

Abstract
Autoantibodies against complement C1q (anti-C1q) have been well described in patients with systemic lupus erythematosus, where they correlate with the occurrence of severe lupus nephritis. However, data on anti-C1q in organ-specific autoimmune diseases are scarce. In order to determine the prevalence of anti-C1q in patients with autoimmune thyroid disorders (AITD) and a possible association with thyroid function, we measured prospectively anti-C1q in 23 patients with Graves' disease (GD) and 52 patients with Hashimoto's thyroiditis (HT). Anti-C1q levels were correlated with parameters of thyroid function and autoantibodies against thyroperoxidase, thyroglobulin and thyroid stimulating hormone (TSH) receptor. Twenty-one patients with multi-nodular goitre and 72 normal blood donors served as controls. We found elevated concentrations of anti-C1q more frequently in patients with AITD than in controls: seven of 23 (30%) patients with GD and 11 of 52 (21%) patients with HT, compared with one of 21 (5%) patients with multi-nodular goitre and six of 72 (8%) normal controls. Anti-C1q levels did not correlate with thyroid autoantibodies. However, in GD absolute levels of anti-C1q correlated negatively with TSH and positively with free thyroxine (FT4) and triiodothyronine (FT3). In contrast, in HT, anti-C1q correlated positively with TSH levels. No correlation between TSH and thyroid autoantibodies was found. In conclusion, we found an increased prevalence of anti-C1q in patients with AITD and their levels correlated with the thyroid function in both GD and HT. This correlation seems to be independent of thyroid autoantibodies. Therefore, anti-C1q might point to a pathogenic mechanism involved in the development of AITD that is independent of classical thyroid autoantibodies.
Keywords: anti-C1q antibodies, complement, Graves' disease, Hashimoto's thyroiditis
Introduction
Complement activation has been implicated in the pathogenesis of autoimmune thyroid disorders (AITD), but the precise role of complement in the pathogenesis of AITD remains to be elucidated. Healthy thyrocytes have been shown to express a number of complement components such as C2, C3, C5, C6 and C9, and the expression differs in thyrocytes from thyroids with AITD [1]. Others have shown C1q to be expressed on thyrocytes from patients with AITD [2] and in intrathyroidal lymph follicles [3]. In addition, terminal complement complexes have been found in autoimmune thyroid disease around thyroid follicles [4]. According to experimental studies, different ways of complement activation might occur in the thyroid tissue. Complement could be activated by immune complexes containing complement-activating autoantibodies against thyroid autoantigens [5–7], by direct binding of C4 to thyroperoxidase (TPO) or by direct complement activation through reactive oxygen radicals [1]. Independently, thyrocytes are known to express inhibitors of the complement cascade [4,8]. Thus, they are relatively resistant to complement attack. However, once injured sublethaly, thyrocytes release proinflammatory cytokines and reactive oxygen radicals and promote the inflammatory process in the thyroid [9,10].
Autoantibodies against C1q (anti-C1q), the first component of the classical pathway of complement, can be found in a number of systemic autoimmune diseases [11–14]. Although not being specific for this entity, anti-C1q have been best described in patients with systemic lupus erythematosus (SLE), where they have an excellent negative predictive value for the occurrence of a proliferative lupus nephritis [15–17]. In a more recent study, anti-C1q were found in more than 97% of patients with biopsy-proven active lupus nephritis supporting the hypothesis of a pathogenic role of anti-C1q in lupus nephritis [18]. The role of anti-C1q in other diseases is less clear. So far, no association of anti-C1q with disease activity in other autoimmune diseases has been described. However, a recent study on children with acute post-streptococcal glomerulonephritis could demonstrate that the presence of anti-C1q was associated with more severe forms of nephritis [19]. Thus, anti-C1q might also play a role in other diseases leading to complement deposition.
Because complement seems to be involved in the pathogenesis of AITD, and because of a lack of data on anti-C1q in organ-specific autoimmune diseases, the aim of the study presented here was to determine the prevalence of anti-C1q and their possible association with disease expression in patients with AITD.
