

Abstract
Systemic lupus erythematosus (SLE) is an autoimmune disease distinguished by great heterogeneity in clinical manifestations and autoantibody expression. While only a handful of autoantibody specificities have proved useful for clinical diagnosis, to characterize complex lupus-associated autoantibody profiles more fully we have applied proteome microarray technology. Our multiplex microarrays included control ligands and 65-autoantigens, which represent diverse nuclear and cytoplasmic antigens recognized by disease-associated and natural autoantibodies. From longitudinal surveys of unrelated SLE patients, we found that autoantibody profile patterns can be patient-specific and highly stable overtime. From profiles of 38 SLE patients that included 14 sets of SLE twins, autoantibodies to the phospholipid neo-determinants, malondialdehyde (MDA) and phosphorylcholine (PC), which are exposed on apoptotic but not healthy cells, were among the most prevalent and highly expressed. We also found that immunoglobulin M (IgM) reactivity to MDA and PC ligands had significant direct correlations with DNA-containing antigens, while such a general relationship was not found with a panel of RNA-related antigens, or for IgG-autoantibodies. Significantly, hierarchical analysis revealed co-distribution/clustering of the IgM autoantibody repertoire patterns for six of 14 twin sets, and such patterns were even more common (10 of 14) for IgG autoantibody profiles. Our findings highlight the potentially distinct roles of IgM and IgG autoantibodies, as we postulate that the direct correlations for IgM autoantibodies to DNA antigens with apoptosis-related determinants may be due to co-expression arising from common pro-homeostatic protective roles. In contrast, the sharing of IgG autoantibody fingerprints by monozygotic twins suggests that lupus IgG autoantibodies can arise in predisposed individuals in genetically determined patterns.
Keywords: apoptosis, autoimmunity, biotechnology, microarray
Introduction
Systemic lupus erythematosus (SLE) is a prototypic systemic autoimmune disease, believed to result from immune recognition of cytoplasmic and nuclear antigens [1]. Among the earliest evidence of immune abnormalities in SLE patients was the identification of anti-nuclear autoantibodies (ANA) [2], which in some cases have been implicated directly in immune complex-mediated pathogenesis and end-organ damage (reviewed in [3]). While clinical diagnosis is aided by the detection of circulating ANA, with special emphasis on antibodies to the ribonucleoprotein (RNP), Sm, and native DNA, the clinical presentation, organ system involvement and range of autoantibodies expressed in SLE patients is highly heterogeneous.
Circulating autoantibodies can be detected years prior to the clinical diagnosis of SLE, and in many SLE patients the number of distinct specificities of lupus autoantibodies were found to increase over time [4]. However, many apparently healthy individuals also have detectable circulating autoantibodies [5]. It therefore remains unclear if clinical features are commonly present, and organ damage may progress for long periods before the practitioner can make the SLE diagnosis [6].
The genetic contribution to lupus pathogenesis has been well established, and there is a greater than 100-fold risk for lupus among family members of affected individuals than in the general population [7–9]. The powerful contribution of inheritance to disease predisposition has been highlighted in studies of twins, as non-identical (i.e. dizygotic, DZ) twins display the same risk as close siblings (i.e. ∼ 2–3%), while identical (monozygotic, MZ) lupus twins display an estimated ∼ 25% or greater concordance for the development of lupus [10] (reviewed in [11]) and concordance increases over time [12]. Although it is difficult to separate out the influence of environmental factors in family studies, MZ twins with different environment histories have been reported to also become disease-concordant [13]. In any case, twin studies can provide invaluable perspectives for studying the potential variations in immune function that can develop in genetically identical humans.
A decade ago, we characterized the cloned antibody repertoires of a set of MZ twins discordant for lupus disease [14]. Despite shared genetic features, only the lupus-affected twin had circulating immunoglobulin G (IgG) anti-native DNA, and recombinant IgG anti-native DNA autoantibodies could be recovered only from the phage-display libraries from the affected twin and another individual with SLE disease activity [14]. However, the healthy (i.e. disease-discordant) twin also had levels of circulating IgM autoantibodies to both denatured DNA and native DNA, which were significantly higher than found in a panel of healthy controls [14]. In light of the seemingly robust health of this unaffected twin, we began to wonder whether her IgM autoantibody responses were not only non-pathogenic but might instead represent a protective response to immune abnormalities that otherwise conveyed autoimmune disease susceptibility.
With the adaptation of technology developed originally for gene transcript surveys and the availability of robust chips for proteomic analyses, antigen microarrays have enabled comprehensive comparisons of highly multiplex surveys of (auto)antibody profiles [15–18]. While a single platform and set of binding conditions may not be optimal for the detection of every antibody-binding specificity, microarray approaches enable the simultaneous analyses of binding interactions with potentially hundreds (or even thousands) of antigenic ligands. Moreover, this approach can have fourfold or greater sensitivity and precision than standard immunoassays, and it is well suited to high-throughput screening with data output compatible with multi-parameter bioinformatics analyses.
To extend our investigations of the interplay of genetic inheritance with the immunological features of SLE, we now report our findings from the application of antigen microarray technology to characterize IgM and IgG lupus autoantibody profiles. Significantly, our studies detected hierarchies in the types of detected autoantibodies, which may reflect distinct immunobiological cellular origins and different potential physiological and pathophysiological roles. We also found that disease-concordant lupus twins commonly share IgG autoantibody expression patterns that are distinct from those of other affected individuals.
Methods
Patients
Blood samples were obtained following informed consent with institutional oversight. The diagnosis of SLE was determined at the time of sample donation based on American College of Rheumatology criteria [1] (Table 1). All twins were female and samples were designated ‘F’ for familial relatedness, with unrelated SLE patients identified by ‘S’. Genotyping confirmed monozygosity (MZ) for F122, F124, F167 and the dizygosity for F7 twins, while others were determined based on patient reporting of a single or dual placentas.
Table 1.
Characteristics of lupus twins.
| Ethnicity† | Zygosity‡ | Subject | Age at Dx | Age at blood draw | SLE Dx§ | Immunoglobulin M (IgM) autoantibody profiles¶ | IgG autoantibody profiles | Medications | |
|---|---|---|---|---|---|---|---|---|---|
| F001†† | c | MZ | AA | 26 | 28 | + | − | − | Prednisone 40 mg/day, hydroxychloroquin |
| BB | − | None | |||||||
| F122 | as | MZ | 744 | 28 | 30 | + | Co-distribute | − | Prednisone 20 mg/day, hydroxychloroquin |
| 745 | − | None | |||||||
| F124 | c | MZ | 751 | 20 | 24 | + | Cluster | Cluster | Prednisone <10 mg/day, hydroxychloroquin, NSAID |
| 765 | ND | + | Unknown | ||||||
| F167 | c | MZ | 927 | 44 | 52 | + | − | − | Unknown |
| 933 | 46 | + | Unknown | ||||||
| F191 | c | MZ | 498 | 48 | 57 | + | Co-distribute | Cluster | Unknown |
| 499 | ND | + | Unknown | ||||||
| F225 | as | MZ | 1218 | 23 | 44 | + | − | Cluster | Unknown |
| 1219 | 30 | + | Unknown | ||||||
| F323 | aa | MZ | 1730 | ND | ND | + | − | Cluster | NSAID |
| 1760 | ND | + | NSAID | ||||||
| F794 | c | MZ | 4213 | 12 | 25 | + | NR | Cluster | Unknown |
| 4214 | ND | + | Prednisone 1 mg/day, hydroxychloroquin, MMF | ||||||
| F986 | c | MZ | 4827 | ND | 34 | + | − | Co-distribute | Unknown |
| 4828 | ND | + | Prednisone 20 mg/day, hydroxychloroquin | ||||||
| F1008 | aa | MZ | 4884 | 15 | 16 | + | Cluster | Cluster | Prednisone, hydroxychloroquin, MMF |
| 4885 | 15 | + | Prednisone, hydroxychloroquin, MMF and i.v. cyclophosphamide | ||||||
| F7 | c | DZ | 31 | 15 | 24 | + | − | − | Prednisone, hydroxychloroquin, methotrexate NSAID, IVIG |
| 34 | 13 | + | Unknown | ||||||
| F285 | c | DZ | 1402 | 38 | 40 | + | − | Co-distribute | Unknown |
| 1403 | ND | + | Unknown | ||||||
| F988 | as | DZ | 4832 | 43 | 46 | + | Cluster | Cluster | Prednisone <10 mg/day, hydroxychloroquin, NSAID |
| 4849 | − | None | |||||||
| F1001 | c | DZ | 4862 | 78 | −(GCA)‡‡ | Co-distribute | Co-distribute | Unknown | |
| 4863 | + | Prednisone, hydroxychloroquin, azathioprine |
Ethnicity was self-determined: aa: African American; as: Asian; c: Caucasian.
