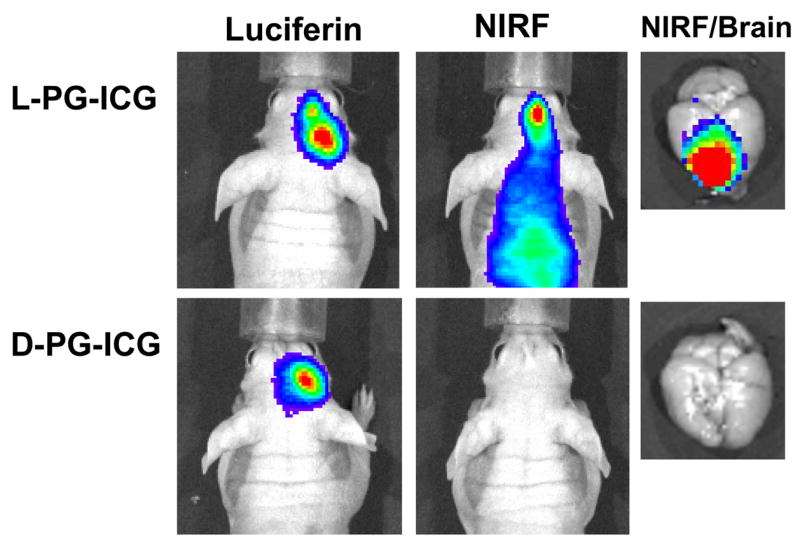
Representative in vivo near-infrared fluorescence (NIRF) images of cathepsin B activity in intracranially inoculated U87/TGL tumors. The mice used in the study had the same tumor burden, as indicated by the bioluminescence signal generated after intravenous injection of luciferin. NIRF Images were acquired 24 h after intravenous injection of L-PG-NIR813 (50 nmol/mouse) or D-PG-NIR813 (50 nmol/mouse) with the same NIR dye loading (10%) and the same molecular weight (17,500). The NIRF signal was visualized in the mouse injected with L-PG-NIR813 but not the mouse injected with D-PG-NIR813 (from reference [25]).
2.3. Clinical Findings
Results of clinical trials of CT-2103 (Xyotax®, PG-TXL) from data available in the literature are summarized in Table 1.
Phase I Studies
In the CT-2103 formulation used in clinical studies, the median molecular weight of PG-TXL is 48,000 Da, and the content of paclitaxel is about 37% by weight, equivalent to approximately one paclitaxel molecule for every 11 glutamic acid units in each PG polymer chain [18]. This formulation eliminated the use of Cremophor and alcohol and allows infusion of paclitaxel as PG-TXL over 30 minutes. In the initial clinical trials in the United Kingdom, the investigators gave PG-TXL (CT-2103) to cancer patients in a 30-minute infusion every 3 weeks at doses ranging from 30 to 720 mg/m2 [27]. They detected CT-2103 in the plasma of all of the patients and observed a long plasma half-life of up to 185 h. CT-2103 could be detected in plasma 3 weeks after administration at a dose of 200 mg/m2. Importantly, the AUC of free paclitaxel was about 1% to 2% of the AUC of CT-2103, which supported the in vivo stability of CT-2103 in the plasma and the slow, prolonged release of the active moiety [28]. The steady-state volume of distribution range was low (Vss = 2.7–13.2 L), which suggests that the distribution of CT-2103 was restricted mainly to the plasma and other extracellular body fluids [29]. The MTD was 233 mg/m2 with a 3-weekly schedule and 177 mg/m2 with a more intense 2-weekly schedule. The dose-limiting toxicity (DLT) was primarily neuropathy. Notably, alopecia was almost absent in patients treated with CT-2103 compared with an incidence of over 50% following Taxol® as a 3- or 24-h infusion.
A second Phase I study evaluated the MTD of CT-2103 in combination with carboplatin in patients with a variety of solid tumors refractory to conventional treatment [30]. The MTD was determined to be 250 mg/m2, and DLTs observed at 250 mg/m2 were neutropenia and thrombocytopenia. Interestingly, the three patients who had partial responses in this series were all patients with ovarian cancer who had previously undergone unsuccessful Taxol® therapy.
Another Phase I study determined the MTD and DLTs of PG-TXL and concurrent radiation (50.4 Gy) in patients with esophageal and gastric cancer [31]. The MTD was found to be 70 mg/m2/week, with gastritis, esophagitis, eutropenia, and dehydration being the DLTs. Strong antitumor activity was observed in this Phase I study, with four (33%) of 12 patients with loco-regional disease undergoing a complete clinical response. An additional seven patients (58%) had partial response (>50% tumor reduction by post-treatment endoscopy). These promising preliminary results with combined PG-TXL and radiation indicate that PG-TXL warrants further evaluation as a radiosensitizer in other solid tumors.
Phase II Studies
A multicenter Phase II trial of PG-TXL to determine the efficacy and toxicity in heavily pretreated patients with advanced ovarian cancer has been completed [18, 32]. Ninety-nine patients registered in this trial received PG-TXL at a dose of 175 mg/m2 every 3 weeks. The toxic effects were mild and consisted of grade 3 neuropathy (15%), grade 3 neutropenia (15%), and grade 4 neutropenia (9%). The observed response rates (28% in platinum-sensitive patients and 10% in platinum-resistant patients with one or two prior regimens) are in line with results seen in studies of other agents.
A recent report on a Phase II study in patients with metastatic breast cancer showed an unexpected incidence (22%) of hypersensitivity reactions [33]. This reaction is puzzling, considering that the approximate incidence of grade 3 or 4 hypersensitivity reaction in over 1,400 patients who have received CT-2103 in clinical trials was only 2%. Because the majority of women were postmenopausal at the study entry, one possible explanation is the differing pharmacokinetics of CT-2103 in this population of patients. Further studies are needed to determine the possible roles of sex, hormonal status, and the duration of treatment in hypersensitivity reactions to CT-2103. Of the 18 patients treated with CT-2103, four (22%) had responses. In comparison, a study with Taxol® in a similar population of patients with largely HER2-negative metastatic breast cancer showed a 14% objective response rate in the first-line setting [33]. Unfortunately, because of the unexpected high incidence of hypersensitivity reaction, this Phase II study was terminated early, and the authors were unable to evaluate the clinical benefit rate or time to progression.
In an ongoing Phase II trial in patients with androgen independent prostate cancer, CT-2103 was found well tolerated and demonstrates activity measured by PSA response and PSA velocity changes [34]. Of the 21 patients enrolled to date (16 previously treated/5 untreated), 2 previously treated patients and 1 previously untreated responded based on a 50% PSA reduction that were maintained for at least 8 wks. In addition, 4 previously treated and 2 untreated patients demonstrated a delay in their PSA velocity. Adverse events included hypersensitivity reaction (1 patient), grade 2/3 neuropathy (7 patients), grade 3/4 neutropenia (8 patients) and grade 3 thrombocytopenia (1 patient). Currently, this trial continues to enroll patients.
Phase II studies in patients with resistant colorectal cancer and in previously untreated high-risk patients with NSCLC have been completed [18].