Patients and methods
Patients
Between January 2004 and August 2005, 76 adult patients with AITD and 21 control patients with non-AITD (multi-nodular goitre or thyroid nodules) were included consecutively into the study. The inclusion criteria were: (i) clinical and laboratory signs of Graves' disease (GD) [thyrotoxicosis associated with increased levels of antibodies against thyroid stimulating hormone (TSH)-receptor] confirmed by thyroid sonography including Doppler imaging (diffuse goitre with increased thyroid blood flow) either at present or in recent history; (ii) clinical and laboratory signs of Hashimoto's thyroiditis (HT) (thyroid dysfunction associated with either antibodies against TPO or thyroglobulin and/or typical changes on thyroid sonography and/or fine needle aspiration biopsy); (iii) isolated thyroid nodule or multi-nodular goitre on thyroid sonography without any clinical evidence of autoimmune disease. Patients were excluded from the study if they were less than 18 years old, if they were seriously ill or if they did not give written informed consent.
Of the 76 patients with AITD included, 23 had GD, 52 had HT and one patient had HT that presented as GD at the time of inclusion. All patients were recruited from the Outpatient Department of the 3rd Clinic of Medicine, General Teaching Hospital and the 1st Medical Faculty, Charles University in Prague/Czech Republic. The patients were evaluated by an endocrinologist and underwent standard clinical and laboratory examination. The clinical evaluation included history, physical examination and thyroid ultrasound. The laboratory examinations included TSH, free thyroxine (FT4), free triiodothyronine (FT3), antibodies against TPO (TPOAb), thyroglobulin (TgAb) and antibodies against TSH-receptor (TRAb) (only in thyrotoxicosis). The following ranges were considered normal: TSH: 0·4–4·0 mIU/l, FT4: 9·8–23·1 pmol/l, FT3: 3·5–6·5 pmol/l, TgAb: < 60·0 kIU/l, TPOAb: < 60·0 kIU/l; TRAb: < 1 IU/l. Patient characteristics are summarized in Table 1. Serum samples were collected at the time of inclusion, and stored in aliquots frozen at −20°C until further use.
Table 1.
Characteristics of the patients.
| n | Female n (%) | Age | TSH (mIU/l) | FT4 (pmol/l) | FT3 (pmol/l) | TPOAb (kIU/l) | TgAb (kIU/l) | TRAb (U/ml) | |
|---|---|---|---|---|---|---|---|---|---|
| GD | 23 | 20 (87) | 46 (32–56) | < 0·02 (0·0–3·4) | 33·3 (12·3–146) | 10·8 (4·3–54·5) | 704·0 (36·8–11 500) | 83·8 (3·7–1695) | 6·4 (4·1–18·2) |
| HT | 52 | 47 (90·3) | 53·5 (41–70) | 6·0 (0·04–135) | 13·0 (1·9–23·9) | 4·6 (0·4–5·4) | 710 (15·8–23 372) | 105·8 (0·0–7338) | – |
| MNG | 21 | 19 (90·5) | 51 (40–74) | 0·89 (0·02–2·9) | 15·5 (10·8–24) | 4·8 (4·4–5·4) | 43·0 (10·4–77·5) | 21·0 (9·5–39·6) | – |
Values are expressed as median and range (in brackets), unless otherwise stated. FT3, free triiodothyronine; FT4, free thyroxin; GD, Graves' disease; HT, Hashimoto's thyroiditis; MNG, multi-nodular goitre/thyroid nodules; TgAb, antibodies against thyroglobulin; TPOAb, antibodies against thyroperoxidase; TRAb, antibodies against TSH receptor; TSH, thyroid stimulating hormone.
Patients with GD were included either during the phase of thyrotoxicosis at the first occurrence of the disease (16 patients), during a relapse (two patients) or in remission (five patients; two had total thyroidectomy for GD more than 1 year before and three were treated chronically with anti-thyroid drugs). Except for the patients in remission, all patients had thyrotoxicosis at the time of the blood sample. Twelve of the 23 patients had signs of thyroid-associated ophthalmopathy (TAO) and none had pretibial myxoedema. One of the patients had Wegener's granulomatosis.
Patients with HT were included during all phases of the disease, ranging from active autoimmune inflammation to long-term hypothyroidism requiring substitution therapy with levothyroxine. At the time of the blood sample, 29 were not treated and 23 were treated by levothyroxine. One patient presented as GD at the time of inclusion but developed HT later. Thus, this patient was analysed separately from the two other cohorts. Eleven of the 52 patients had other autoimmune disease. Four patients had coeliac disease, one was suspected to have a combination of coeliac disease with pernicious anaemia, two patients had diabetes mellitus type 1 (DM1), one non-differentiated arthritis, one vitiligo, one combination of DM1 and rheumatoid arthritis and one patient had pernicious anaemia.