Zygosity: MZ, monozygotic; DZ, dizygotic.
Systemic lupus erythematosus (SLE) diagnosis was determined based on American College of Rheumatology criteria [1].
Based on comparisons of autoantibody profiles determined by microarray, twin sets that distributed into paired terminal dendrogram were termed a ‘cluster’, twins that distributed adjacent to one another but not in the same dendrogram were termed ‘co-distribute’. If the profiles of twins neither clustered nor co-distributed, comparisons for a twin set were designated negative (–). Only the F794 were found to be non-reactive (NR) for all IgM autoantibodies, even though found to be positive in control assays for detectable total IgM and control antigens on slides.
Detailed Ig repertoire analyses of the F001 twins, AA and BB, have been reported previously [14].
At time of blood draw this dizygotic twin fulfilled criteria for giant cell arteritis (GCA) but not SLE. NSAID, non-steroidal anti-inflammatory drug; MMF, mycophenolate mofetil. ND, not determined.
Antigens and antibodies
A panel of 65 self-antigens was selected for their relevance to autoimmune disease diagnosis or reported reactivity with natural antibodies (see Table 2). In particular, this panel included human low-density lipoprotein (LDL) that was modified chemically by oxidation [i.e. oxidized LDL (OxLDL)] on which phosphorylcholine (PC) is reported to be a dominant epitope [19], and also malondialdehyde (MDA)-modified LDL on which MDA is a dominant epitope [20]. Because of evidence of roles in pathogenesis and utility for diagnosis [21,22], several RNP-related antigens were also included. Antigens in these studies were of the highest quality available (Academy Bio-Medical, Houston, TX, USA; AXXORA, San Diego, CA, USA; BD Bioscience, San Diego, CA, USA; INOVA Diagnostics, San Diego, CA, USA; SurModics, Eden Prairie, MN, USA; US Biological, Swampscott, MA, USA; Fisher Scientific, Houston, TX, USA; MP Biomedical, Solon, OH, USA). Nucleolar extracts, isolated as described previously [23], were provided by INOVA Diagnostics). Antigenic reactivity was confirmed with positive control antibody standards and using individual and pooled sera samples from patients with well-characterized disease (not shown). As controls, anti-IgM, anti-IgG and purified Ig samples were also printed. Control studies documented less than 1% cross-reactivity between the IgM- and IgG-specific detection reagents (not shown). In control studies, our methods were refined for consistent antigen spot characteristics, antibody signal and by correlations of results from enzyme-linked immunosorbent assay (ELISA) and Luminex antibody assays (data not shown).
Table 2.
Immunoglobulin M (IgM) and IgG autoantigen reactivities of 38 systemic lupus erythematosus (SLE) patients and four disease-discordant lupus twins.
| Antigen | Percentage of IgM-positive individuals | Antigen | Percentage of IgG-positive individuals |
|---|---|---|---|
| ssDNA | 93% | MDA-LDL | 95% |
| MDA-LDL | 88% | snRNP 68/70 | 79% |
| Chromatin | 64% | ssDNA | 76% |
| dsDNA | 57% | Laminin | 64% |
| Nucleolar extract | 48% | Chromatin | 62% |
| Tubulin | 40% | Collagen type IV | 57% |
| SSA-52 | 38% | SSA-52 | 48% |
| OxLDL | 38% | dsDNA | 43% |
| PL-12 | 38% | PL-12 | 43% |
| SnRNP 68/70 | 31% | Sm RNP | 43% |
| snRNP BB′ | 29% | C1q | 43% |
| Sm RNP | 24% | Nucleolar extract | 40% |
| Sm | 24% | snRNP BB′ | 36% |
| Tissue transglutaminase (HtTG) | 21% | Entactin | 33% |
| β2 glycoprotein I (β2 gp I) | 21% | Collagen type II | 33% |
| GPI | 19% | CENP-B | 31% |
| CENP-B | 17% | HtTG | 29% |
| Glomerular extract | 17% | GPI | 24% |
| HDL | 17% | SSA 60 | 24% |
| Laminin | 17% | OxLDL | 21% |
| Actin | 14% | Sm | 21% |
| C1q | 14% | Ribosomal-P | 19% |
| Histone | 14% | SSB | 19% |
| Insulin | 12% | Thyroglobulin | 14% |
| Lipoprotein lipase | 12% | snRNP A | 14% |
| Entactin | 12% | Ann V | 14% |
| Ribosomal-P | 10% | Glomerular ext. | 12% |
| PL-7 | 7% | CENP-A | 12% |
| Aggrecan | 7% | Actin | 10% |
| Cardiolipin + β2 gp I | 7% | Lipoprotein lipase | 10% |
| M2 | 7% | M2 | 10% |
| Hep 2 extract | 7% | Hep 2 | 10% |
| gpIIb IIIa | 7% | Scl-70 | 10% |
| Thyroglobulin | 5% | HDL | 7% |
| snRNP A | 5% | Histone | 7% |
| Protein S | 5% | Jo-1 | 7% |
| SSB | 5% | Tubulin | 5% |
| SSA 60 | 5% | Insulin | 5% |
| TPO | 5% | TPO | 5% |
| Jo-1 | 5% | Ku | 5% |
| Myosin | 5% | β2 gp I | 2·4% |
| Proteoglycan | 5% | PL-7 | 2·4% |
| Collagen type II | 5% | Aggrecan | 2·4% |
| Collagen type IV | 5% | Cardiolipin + β2 gp I | 2·4% |
| Annexin V | 5% | Proteoglycan | 2·4% |
| LAP | 2·4% | LAP | 2·4% |
| Scl-70 | 2·4% | Apolipoprotein A1 | 2·4% |
| Cardiolipin + annexin V | 2·4% | Gliadin | 2·4% |
| Ku | 2·4% | Phosphatidyl serine | 2·4% |
| β Thrombin | 2·4% | Fibrinogen type 1-S | 2·4% |
| Vimentin | 2·4% | ||
| MPO | 2·4% | ||
| Plasmin | 2·4% | ||
| Apo A1 | 2·4% | ||
| Gliadin | 2·4% | ||
| Prothrombin | 2·4% |
Results are depicted for the percentage of the 42 tested subjects who were reactive with each autoantigen. For IgM proteomic array assays, all subjects were also non-reactive for: unmodified fibrinogen, citrullinated fibrinogen, CCP3, enolase, phosphatidyl serine, fibrinogen type I-S, CENP-A. For IgG proteomic arrays assays, all subjects were also non-reactive for: unmodified fibrinogen, citrullinated fibrinogen, CCP3, enolase, gpIIb IIIa, protein S, myosin, cardiolipin + annexin V, β thrombin, vimentin, myeloperoxidase (MPO), plasmin and prothrombin. MDA-LDL: malondialdehyde-low-density lipoprotein; OxLDL: oxidated LDL. MPO, myeloperoxidase; LAP, leukocyte alkaline phosphatase; snRNP, small nucleolar ribonucleoprotein; Sm, Smith antigen; PL, phospholipid; CENP-B, centromere protein-B; TPO, thrombopoietin.
Proteomic arrays
For our surveys, we adapted methods reported earlier [15]. Proteomic microarrays were printed onnitrocellulose-coated FAST slides (Whatman, Florham Park, NJ, USA) with a QSoft QArray Mini, using QSoft microarray software (Genetix USA, Boston, MA, USA). On each slide were two nitrocellulose pads, with most antigens printed at 200 μg/ml on each pad in six replicates at known positions that alternated with printing of buffer alone to eliminate carry-over. Slides were then blocked (Whatman blocking buffer) and incubated for 2 h with 400 μl of serum diluted at 1 : 200 in phosphate-buffered saline (PBS), pH 7·4. After washing with Tris-buffered saline/0·1% Tween 20, the slides were reacted with Cy-5 labelled anti-human IgG and Cy-3-labelled anti-human IgM (Jackson Immunoresearch, West Grove, PA, USA) for 1 h incubation, then washed with PBS/0·05% Tween20. Slides were washed with PBS, water-rinsed and then scanned (Genepix 4000B; Molecular Devices, Sunnyvale, CA, USA) using Genepix Pro version 6·0 software at 532 and 635 nm to detect IgG and IgM reactivity, respectively, with images saved as tiff files.
