

Abstract
Ku is a heterodimeric protein comprising 70- and 80-kDa subunits that participate in the non-homologous end-joining (NHEJ) repair pathway for rejoining DNA double strand breaks. We have analyzed the pre-steady state binding of Ku with various DNA duplex substrates and identified a redox-sensitive Ku-DNA interaction. Pre-steady state analysis of Ku DNA binding was monitored via intrinsic Ku quenching upon binding DNA and revealed that, under fully reduced conditions, binding occurred in a single-step process. Reactions performed under limited reduction revealed a two-step binding process, whereas under fully oxidized conditions, we were unable to detect quenching of Ku fluorescence upon binding DNA. The differential quenching observed under the different redox conditions could not be attributed to two Ku molecules binding to a single substrate or Ku sliding inward on the substrate. Although only modest differences in Ku DNA binding activity were observed in the stoichiometric anisotropy and electrophoretic mobility shift assay studies, as a function of redox conditions, a dramatic difference in the rate of Ku dissociation from DNA was observed. This effect was also induced by diamide treatment of Ku and could be abrogated by dithiothreitol treatment, demonstrating a reversible redox effect on the stability of the Ku-DNA complex. The redox-dependent alteration in Ku-DNA interactions is manifested by a redox-dependent alteration in Ku structure, which was confirmed by limited proteolysis and mass spectrometry analyses. The results support a model for the interaction of Ku with DNA that is regulated by redox status and is achieved by altering the dissociation of the Ku-DNA complex.
DNA double strand breaks (DSBs)2 can be a lethal type of DNA damage. Cells have developed two major pathways in which to remove such damage, homologous recombination and NHEJ (1). The Ku autoantigen is an essential component of the NHEJ pathway. Ku shows a high affinity for double strand DNA termini (2), and once bound to the DNA end, increases the affinity of the DNA-PK catalytic subunit (DNA-PKcs) for the DNA termini (3). To accommodate the large 469-kDa DNA-PKcs, Ku translocates inward on the DNA in an ATP-independent manner, ultimately forming the active DNA-PK complex (3, 4). Activated DNA-PK then phosphorylates target proteins, including Artemis (5), Mre11/Rad50/NBS1 complex (6), and DNA ligase IV/XRCC4 (7), all of which participate in either signaling events in response to DNA damage or completing the repair process.
The crystal structure of free Ku and the Ku-DNA complex has been solved and reveals a novel DNA binding structure comprising a base, two pillars, and a bridge through which the duplex DNA can thread (8). The Ku70 and -80 subunits share minimal sequence homology, yet adopt similar three-dimensional structures, with each subunit contributing to the base, pillars, and bridge (8). This ring-like structure of Ku provides the structural basis for the DNA termini requirement for binding as well as the energy-independent ability of Ku to slide along a duplex DNA. In the absence of DNA, the same general structure is observed, suggesting that the ring is preformed and not generated as a result of binding to the DNA termini (8). The Ku-DNA co-crystal structure reveals no direct contact with the nucleobases but contain a number of hydrophobic amino acids positioned within the major groove of the duplex DNA termini and basic amino acids that interact with the phosphate backbone (8). Photo cross-linking studies of Ku with duplex DNA have revealed a series of potential contacts that orient the Ku dimer with the 70-kDa subunit positioned closer to the terminus and the 80-kDa subunits positioned internally (3, 9). This geometry of the Ku dimer can be explained by the Ku70 subunit acidic region that potentially restricts the duplex DNA from entering the Ku dimer from the Ku70 face. This region may also serve to limit the internal sliding activity of Ku.
In vitro analysis of Ku binding has been assessed using a variety of assay systems. The requirement for DNA termini was established relatively early and was demonstrated in filter binding assays (10). Analysis of a large number of DNA structures is consistent with Ku displaying a high affinity for duplex DNA termini (11–13). Once bound to a DNA terminus, Ku also has the ability to slide or translocate along the DNA duplex (2). In the absence of the DNA-PKcs subunit, multiple Ku molecules can thread onto a single DNA duplex, and the number of Ku dimers bound is a function of DNA length. Once bound, the dissociation from the duplex, as a result of the ring structure, requires Ku to slide off the end of the DNA. The mechanism and regulation of this dissociation is largely unclear, especially in the context of NHEJ, where dissociation must occur prior to ligation of the DNA termini. Using a series of fluorescence-based assays, there was limited cooperativity observed in the Ku-DNA binding interaction (14). These results suggest that there is minimal interaction of the Ku dimers when bound to a DNA. These solution-based assays also demonstrate that, under moderate ionic strength, binding was stoichiometric such that minimal dissociation was observed (15). ATP and Mg2+ were found not to have major influences on binding (15), yet dithiothreitol (DTT) has been demonstrated to increase binding activity in EMSAs, suggesting that the interaction with DNA may be regulated by redox conditions (16).