Patients with AITD were compared with 72 healthy blood donors without clinical evidence of thyroid disease and with 21 patients with non-AITD (13 patients with multi-nodular goitre and eight patients with isolated thyroid nodules). Of the 21 control patients, 19 were euthyroid, one had subclinical hyperthyroidism based on autonomous hyperfunction of the nodule and one was treated for autonomous hyperthyroidism with anti-thyroid drugs. None of the control patients received levothyroxine and all were negative for TPOAb and TgAb.
The study was approved by the Ethical Committee of the General Teaching Hospital and the 1st Medical Faculty of the Charles University in Prague.
Autoantibodies
Anti-C1q were measured in serum using a commercially available enzyme-linked immunosorbent assay kit (Bühlmann Laboratories, Schönenbuch, Switzerland), as described previously [18]. In this assay, undigested purified human C1q served as antigen, and sera were diluted and incubated in a high-salt buffer (1 M NaCl) in order to prevent the binding of immune complexes. The optical densities were measured at 450 nm converted into units (U/ml) by being plotted against the autoantibody concentration of the standards given by the manufacturer. The upper limit of detection of the assay was at 400 U/ml. The technical cut-off for a positive test result as determined by the manufacturer (15 U/ml) was obtained by testing samples from 220 normal blood donors.
Antibodies against TPO and TgAb were measured using chemiluminescence on a Centaur analyser (Bayer, Germany) and TRAb were measured using a radioimmunoassay DYNO test on a STRATEC SR 300 analyser (Brahms, Germany).
Statistics
Statistical analysis was conducted using GraphPad Prism 3·2 (Graphpad Software, San Diego, CA, USA). Unless stated otherwise, all values described in the text and figures are expressed as median and range. A one-tailed χ2 test and non-parametric tests (one-tailed Mann–Whitney U-test and Spearman's rank correlation test) were applied throughout, with differences being considered significant for P < 0·05.
Results
Patients with GD (seven of 23) and HT (11 of 52) were positive more frequently for anti-C1q than controls [one of 21 patients with multi-nodular goitre/thyroid nodules (MNG) and six of 72 of normal controls]. Absolute values of serum anti-C1q levels in the four groups are demonstrated in Fig. 1.
Fig. 1.
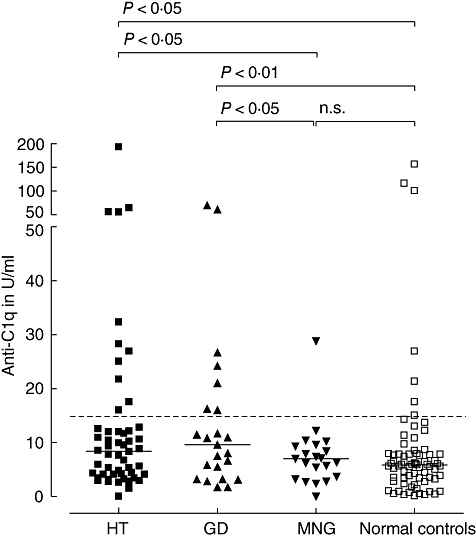
Serum anti-C1q concentrations in patients with thyroid disorders compared with normal controls. HT, Hashimoto's thyroiditis; GD, Graves' disease; MNG, multi-nodular goitre/thyroid nodules. The horizontal lines represent the median. The dotted line indicates the cut-off for a positive test result. Median (range) concentrations of anti-C1q were 8·4 U/ml (0·0–192·5) in HT, 9·6 U/ml (1·7–69·8) in GD and 7·0 U/ml (0·0–28·8) in MNG.