Data analysis
Genepix Pro version 6·0 software was used to analyse the intensity of each spot with foreground and background fluorescence intensities determined for each spot on the slide, with output as a gpr file. Data were extracted from the gpr files with r software (http://www.r-project.org). Background correction used a Matlab script, and the mean values for replicate spots determined with jmp version 7·0 software (http://www.jmp.com). Comparisons of array data for subjects and controls were performed and visual depictions generated with Cluster version 3·0 (Stanford University) and Java TreeView (http://rana.lbl.gov/EisenSoftware.htm). Final analyses included data for lupus patients or their twins, which were considered potentially immunologically abnormal. Background levels were therefore determined based on replicate slides developed without sera, and with sera from healthy adult controls (not shown). In these studies, a level of 600 digital fluorescence intensity units was set as a threshold for significant reactivity, which was greater than the mean background plus 2 standard deviations (s.d.). Independent analyses with duplicate arrays were performed with samples from lupus twins, with equivalent results obtained each time. From control studies, statistically significant (P < 0·0001) Pearson's correlation coefficients (R) were found for IgG binding results from microarray analyses from comparisons with approved clinical pathology laboratory tests for anti-dsDNA (R = 0·97) determined by the fluorescence enzyme immunoassay method (Phadias Cap-system, Uppsala, Sweden), for SSA52 (R = 0·96) and for SSA60 (R = 0·82) by ELISA (QUANTA LiteTM; INOVA Diagnostics).
To determine whether there were correlations of reactivity levels within an individual sample between different fine specificities of autoantibodies, the Matlab toolbox was used for regression analyses and determination of P-values. To identify data sets with similar reactivity patterns from more limited numbers of antigens, the entire antibody data sets for each serum sample were transformed into a vector, and analyses performed on vectors with data sets as antigenic reactivities were removed sequentially one by one, using the leave-one-out cluster algorithm (http://www.biomedcentral.com/1471-2105/7/95). Smaller antigen sets were thereby identified that represented the entire data set most closely.
Significance was assigned for P < 0·05, using the statistical model as indicated.
Results
Autoantibody profiles may be stable over time
As our primary goal was to evaluate whether SLE patients express autoantibody patterns that can distinguish affected individuals, we first performed longitudinal surveys with a panel of 65 ‘self’-antigens selected for their proven utility in confirming diagnoses of autoimmune diseases or for reported reactivity with natural autoantibodies (Table 2). In these studies, we assessed IgG autoantibody profiles in four or five sera samples of five unrelated SLE patients obtained during out-patient visits over 10–18 months. Results shown in Fig. 1 depict heatmaps of relative reactivities, which are organized based on unsupervised clustering by most similar sera samples and by autoantigen reactivities. For three of these SLE patients (S101, S104 and S105) each of the longitudinal samples clustered separately by donor, even to the terminal cluster. The different samples obtained over time also distributed in the dendrogram together for the other two patients, S102 and S103, although the ‘10-month’ sample from S103 was not in the same terminal cluster (which is most affected by noise from different sources) but instead was only adjacent (or ‘co-distributed’) with other S103 time-point samples. These findings demonstrate the stability of the IgG autoantibody profiles of these SLE patients during the 10–18-month periods studied.
Fig. 1.
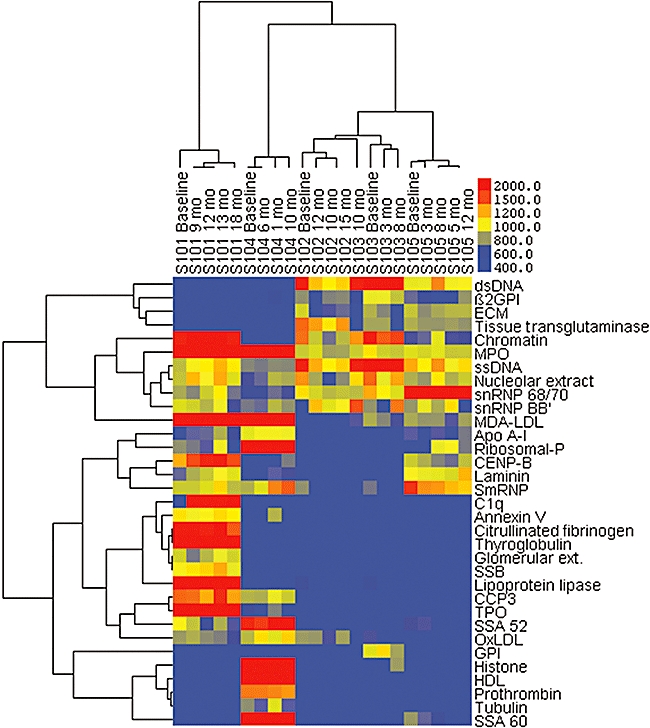
Longitudinal analyses of immunoglobulin G autoantibody profiling of five unrelated systemic lupus erythematosus (SLE) patients. These results represent a two-dimensional cluster analysis, in which sera samples with similar antibody response patterns were placed adjacent to one another, and the clustering of nested sets are indicated. In the second dimension a similar computational clustering of antigenic reactivities is shown. Heat maps are depicted with bright red, representing the highest relative activity level, and bright blue, representing the absence of relative antibody activity level, as indicated in the legend. Sera samples are labelled by SLE donor and time-point during longitudinal surveillance. Results from analysis with a hierarchical cluster algorithm are shown for immunoglobulin G reactivity after removal of antigens for which two or fewer sera samples were reactive, which yielded a panel of 33 antigens. During the period of surveillance, S101 received daily prednisone 10–25 mg, azathioprine 50–100 mg and dapsone 100 mg; S102 received only daily prednisone 0–5 mg; S103 received daily prednisone 4–15 mg and azathioprine 50 mg; S104 received daily prednisone 10–20 mg, azathioprine 50–100 mg, with dapsone 100 mg added only for the 8-month time-point; and S105 received daily prednisone 7·5–15 mg, except at 8 months when this was increased to 60 mg.
Autoantibody profiles of a panel of SLE patients that include SLE twins
Studies were then performed of the autoantibody profiles of a total of 42 individuals, which included 14 sets of lupus twins (Table 1) in comparisons with 14 unrelated SLE patients. Of the lupus twins, there were 10 sets of MZ twins that included seven concordant for the SLE diagnosis, and four sets of DZ twins that included two who were SLE concordant. Hence, these studies included 38 SLE patients and four unaffected twins, which were evaluated by autoantigen microarray surveys. IgM and IgG binding was assessed with a panel of 65 ‘self’-antigens selected for their proven utility in confirming diagnoses of autoimmune diseases or for reported reactivity with natural autoantibodies (Table 2).
Immunoglobulin M autoantibody analyses
From our surveys, we identified the self-antigens that were recognized most commonly by IgM antibodies, which in descending order included: denatured DNA (39 reactive subjects), MDA-LDL (37), chromatin (27), native DNA (24), nucleolar extract (20), tubulin (17), SSA52 (16), OxLDL (16), PL-12 (16 snRNP 68/70 (13), snRNP BB′ (12), SmRNP (10) and Sm (10), with the remainder of the antigens in the panel recognized by the IgM of nine or fewer individuals (Table 2).
Systemic lupus erythematosus patients displayed a somewhat greater mean number of IgM autoantibody specificities (10·6 ± 8·3, mean ± s.d.), which was not significantly different from the five subjects without the SLE diagnosis (8·4 ± 6·8). Notably, within each SLE twin set there was a sharing of a mean of 4·2 ± 4·0 IgM autoreactive binding specificities.
We also assessed whether there was a relationship between the levels of different IgM autoantibody specificities, with special interest in the immune response to MDA-LDL and OxLDL, which express neo-determinants (not on native LDL) that are also exposed on apoptotic cells and atherosclerotic plaques [24,25] (reviewed in [26]). Significantly, by linear regression analysis we found significant Pearson's correlation coefficients between IgM antibodies to MDA-LDL and OxLDL (P < 5·0 × 10−9) (Table 3) which, in part, was predictable due to an overlap of the epitopes expressed in these forms of modified human LDL. To provide comparisons with simpler epitopic ligands, we also assessed binding to an albumin conjugate of MDA, and a conjugate of PC that is reported to be a dominant epitope on OxLDL [19]. Predictably, IgM binding to MDA-LDL and OxLDL correlated significantly with MDA and PC albumin reactivity (Table 3). However, IgM binding to MDA albumin did not correlate with binding to PC albumin, suggesting that these epitopes are not recognized commonly by cross-reactive IgM antibodies. Strikingly, we found that the levels of the IgM antibodies to MDA-LDL and to OxLDL also displayed independently significant direct correlations with IgM antibodies to each of the DNA-containing antigens: denatured DNA (P < 0·02), native DNA (P < 0·02), chromatin (P < 0·008) and nucleolar extract (P < 0·0001) (Table 3). In contrast, despite the fact that many individuals exhibited high autoantibody levels to these autoantigens, we found that there were no general correlations of levels of IgM antibodies to MDA-LDL/OxLDL with autoantibodies to eight purified RNA-related antigens (SSA60, SSB, Sm, SmRNP, snRNP68/70, ribosomal-P and the recombinant RNA-binding proteins; snRNP-A, snRNP-BB). In only a few cases did the levels of IgM antibodies to a DNA-containing antigen correlate with IgM antibodies to a RNA-related antigen (Table 3). Hence, we found a consistent relationship only between levels of IgM autoantibodies to LDL-associated antigens, which express the PC and MDA epitopes also exposed on apoptotic cell membranes, with levels of IgM to DNA-containing antigens. These findings may suggest that there is co-regulation and/or cross-reactivity of IgM antibody responses between phospholipid apoptosis-related epitopes and DNA-containing self-antigens (see Discussion).