Recent reports have demonstrated a link between redox stress and the NHEJ pathway. This connection has been demonstrated in cells where increased oxidative stress results in decreased DNA-PK activity and where decreased oxidative stress increased DNA-PK activity (17). The redox effect on NHEJ was later shown to involve autophosphorylation of DNA-PKcs (18), consistent with the importance of autophosphorylation on DNA-PK activity and NHEJ (19, 20). Regulation of redox stress via genetic mutation has also been found to influence DNA DSB repair (21). Analysis of the Ku protein reveals a decrease in DNA-binding activity as a function of oxidative stress (22).
To further investigate the effect of redox on Ku structure and function, we employed the intrinsic fluorescence of Ku and the subsequent quenching that occurs upon binding to DNA to assess the kinetics of binding. In addition to defining the pre-steady state kinetic interaction of Ku with duplex DNA, we identified a mechanism of redox regulation of Ku binding and demonstrated that the regulation is dependent on a conformational change that influences the Ku-DNA fluorescence and dissociation rate.
EXPERIMENTAL PROCEDURES
DNA Substrates and Protein
Oligonucleotides used in this study are presented in Table 1. The oligonucleotides were purchased from Integrated DNA Technologies, Inc. (Coralville, IA), gel-purified by preparative denaturing polyacrylamide gel electrophoresis, and annealed to the appropriate complement. Human Ku was purified from recombinant baculovirus-infected Sf9 insect cells as previously described (23).
TABLE 1.
Oligonucleotides used in this study
| Name | Sequence 5′–3′ | Reference |
|---|---|---|
| 30a | 5′-CCCCTATCCTTTCCGCGTCCTTACTTCCCC-3′ | 17 |
| 30C | 5′-GGGGAAGTAAGGACGCGGAAAGGATAGGGG-3′ | 17 |
| 21a | 5′-GTTTTTAGTTTATTGGGCGCG-3′ | 8 |
| 34 | 5′-CGCGCCCAGCTTTCCCAGCTAATAAACTAAAAAC-3′ | 8 |
| 75 | 5′-TACCCGGGGATCCTCTAGAGTCGACCTGCAGGCATGCAAGCTTTTGTTCCCTTTAGTGAGGGTTAATTCCGAGCT-3′ | 4 |
| 75C | 5′-AGCTCGGAATTAACCCTCACTAAAGGGAACAAAAGCTTGCATGCCTGCAGGTCGACTCTAGAGGATCCCCGGGTA-3′ | 4 |
Labeled with fluorescein for anisotropy studies.
Stopped-flow Kinetic Experiments
Stopped-flow traces were obtained using an SX.18MV stopped-flow reaction analyzer (Applied Photophysics). Reactions were excited at 280 nm, and fluorescence emission was measured using a 350-nm-long pass cut on filter (LG-350 from Corion, Franklin, MA). A constant Ku concentration of 25 nM was used with varying concentrations of DNA (as indicated in figure legends) to determine kinetic binding. Equal volumes of Ku and DNA were prepared in buffer containing 50 mM Tris-Cl, pH 7.8, 10 mM MgCl2, and 50 mM NaCl and were rapidly mixed at 24 °C. Traces represent an average of 12–14 individual shots. Kinetic data were fit to either a single exponential or double exponential decay using Sigmaplot software to determine kobs values. Rate constants were determined by plotting kobs values and using linear regression analysis in Sigmaplot, where the slope is the rate of association (kon), and the y-intercept is the rate of dissociation (koff). Each point represents the average ± S.D. from five individual experiments.
EMSAs
EMSAs were performed in 20-μl reactions in a buffer containing 50 mM Tris-Cl, pH 7.8, 10 mM MgCl2, and 50 mM NaCl. Each reaction contained 500 fmol of 32P-labeled 30-bp duplex DNA with the indicated concentrations of Ku that had been dialyzed either in the presence or absence of DTT, Triton X-100, and protease inhibitors. Reactions were mixed and loaded onto a 6% native polyacrylamide gel. The gels were dried and exposed to a PhosphorImager (Amersham Biosciences) screen and quantified using ImageQuant software.