Graves' disease
All seven of 23 patients with GD positive for anti-C1q had active thyrotoxicosis, whereas the five patients in remission were anti-C1q-negative. Levels of anti-C1q correlated negatively with TSH (r =−0·43, P = 0·022) and positively with FT4 (r = 0·44, P = 0·019) and FT3 (r = 0·47, P = 0·014). In contrast, there was no correlation of anti-C1q with any of the antibodies against thyroid antigens. Of these thyroid autoantibodies (TPOAb, TgAb, TRAb), none correlated negatively with TSH. However, TRAb showed a positive correlation with FT4 (r = 0·55, P = 0·004) and FT3 (r = 0·64, P = 0·001). The data are demonstrated in Fig. 2.
Fig. 2.
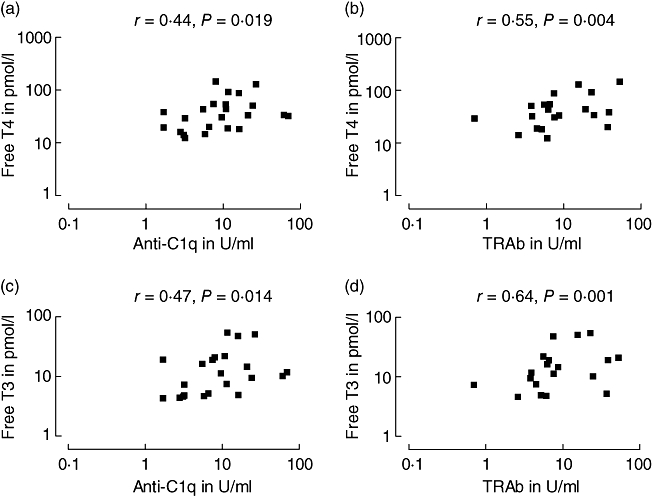
Correlation of anti-C1q with thyroid hormones (a and c) in comparison with TRAb (b and d) in patients with Graves' disease. TRAb, antibodies against receptor for thyroid stimulating hormone; free T4, free thyroxine; free T3, free triiodothyronine.
Of the 23 patients with GD, 12 had signs of TAO. Of these 12 patients, four were positive for anti-C1q. The patient with concomitant Wegener's granulomatosis was anti-C1q negative.
Hashimoto's thyroiditis
Eleven of the 52 patients with HT were positive for anti-C1q (21%). All but one patient with HT were positive for TPOAb and TgAb, the negative patient also being negative for anti-C1q. Of the 11 anti-C1q-positive patients, seven had hypothyroidism at the time of the blood sample and four were euthyroid on substitution with levothyroxine. None of the five patients in the HT group, who were euthyroid without need of therapy, were anti-C1q-positive. Three patients had signs of TAO and all were positive for anti-C1q.
Overall, in patients with HT, anti-C1q showed a positive correlation with TSH (r = 0·29; P = 0·02) and a trend towards a negative correlation with FT3 (P = 0·08). In the subgroup of 29 patients with HT who were not treated with levothyroxine at the time of the blood sampling, the correlation of anti-C1q with TSH was distinct (r = 0·42, P = 0·01). However, such a correlation was not found in the 23 patients who were substituted with levothyroxine (r = 0·08, P = 0·36). Despite a positive correlation of anti-C1q with TgAb (r = 0·35, P = 0·005), TgAb themselves did not correlate with TSH levels. No correlation was found between levels of TPOAb and anti-C1q, nor between TPOAb and TSH.
In 11 of the 52 patients with HT, other autoimmune disorders were present. From these patients, only two were anti-C1q-positive: one with established and one with suspected coeliac disease. Excluding these 11 patients with other autoimmune disorders from the overall analysis improved the correlation slightly between TSH and anti-C1q (r = 0·33, P = 0·016), but did not affect the lack of a correlation between TSH and TPOAb or TgAb respectively.
Neither in HT nor in GD patients could we identify consistent relationships between the presence of anti-C1q and general markers of inflammation in the peripheral blood, such as leucocytes and CRP concentrations.
Discussion
We found an increased prevalence of anti-C1q in GD and HT compared with normal controls and euthyroid patients with thyroid nodules. Absolute levels of anti-C1q correlated with parameters of the thyroid function but not with thyroid autoantibodies in both patients with HT and GD.