Table 3.
Correlations between immunoglobulin M (IgM)-autoantibody levels
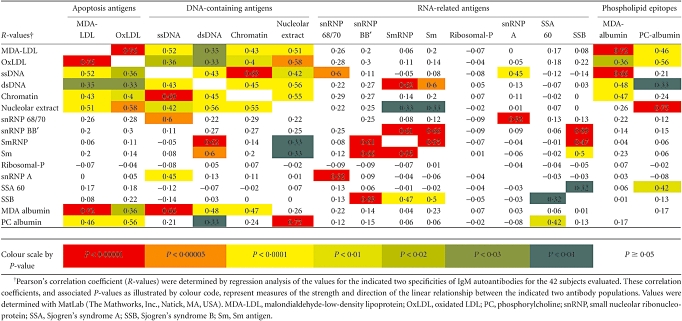 |
In order to identify individuals with the most similar IgM autoantibody profiles, we next performed unsupervised hierarchical cluster analysis for the IgM autoantibody profiles of each individual in comparison with the 41 other samples. Because of the documented importance of autoantibody responses to these nuclear antigens, we first assessed the patterns of IgM reactivity against the subset of autoantigens with DNA-containing antigens. However, presumably because of limited differences between individuals, this yielded clustering of only two twin sets (F167 and F988), and the addition of histones to this antigen panel yielded the same results (not shown). Similar analyses with the panel of RNA-related antigens identified four clustered twin sets (F001, F191, F323 and F1008). When analyses were performed with a panel that included all these DNA- and RNA-related antigens, four twin pairs (F001, F191, F988, F1008) were identified (not shown). Hence, analyses restricted to these lupus-associated binding self-specificities were inadequate to discriminate between the autoantibody profiles of different lupus-affected individuals.
With the goal of improving the identification of individual-specific reactivity profiles, hierarchical clustering analyses for all subjects were then performed for the IgM reactivities with all the 65 autoantigens in the panel. A subset of 56 antigens were recognized by the IgM by one or more of the subjects (Table 2). Only two twin sets were found to cluster (F124, F988), while an additional two sets of twins (F122 and F1008) distributed adjacent (i.e. co-distribute) to each other in the dendrogram. Although the F794 twin set was also found to co-distribute, we did not designate them as an IgM clustered set because they displayed IgM non-reactivity with all the self-antigens in the panel (Table 1). In explanation, the F794 twins were also both within the lowest quintile of total IgM levels detected in these studies (not shown).
These IgM analyses were repeated after removal of data for antigens for which five or fewer individuals were reactive. With the resulting smaller panel of 26 antigens, we found the same four twin sets (F124, F988, F1001 and F1008) that clustered, while two additional (F122 and F191) twin sets co-distributed in the dendrogram adjacent to one another (Fig. 2). Additional leave-one-out cluster analyses did not identify clustering of additional twin sets (not shown). Hence, hierarchical cluster analyses identified shared twin-specific IgM autoantibodies in six of 14 cases.
Fig. 2.
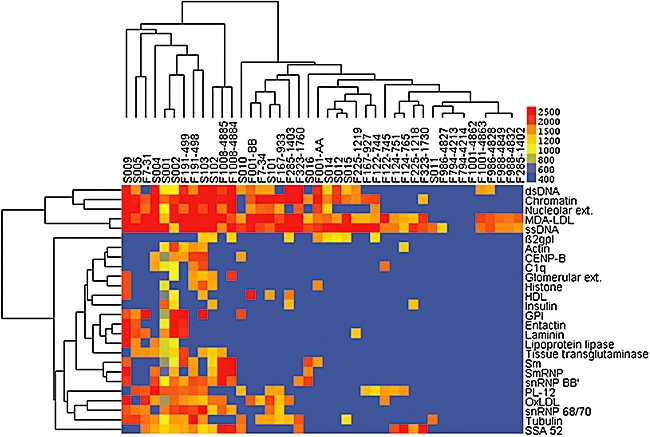
Immunoglobulin M (IgM) autoantibody profiling of sera samples from systemic lupus erythematosus (SLE) patients. Hierarchical cluster analysis results were performed shown for IgM reactivity with the panel of 14 twin sets and 14 unrelated SLE patients. Results are depicted after removal of antigens for which five or fewer individuals were reactive, which yielded a smaller panel of 26 antigens. Twins are identified by shared ‘F’ naming, while unrelated SLE patients are identified by ‘S’. Based on hierarchical cluster analyses, related IgM autoantibody profiles were identified for monozygotic (MZ) twins (F122, F124, F191 and F1008) and for two dizygotic (DZ) twins (F988 and F1001). Lupus-discordant twin sets were F001, F122, F988 and F1001. Control array studies demonstrated a trend in SLE patients (n = 10) towards higher IgM autoantibodies than in healthy controls (n = 10) to: oxidated low-density lipoprotein (OxLDL) (mean ± standard deviation) 5049·6 ± 3575 versus 2239 ± 2840; malondialdehyde (MDA)-LDL, 4607 ± 3406 versus 1736 ± 1379; phosphorylcholine-albumin, 16760 ± 14501 versus 12936 ± 10286; MDA-albumin, 3289 ± 3338 versus 904 ± 942.
Immunoglobulin G autoantibody analyses
We next identified the self-antigens recognized most commonly by IgG; MDA-LDL (40 reactive subjects), snRNP 68/70 (33), laminin (27), chromatin (26) collagen IV (24), SSA52 (20), native DNA (18PL-12 (18), SmRNP (18) and C1q (18), with the remainder recognized by 17 or fewer sera samples (Table 2). Hence, we found that many more IgG autospecificities were detected with our antigen panel than for IgM autospecificities. Notably, within the 10 antigens recognized most commonly by IgG there were seven antigens (denatured DNA, MDA-LDL, chromatin, native DNA, SSA52, PL-12 and snRNP 68/70) that were among the 10 antigens recognized most commonly by IgM autoantibodies. There was also a greater number of IgG autoantibody specificities among SLE patients (14·2 ± 6·7) than for disease-unaffected individuals (11·4 ± 8·2), although this trend was not statistically significant. Significantly, twin sets shared a greater number of IgG autoantibody types (8·9 ± 5·8) than of IgM autoantibodies (4·2 ± 4·0) (P < 0·009, two-tailed t-test).
Reiterating the above-described linear regression analyses, we also looked for correlations in the expression patterns of IgG autoantibodies between individuals (Table 4), and found a correlation for IgG binding between each of the DNA-containing antigens. However, distinct from the conserved patterns seen among IgM antibodies, we did not find a general relationship between levels of IgG reactivity to MDA-LDL and OxLDL and to the DNA-containing antigens (Table 4). Also, unlike the patterns for IgM antibodies, there was a significant correlation for IgG antibodies to MDA albumin and the PC-albumin conjugate. These findings may suggest that there is unlinked regulation of IgM and IgG responses to these apoptosis-related phospholipid determinants.
Table 4.
Correlation between IgG-autoantibody levels
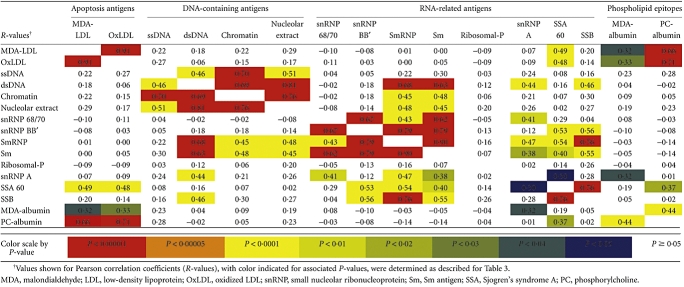 |
We also performed hierarchical cluster analyses first for the panel of DNA-containing antigens and found clustering of three twin sets (F124, F794 and F1008) (not shown). Analyses of a panel of purified RNA-related antigens yielded four twin sets (F124, F323, F988 and F1008) (not shown). When hierarchical analyses were performed on a panel that included the DNA- and RNA-related antigens we identified the same four twin pairs (F124, F323, F988 and F1008) (not shown). Hence, IgG reactivity patterns for the DNA- and RNA-related antigens provided twin-specific immune profiles in only a limited number of cases.