Fluorescence Polarization
Fluorescence polarization experiments were performed using a Cary Eclipse Fluorescence Spectrophotometer (Varian). Reactions were performed in a volume of 0.5 ml in a buffer containing 50 mM Tris-Cl, pH 7.8, 10 mM MgCl2, 50 mM NaCl, and DTT as indicated. Saturation curves were obtained by titrating Ku into reactions containing 10 nM fluorescein-labeled duplex DNA ends in the presence or absence of 1 mM DTT and measuring anisotropy at each Ku concentration. Fluorescence excitation was at 495 nm and emission was read at 515 nm, both having bandwidths set to 5 nm. The Ku-DNA binding is represented as the r value and was calculated as described previously (24).
Heparin Trap
To determine rates of Ku dissociation from the DNA duplex, we employed a heparin trap assay. The Ku preparation was added to 0.5-ml reactions containing the fluorescein-labeled DNA substrate. The DNA concentration used was based on DNA termini and was 10 nM for all dissociation experiments. Pre-treatment of Ku with 2 mM diamide or 5 mM DTT was performed for 15 min at room temperature. Following formation of the Ku-DNA complex, heparin (10 μg) was added, and the change in anisotropy was measured at fixed intervals over a 15-min time frame. At each time point, 2–5 independent readings were taken with parallel and perpendicular polarizers, and the calculated r values were averaged to give the value for that time point. Each dissociation experiment was performed three times, and the average ± S.D. is presented in Fig. 6.
FIGURE 6. Redox alters the dissociation of Ku from DNA.
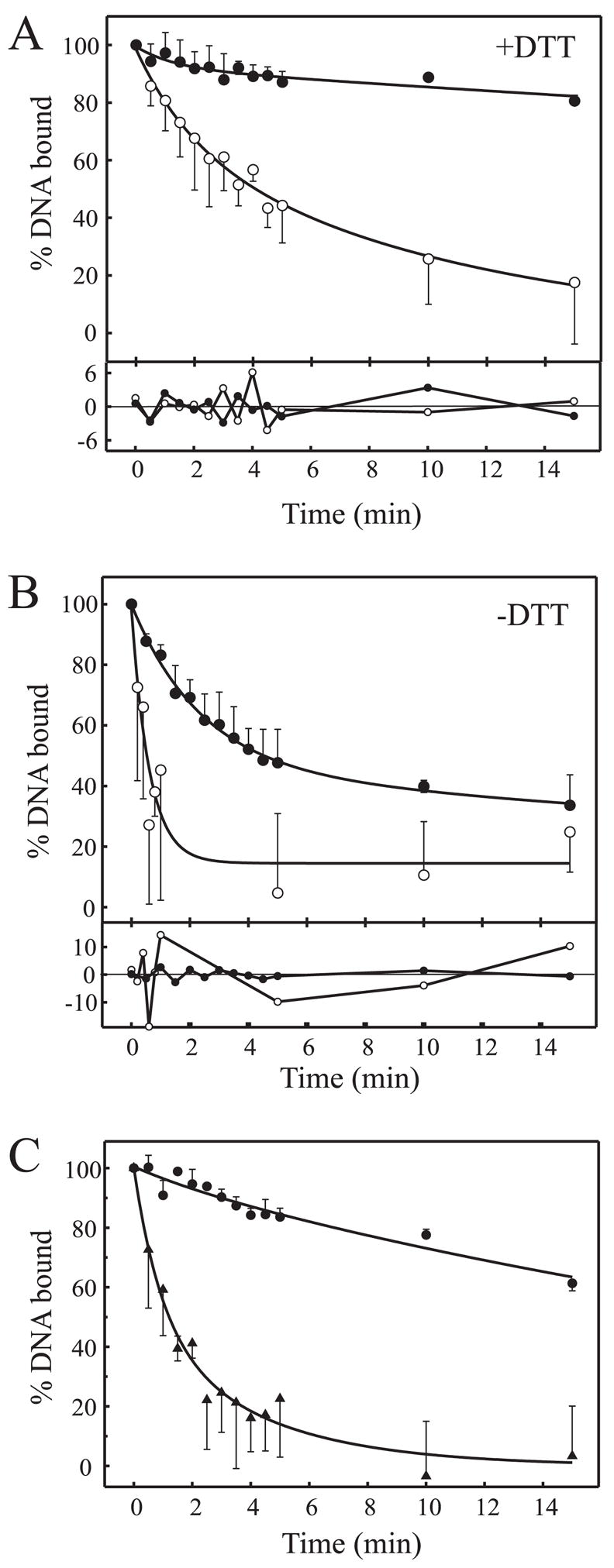
A, Ku-DNA dissociation performed in the presence of 1 mM DTT. Rates of Ku dissociation from duplex DNA was performed using the heparin trap assay system described under ”Experimental Procedures.“ Dissociation of Ku was measured from the 30-bp duplex DNA (filled circles) or 21/34 hairpin DNA (open circles). The percent of DNA bound was determined and plotted versus time. The data were fit to double exponential decays, and residual values are presented below each panel. B, Ku-DNA dissociation reactions performed in the absence of DTT. Reactions were performed exactly as described for A. C, effect of diamide oxidation and DTT reversal on Ku-DNA (30-bp) dissociation. Ku was treated with 2 mM diamide for 15 min (filled triangles) and subsequently re-reduced with 5 mM DTT for 15 min (filled circles) and dissociation measured as described for A.