Phase III Studies
More than 1,700 patients with NSCLC have participated in Phase III trials of PG-TXL (CT-2103), which included two Phase III trials of CT-2103 as first-line treatment in performance status-2 (PS2) patients (STELLAR 3 and STELLAR 4) and one Phase III trial of CT-2103 as second-line treatment in PS0-2 patients (STELLAR 2) [18, 35]. In the STELLAR 3 trial, CT-2103 plus carboplatin AUC 6 was compared with Taxol® plus carboplatin; in the STELLAR 4 trial, CT-22103 used as a single agent was compared with gemcitabine or vinorelbine; in STELLAR 2, CT-2103 used as a single agent was compared with docetaxel. In all studies, the primary efficacy end point was the duration of overall survival. The total numbers of patients in the STELLAR 2, STELLAR 3, and STELLAR 4 trials were 850, 400, and 477, respectively. As the results of the Phase III study completed in 2005, there was no significant overall improvement in survival of the patients evaluated. However, these studies did demonstrate similar efficacy, reduced side effects, and more convenient administration of CT-2103 compared with the control treatment arms [18, 35].
Interestingly, in a subset evaluation of women with NSCLC, the premenopausal women faired significantly better as long as their estrogen levels were normal. Clinical data from a pooled analysis of CTI’s STELLAR 3 and 4 trials showed that in the 198 women treated on those trials, superior survival was observed in those who received CT-2103 (p = 0.03). The most notable effect was among women less than 55 years old and presumably pre-menopausal who were treated with CT-2103 compared with standard chemotherapy (median survival 10.0 vs. 5.3 months, hazard ratio = 0.51, log rank p = 0.038), while a survival trend (p = 0.134) was observed in women 55 years of age and older (post-menopausal) [36]. The favorable antitumor activity among women less than 55 years stimulated the initiation of the PIONEER 1 clinical Phase III trial to evaluate prospectively CT-2103 given at a dose of 175 mg/m2 once every 3 weeks in women with advanced NSCLC and PS2 status [37].
In general, CT-2103-related nonhematological toxicities were similar to those for other agents, with dose-related neurotoxicity as the most important clinical issue [35]. Neuropathy was a common toxicity associated with exposure to taxanes. In STELLAR 3, the frequency of neuropathy (all grades) was similar between treatment arms; grade 3 neuropathy occurred in 17% of patients receiving CT-2103 (210 mg/m2) versus 10% of patients receiving Taxol®. CT-2103-induced neuropathy seemed to be dose dependent. Grade 3 neuropathy was limited to 4% of patients receiving single-agent CT-2103 175 mg/m2 in STELLAR 4. Neurotoxicity occurred significantly later with CT-2103 (210 mg/m2) compared with Taxol® in STELLAR 3, suggesting that there might be slow accumulation of CT-2103 in peripheral nerves at doses greater than (175 mg/m2). Alternatively, the longer terminal half-life of CT-2103 might be related to increased neurotoxicity at higher CT-2103 doses. These observations suggest that significant neurotoxicity can be minimized by administering CT-2103 at 175 mg/m2. Consequently, this dose will be used in subsequent trials.
A randomized Phase III trial of maintenance chemotherapy comparing 12 monthly cycles of single agent paclitaxel or Xyotax™ versus no treatment in women with stage III/IV ovarian epithelial or peritoneal cancer is currently recruiting participants (www.cancer.gov). This trial, sponsored by the National Cancer Institute and the Gynecologic Oncology Group (GOG), is projected to recruit 1550 eligible participants. The primary objective of this study is to determine whether Xyotax™ or paclitaxel, administered to women with advance ovarian cancer who have attained a clinically-defined complete response to primary platinum/taxane chemotherapy, will reduce the death rate, compared to retreatment at the time of documented disease progression. The secondary objective is to determine if, in this clinical setting, Xyotax™ produces a more favorable toxicity profile and superior quality of life compared to paclitaxel.
Summary
Compared with conventional paclitaxel-based treatment, PG-TXL showed three safety-related advantages. First, alopecia was rare, and complete hair loss was not observed. Second, nausea and vomiting were uncommon. Third, hypersensitivity reactions were rarely observed, and those that did occur were usually mild to moderate; thus, routinely used prophylactic premedications were not required. CT-2103 is active against a variety of cancers. Further studies to define toxicity and efficacy in patients with less prior therapy are needed to determine the role of CT-2103 in these cancers as first-line and second-line therapy. Of note is the observed improvement in overall survival for women receiving CT-2103 vs. control in combined log-rank analysis of STELLAR 3 and 4 Phase III trials. Consequently, a request for approval of CT-2103 for the treatment of NSCLC in women with normal estrogen levels (premenopausal) based on non-inferiority will be submitted to the EMEA, the European equivalent of the FDA, in 2008. In the United States, a confirmatory Phase III trial was initiated in 2007 on 450 women (normal premenopausal estrogen levels) for the treatment of NSCLC, comparing CT-2103 and carboplatin with paclitaxel and carboplatin. If results are favorable, CT-2103 will be submitted to the FDA for approval in 2009.
2.4. Modulation of Antitumor Therapy with External Radiation
Combining chemotherapy and radiotherapy has significantly improved response and survival rates in patients with many solid tumors. Many chemotherapeutic agents can increase the radiosensitivity of tumors, thus potentiating the tumor response to radiation-caused damage. We hypothesized that combining radiotherapy and chemotherapy using a polymer-drug conjugate could lead to a stronger radiosensitizing effect than using the parent drug owing to sustained release of the free drug from polymer-bound drug in the tumor. Irradiation can in turn potentiate the tumor response to polymer-drug conjugates by increasing tumor vascular permeability and thus the uptake of these conjugates into solid tumors. To test this hypothesis, PG-TXL was used as a model polymer-drug conjugate. Administration of PG-TXL delayed the growth of OCa-1 syngeneic murine ovarian tumors in C3Hf/Kam mice. However, when PG-TXL was given in combination with tumor irradiation, antitumor activity was significantly enhanced. Using tumor growth delay as an end point, enhancement factors ranging from 1.36 to 4.4 were observed; these values depended on the doses of PG-TXL and radiation delivered. Complete tumor regression was found with the use of increased radiation doses (>10 Gy) and PG-TXL doses (>80 mg/kg equivalent paclitaxel) [38, 39]. Similar results were observed in a mammary MCa-4 carcinoma model [40]. In contrast, combined radiotherapy and paclitaxel treatment yielded an enhancement factor of less than 1.0 in MCa-4 tumors, indicating that conjugation of paclitaxel with PG is necessary to improve the radiosensitization effect of paclitaxel. When the treatment end point was tumor cure, the enhancement factors were as high as 8.4 and 7.2 after fractionated and single-dose radiotherapy, respectively [41, 42].
To determine whether prior irradiation affects tumor uptake of PG-TXL, [3H]PG-TXL was injected into mice with OCa-1 ovarian tumors 24 h after local irradiation at 15 Gy [39]. The uptake of [3H]PG-TXL in irradiated tumors was 28% to 38% higher than that in nonirradiated tumors at different times after injection of [3H]PG-TXL, suggesting that irradiation increased the accumulation of PG-TXL in the tumors. Thus, the super-synergistic effect of combined radiotherapy and PG-TXL–based chemotherapy is partly ascribed to the enhanced permeability and retention effect of macromolecules caused by irradiation.
These promising preclinical studies have prompted the initiation of clinical trials combining PG-TXL with concurrent radiation in patients with esophageal and gastric cancer [31]. Data from this Phase I trial demonstrated major tumor responses in 91% of patients.