Hierarchical analyses of IgG responses were then performed with the entire antigen panel, which identified five clustered twin sets (F124, F225, F323, F986 and F1008) (not shown). Reiterating the above described approach, analyses were then repeated after removal of antigens for which five or fewer individuals were IgG reactive. With this reduced set of 31 antigens, we found clustering and/or co-distribution of an overlapping five twin sets within the dendrogram (F7, F124, F323, F1001 and F1008) (Fig. 3a and data not shown).
Fig. 3.
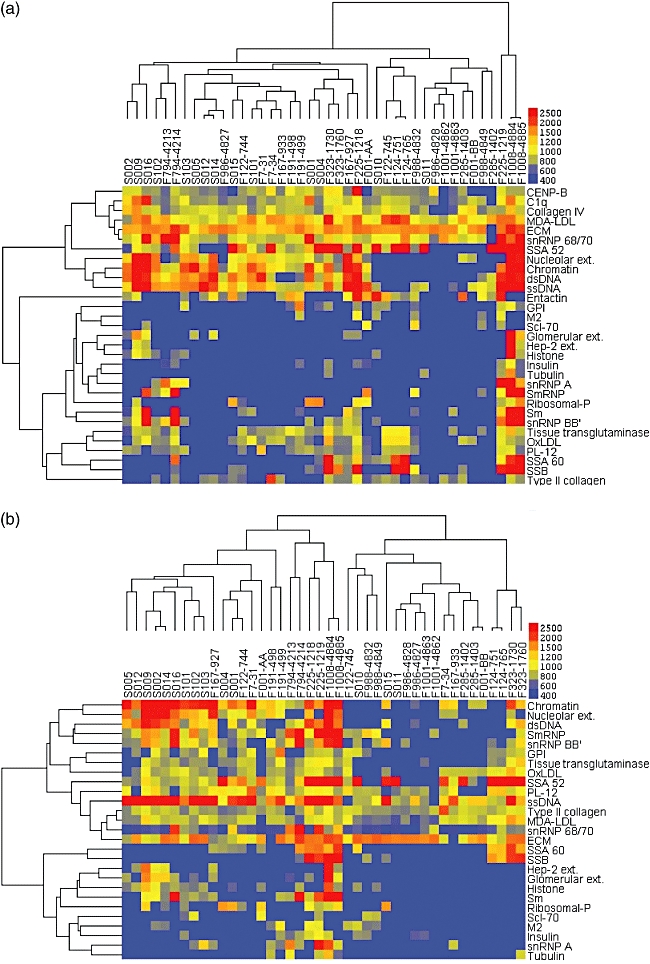
Immunoglobulin G (IgG) autoantibody profiling of sera samples from systemic lupus erythematosus (SLE) patients. Akin to the analysis in Fig. 2, these results represent a two-dimensional cluster analysis, in which individuals with similar antibody response patterns were distributed adjacent to one another, and clustering of nested sets are indicated. (a) Results are depicted for IgG reactivity patterns with 31 antigens, which represented the panel of reactive autoantigens after removal of antigens for which five or fewer individuals were reactive. (b) Results from the same analysis after further reduction of the antigen panel to 27 antigens based on leave-one-out cluster analysis. Significantly, eight of the 10 known disease diagnosis-concordant pair were shown to cluster or co-distribute adjacent to one another (i.e. ‘co-distribute’) (see Table 1). These IgG autoantibody profile results suggest that genetic inheritance is a major determinant of autoantibody response patterns. Control array studies demonstrated a trend in SLE patients (n = 38) towards higher IgG autoantibodies than in healthy controls (n = 16) to: oxidated low-density lipoprotein (OxLDL) (mean ± standard deviation) 1484 ± 264 versus 816 ± 150; malondialdehyde (MDA)-LDL, 3269 ± 355 versus 1838 ± 173; phosphorylcholine-albumin, 4363 ± 719 versus 3082 ± 943; MDA-albumin, 2456 ± 460 versus 1121 ± 447.
We therefore also performed leave-one-out cluster analysis to reveal shared patterns associated with a smaller panel of self-antigens. Using this approach, we identified a minimal panel of 27 antigens for IgG reactivities that resulted in the clustering/co-distribution of 10 pairs of lupus twin sets (F124, F191, F225, F285, F323, F794, F986, F988, F1001 and F1008) (Fig. 3b). Notably, this list included the clustering of the F794 twins based on similar IgG autoantibody reactivity profiles, even though they were devoid of IgM reactivity with this same autoantigen panel.
Taken together, these findings demonstrate the range of expressed autoreactive specificities in different SLE patients, but most importantly that genetically identical individuals often express very similar IgG autoantibody profiles. Significantly, profiles of expressed IgG autoantibodies appeared to be more individualized and characteristic for each twin sets than found for IgM autoantibodies.
Comparisons of IgM autoantibody repertoires in disease-discordant twins
As described above, of the four twin sets discordant for the SLE diagnosis, one of the lupus-unaffected twins (F1001-4862) was found to be devoid of detectable IgM autoantibodies (Table 1). For the other three SLE-discordant twin sets, comparisons between their IgM autoantibody repertoires were performed, and are depicted in Fig. 4. We were surprised to discover that in each case the detected levels of IgM antibodies to MDA-LDL were higher in the ‘healthy’ twin than in their twin sisters. Notably, the same trend was also found for IgM anti-OxLDL antibodies in two (F001 and F122) of these same twin sets. While IgM reactivity was also detected with some DNA-containing antigens and several other autoantibodies, these did not display the same relationships for relative reactivity within the discordant twin pairs. Although there were few discordant twins available for these surveys, these findings support the hypothesis that IgM autoantibodies to apoptosis-associated phospholipid antigens are not necessarily part of the pathogenic autoimmune response in predisposed individuals but may, in fact, play other roles (discussed further below).
Fig. 4.
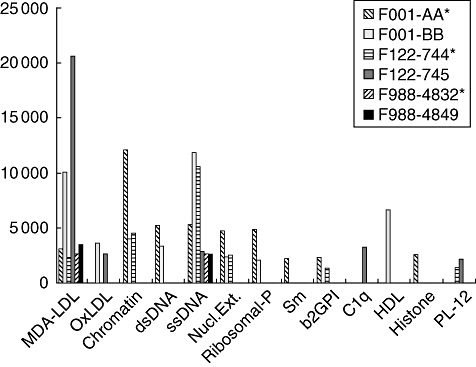
Immunoglobulin M (IgM) autoantibody repertoires in disease-discordant twins. Values are depicted for IgM autoantibody reactivity levels detected by proteomic microarrays. Twin sets are depicted as adjacent bars, with lupus-unaffected twin indicated as a solid bar, and the affected twins are marked by patterned bars and are also marked by * in the figure legend.
Discussion
In the current studies we have used multiplex proteome microarrays to characterize and compare the autoantibody responses of one of the largest panels of lupus twins reported to date. From these studies, we found that individual IgG autoantibody profiles often clustered according to natural twin pairs, while similar patterns were not found with unrelated SLE patients. Notably, we found that six of eight disease-concordant MZ twin sets displayed hierarchical clustering of their IgG patterns, while an additional twin pair also co-distributed in the dendrogram because of very similar patterns. Neither of two SLE disease non-concordant MZ twins displayed clustered IgG patterns (Table 1). Of the four DZ twin sets studied, only one displayed true IgG autoantibody pattern-clustering, although these twins were among the two disease-discordant DZ pairs. By contrast, altogether only two of 10 MZ and one of four DZ twin sets were found to have true clustering and two additional MZ and one DZ twin sets had related/co-distribution of their IgM autoantibody profiles (total of six of 14), and these profiles did not appear to correlate rigorously with SLE disease concordance (Table 1).
Our antigen panel included disease-associated autoantigens that are used routinely to aid in the diagnosis of SLE and other autoimmune diseases. Despite methodological differences that can affect sensitivity and specificity of detection, we detected autoantibody reactivity patterns that reiterated results reported commonly using more conventional immunoassay approaches (i.e. high prevalence of autoantibodies to DNA- and RNA-related antigens). We also studied autoantigens recognized by ‘natural’ autoantibodies, and found high reactivity levels and great prevalence (i.e. 37 of 42 IgM-reactive and 40 of 42 IgG-reactive) to the phospholipid-containing antigens, MDA-LDL, and 16 of 42 were IgM-reactive and nine of 42 IgG-reactive for OxLDL (Figs 1 and 2, Table 2). These specificities of autoantibodies were first shown to be elevated in hyperlipidaemic mice and humans with levels correlated directly with severity of atherosclerosis (reviewed in [26]).