Limited Proteolysis
7.5 μg of purified Ku was analyzed in reactions containing 25 mM Tris-Cl, pH 8.0, 100 mM KCl with or without the addition of 1 mM DTT. The reactions were incubated on ice for 10 min prior to the addition of 12.8 or 32 ng of bovine trypsin (Roche Diagnostics) and then incubated at 37 °C for 45 min. The reactions were terminated by the addition of SDS to a concentration of 1%, and the samples were heated to 90 °C for 5 min and loaded onto 10% SDS-polyacrylamide gel. Protein was detected with Coomassie Blue.
Mass Spectrometry and Peptide Mapping
7.5 μg of Ku was subjected to limited proteolysis as described above. The reactions were terminated by heating to 90 °C for 5 min, and the samples were processed via PepClean™ C-18 spin columns (Pierce) according to the manufacturer’s protocol. Samples were dried and suspended in 20 μl of 50% acetonitrile and 0.01% trifluoroacetic acid. 2 μl of each sample was mixed with 100 fmol of a standard, insulin-oxidized B chain (3,494.65 Da mass) prior to application onto a gold ProteinChip® array (Ciphergen). CHCA was used as the matrix and was prepared as a saturated, aqueous solution that contained 50% acetonitrile and 0.01% trifluoroacetic acid. A total of 1.5 μl of CHCA matrix was applied to the chip and allowed to completely dry. A trypsin control was spotted alone to identify any nonspecific peaks associated from autocatalysis during each experiment. A Protein Biology System II mass spectrometer (Ciphergen) was used for MALDI-TOF mass spectrometry. The machine was externally calibrated each time prior to use with the following peptides (Sigma) and their corresponding m/z values: human angiotensin II (1,046.54), P14R synthetic peptide (1,533.86), human ACTH-(18–39) (2,465.20), and bovine insulin-oxidized B chain (3,494.65). Each individual chip spot was analyzed with a protocol that ionized five times over 20 different areas. Spot positions were warmed with two laser shots at an intensity of 145 with the sensitivity set at 10, followed by four laser hits at an intensity of 150. The source voltage was set at 20,000 V and detector voltage at 1,900 V. Masses were collected up to 8000 Da with optimization from 1000 to 5000 Da. All mass spectra collected represent the sum of the data from 100 laser shots. Selection of peaks from raw spectra was done qualitatively using a cutoff of 2.5 for the signal-to-noise ratio. Peptide masses from data were matched with expected masses from the Ku80 (P13010) or Ku70 (P12956) subunit on the ExPASy Peptide Mass program. Matches were made up to ±3 Da relative to the observed peptide mass, taking into account enzymatic missed cut sites.
LC-Q MS/MS Analysis
Following limited proteolysis, peptides were processed through C-18 spin columns, dried down, and suspended in 10 μl of 1% formic acid. Peptides were separated via reverse-phase high performance liquid chromatography on a 0.1 × 150-mm EVEREST 5 μm, 30-nm C-18 capillary column (Vydac). The column was equilibrated and samples loaded on the column in buffer A (5% acetonitrile, 0.2% formic acid). Peptides were eluted with linear gradient from 0 to 60% buffer B (95% acetonitrile, 0.2% formic acid) over 60 min at 0.5 microliters/min. The eluate was analyzed directly using a standard orthogonal μ electrospray source and peptides detected with an LCQAdvantage ion trap mass spectrometer (ThermoFinnigan). Individual Ku peptides were identified from their MS/MS spectra using the MASCOT search algorithm.