3. Other Polymer-Drug Conjugates in the Clinic
Over the last decade, nearly a dozen polymer-drug conjugates have entered clinical studies (Table 2). Early results have generally been promising, and antitumor activity has been seen in chemotherapy-refractory patients. On the basis of the current status in clinical studies, these conjugates can be categorized into three groups: 1) Conjugates that have progressed further to Phase II/III trials or conjugates for which further studies are planned. Polymer-drug conjugates belonging to this category include PK1, PK2, HPMA copolymer-platinate, polymeric micelle NK911, carboxymethyldextran-bound CPT DE-310, and poly(L-glutamic acid)-paclitaxel Xyotax™. 2) Conjugates that further clinical development has been either discontinued or suspended because of unacceptable toxicity, inadequate linker chemistry, or pharmacoeconomic considerations. HMPA copolymer-camptothecin, HPMA copolymer paclitaxel, PEG-camptothecin, PEG-paclitaxel, and PG-CPT (CT-2106) belong to this group. 3) Conjugates just entered into clinical trial studies, such as PEG-SN38 (EZN-2208) and a linear, cyclodextrin-bound CPT (IT101).
Table 2.
Polymer-drug conjugates in clinical trials
| Conjugates | Indication | Status | Company | References |
|---|---|---|---|---|
| HPMA-Doxorubicin(PK1; FCE28068) | Lung and breast cancers | Phase II as of 2002 | Pfizer; Cancer Research Campaign UK | [47, 48] |
| HPMA-doxorubicin- galactosamine (PK2, FCE28069) | Hepatocellular carcinoma | Phase I/II | Pfizer; Cancer Research Campaign UK | [52] |
| HPMA-camptothecin (PNU166148) | Solid tumors | Phase I; discontinued | Pfizer; Cancer Research Campaign UK | [54] |
| HPMA-paclitaxel (PNU166945) | Solid tumors | Phase I; discontinued | Pfizer; Cancer Research Campaign UK | [55] |
| HPMA-platinate (AP5346, ProLindac) | Ovarian, melanoma, and colorectal cancers | Phase I | Access Pharmaceutical | [56] |
| PEG-Camptothecin(Pegamotecan) | Solid tumors | Phase I; discontinued | Enzon | [58] |
| PEG-SN38 (EZN-2208) | Solid tumors | Phase I; initiated as of October 2007 | Enzon | [59] |
| Polymeric Micelles(NK911) | Pancreatic cancer | Phase II | Nippon Kayaku, Japan | [60] |
| Cyclodextrin-Based Polymer-CPT (IT-101) | Solid tumors | Phase I | Insert Therapeutics | [62] |
| Carboxymethyldextra n-Exatecan (DE-310) | Solid atumors | Phase I | Daiichi Pharmaceuticals, Japan | [63] |
| PG-TXL (CT-2103, Xyotax) | Lung, ovarian, colorectal, breast, and esophageal cancers | Phase III | Cell Therapeutics | [12, 18, 29–37] |
| PG-camptothecin(CT2106) | Colorectal, lung, and ovarian cancers | Phase I | Cell Therapeutics | [69, 70] |
3.1. HMPA Copolymer-Doxorubicin (PK1)
Doxorubicin is an anthracycline cytotoxic agent widely used in the treatment of solid tumors, lymphoma, and leukemia. The DLTs of doxorubicin include bone marrow suppression, mucositis, and cardiotoxicity. In an attempt to reduce its toxicity, doxorubicin was conjugated to a water-soluble synthetic polymer, N-(2-hydroxypropyl) methacrylamide (HMPA)/methacylic acid copolymers through a tetrapeptide linker (Gly-Phe-Leu-Gly) [43, 44]. The tetrapeptide linker allowed selective release of the active drug in the tumors through the action of lysosomal enzymes. The conjugate, known as PK1, was found to be more potent than free doxorubicin in antitumor activity in preclinical animal studies [45, 46]. PK1 was the first synthetic polymer-anticancer drug conjugate that entered clinical trials (in 1994) [47]. Data from Phase II trials for the treatment of breast, NSCLC, and colon cancers were presented in 2002 and showed positive responses in six of 63 patients, with manageable side effects [48]. The subject of development history, mechanism of action, and pharmacology for PK1 has been reviewed extensively [48–51].
3.2. HMPA Copolymer-Doxorubicin-Galactosamine (PK2)
PK2 is a 27-kDa HMPA copolymer derivatized with 6.5% mol/wt, <2% free doxorubicin, and 2 % mol/wt galactose, with efficient targeting of the asialoglycoprotein receptor selectively expressed on hepatocytes and in hepatomas. In preclinical murine models, 80% of an administered dose targeted the liver, associated with high anticancer activity. PK2 remained the only targeted polymeric conjugate to be tested clinically to date [52]. In Phase I/II trial, the maximum tolerated dose of PK2 was 160 mg/m2, with neutropenia being the DLT. Hepatic targeting was confirmed by planar imaging and single photon emission computed tomography (SPECT) with 123I-labeled PK2 co-injected with unlabeled PK2. The majority of conjugate was present in normal liver (16.9% after 24 h), with lower accumulations within the hepatic tumors (3.2%).
3.3. HMPA Copolymer-Camptothecin (MAG-CPT, PNU166148) and HMPA Copolymer-Paclitaxel (PNU166945)
The camptothecins (CPTs) are a family of synthetic and semisynthetic analogues of 20(S)-camptothecin that exhibit a broad range of anticancer activity by inhibiting topoisomerase-1 activity. Two properties of CPT compounds limit their therapeutic efficacy in humans: instability of the lactone form because of preferential binding of the carboxylate to serum albumin, and a lack of aqueous solubility. Conjugation of CPT to water-soluble polymeric carriers has in general resulted in improved stability of the lactone ring and increased aqueous solubility.
In HPMA-CPT (MAG-CPT, PNU166148), CPT is attached to the copolymer at the C-20 hydroxyl group of CPT through an ester linkage. A Phase I study in a total of 23 patients showed DLTs of myelosuppression, neutropaenic sepsis, and diarrhoea. The maximum tolerated dose and dose recommended for further clinical study was 200 mg equivalent CPT/m2. The plasma half-life of both MAG-CPT and released CPT was extended to > 6 days, indicating that the kinetics of free CPT was release rate dependent [53]. Results presented in an earlier clinical study showed serious bladder toxicity which became dose limiting. Patients with higher plasma AUC values had the more severe symptoms of renal toxicity [54].
In HPMA copolymer-paclitaxel (PNU166945), paclitaxel was linked to the same tetrapeptide linker used to create PK1 and PK2. In a Phase I clinical study in a small patient cohort (12 patients), one patient with advanced breast cancer had a partial response. PNU166945 showed toxicity consistent with commonly observed toxicities associated with paclitaxel [55]. One patient developed grade 3 neurotoxicity. Dose escalation was discontinued prematurely due to concerns about potential clinical neurotoxicity.