Our microarray surveys found consistent trends towards higher levels of both IgM and IgG autoantibodies to MDA-LDL and OxLDL, as well as to PC- and MDA-protein conjugates in SLE patients compared with healthy adults (Figs 2 and 3), and these findings reiterated an earlier report of significantly increased levels of IgM and IgG antibodies to MDA-LDL and OxLDL in 157 SLE patients compared with 60 age- and gender-matched healthy controls, using chemiluminescent immunoassays [27]. More recently, from a microarray panel of over 300 antigens, surveys of human umbilical cord blood (i.e. neonatal blood) found that the highest relative IgM antibody reactivities were to LDL (which undergoes spontaneous oxidative changes), demonstrating that these IgM natural autoantibodies are also common from very early in human immune development [28]. Our studies therefore support the notion that autoantibodies to these phospholipid-related antigens may, in fact, be among the most prevalent types of autoantibodies in SLE patients (discussed further below).
A recent meta-analysis found that there are at least four different genes/loci that can contribute to lupus predisposition in different populations [29]. Notably, genome-wide scans in 229 SLE multiplex pedigrees reportedly found genetic linkages with conventional immunochemical assays for seven types of IgG autoantibody fine binding specificities [30]. However, the mechanistic bases by which such genetic variations lead to specific autoantibody expression remain poorly explained.
Our findings suggest that there can be a common conservation of IgG autoantibody profiles that arise because of shared genetic inheritance of twins. From one perspective, these findings were unexpected, as autoantibody responses are generated somatically from recombination of diverse sets of antibody minigenes, and which are affected by random environmental and stochastic influences. In fact, the conservation of expressed IgG responses probably results in part from shared genetic inheritance of the same sets of histocompatibility immune-responsiveness elements [e.g. major histocompatibility complex (MHC)] that define and restrict the range of minimal peptides that can be recognized by T cells. Hence, if immune tolerance is broken, MHC largely determines the peptides from autoantigens that can be recognized by autoreactive T cells, and this autoreactive T cell repertoire is then intertwined in T dependent IgG autoantibody responses [31]. We therefore believe that our results reflect the influence of MHC, as well as immunoregulatory genes such as PTPN22, CTLA4, PDCD1, IRF5, Ox40L [32] and STAT4 [33] (reviewed in [30]), which together restrict the IgG repertoire that is shared by each lupus twin set. We speculate that these major response factors determine the specific immunological path that an individual may be destined to travel, with the associated induction of a characteristic autoantibody profile, as they progress towards overt lupus disease.
At the time of blood sampling for our panel, 10 of the 14 twin sets were known to be concordant for SLE − a rate of disease concordance for MZ and DZ twins higher than found generally by other investigators that we assume resulted from unintended biases in patient referral and enrolment. While we were surprised to find conserved IgG autoantibody profiles in twin sets that were identified as MZ and also as DZ, it should be mentioned that the zygosity status of only four of the twin sets was determined by genotyping. For the remaining 10 twins, status was assigned based on a history of the presence of single or dual placentas. We may be speculate that in such cases there may be errors such that one or more of the DZ twins, F285, F988 and F1001, which each displayed features of autoantibody repertoire concordance, were actually MZ sets.
Our studies extend earlier reports of conserved IgG autoantibody reactivity among related individuals, including twins (reviewed in [11]). However, these earlier studies were of much more limited scope with findings for, at most, a few autoantibody specificities. We believe that ours is the first to report broad surveys of IgM and IgG autoreactivity patterns for natural and disease-associated autoantibodies in lupus family members. Hence, these earlier studies do not diminish the significance of our findings of the conservation of shared IgG autoantibody reactivity patterns by related twins, while shared twin-specific IgM autoantibody profiles are less common.
The contribution of circulating IgM to lupus pathogenesis is not well understood. SLE patients have been reported to have defects in the capacity to make polymeric IgM [34], which could impair the strength of the antigen-binding interactions. Moreover, mice unable to make polymeric IgM are reported to be predisposed to lupus and display accelerated disease [35]. Similarly, mice deficient in circulating IgM develop IgG autoantibodies and lupus-like disease spontaneously [36,37]. In our clinical studies, we found that the F794 set of disease-concordant MZ twins had an absence of detectable IgM autoantibodies, which is consistent with the hypothesis that certain clones within circulating polymeric IgM protects from lupus pathogenesis. The only other individual that we found with this IgM-deficient pattern (F1001-4862) was a DZ twin diagnosed with giant cell arteritis, an idiopathic inflammatory disease, who was also SLE discordant.
Circulating IgM and IgG may have very distinct cellular immunobiological origins and play different functional roles. In mice, most circulating IgM comes from B-1 cells [19,38], a distinct subset of mature B cells with differences in surface phenotype and activation thresholds than the follicular B cells that are the predominant source of T cell-dependent IgG responses. Significantly, B-1 cells are the major source of murine antibodies to OxLDL, and this type of IgM natural autoantibody can protect from the progression of atherosclerosis [39]. IgM autoantibodies to OxLDL and MDA-LDL cross-react with determinants on cells undergoing apoptotic death, but do not recognize healthy cells [19,40]. Levels of these autoantibodies are increased greatly following intravenous infusions of apoptotic cells [41], which suggests that lupus-associated increases in anti-MDA and anti-PC antibodies could reflect direct T cell-independent B cell immune responses to apoptotic cells.
Apoptosis is an obligatory outcome of development, proliferation and cell differentiation that continues throughout the life of the host. Every day, more than 1011 cells in our bodies die by apoptosis. Apoptotic cells are therefore ubiquitous, and in health they themselves do not pose an immediate threat to the host. Presumably for this reason, under certain conditions apoptotic cells and their membranes have anti-inflammatory influences. However, if apoptotic cells are not cleared quickly there can be cellular changes with progression to necrosis with release proinflammatory substances. This instead induces potentially damaging inflammatory responses, and may also enhance the immunogenicity of self-antigens that results in selection and expansion of potentially pathogenic B cell and T cell clones. To relate these events to pathogenesis, Walport and colleagues have described the ‘waste disposal’ hypothesis, in which defects in the removal of dying cells and cell debris have been linked to breaches in immune tolerance and a predisposition to autoimmune disease [42].
In recent studies, infusions of a B-1 cell-derived monoclonal IgM anti-OxLDL antibody was shown to enhance in vivo macrophage-mediated phagocytic clearance of apoptotic cells, and blunted responses to Toll-like receptor-mediated inflammatory responses [43]. Evidence that such treatments can also improve the survival of lupus-prone mice [43] supports the hypothesis that there are certain specificities of IgM autoantibodies that play protective roles in individuals predisposed to autoimmune disease. Moreover, other investigators have shown that infusions of an IgM anti-native DNA antibody was also protective in another murine lupus model [44]. Such natural antibody regulatory protective roles may therefore explain our finding of significant direct correlations in the levels of IgM anti-MDA and anti-OxLDL antibodies with IgM anti-DNA responses (Table 3). Notably, in the current studies three of the lupus-discordant twins had significantly elevated levels of IgM autoantibodies to MDA-LDL and/or to DNA antigens (Figs 2 and 4) and two also had IgM autoantibodies to OxLDL, which included the discordant F001-BB subject, who was a major focus of our previous lupus-repertoire cloning studies that prompted the current investigations [14]. Also consistent with this hypothesis, in their autoantigen microarray surveys Olsen and co-workers found higher levels of IgM autoantibody in patients with incomplete lupus syndromes (ILE) compared with healthy controls [16]. These ILE patients were also found to have diminished clinical disease activity [18], which was postulated to explain the better prognosis associated with such patients [45].
While we found an overall correlation in SLE patients between levels of IgM autoantibodies to apoptosis-related phospholipid and DNA-containing antigens, a similar relationship was not found for IgM autoantibodies to RNA-containing antigens. While we wondered whether these differences may reflect the in vivo accessibility of these different autoantigens, Casciola-Rosen et al. reported that DNA- and RNA-related antigens may become co-clustered in the blebs on the surface of cells undergoing apoptotic death [46], and later studies have shown that Ro60 and La (SSB) antigens may become surface exposed and accessible to recognition by specific IgG autoantibodies [47,48]. In fact, interactions of IgG autoantibody with exposed Ro antigens on apoptotic cardiac myocytes are reported to interfere with the efficient phagocytic clearance of these cells, and may contribute to proinflammatory pathological autoimmune responses [49]. However, it remains possible that these autoantigen expression patterns, described for in vitro-induced apoptosis, may differ in other cell types and during the pathways of apoptotic death occurring in vivo. The capacity of IgM autoantibodies for immune recognition of these RNA-related antigens may also differ. Alternatively, our findings of correlation of IgM autoantibodies to apoptosis-associated antigens with DNA-related but not with RNA-related antigens could also be explained if different inherited immune response elements are required for anti-DNA and for anti-RNA-related antibody responses. Based in part on evidence of higher IgG autoantibody responses in the affected twins of the disease discordant pairs, we also wonder whether otherwise protective IgM autoantibody responses may at times undergo disease-associated dysregulated IgG class-switch to subclasses capable of proinflammatory FcγR-mediated interactions, which then contribute to lupus pathogenesis.