RESULTS
The interaction of Ku with DNA has been analyzed using a number of systems including EMSA, surface plasmon resonance, and fluorescence-based assays. The Ku structure provides a unique model for studying DNA-protein interactions, where a preformed ring in Ku encounters a DNA terminus and the duplex DNA threads through the ring. The kinetics of this interaction has not been studied and presents a novel model system for analysis of protein-DNA interactions. Ku was purified to apparent homogeneity from insect cells infected with recombinant baculovirus (25, 26). Intrinsic Ku fluorescence was assessed and revealed an excitation maximum at 280 nm and a broad emission centered at 350 nm, consistent with Trp fluorescence (data not shown). These data are consistent with a previous report of Ku fluorescent properties and demonstrate that the presence of the N-terminal His tag on the p70 subunit does not alter the intrinsic Trp fluorescence (14). Ku fluorescence was reduced ~20% upon binding to DNA, and this signal was monitored to determine the kinetics of the Ku-DNA interaction.
Pre-steady state kinetic analysis of the Ku-DNA binding reactions was assessed by stopped-flow analysis. In these experiments, a fixed concentration of Ku is rapidly mixed with DNA, and the change in Ku fluorescence is monitored on a millisecond time frame. Preliminary experiments were performed to determine the optimal level of Ku and the time base in which to measure the interaction (data not shown). From these experiments, it became apparent that a change in the initial steady state level of Ku fluorescence was observed over time in dilute solutions. To circumvent this variation, it was determined that Ku must be diluted immediately prior to use, after which the signal was stable. Under the proper conditions, the Ku fluorescence signal was stable over a 20-s time frame when mixed with buffer alone (data not shown). During these experiments, it also became evident that the addition of DTT to the reactions resulted in a dramatically different quenching effect observed upon the interaction of Ku with DNA. In reactions supplemented with 1 mM DTT, the addition of a double strand 30-bp DNA resulted in a rapid quenching of fluorescence within 100 ms and was observed to fit to a single exponential decay, suggesting that binding under these conditions is a simple one-step interaction (Fig. 1, top trace). In DNA binding reactions performed, where the only reducing agent present was contributed from the buffer in which Ku was dialyzed such that the concentration was below 120 μ M DTT, a more complex interaction was observed (Fig. 1, middle trace). Binding of Ku to DNA observed under these limited reducing conditions did not fit a single exponential decay but instead fit a double exponential decay. To