3.4. HMPA Copolymer-Platinate (AP5346)
The platinum-based compounds, cisplatin and carboplatin, are standard treatment regimens for a wide range of cancers. The DLTs of these drugs include nephrotoxicity, neurotoxicity, and myelosuppression. 1,2-Diaminocyclohexane (DACH) has received increasing attention in recent years because of its activity against cisplatin-resistant cancer cells. In AP5346, DACH-platinum was bound to HPMA via a pH-sensitive chelate. A methacrylamide monomer substituted with a triglycine aminomalonate group provided the primary binding site for the DACH-platinum moiety [56]. The conjugate contained approximately 10% platinum by weight and had a molecular weight of 25 kDa, which was designed to allow glomerular filtration while also being large enough to benefit from an EPR effect. While negligible in neutral solutions, release of the DACH-platinum moiety was increased at low pH so that platinum release was favored in environments such as the extracellular space of hypoxic tumors and the intracellular lysosomal compartment. In a clinical Phase I study, AP5346 was administered as a 1-hour intravenous infusion on days 1, 8, and 15 of a 28-day cycle. Twenty-six patients received 41 cycles. Antitumor activity included two partial responses in metastatic melanoma and ovarian cancer and an additional CA-125 normalization in a suspected ovarian cancer. AP5346 administered weekly for 3 weeks out of every 4 weeks was tolerated up to a dose of 640 mg Pt/m2 on the first cycle. The pharmacokinetics of AP5346 indicated a prolonged half-life (mean terminal half-life t1/2 = 72 h) and evidence of antitumor activity. Additional human clinical trials are planned for this agent.
3.5. PEG-Camptothecin (Pegamotecan) and PEG-SN38 (EZN-2208)
Pegamotecan was developed by Enzon Pharmaceuticals, Inc. The conjugate consists of two CPT molecules conjugated to a 40-kDa PEG using an alaninate ester linkage. Free CPT must be cleaved from the PEG to be pharmacologically active. The hydroxyl group (-OH) at the 20-position of CPT is the active portion of the molecule responsible for the conformational changes between the active lactone and relatively inactive carboxylate forms. The 20-OH of CPT in Pegamotecan, is blocked by the alaninate linker, which stabilizes the CPT molecule into its active lactone conformation [57]. The MTD of Pegamotecan when administered weekly for 3 of 4 weeks was 3,240 mg/m2, with neutropenia being its DLT [58]. Other grade 3 and 4 toxic effects were anemia, thrombocytopenia, fatigue, prolonged partial thromboplastin time, hemorrhagic cystitis, dysuria, and urinary frequency. Of the 27 patients enrolled, two patients had unconfirmed partial responses. Further development of Pegamotecan appears to be discontinued; the drug is no longer listed in the company’s drug portfolio/pipeline (www.enzon.com).
The main limitation of PEG as drug carrier is the presence of only two reactive groups per polymer chain, which leads to an intrinsically low drug payload. To overcome this limitation, the construction of a dendron structure at the PEG’s end chain has been proposed. Enzon is currently developing a conjugate of SN38, an active metabolite of CPT, with a 40-kDa PEG containing four arms. EZN-2208 has shown activity in a panel of human tumor xenografts [59]. A Phase I study of EZN-2208 to evaluate the safety and tolerability of intravenous EZN-2208 in patients with advanced solid tumors or lymphoma is currently enrolling participants (http://clinicaltrials.gov).
3.6. PEG-poly(Aspartic Acid)-Doxorubicin Micelles (NK911)
The polymeric micelle consists of a PEG-poly(aspartic acid) block copolymer conjugated with doxorubicin (NK911) [60]. PEG is believed to form the outer shell of the micelle, producing a ‘stealth’ effect that prevents NK911 from being captured by the liver and spleen. The doxorubicin-conjugated poly(aspartic acid) chain is hydrophobic and is believed to form the hydrophobic inner core of the micelles in aqueous media. The hydrophobic inner core enables NK911 to entrap a sufficient amount of doxorubicin. NK911 has a diameter of about 40 nm. The plasma AUC and the tumor AUC of NK911 were lower than those of liposomal doxorubicin because NK911 was less stable in the bloodstream than doxorubicin [60]. Twenty-three patients participated in a Phase I study performed at the National Cancer Center Hospital, Tokyo, Japan. Neutropenia was the predominant hematological toxicity, and grade 3 or 4 neutropenia was observed at doses of 50 and 67 mg/m2. The DLT occurred at a dose of 67 mg/m2 (grade 4 neutropenia lasting more than 5 days). A partial response was obtained in one patient with metastatic pancreatic cancer among the 23 patients. A Phase II trial of NK911 for the treatment of metastatic pancreatic cancer is ongoing.
3.7. Cyclodextrin-Based Polymer-CPT (IT-101)
IT101 is a conjugate of CPT and a linear, cyclodextrin-based polymer (CDP) [61, 62]. The components of CDP are beta-cyclodextrin and polyethylene-glycol (PEG). Camptothecin is covalently attached to CDP through a glycine linker, which preserves CPT in its active form and increases its water solubility. Preclinical efficacy of i.v. IT-101 (molecular weight ~90 KDa) demonstrated a significant antitumor effect with IT-101 in a range of mouse xenografts, including several tumors that were resistant to CPT [62]. Pharmacokinetic and biodistribution studies indicated that CDP–CPT conjugate gave prolonged plasma half-life, which led to enhanced distribution to tumor when compared to CPT alone [61]. These favorable results prompted the initiation of a Phase I trial designed to determine the safety, MTD, and the pharmacokinetics of IT-101 when administered intravenously to patients with relapsed or refractory cancer. A total enrollment of 36 patients is expected to complete by first quarter of 2008.
3.8. Carboxymethyldextran-Exatecan (DE-310)
In DE-310, DX-8951 (exatecan mesylate, a CPT analogue) is linked to carboxymethyldextran (Mw = 340 kDa) via a glycyl-glycyl-phenylalanyl-glycyl-peptidyl spacer. This peptide spacer is intended to provide sustained release of the active moiety DX-8951 within the tumor as a result of enzymatic cleavage of the peptide by cathepsin B and cathepsin L [63]. In Phase I trial, patients received DE-310 as a 3-hour infusion once every 2 weeks (dose, 1.0–2.0 mg/m2) or once every 6 weeks (dose, 6.0–9.0 mg/m2) [64]. Neutropenia and grade 3 thrombocytopenia, and grade 3 hepatotoxicity with veno-occlusive disease, were dose-limiting toxicities. Other hematologic and nonhematologic toxicities were mild to moderate and reversible. The apparent half-life of conjugated DX-8951, glycyl-DX-8951, and DX-8951 was 13 days. The observed long blood half-lives are no surprising considering that DE-310 had the highest molecular weight among all polymer-drug conjugates tested so far. Of the 27 patients enrolled into the study, 1 patient achieved a histologically proven complete remission, 1 patient confirmed partial remission, and 14 additional patients showed disease stabilization.
3.9. PG-Gly-CPT (CT-2106)
PG was an effective solubilizing carrier of CPT and stabilized the E-ring lactone structure in CPT. PG-CPT conjugate was obtained by directly coupling the hydroxy group at the C20(S)-position of CPT with the carboxylic acid of PG [65]. When given intravenously in four doses every 4 days at an equivalent CPT of 40 mg/kg, PG-CPT delayed the growth of established H322 human lung tumors grown subcutaneously in nude mice. In mice that received intratracheal inoculation of H322 cells, the same treatment prolonged the median survival duration in mice by four-fold when compared with that in untreated control mice. These results showed that PG was as efficient carrier of CPT [65].