In conclusion, among the great variation in specific autoantibody profiles expressed by unrelated SLE individuals, our findings demonstrate that autoantigen microarrays can be used to identify characteristic autoantibody fingerprints. Although our studies could not consider independently the influence of environmental factors, the current surveys provide strong evidence that there is genetic determination of expressed IgG autoantibody profiles. We have also found evidence that the mostly highly expressed autoantibody specificities may be for phospholipid-related determinants expressed on apoptotic cells. While our studies were inadequate to consider directly the potential protective roles of such natural antibodies in genetically predisposed but unaffected individuals, this issue should be addressed in larger family-based studies.
Our longitudinal surveys demonstrated that IgG autoantibody patterns were generally stable in clinically stable patients on oral immunosuppressives (Fig. 1). Clustering of autoantibody profiles was also found in twins on oral immunosuppressives and in a twin set (F1008) that both received mycophenolate mofetil, while one was also treated with intravenous cyclophosphamide (Figs 2 and 3). This methodological array approach therefore appears well suited for investigations of the impact on the B cell compartment of therapeutic interventions, including agents in development that are designed to induce autoantigen-specific immune tolerance (e.g. abetimus, synthetic DNA ligand) [50], or affect specific autoreactive responses by interfering with specific Toll-like receptors [51] or cytokine responses (e.g. type I interferon) [52,53].
Acknowledgments
We thank Drs P. J. Utz and W. Robinson for technical advice. We also thank the UCSD BIOGEM Core for assisting our initial printing efforts, Dr Gary Gilkesen (MUSC) for human glomerular extract, Dr X. Wu for early contributions to assay analyses and Dr Rufus Burlingame and INOVA Diagnostics for materials and technical advice. We acknowledge the institutional contributions of UCLA and SUNY Downstate that support the lupus clinical investigations. This work was supported by grants AI40305, AR47360, AR50659 and AI46637 from NIH and the Alliance for Lupus Research (to G. J. S.), AR43814 (to B. P. T.) and a summer fellowship from the Lupus Foundation of America (to R. S.).
References
- 1.Tan EM, Cohen AS, Fries JF, et al. The 1982 revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1982;25:1271–7. doi: 10.1002/art.1780251101. [DOI] [PubMed] [Google Scholar]
- 2.Holborow EJ, Weir DM, Johnson GD. A serum factor in lupus erythematosus with affinity for tissue nuclei. BMJ. 1975;2:732–4. doi: 10.1136/bmj.2.5047.732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lefkowith JB, Gilkeson GS. Nephritogenic autoantibodies in lupus: current concepts and continuing controversies. Arthritis Rheum. 1996;39:894–903. doi: 10.1002/art.1780390605. [DOI] [PubMed] [Google Scholar]
- 4.Arbuckle MR, McClain MT, Rubertone MV, et al. Development of autoantibodies before the clinical onset of systemic lupus erythematosus. N Engl J Med. 2003;349:1526–33. doi: 10.1056/NEJMoa021933. [DOI] [PubMed] [Google Scholar]
- 5.Heinlen LD, McClain MT, Kim X, et al. Anti-Ro and anti-nRNP response in unaffected family members of SLE patients. Lupus. 2003;12:335–7. doi: 10.1191/0961203303lu377xx. [DOI] [PubMed] [Google Scholar]
- 6.Heinlen LD, McClain MT, Merrill J, et al. Clinical criteria for systemic lupus erythematosus precede diagnosis, and associated autoantibodies are present before clinical symptoms. Arthritis Rheum. 2007;56:2344–51. doi: 10.1002/art.22665. [DOI] [PubMed] [Google Scholar]
- 7.Kokori SI, Ioannidis JP, Voulgarelis M, Tzioufas AG, Moutsopoulos HM. Autoimmune hemolytic anemia in patients with systemic lupus erythematosus. Am J Med. 2000;108:198–204. doi: 10.1016/s0002-9343(99)00413-1. [DOI] [PubMed] [Google Scholar]
- 8.McCarty DJ, Manzi S, Medsger TA, Ramsey-Goldman R, LaPorte RE, Kwoh CK. Incidence of systemic lupus erythematosus. Race and gender differences. Arthritis Rheum. 1995;38:1260–70. doi: 10.1002/art.1780380914. [DOI] [PubMed] [Google Scholar]
- 9.Michel M, Johanet C, Meyer O, et al. Familial lupus erythematosus. Clinical and immunologic features of 125 multiplex families. Medicine (Balt) 2001;80:153–8. doi: 10.1097/00005792-200105000-00001. [DOI] [PubMed] [Google Scholar]
- 10.Block SR, Lockshin MD, Winfield JB, et al. Immunologic observations on 9 sets of twins either concordant or discordant for SLE. Arthritis Rheum. 1976;19:545–54. doi: 10.1002/art.1780190306. [DOI] [PubMed] [Google Scholar]
- 11.Block SR. A brief history of twins. Lupus. 2006;15:61–4. doi: 10.1191/0961203306lu2263ed. [DOI] [PubMed] [Google Scholar]
- 12.Deapen D, Escalante A, Weinrib L, et al. A revised estimate of twin concordance in systemic lupus erythematosus. Arthritis Rheum. 1992;35:311–18. doi: 10.1002/art.1780350310. [DOI] [PubMed] [Google Scholar]
- 13.Koroleva NI, Kaladze NN, Kirichenko AI. [Collagen diseases in twins] Vopr Revm. 1973;13:53–5. [PubMed] [Google Scholar]
- 14.Roben P, Barbas SM, Sandoval L, et al. Repertoire cloning of lupus anti-DNA autoantibodies. J Clin Invest. 1996;98:2827–37. doi: 10.1172/JCI119111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Robinson WH, DiGennaro C, Hueber W, et al. Autoantigen microarrays for multiplex characterization of autoantibody responses. Nat Med. 2002;8:295–301. doi: 10.1038/nm0302-295. [DOI] [PubMed] [Google Scholar]
- 16.Li QZ, Zhou J, Wandstrat AE, et al. Protein array autoantibody profiles for insights into systemic lupus erythematosus and incomplete lupus syndromes. Clin Exp Immunol. 2007;147:60–70. doi: 10.1111/j.1365-2249.2006.03251.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Graham KL, Robinson WH, Steinman L, Utz PJ. High-throughput methods for measuring autoantibodies in systemic lupus erythematosus and other autoimmune diseases. Autoimmunity. 2004;37:269–72. doi: 10.1080/08916930410001710686. [DOI] [PubMed] [Google Scholar]
- 18.Li QZ, Xie C, Wu T, et al. Identification of autoantibody clusters that best predict lupus disease activity using glomerular proteome arrays. J Clin Invest. 2005;115:3428–39. doi: 10.1172/JCI23587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shaw PX, Horkko S, Chang MK, et al. Natural antibodies with the T15 idiotype may act in atherosclerosis, apoptotic clearance, and protective immunity. J Clin Invest. 2000;105:1731–40. doi: 10.1172/JCI8472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Palinski W, Yla-Herttuala S, Rosenfeld ME, et al. Antisera and monoclonal antibodies specific for epitopes generated during oxidative modification of low density lipoprotein. Arteriosclerosis. 1990;10:325–35. doi: 10.1161/01.atv.10.3.325. [DOI] [PubMed] [Google Scholar]
- 21.Shoenfeld Y, Gershwin ME, Meroni PL. Autoantibodies. 2. Amsterdam: Elsevier; 2007. [Google Scholar]
- 22.von Mühlen CA, Tan EM. Autoantibodies in the diagnosis of systemic rheumatic diseases. Semin Arthritis Rheum. 1995;24:323–58. doi: 10.1016/s0049-0172(95)80004-2. [DOI] [PubMed] [Google Scholar]
- 23.Ochs RL. Methods used to study structure and function of the nucleolus. Methods Cell Biol. 1998;53:303–21. doi: 10.1016/s0091-679x(08)60884-5. [DOI] [PubMed] [Google Scholar]