In GD, anti-C1q were present only in patients with active disease accompanied by active thyrotoxicosis. None of the patients with GD in remission were positive for anti-C1q. These observations point to a pathogenic mechanism that is independent of classical thyroid autoimmunity. Furthermore, in this cohort we did not observe a correlation of TPOAb or TgAb with TSH or thyroid hormones in patients with HT. These findings are in concordance with previous analyses [20]. Although TPOAb are increased in patients with both autoimmune hypo- and hyperthyroidism [21], a correlation with TSH levels has been described only in healthy euthyroid subjects [22]. Thus, to our knowledge, this is the first description of a correlation between an autoantibody and parameters of the thyroid function in patients with HT.
Anti-C1q levels in patients with AITD were relatively low when compared with those observed in patients with SLE and appear to be of limited diagnostic help. One might speculate that these relatively low levels are due to the more local inflammatory process when compared with chronic and severe systemic autoimmunity occurring in patients with SLE. Therefore, we decided to use a lower cut-off (15 mU/l) for a positive test result than in a previous study on SLE patients using the same assay [18]. However, the lower cut-off used in this study neither affected the differences in absolute levels of anti-C1q between the patient groups (see Fig. 1) nor the correlations between anti-C1q levels and the parameters of thyroid function in patients with AITD. Furthermore, the number of positive individuals in the control groups (8% of normal blood donors and 5% in patients with MNG) remained low. However, more extensive analyses of patients with active thyrotoxicosis in particular are required to determine the true diagnostic value of anti-C1q in AITD.
Independently, our observations might lead to a better understanding of mechanisms leading to the development of AITD. It is well known that cytotoxic T cells and antibodies directed against TSH-receptor are the primary causal immune factors responsible for HT and GD respectively [23,24]. Accordingly, our findings suggest that anti-C1q are not a primary event but might be a secondary phenomenon following localized complement activation during the underlying inflammatory process. In lupus nephritis, anti-C1q seems to have a secondary disease-altering effect [25]. However, the precise mechanism of this disease-altering effect remains speculative. Anti-C1q could lead to a potentiation of complement activation or, in contrast, lead to a blockade of advantageous functions of deposited C1q mediated by C1q receptors. Our observations would be in concordance with those mechanisms in patients with AITD. In both GD and HT, anti-C1q might have a disadvantageous effect by an altered complement activation leading to increased hyperthyroidism in GD and hypothyroidism in HT.
Complement has been shown to be involved in the pathogenesis of AITD, but data on the role of C1q antigen in AITD are scarce [2]. However, Nielsen et al. have shown that B cells of patients with HT bind increased amounts of complement-activating immune complexes of Tg/TgAb and thus induce proliferation of B and T cells subsets in a complement-dependent manner [6,26]. Furthermore, complement-activating TPOAb, so-called bioactive TPOAb, have been described in postpartum thyroiditis [27]. They were found in TPOAb-positive women who developed thyroiditis within 6 months after delivery [7], and their levels increased with the severity of thyroiditis [28]. Also, levels of bioactive TPOAb correlated with the degree of hypoechogenicity on thyroid ultrasound and abnormal thyroid status [29]. In addition to immune complex-mediated complement activation, a recent study found that complement C4 binds to a part of the TPO molecule, which leads to a direct activation of complement via the classical pathway [1]. Activation of complement during AITD leads to the formation of membrane attack complexes around thyroid follicles. They have been observed histologically in thyroids of patients with GD and their serum concentration has been shown to decrease with successful treatment of the disease [4]. Although thyrocytes are protected against complement by expression of a number of complement inhibitory molecules [2,8], sublethal attack by complement leads to a release of a number of cytokines and reactive oxygen species by thyrocytes and aggravation of the autoimmune process [9,10].
In conclusion, we found an increased prevalence of anti-C1q autoantibodies in AITD compared with non-autoimmune controls. Anti-C1q correlated with parameters of the thyroid function in both GD and HT but not with classical thyroid autoantibodies. Therefore, anti-C1q might point to an as yet unrecognized pathogenic mechanism involved in AITD that is independent of classical thyroid autoantibodies. However, further studies are required to establish the role of anti-C1q in AITD.
Acknowledgments
We thank Professor J. A. Schifferli from the University Hospital Basel for his continuing support and counselling. We would also like to thank Mrs J. Tomesova from the General Teaching Hospital in Prague, who has been a great help in the administrative issues of the study. This work was supported by the Czech Ministry of Health (grant IGA no. 8352-3). Dr Trendelenburg is a recipient of a SCORE fellowship from the Swiss National Foundation (3232B0-107248/1 and 3200B0-107249/1).