Studies have systematically investigated the structural effects of the antitumor efficacy of PG-bound CPT, including linkers between PG and CPT, the point of attachment of PG on the CPT molecule, the polymer molecular weight, and drug loading [66–68]. Coupling through the 20(S)-hydroxy group of CPT with or without a glycine linker yielded the most active conjugates, because this site is located in close proximity to the lactone ring; thus, the E-ring lactone is better protected by the linked PG chains. However, the linkers affected the maximum drug payload; for example, only 15% (by weight) of CPT loading could be achieved for direct conjugation of PG-CPT by the ester linkage because of steric hindrance, whereas up to 50% CPT loading could be achieved for the case with a glycine as linker [67]. Increasing the molecular weight of PG from 33 to 50 kDa improved the antitumor efficacy of PG-Gly-CPT, probably because of an increased plasma half-life and reduced renal clearance [66]. Considering its ease of synthesis, superior aqueous solubility and stability, and marked efficacy in various in vivo tumor models, PG-Gly-CPT with 30–35% CPT was selected for further study.
CT-2106 (PG-Gly-CPT) entered clinical Phase I trials in April 2004. Preliminary results on pharmacokinetics in 19 patients with CT-2106 administered as a 10 minute intravenous infusion every 3 weeks were recently presented [69, 70]. The apparent termination t½ of unconjugated CPT is quite long—approximately 40 h. Five days after the first administration (cycle 1), the excretion in urine of CT-2106 and unconjugated CPT accounted for 27.9% and 5.1% of the administered dose, respectively. Thus, CT-2106 generates prolonged systemic exposures to unconjugated CPT in plasma, and the urine excretion levels of conjugated camptothecin are substantial. A major CPT conjugated species found in urine was glu-gly-CPT. DLs included grade 3/4 neutropenia with concomitant grade 3 thrombocytopenia (3 patients, 105 mg/m2) and grade 2 hematuria (1 patient, 25 mg/m2). Most adverse events have been mild or moderate. Four patients had stable disease (SD) for >9 weeks; 2 lung cancer patients (25 mg/m2) had SD for >27 weeks. In a second Phase I trial in 26 patients, the MTD, toxicity, pharmacokinetics, and response rate of CT-21206 when administered weekly for 3 consecutive weeks of every 4-week cycle were determined [71]. With this schedule, the MTD was 75 mg/m2. DLTs observed were grade 3 or 4 neutropenia and concomitant grade 3 or 4 thrombocytopenia in the three patients treated at 105 mg/m2 and grade 2 hematuria in a patient treated at 25 mg/m2. Of the 25 patients assessable for response, no objective responses (complete or partial) were noted. Three patients had stable disease. The absence of objective responses (complete or partial) was not surprising in this heavily treated population.
Presently, Cell Therapeutics has no clinical trial material for additional trials. Further development of CT-2106 is delayed so that the company can focus resources on the development of Xyotax™ and other agents in the pipeline.
4. Conclusions
Results from early clinical trials of about a dozen polymer-drug conjugates have demonstrated several advantages over the corresponding parent drugs, including fewer side effects, enhanced therapeutic efficacy, ease of drug administration, and improved patient compliance. Owing to its favorable physicochemical properties, PG has been shown to be an excellent polymeric carrier of therapeutic agents. Xyotax™ (PG-TXL) has become the first polymer-drug conjugate for the delivery of cytotoxic chemotherapeutic agents to advance to clinical Phase III trials. Clinical data collected over the last decade warrant further development of polymer-drug conjugates as a new class of anticancer agents. Future generation of polymer-drug conjugates will have to meet a number of challenges, including the development of novel polymers with modulated rates of degradation, versatile conjugation chemistry allowing site-specific attachment of targeting moieties, and polymerization methods that allow accurate control of polymer molecular weights and molecular weight distributions. The incorporation of novel imaging techniques into the clinical trial design of new polymer-drug conjugates is also expected to facilitate noninvasive monitoring of drug delivery efficiency in the era of individualized therapy.
Acknowledgments
This work was supported in part by the National Cancer Institute (R29 CA74819), the Department of Defense (BC960384), and the John S. Dunn Foundation. We thank Michael Worley for editing the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ringsdorf H. Structure and properties of pharmacologically active polymers. J Polym Sci Symp. 1975;51:135–153. [Google Scholar]
- 2.Maeda H, Wu J, Sawa T, Matsumura Y, Hori K. Tumor vascular permeability and the EPR effect in macromolecular therapeutics: a review. J Control Release. 2000;65:271–284. doi: 10.1016/s0168-3659(99)00248-5. [DOI] [PubMed] [Google Scholar]
- 3.Li C. Poly(L-glutamic acid)--anticancer drug conjugates. Adv Drug Deliv Rev. 2002;54:695–713. doi: 10.1016/s0169-409x(02)00045-5. [DOI] [PubMed] [Google Scholar]
- 4.Maeda H, Kono T. Neocarzinostatin: The Past, Present, and Future of an Anticancer Drug. Springer; Berlin: 1997. pp. 227–267. [Google Scholar]
- 5.Graham LM. PEGASPARAGINASE: a review of clinical studies. Adv Drug Deliv Rev. 2003;10:1293–1302. doi: 10.1016/s0169-409x(03)00110-8. [DOI] [PubMed] [Google Scholar]
- 6.Wang YS, Youngster S, Grace M, Bausch J, Bordens R, Wyss DF. Structural and biological characterization of pegylated recombinant interferon alpha-2b and its therapeutic implications. Adv Drug Deliv Rev. 2002;54:547–570. doi: 10.1016/s0169-409x(02)00027-3. [DOI] [PubMed] [Google Scholar]
- 7.Kinstler O, Molineux G, Treuheit M, Ladd D, Gegg C. Mono-N-terminal poly(ethylene glycol)-protein conjugates. Adv Drug Deliv Rev. 2002;54:477–485. doi: 10.1016/s0169-409x(02)00023-6. [DOI] [PubMed] [Google Scholar]