- 24.Chang MK, Bergmark C, Laurila A, et al. Monoclonal antibodies against oxidized low-density lipoprotein bind to apoptotic cells and inhibit their phagocytosis by elicited macrophages: evidence that oxidation-specific epitopes mediate macrophage recognition. Proc Natl Acad Sci USA. 1999;96:6353–8. doi: 10.1073/pnas.96.11.6353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Horkko S, Bird DA, Miller E, et al. Monoclonal autoantibodies specific for oxidized phospholipids or oxidized phospholipid-protein adducts inhibit macrophage uptake of oxidized low-density lipoproteins. J Clin Invest. 1999;103:117–28. doi: 10.1172/JCI4533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Binder CJ, Silverman GJ. Natural antibodies and the autoimmunity of atherosclerosis. Springer Semin Immunopathol. 2005;26:385–404. doi: 10.1007/s00281-004-0185-z. [DOI] [PubMed] [Google Scholar]
- 27.Frostegard J, Svenungsson E, Wu R, et al. Lipid peroxidation is enhanced in patients with systemic lupus erythematosus and is associated with arterial and renal disease manifestations. Arthritis Rheum. 2005;52:192–200. doi: 10.1002/art.20780. [DOI] [PubMed] [Google Scholar]
- 28.Merbl Y, Zucker-Toledano M, Quintana FJ, Cohen IR. Newborn humans manifest autoantibodies to defined self molecules detected by antigen microarray informatics. J Clin Invest. 2007;117:712–18. doi: 10.1172/JCI29943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Harley JB, Kelly JA, Kaufman KM. Unraveling the genetics of systemic lupus erythematosus. Springer Semin Immunopathol. 2006;28:119–30. doi: 10.1007/s00281-006-0040-5. [DOI] [PubMed] [Google Scholar]
- 30.Ramos PS, Kelly JA, Gray-McGuire C, et al. Familial aggregation and linkage analysis of autoantibody traits in pedigrees multiplex for systemic lupus erythematosus. Genes Immun. 2006;7:417–32. doi: 10.1038/sj.gene.6364316. [DOI] [PubMed] [Google Scholar]
- 31.Kuwana M, Feghali CA, Medsger TA, Wright TM. Autoreactive T cells to topoisomerase I in monozygotic twins discordant for systemic sclerosis. Arthritis Rheum. 2001;44:1654–9. doi: 10.1002/1529-0131(200107)44:7<1654::AID-ART288>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 32.Graham DS, Graham RR, Manku H, et al. Polymorphism at the TNF superfamily gene TNFSF4 confers susceptibility to systemic lupus erythematosus. Nat Genet. 2008;40:83–9. doi: 10.1038/ng.2007.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Remmers EF, Plenge RM, Lee AT, et al. STAT4 and the risk of rheumatoid arthritis and systemic lupus erythematosus. N Engl J Med. 2007;357:977–86. doi: 10.1056/NEJMoa073003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kaufman LD, Heinicke MH, Hamburger M, Gorevic PD. Male lupus: prevalence of IgA deficiency, 7S IgM and abnormalities of reticuloendothelial system Fc-receptor function. Clin Exp Rheumatol. 1991;9:265–9. [PubMed] [Google Scholar]
- 35.Youd ME, Luus L, Corley RB. IgM monomers accelerate disease manifestations in autoimmune-prone Fas-deficient mice. J Autoimmun. 2004;23:333–43. doi: 10.1016/j.jaut.2004.09.001. [DOI] [PubMed] [Google Scholar]
- 36.Boes M, Schmidt T, Linkemann K, Beaudette BC, Marshak-Rothstein A, Chen J. Accelerated development of IgG autoantibodies and autoimmune disease in the absence of secreted IgM. Proc Natl Acad Sci USA. 2000;97:1184–9. doi: 10.1073/pnas.97.3.1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ehrenstein MR, Cook HT, Neuberger MS. Deficiency in serum immunoglobulin (Ig) M predisposes to development of IgG autoantibodies. J Exp Med. 2000;191:1253–8. doi: 10.1084/jem.191.7.1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Baumgarth N, Herman OC, Jager GC, Brown LE, Herzenberg LA, Chen J. B-1 and B-2 cell-derived immunoglobulin M antibodies are nonredundant components of the protective response to influenza virus infection. J Exp Med. 2000;192:271–80. doi: 10.1084/jem.192.2.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Binder C, Hörkkö S, Dewan A, et al. Pneumococcal vaccination decreases atherosclerotic lesion formation: molecular mimicry between oxidized LDL and Streptococcus pneumoniae. Nat Med. 2003;9:736–43. doi: 10.1038/nm876. [DOI] [PubMed] [Google Scholar]
- 40.Shaw PX, Goodyear CS, Chang M-K, Witztum J, Silverman GJ. The autoreactivity of anti-phosphorylcholine antibodies for atherosclerosis-associated neo-antigens and apoptotic cells. J Immunol. 2003;170:6151–7. doi: 10.4049/jimmunol.170.12.6151. [DOI] [PubMed] [Google Scholar]
- 41.Chang MK, Binder CJ, Miller YI, et al. Apoptotic cells with oxidation-specific epitopes are immunogenic and proinflammatory. J Exp Med. 2004;200:1359–70. doi: 10.1084/jem.20031763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Manderson AP, Botto M, Walport MJ. The role of complement in the development of systemic lupus erythematosus. Annu Rev Immunol. 2004;22:431–56. doi: 10.1146/annurev.immunol.22.012703.104549. [DOI] [PubMed] [Google Scholar]
- 43.Silverman GJ, Haraldsson MK, Goodyear CS, Chen YF, Kono DH. Passive monoclonal antibody treatment that enhances clearance of apoptotic particles improves survival in lupus-prone (NZW×BXSB) F1 mice. Arthritis Rheum. 2005;52(Suppl. 89):s699. [Abstract] [Google Scholar]
- 44.Werwitzke S, Trick D, Kamino K, et al. Inhibition of lupus disease by anti-double-stranded DNA antibodies of the IgM isotype in the (NZB × NZW) F1 mouse. Arthritis Rheum. 1891 2005;52:3629–38. doi: 10.1002/art.21379. [DOI] [PubMed] [Google Scholar]
- 45.Swaak AJ, van de Brink H, Smeenk RJ, et al. Incomplete lupus erythematosus: results of a multicentre study under the supervision of the EULAR Standing Committee on International Clinical Studies Including Therapeutic Trials (ESCISIT) Rheumatology (Oxford) 2001;40:89–94. doi: 10.1093/rheumatology/40.1.89. [DOI] [PubMed] [Google Scholar]
- 46.Casciola-Rosen LA, Anhalt G, Rosen A. Autoantigens targeted in systemic lupus erythematosus are clustered in two populations of surface structures on apoptotic keratinocytes. J Exp Med. 1994;179:1317–30. doi: 10.1084/jem.179.4.1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Miranda-Carus ME, Askanase AD, Clancy RM, et al. Anti-SSA/Ro and anti-SSB/La autoantibodies bind the surface of apoptotic fetal cardiocytes and promote secretion of TNF-alpha by macrophages. J Immunol. 2001;165:5345–51. doi: 10.4049/jimmunol.165.9.5345. [DOI] [PubMed] [Google Scholar]
- 48.Reed JH, Neufing PJ, Jackson MW, et al. Different temporal expression of immunodominant Ro60/60 kDa-SSA and La/SSB apotopes. Clin Exp Immunol. 2007;148:153–60. doi: 10.1111/j.1365-2249.2007.03331.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Clancy RM, Neufing PJ, Zheng P, et al. Impaired clearance of apoptotic cardiocytes is linked to anti-SSA/Ro and -SSB/La antibodies in the pathogenesis of congenital heart block. J Clin Invest. 2006;116:2413–22. doi: 10.1172/JCI27803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mosca M, Baldini C, Bombardieri S. LJP-394 (abetimus sodium) in the treatment of systemic lupus erythematosus. Expert Opin Pharmacother. 2007;8:873–9. doi: 10.1517/14656566.8.6.873. [DOI] [PubMed] [Google Scholar]
- 51.Pawar RD, Ramanjaneyulu A, Kulkarni OP, Lech M, Segerer S, Anders HJ. Inhibition of Toll-like receptor-7 (TLR-7) or TLR-7 plus TLR-9 attenuates glomerulonephritis and lung injury in experimental lupus. J Am Soc Nephrol. 2007;18:1721–31. doi: 10.1681/ASN.2006101162. [DOI] [PubMed] [Google Scholar]
- 52.Hua J, Kirou K, Lee C, Crow MK. Functional assay of type I interferon in systemic lupus erythematosus plasma and association with anti-RNA binding protein autoantibodies. Arthritis Rheum. 2006;54:1906–16. doi: 10.1002/art.21890. [DOI] [PubMed] [Google Scholar]
- 53.Strandberg L, Ambrosi A, Espinosa A, et al. Interferon-alpha induces up-regulation and nuclear translocation of the Ro52 autoantigen as detected by a panel of novel Ro52-specific monoclonal antibodies. J Clin Immunol. 2007 doi: 10.1007/s10875-007-9157-0. epub ahead of press. [DOI] [PubMed] [Google Scholar]