References
- 1.Blanchin S, Estienne V, Durand-Gorde J, et al. Complement activation by direct C4 binding to thyroperoxidase in Hashimoto's thyroiditis. Endocrinology. 2003;144:5422–9. doi: 10.1210/en.2003-0918. [DOI] [PubMed] [Google Scholar]
- 2.Nakai H, Hirakawa S, Hayakawa N, et al. Enhanced expression of complement regulatory proteins on thyroid epithelial cells of Graves' disease. Acta Med Okayama. 1992;46:323–30. doi: 10.18926/AMO/32658. [DOI] [PubMed] [Google Scholar]
- 3.Kasajima T, Yamakawa M, Imai Y. Immunohistochemical study of intrathyroidal lymph nodes. Clin Immunol Immunopathol. 1987;43:117–28. doi: 10.1016/0090-1229(87)90163-2. [DOI] [PubMed] [Google Scholar]
- 4.Weetman A, Cohen SB, Oleesky DA, et al. Terminal complement complexes and C1/C1 inhibitor complexes in autoimmune thyroid disease. Clin Exp Immunol. 1989;77:25–30. [PMC free article] [PubMed] [Google Scholar]
- 5.Inoue K, Niesen N, Milgrom F, et al. Transfer of experimental autoimmune thyroiditis by in situ perfusion of thyroids with immune sera. Clin Immunol Immunopathol. 1993;66:11–17. doi: 10.1006/clin.1993.1002. [DOI] [PubMed] [Google Scholar]
- 6.Nielsen CH, Hegedus L, Leslie RG. Autoantibodies in autoimmune thyroid disease promote immune complex formation with self antigens and increase B cell and CD4+ T cell proliferation in response to self antigens. Eur J Immunol. 2004;34:263–72. doi: 10.1002/eji.200324413. [DOI] [PubMed] [Google Scholar]
- 7.Parkes A, Othman S, Hall R, et al. The role of complement in the pathogenesis of postpartum thyroiditis. J Clin Endocrinol Metab. 1994;79:395–400. doi: 10.1210/jcem.79.2.8045954. [DOI] [PubMed] [Google Scholar]
- 8.Tandon N, Yan SL, Morgan BP, et al. Expression and function of multiple regulators of complement activation in autoimmune thyroid disease. Immunology. 1994;81:643–7. [PMC free article] [PubMed] [Google Scholar]
- 9.Weetman A, Freeman M, Morgan BP. Thyroid follicular cell function after non-lethal complement membrane attack. Clin Exp Immunol. 1990;82:69–74. doi: 10.1111/j.1365-2249.1990.tb05405.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Weetman A, Tandon N, Morgan BP. Antithyroid drugs and release of inflammatory mediators by complement-attacked thyroid cells. Lancet. 1992;340:633–6. doi: 10.1016/0140-6736(92)92171-b. [DOI] [PubMed] [Google Scholar]
- 11.Potlukova E, Kralikova P. Complement component C1q and anti-C1q antibodies in theory and in clinical practice. Scand J Immunol. 2008;20:13. doi: 10.1111/j.1365-3083.2008.02089.x. [DOI] [PubMed] [Google Scholar]
- 12.Siegert CEH, Kazatchkine MD, Sjöholms A, et al. Autoantibodies aganst C1q: view on clinical relevance and pathogenic roles. Clin Exp Immunol. 1999;116:4–8. doi: 10.1046/j.1365-2249.1999.00867.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Trendelenburg M. Antibodies against C1q in patients with systemic lupus erythematosus. Springer Semin Immunopathol. 2005;27:276–85. doi: 10.1007/s00281-005-0007-y. [DOI] [PubMed] [Google Scholar]
- 14.Wisnieski J, Jones SM. IgG autoantibody to the collagen-like region of C1q in hypocomplementemic urticarial vasculitis syndrome, systemic lupus erythematosus and six other skeletal or rheumatic diseases. J Rheumatol. 1992;19:884–8. [PubMed] [Google Scholar]
- 15.Siegert CEH, Daha MR, Westedt ML, et al. IgG autoantibodies against C1q are correlated with nephritis, hypocomplementemia, and dsDNA antibodies in systemic lupus erythematosus. J Rheumatol. 1991;18:230–4. [PubMed] [Google Scholar]