- 8.Harris JM, Chess RB. Effect of pegylation on pharmaceuticals. Nat Rev Drug Discov. 2003;2:214–221. doi: 10.1038/nrd1033. [DOI] [PubMed] [Google Scholar]
- 9.Greenwald RB. PEG drugs: an overview. J Control Release. 2001;74:159–171. doi: 10.1016/s0168-3659(01)00331-5. [DOI] [PubMed] [Google Scholar]
- 10.Horwitz SB. Taxol (Paclitaxel): mechanisms of action. Ann Oncol. 1994;5:S3–S6. [PubMed] [Google Scholar]
- 11.Allouache D, Gawande SR, Tubiana-Hulin M, Tubiana-Mathieu N, Piperno-Neumann S, Mefti F, Bozec L, Genot JY. First-line therapy with gemcitabine and paclitaxel in locally, recurrent or metastatic breast cancer: a phase II study. BMC Cancer. 2005;5:151. doi: 10.1186/1471-2407-5-151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li C, Yu DF, Newman RA, Cabral F, Stephens LC, Hunter N, Milas L, Wallace S. Complete regression of well-established tumors using a novel water-soluble poly(L-glutamic acid)-paclitaxel conjugate. Cancer Res. 1998;58:2404–2409. [PubMed] [Google Scholar]
- 13.Li C, Price JE, Milas L, Hunter NR, Ke S, Yu DF, Charnsangavej C, Wallace S. Antitumor activity of poly(L-glutamic acid)-paclitaxel on syngeneic and xenografted tumors. Clin Cancer Res. 1999;5:891–897. [PubMed] [Google Scholar]
- 14.Auzenne E, Donato NJ, Li C, Leroux E, Price RE, Farquhar D, Klostergaard J. Superior therapeutic profile of poly-L-glutamic acid-paclitaxel copolymer compared with taxol in xenogeneic compartmental models of human ovarian carcinoma. Clin Cancer Res. 2002;8:573–581. [PubMed] [Google Scholar]
- 15.Multani AS, Li C, Ozen M, Imam AS, Wallace S, Pathak S. Cell-killing by paclitaxel in a metastatic murine melanoma cell line is mediated by extensive telomere erosion with no decrease in telomerase activity. Oncol Rep. 1999;6:39–44. doi: 10.3892/or.6.1.39. [DOI] [PubMed] [Google Scholar]
- 16.Oldham EA, Li C, Ke S, Wallace S, Huang P. Comparison of action of paclitaxel and poly(L-glutamic acid)-paclitaxel conjugate in human breast cancer cells. Int J Oncol. 2000;16:125–132. [PubMed] [Google Scholar]
- 17.Li C, Newman RA, Wu QP, Ke S, Chen W, Hutto T, Kan Z, Brannan MD, Charnsangavej C, Wallace S. Biodistribution of paclitaxel and poly(L-glutamic acid)-paclitaxel conjugate in mice with ovarian OCa-1 tumor. Cancer Chemother Pharmacol. 2000;46:416–422. doi: 10.1007/s002800000168. [DOI] [PubMed] [Google Scholar]
- 18.Singer JW, Shaffer S, Baker B, Bernareggi A, Stromatt S, Nienstedt D, Besman M. Paclitaxel poliglumex (XYOTAX; CT-2103): an intracellularly targeted taxane. Anticancer Drugs. 2005;16:243–254. doi: 10.1097/00001813-200503000-00003. [DOI] [PubMed] [Google Scholar]
- 19.Shaffer S, Baker-Lee C, Kennedy J, Lai M, de Vries P, Buhler K, Singer J. In vitro and in vivo metabolism of paclitaxel poliglumex: identification of metabolites and active proteases. Cancer Chemother and Pharmacol. 2007;59:537–548. doi: 10.1007/s00280-006-0296-4. [DOI] [PubMed] [Google Scholar]
- 20.Jackson EF, Esparza-Coss E, Wen X, Ng CS, Daniel SL, Price RE, Rivera B, Charnsangavej C, Gelovani JG, Li C. Magnetic resonance imaging of therapy-induced necrosis using gadolinium-chelated polyglutamic acids. Int J Radiat Oncol Biol Phys. 2007;68:830–838. doi: 10.1016/j.ijrobp.2007.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chipman S, Rosler R, Bonham L, Nudelman E, Singer J. Energy dependent uptake of paclitaxel poliglumex by human NSCLC tumor and murine macrophase-like cell lines. 18th EORTC-NCI-AACR Symposium on Molecular Targets and Cancer Therapeutics; Prague, Czech Republic. November 7–10, 2006; 2006. Abstract #643. [Google Scholar]
- 22.Fornasier M, Bergottini R, Radaelli E, Cavagnoli R, Di Giovine S, De Feudis P, Chipman S, Pezzoni G, Singer J. Paclitaxel poliglumex cellular uptake by normal tissues and human tumor xenograft: an IHC study in nude mice. 18th EORTC-NCI-AACR Symposium on Molecular Targets and Cancer Therapeutics; Prague, Czech Republic. November 7–10, 2006; 2006. Abstract #637. [Google Scholar]
- 23.Jain RK. Barriers to drug delivery in solid tumors. Sci Am. 1994;271:58–65. doi: 10.1038/scientificamerican0794-58. [DOI] [PubMed] [Google Scholar]
- 24.Lu D, Wientjes MG, Lu Z, Au JL. Tumor priming enhances delivery and efficacy of nanomedicines. J Pharmacol Exp Ther. 2007;322:80–88. doi: 10.1124/jpet.107.121632. [DOI] [PubMed] [Google Scholar]
- 25.Melancon MP, Wang W, Wang Y, Shao R, Ji X, Gelovani JG, Li C. A novel method for imaging in vivo degradation of poly(L-glutamic acid), a biodegradable drug carrier. Pharm Res. 2007;24:1217–1224. doi: 10.1007/s11095-007-9253-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Roshy S, Sloane BF, Moin K. Pericellular cathepsin B and malignant progression. Cancer Metastasis Rev. 2003;22:271–286. doi: 10.1023/a:1023007717757. [DOI] [PubMed] [Google Scholar]
- 27.Todd R, Sludden J, Boddy AV, Griffin MJ, Robson L, Cassidy J, Bissett D, Main M, Brannan MD, Elliott S, Fishwick K, Verrill M, Calvert H. Phase I and pharmacological study of CT-2103, a poly(L-glutamic acid)-paclitaxel conjugate. ASCO. 2001:439. [Google Scholar]
- 28.Bernareggi A, Oldham F, Barone C. XYOTAX (paclitaxel poliglumex; CT-2103): pharmacokinetic evidence for an enhanced permeability and retention effect. 29th Congress of the European Society for Medical Oncology; 2004. [Google Scholar]
- 29.Boddy AV, Plummer ER, Todd R, Sludden J, Griffin M, Robson L, Cassidy J, Bissett D, Bernareggi A, Verrill MW, Calvert AH. A phase I and pharmacokinetic study of paclitaxel poliglumex (XYOTAX), investigating both 3-weekly and 2-weekly schedules. Clin Cancer Res. 2005;11:7834–7840. doi: 10.1158/1078-0432.CCR-05-0803. [DOI] [PubMed] [Google Scholar]
- 30.Nemunaitis J, Cunningham C, Senzer N, Gray M, Oldham F, Pippen J, Mennel R, Eisenfeld A. Phase I study of CT-2103, a polymer-conjugated paclitaxel, and carboplatin in patients with advanced solid tumors. Cancer Invest. 2005;23:671–676. doi: 10.1080/07357900500359935. [DOI] [PubMed] [Google Scholar]