determine whether this phenomenon was a result of the reducing activity of DTT, we substituted β-mercaptoethanol for DTT and analyzed the kinetics of Ku binding. The result from this experiment is presented in Fig. 1, bottom trace, and shows a single exponential decay similar to that observed for DTT-containing reactions. These results suggest that the oxidation-reduction state of Ku influences its interaction with DNA or the ability to detect the interaction via intrinsic fluorescence.
FIGURE 1. Stopped-flow kinetic analysis of Ku binding a duplex 30-bp DNA.
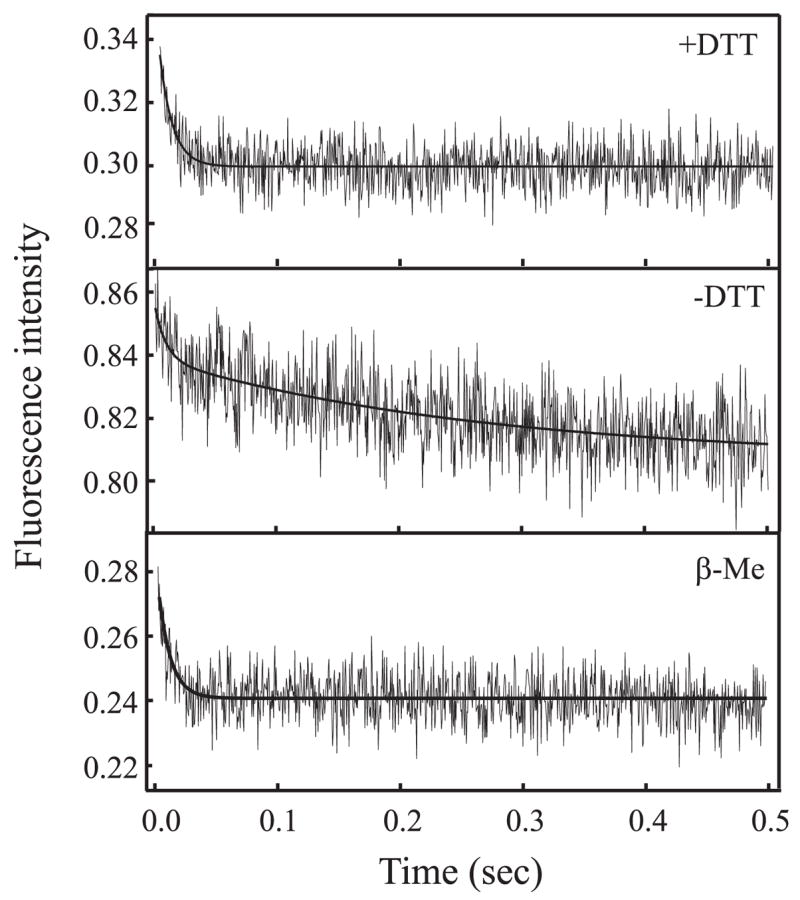
Kinetic traces were measured at a concentration of 25 nM Ku and 100 nM DNA (2.1/2.2) as described under ”Experimental Procedures.“ Reactions were performed in the presence of 2.5 mM DTT (top trace), in the absence of DTT (middle trace), and in the presence of 5 mm β-mercaptoethanol (bottom trace). Reactions performed in the presence of DTT and β-mercaptoethanol fit to a single exponential decay, and in the absence of DTT, fit a double exponential decay. The traces represent the average of 12–14 individual shots. Control traces were performed in the absence of DNA and showed no decrease in intrinsic fluorescence (data not shown).
One interpretation of the two-step process observed in the binding reactions performed under the limited reducing conditions is that the second step represents a second binding event on the same DNA. Considering Ku can encompass 20 bp of duplex DNA, the second event on the 30 bp would be expected to be slower as a result of steric hindrance on such a short DNA substrate. To test this hypothesis, reactions were performed using a 75-bp duplex DNA to which a second binding event would be expected to occur at the same rate as the first event, and therefore quenching under both conditions would be expected to fit a single exponential. The results presented in Fig. 2A demonstrate that, under the fully reduced conditions (top trace), binding does fit a single exponential. However, in the limited reducing reaction conditions (Fig. 2A, bottom trace), the data again fit a double exponential decay.
FIGURE 2. Stopped-flow kinetic analysis of Ku binding duplex DNA substrates.
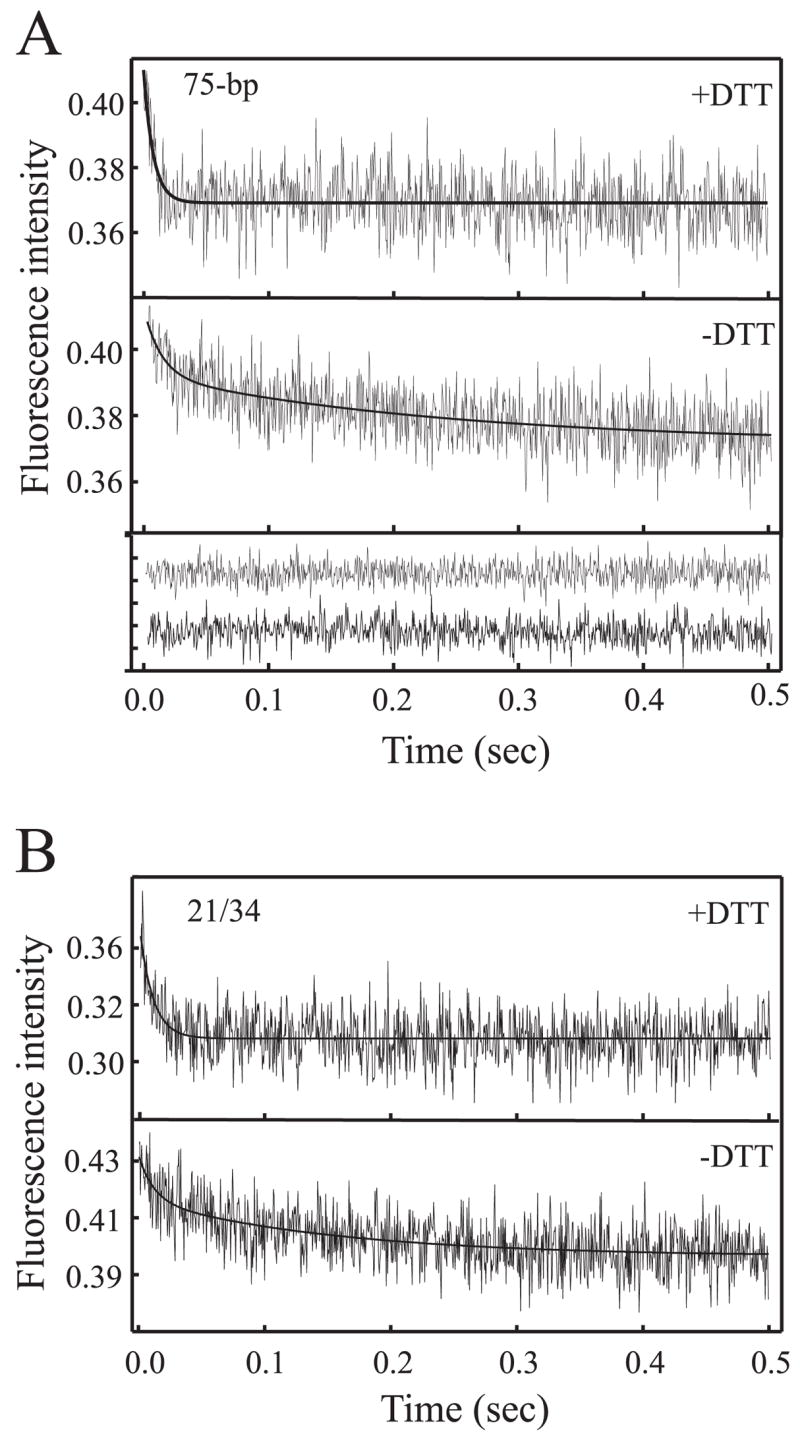
A, kinetics of Ku binding a 75-bp duplex DNA. Kinetic traces were measured at a concentration of 25 nM Ku and 100 nM duplex DNA. Binding reactions were performed in the presence of 2.5 mM DTT (top trace) and in the absence of DTT (bottom trace). Measurements taken in the presence of DTT fit a single exponential decay, and in the absence of DTT, fit a double exponential decay. Each trace is the average of 12–14 individual shots. The residual values obtained for the fits are presented at the bottom of the panel. B, kinetics of Ku binding to the 21/34 hairpin DNA. Reactions and analyses were performed exactly as described in A.