- 16.Siegert CEH, Daha MR, Tseng CMES, et al. Predictive value of IgG autoantibodies against C1q for nephritis in systemic lupus erythematosus. Ann Rheum Dis. 1993;52:851–6. doi: 10.1136/ard.52.12.851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Trendelenburg M, Courvoisier S, Spaeth PJ, et al. Hypocomplementemic urticarial vasculitis or systemic lupus erythematosus? Am J Kidney Dis. 1999;34:745–51. doi: 10.1016/S0272-6386(99)70402-6. [DOI] [PubMed] [Google Scholar]
- 18.Trendelenburg M, Lopez-Trascasa M, Potlukova E, et al. High prevalence of anti-C1q antibodies in biopsy-proven active lupus nephritis. Nephrol Dial Transplant. 2006;21:3115–21. doi: 10.1093/ndt/gfl436. [DOI] [PubMed] [Google Scholar]
- 19.Kozyro I, Perahud I, Sadallah S, et al. Clinical value of autoantibodies against C1q in children with glomerulonephritis. Pediatrics. 2006;117:1663–8. doi: 10.1542/peds.2005-1148. [DOI] [PubMed] [Google Scholar]
- 20.Carle A, Laurberg P, Knudsen N, et al. Thyroid peroxidase and thyroglobulin auto-antibodies in patients with newly diagnosed overt hypothyroidism. Autoimmunity. 2006;39:497–503. doi: 10.1080/08916930600907913. [DOI] [PubMed] [Google Scholar]
- 21.Hooogendoorn EH, Hermus AR, De Vegt F, et al. Thyroid function and prevalence of anti-thyroperoxidase antibodies in a population with borderline sufficient iodine intake: influences of age and sex. Clin Chem. 2006;52:104–11. doi: 10.1373/clinchem.2005.055194. [DOI] [PubMed] [Google Scholar]
- 22.Strieder TG, Prummel MF, Tijssen JG, et al. Risk factors for and prevalence of thyroid disorders in a cross-sectional study among healthy female relatives of patients with autoimmune thyroid disease. Clin Endocrinol. 2003;59:396–401. doi: 10.1046/j.1365-2265.2003.01862.x. [DOI] [PubMed] [Google Scholar]
- 23.Weetman AP. Graves' disease. N Engl J Med. 2000;343:1236–48. doi: 10.1056/NEJM200010263431707. [DOI] [PubMed] [Google Scholar]
- 24.Weetman AP. Cellular immune responses in autoimmune thyroid disease. Clin Endocrinol. 2004;61:405–13. doi: 10.1111/j.1365-2265.2004.02085.x. [DOI] [PubMed] [Google Scholar]
- 25.Flierman R, Daha MR. Pathogenic role of anti-C1q autoantibodies in the development of lupus nephritis − a hypothesis. Mol Immunol. 2007;44:133–8. doi: 10.1016/j.molimm.2006.06.010. [DOI] [PubMed] [Google Scholar]
- 26.Nielsen CH, Moeller AN, Hegedus L, et al. Self-reactive CD4+ T cells and B cells in the blood in health and autoimmune disease: increased frequency of thyroglobulin-reactive cells in Graves' disease. J Clin Immunol. 2006;26:126–36. doi: 10.1007/s10875-006-9000-z. [DOI] [PubMed] [Google Scholar]
- 27.Okosieme O, Parkes AB, McCullough B, et al. Complement activation in postpartum thyroiditis. Q J Med. 2002;95:173–9. doi: 10.1093/qjmed/95.3.173. [DOI] [PubMed] [Google Scholar]
- 28.Parkes A, Othman S, Hall R, et al. Role of complement in the pathogenesis of postpartum thyroiditis: relationship between complement activation and disease presentation and progression. Eur J Endocrinol. 1995;133:210–15. doi: 10.1530/eje.0.1330210. [DOI] [PubMed] [Google Scholar]
- 29.Parkes A, Adams H, Othman S, et al. The role of complement in the pathogenesis of postpartum thyroiditis: ultrasound echogenicity and the degree of complement-induced thyroid damage. Thyroid. 1996;6:177–82. doi: 10.1089/thy.1996.6.177. [DOI] [PubMed] [Google Scholar]