- 31.Dipetrillo T, Milas L, Evans D, Akerman P, Ng T, Miner T, Cruff D, Chauhan B, Iannitti D, Harrington D, Safran H. Paclitaxel poliglumex (PPX-Xyotax) and concurrent radiation for esophageal and gastric cancer: a phase I study. Am J Clin Oncol. 2006;29:376–379. doi: 10.1097/01.coc.0000224494.07907.4e. [DOI] [PubMed] [Google Scholar]
- 32.Sabbatini P, Aghajanian C, Dizon D, Anderson S, Dupont J, Brown JV, Peters WA, Jacobs A, Mehdi A, Rivkin S, Eisenfeld AJ, Spriggs D. Phase II study of CT-2103 in patients with recurrent epithelial ovarian, fallopian tube, or primary peritoneal carcinoma. J Clin Oncol. 2004;22:4523–4531. doi: 10.1200/JCO.2004.12.043. [DOI] [PubMed] [Google Scholar]
- 33.Lin NU, Parker LM, Come SE, Burstein HJ, Haldoupis M, Ryabin N, Gelman R, Winer EP, Shulman LN. Phase II study of CT-2103 as first- or second-line chemotherapy in patients with metastatic breast cancer: unexpected incidence of hypersensitivity reactions. Invest New Drugs. 2007;25:369–375. doi: 10.1007/s10637-007-9034-y. [DOI] [PubMed] [Google Scholar]
- 34.Amato RJ, Khan M, MT Phase II study of paclitaxel poliglumex (PPX) for androgen independent prostate cancer (AIPC). American Society of Clinial Oncology (ASCO), 2007 Prostate Cancer Symposium; 2007. Abstract 243. [Google Scholar]
- 35.Bonomi P. Paclitaxel poliglumex (PPX, CT-2103): macromolecular medicine for advanced non-small-cell lung cancer. Expert Rev Anticancer Ther. 2007;7:415–422. doi: 10.1586/14737140.7.4.415. [DOI] [PubMed] [Google Scholar]
- 36.Ross H, Bonomi P, Langer C, O’Brien M, O’Byrne K, Paz-Ares L, Sandler A, Socinski M, Oldham F, Singer J. Effect of gender on outcome in two randomized phase III trials of paclitaxel poliglumex (PPX) in chemo-naïve pts with advanced NSCLC and poor performance status (PS2) J Clin Oncol. 2006;24 Abstract #7039. [Google Scholar]
- 37.Albain KS, Belani CP, Bonomi P, O’Byrne KJ, Schiller JH, Socinski M. PIONEER: a phase III randomized trial of paclitaxel poliglumex versus paclitaxel in chemotherapy-naive women with advanced-stage non-small-cell lung cancer and performance status of 2. Clin Lung Cancer. 2006;7:417–419. doi: 10.3816/CLC.2006.n.027. [DOI] [PubMed] [Google Scholar]
- 38.Li C, Ke S, Wu QP, Tansey W, Hunter N, Buchmiller LM, Milas L, Charnsangavej C, Wallace S. Potentiation of ovarian OCa-1 tumor radioresponse by poly (L-glutamic acid)-paclitaxel conjugate. Int J Radiat Oncol Biol Phys. 2000;48:1119–1126. doi: 10.1016/s0360-3016(00)00757-4. [DOI] [PubMed] [Google Scholar]
- 39.Li C, Ke S, Wu QP, Tansey W, Hunter N, Buchmiller LM, Milas L, Charnsangavej C, Wallace S. Tumor irradiation enhances the tumor-specific distribution of poly(L-glutamic acid)-conjugated paclitaxel and its antitumor efficacy. Clin Cancer Res. 2000;6:2829–2834. [PubMed] [Google Scholar]
- 40.Ke S, Milas L, Charnsangavej C, Wallace S, Li C. Potentiation of radioresponse by polymer-drug conjugates. J Control Release. 2001;74:237–242. doi: 10.1016/s0168-3659(01)00322-4. [DOI] [PubMed] [Google Scholar]
- 41.Milas L, Mason KA, Hunter N, Li C, Wallace S. Poly(L-glutamic acid)-paclitaxel conjugate is a potent enhancer of tumor radiocurability. Int J Radiat Oncol Biol Phys. 2003;55:707–712. doi: 10.1016/s0360-3016(02)04153-6. [DOI] [PubMed] [Google Scholar]
- 42.Tishler RB. Polymer-conjugated paclitaxel as a radiosensitizing agent--a big step forward for combined modality therapy? Int J Radiat Oncol Biol Phys. 2003;55:563–564. doi: 10.1016/s0360-3016(02)04154-8. [DOI] [PubMed] [Google Scholar]
- 43.Duncan R, Cable HC, Lloyd JB, Rejmanova P, Kopecek J. Degradation of side-chains of N-(2-hydroxypropyl)methacrylamide copolymers by lysosomal thiol-proteinases. Biosci Rep. 1982;2:1041–1046. doi: 10.1007/BF01122173. [DOI] [PubMed] [Google Scholar]
- 44.Duncan R, Rejmanova P, Kopecek J, Lloyd JB. Pinocytic uptake and intracellular degradation of N-(2-hydroxypropyl)methacrylamide copolymers. A potential drug delivery system. Biochim Biophys Acta. 1981;678:143–150. doi: 10.1016/0304-4165(81)90058-1. [DOI] [PubMed] [Google Scholar]
- 45.Seymour LW, Ulbrich K, Steyger PS, Brereton M, Subr V, Strohalm J, Duncan R. Tumour tropism and anti-cancer efficacy of polymer-based doxorubicin prodrugs in the treatment of subcutaneous murine B16F10 melanoma. Br J Cancer. 1994;70:636–641. doi: 10.1038/bjc.1994.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Minko T, Kopeckova P, Kopecek J. Efficacy of the chemotherapeutic action of HPMA copolymer-bound doxorubicin in a solid tumor model of ovarian carcinoma. Int J Cancer. 2000;86:108–117. doi: 10.1002/(sici)1097-0215(20000401)86:1<108::aid-ijc17>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 47.Vasey PA, Kaye SB, Morrison R, Twelves C, Wilson P, Duncan R, Thomson AH, Murray LS, Hilditch TE, Murray T, Burtles S, Fraier D, Frigerio E, Cassidy J. Phase I clinical and pharmacokinetic study of PK1 [N-(2-Hydroxypropyl)methacrylamide copolymer doxorubicin]: first member of a new class of chemotherapeutic agents--drug-polymer conjugates. Clin Cancer Res. 1999;5:83–94. [PubMed] [Google Scholar]
- 48.Bilim V. Technology evaluation: PK1, Pfizer/Cancer Research UK. Curr Opin Mol Ther. 2003;5:326–330. [PubMed] [Google Scholar]
- 49.Duncan R. Drug-polymer conjugates: potential for improved chemotherapy. Anticancer Drugs. 1992;3:175–210. doi: 10.1097/00001813-199206000-00001. [DOI] [PubMed] [Google Scholar]
- 50.Duncan R. The dawning era of polymer therapeutics. Nat Rev Drug Discov. 2003;2:347–360. doi: 10.1038/nrd1088. [DOI] [PubMed] [Google Scholar]
- 51.Duncan R, Vicent MJ, Greco F, Nicholson RI. Polymer-drug conjugates: towards a novel approach for the treatment of endrocine-related cancer. Endocr Relat Cancer. 2005;12 Suppl 1:S189–199. doi: 10.1677/erc.1.01045. [DOI] [PubMed] [Google Scholar]
- 52.Seymour LW, Ferry DR, Anderson D, Hesslewood S, Julyan PJ, Poyner R, Doran J, Young AM, Burtles S, Kerr DJ. Hepatic drug targeting: phase I evaluation of polymer-bound doxorubicin. J Clin Oncol. 2002;20:1668–1676. doi: 10.1200/JCO.2002.20.6.1668. [DOI] [PubMed] [Google Scholar]