An alternative explanation for the double exponential decay is that the slow step may represent Ku sliding along the DNA duplex. To determine whether sliding accounts for the double exponential decay, a duplex hairpin DNA substrate (21/34) identical to that used to determine the Ku DNA co-crystal was employed (8). The interaction of Ku with this DNA substrate is restricted to a single terminus, and the limited length restricts sliding along the duplex. Using this substrate in pre-steady state quenching revealed again that, under limited reducing conditions (Fig. 2B, bottom trace), the data fit well to a double exponential decay, whereas in fully reduced conditions (Fig. 2B, top trace), a single exponential decay was observed. This data demonstrates that the Ku sliding cannot account for the different kinetic patterns observed as a function of redox state. This substrate also restricted binding to a single Ku molecule and confirms that the second event observed in the kinetic analyses is not a result of a second Ku molecule binding to the DNA.
In our standard Ku preparations, the final pool is dialyzed versus buffer containing 1 mM DTT. Depending on the concentration and amount required for preparation of the binding reactions, the final DTT concentration in those reactions that were not supplemented ranged from 27.2 to 109.2 μM depending on the preparation. To determine whether this low concentration of DTT was having any effect on the binding, we purified Ku and split the final pool, half being dialyzed versus standard buffer and the other half versus buffer without DTT. These preparations were employed in the binding reactions, and in reactions with no DTT present, there was no quenching observed upon mixing Ku with the DNA (data not shown). The addition of DTT to a concentration of 1 mM restored the quenching, with the interaction fitting a single exponential decay (data not shown).
The inability to detect quenching of Ku under the fully oxidized conditions could either be a result of Ku not binding the DNA or binding occurring but not quenching the fluorescent signal. Therefore, EMSAs were performed to assess binding (Fig. 3). Although decreased binding was observed under the oxidized conditions, binding was still easily detected. Quantification of the results revealed a 60% decrease in binding under the fully oxidized conditions compared with the moderately reduced conditions (data not shown). The DNA binding activity of the oxidized Ku could be restored by the addition of 1 mM DTT to the binding reaction, whereas the addition of DTT to the moderately reduced Ku had a negligible effect in the EMSA assay (data not shown).
FIGURE 3. DTT stimulation of Ku-DNA binding as assessed by EMSA.