- 53.Bissett D, Cassidy J, de Bono JS, Muirhead F, Main M, Robson L, Fraier D, Magne ML, Pellizzoni C, Porro MG, Spinelli R, Speed W, Twelves C. Phase I and pharmacokinetic (PK) study of MAG-CPT (PNU 166148): a polymeric derivative of camptothecin (CPT) Br J Cancer. 2004;91:50–55. doi: 10.1038/sj.bjc.6601922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schoemaker NE, van Kesteren C, Rosing H, Jansen S, Swart M, Lieverst J, Fraier D, Breda M, Pellizzoni C, Spinelli R, Grazia Porro M, Beijnen JH, Schellens JH, ten Bokkel Huinink WW. A phase I and pharmacokinetic study of MAG-CPT, a water-soluble polymer conjugate of camptothecin. Br J Cancer. 2002;87:608–614. doi: 10.1038/sj.bjc.6600516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Meerum Terwogt JM, ten Bokkel Huinink WW, Schellens JH, Schot M, Mandjes IA, Zurlo MG, Rocchetti M, Rosing H, Koopman FJ, Beijnen JH. Phase I clinical and pharmacokinetic study of PNU166945, a novel water-soluble polymer-conjugated prodrug of paclitaxel. Anticancer Drugs. 2001;12:315–323. doi: 10.1097/00001813-200104000-00003. [DOI] [PubMed] [Google Scholar]
- 56.Campone M, Rademaker-Lakhai JM, Bennouna J, Howell SB, Nowotnik DP, Beijnen JH, Schellens JH. Phase I and pharmacokinetic trial of AP5346, a DACH-platinum-polymer conjugate, administered weekly for three out of every 4 weeks to advanced solid tumor patients. Cancer Chemother Pharmacol. 2007;60:523–533. doi: 10.1007/s00280-006-0397-0. [DOI] [PubMed] [Google Scholar]
- 57.Greenwald RB, Pendri A, Conover C, Gilbert C, Yang R, Xia J. Drug delivery systems. 2. Camptothecin 20-O-poly(ethylene glycol) ester transport forms. J Med Chem. 1996;39:1938–1940. doi: 10.1021/jm9600555. [DOI] [PubMed] [Google Scholar]
- 58.Posey JA, III, Saif MW, Carlisle R, Goetz A, Rizzo J, Stevenson S, Rudoltz MS, Kwiatek J, Simmons P, Rowinsky EK, Takimoto CH, Tolcher AW. Phase 1 study of weekly olyethylene glycol-camptothecin in patients with advanced solid tumors and lymphomas. Clin Cancer Res. 2005;11:7866–7871. doi: 10.1158/1078-0432.CCR-05-0783. [DOI] [PubMed] [Google Scholar]
- 59.Sapra P, Mehlig M, Malaby J, Kraft P, Zhang Z, Longley C, Zhao H, Rubio B, Wu D, Greenberger LM, Horak ID. EZN-2208, a novel polyethyleneglycol-SN38 conjugate, has potent antitumor activity in a panel of human tumor xenografts, American Association of Cancer Research Annual Meeting Poster No. 1494; 2007. [Google Scholar]
- 60.Nakanishi T, Fukushima S, Okamoto K, Suzuki M, Matsumura Y, Yokoyama M, Okano T, Sakurai Y, Kataoka K. Development of the polymer micelle carrier system for doxorubicin. J Control Release. 2001;74:295–302. doi: 10.1016/s0168-3659(01)00341-8. [DOI] [PubMed] [Google Scholar]
- 61.Schluep T, Cheng J, Khin K, Davis M. Pharmacokinetics and biodistribution of the camptothecin–polymer conjugate IT-101 in rats and tumor-bearing mice. Cancer Chemotherapy and Pharmacology. 2006;57:654–662. doi: 10.1007/s00280-005-0091-7. [DOI] [PubMed] [Google Scholar]
- 62.Schluep T, Hwang J, Cheng J, Heidel JD, Bartlett DW, Hollister B, Davis ME. Preclinical efficacy of the camptothecin-polymer conjugate IT-101 in multiple cancer models. Clin Cancer Res. 2006;12:1606–1614. doi: 10.1158/1078-0432.CCR-05-1566. [DOI] [PubMed] [Google Scholar]
- 63.Inoue K, Kumazawa E, Kuga H, Susaki H, Masubuchi N, Kajimura T. CM-dextran-polyalcohol-camptothecin conjugate: DE-310 with a novel carrier system and its preclinical data. Adv Exp Med Biol. 2003;519:145–153. doi: 10.1007/0-306-47932-X_9. [DOI] [PubMed] [Google Scholar]
- 64.Soepenberg O, de Jonge MJA, Sparreboom A, de Bruin P, Eskens FALM, de Heus G, Wanders J, Cheverton P, Ducharme MP, Verweij J. Phase I and pharmacokinetic study of DE-310 in patients with advanced solid tumors. Clin Cancer Res. 2005;11:703–711. [PubMed] [Google Scholar]
- 65.Zou Y, Wu QP, Tansey W, Chow D, Hung MC, Charnsangavej C, Wallace S, Li C. Effectiveness of water soluble poly(L-glutamic acid)-camptothecin conjugate against resistant human lung cancer xenografted in nude mice. Int J Oncol. 2001;18:331–336. doi: 10.3892/ijo.18.2.331. [DOI] [PubMed] [Google Scholar]
- 66.Singer JW, De Vries P, Bhatt R, Tulinsky J, Klein P, Li C, Milas L, Lewis RA, Wallace S. Conjugation of camptothecins to poly-(L-glutamic acid) Ann N Y Acad Sci. 2000;922:136–150. doi: 10.1111/j.1749-6632.2000.tb07032.x. [DOI] [PubMed] [Google Scholar]
- 67.Singer JW, Bhatt R, Tulinsky J, Buhler KR, Heasley E, Klein P, de Vries P. Water-soluble poly-(L-glutamic acid)-Gly-camptothecin conjugates enhance camptothecin stability and efficacy in vivo. J Control Release. 2001;74:243–247. doi: 10.1016/s0168-3659(01)00323-6. [DOI] [PubMed] [Google Scholar]
- 68.Bhatt R, de Vries P, Tulinsky J, Bellamy G, Baker B, Singer JW, Klein P. Synthesis and in vivo antitumor activity of poly(l-glutamic acid) conjugates of 20S-camptothecin. J Med Chem. 2003;46:190–193. doi: 10.1021/jm020022r. [DOI] [PubMed] [Google Scholar]
- 69.Springett GM, Takimoto C, McNamara M, Doroshow JH, Syed S, Eastham E, Spriggs D, Pezzulli S, Michelson G, DJ Phase I study of CT-2106 (polyglutamate camptothecin) in patients with advanced malignancies. Journal of Clinical Oncology, 2004 ASCO Annual Meeting Proceedings 22 Abstract #3127; 2004. [Google Scholar]
- 70.McNamara MV, Doroshow JH, Dupont J, Spriggs D, Eastham E, Pezzulli S, Syed S, Bernareggi A, Takimoto C. Preliminary pharmacokinetics of CT-2106 (polyglutamate camptothecin) in patients with advanced malignancies. J Clin Oncol (Meeting Abstracts) 2004;22:2073. [Google Scholar]
- 71.Homsi J, Simon GR, Garrett CR, Springett G, De Conti R, Chiappori AA, Munster PN, Burton MK, Stromatt S, Allievi C, Angiuli P, Eisenfeld A, Sullivan DM, Daud AI. Phase I Trial of Poly-L-Glutamate Camptothecin (CT-2106) Administered Weekly in Patients with Advanced Solid Malignancies. Clin Cancer Res. 2007;13:5855–5861. doi: 10.1158/1078-0432.CCR-06-2821. [DOI] [PubMed] [Google Scholar]