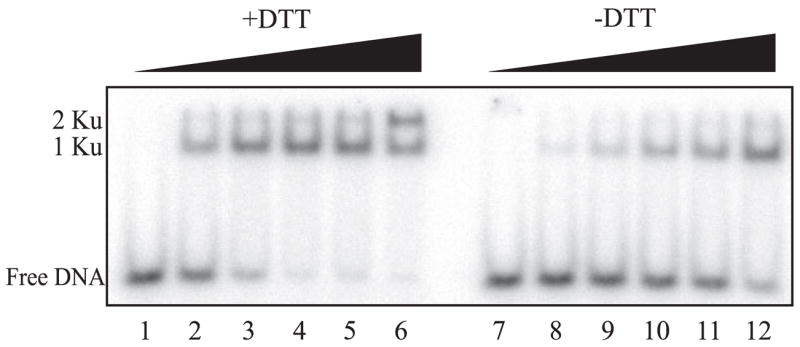
Increasing concentrations of Ku that were dialyzed in buffer with 1 mM DTT (lanes 1– 6) or without DTT (lanes 7–12) were mixed with 25 nM 32P-labeled 30-bp DNA without added DTT, as described under ”Experimental Procedures.“ Reactions were performed with no Ku (lanes 1 and 7) and 12.5, 25, 37.5, 50, and 100 nM Ku (lanes 2– 6 and 8–12, respectively). Samples were separated on a 6% native polyacrylamide gel, dried, and exposed to a PhosphorImager screen and quantified using ImageQuant software. The positions of free DNA and the Ku-DNA complexes are indicated in the left margin.
The ability to detect Ku binding DNA under the fully oxidized conditions in a solution-based assay was confirmed by measuring anisotropy of a fluorescein-labeled DNA as a function of Ku binding. Results presented in Fig. 4A show anisotropy of reactions with (open circles) and without (closed circles) DTT. The results again demonstrate that, in the absence of DTT, there is a modest decrease in Ku binding and a smaller maximum r value obtained. These data demonstrate that the inability to detect Ku quenching when mixed with DNA under non-reducing conditions is not a result of inefficient DNA binding to Ku. The anisotropy data also suggest that the size of the Ku-DNA complexes formed under the non-reducing conditions is smaller. Considering the 30-bp substrate can accommodate two Ku molecules per DNA, the difference in size could be a single Ku bound to the DNA under the non-reducing conditions and two Ku molecules bound under the fully reduced conditions. To determine whether this was in fact the case, binding analyses were performed with the 21/34 hairpin DNA substrate to which only one Ku molecule can bind. The results are presented in Fig. 4B and demonstrate, under non-reducing conditions (filled circles), the r maximum is larger than that observed under fully reduced conditions (open circles). These data are consistent with the oxidized Ku occupying more space, and the roughly 2-fold increase in r maximum observed with the 30 bp compared with the 21/34 hairpin DNA under reduced conditions is consistent with 2 Ku molecules being bound to a single 30-bp DNA, whereas only a single Ku is bound to the 21/34 hairpin DNA substrate under saturating protein levels. The data also demonstrate that, under fully reduced conditions, binding to the 21/34 hairpin DNA is stoichiometric, consistent with previous analysis on short DNA duplex substrates (14). By definition, under stoichiometric binding conditions, the unimolecular dissociation rate was negligible, and therefore analyses performed to this point did not allow the assessment of the rate of dissociation. The slow dissociation was confirmed in the stopped-flow analysis, where titrations of either the 21/34 hairpin or 30-bp DNA into reactions containing constant Ku were performed and the observed rate of quenching was plotted versus DNA concentration (Fig. 5, A and B, respectively). Under the pseudo first-order conditions employed, the slope of the line generated indicates the bimolecular association rate, and the y-intercept indicates the unimolecular dissociation rate. The error of the y-intercept values was extremely large, and in some cases, negative values were obtained. This is often observed where the off rate is considerably less than the kon × protein concentration (27, 28), again consistent with an extremely slow rate of dissociation. The slopes, however, do provide an accurate measure of the bimolecular rates of association. The kon rates for the 21/34 hairpin DNA and 30-bp duplex were calculated to be 0.64 ± 0.06 and 0.37 ± 0.08 nM−1 s−1, respectively. The kon for a duplex blunt end terminus is 0.64 nM −1s−1 which is only slightly slower than diffusion but suggests that a proper orientation must be obtained to achieve high affinity binding. This is consistent with the crystal structure, where numerous other interactions or contacts with DNA may occur, but only one orientation, with the DNA threading through the pillar and which will result in high affinity binding and quenching.
FIGURE 4. Influence of redox on Ku-DNA binding in solution.
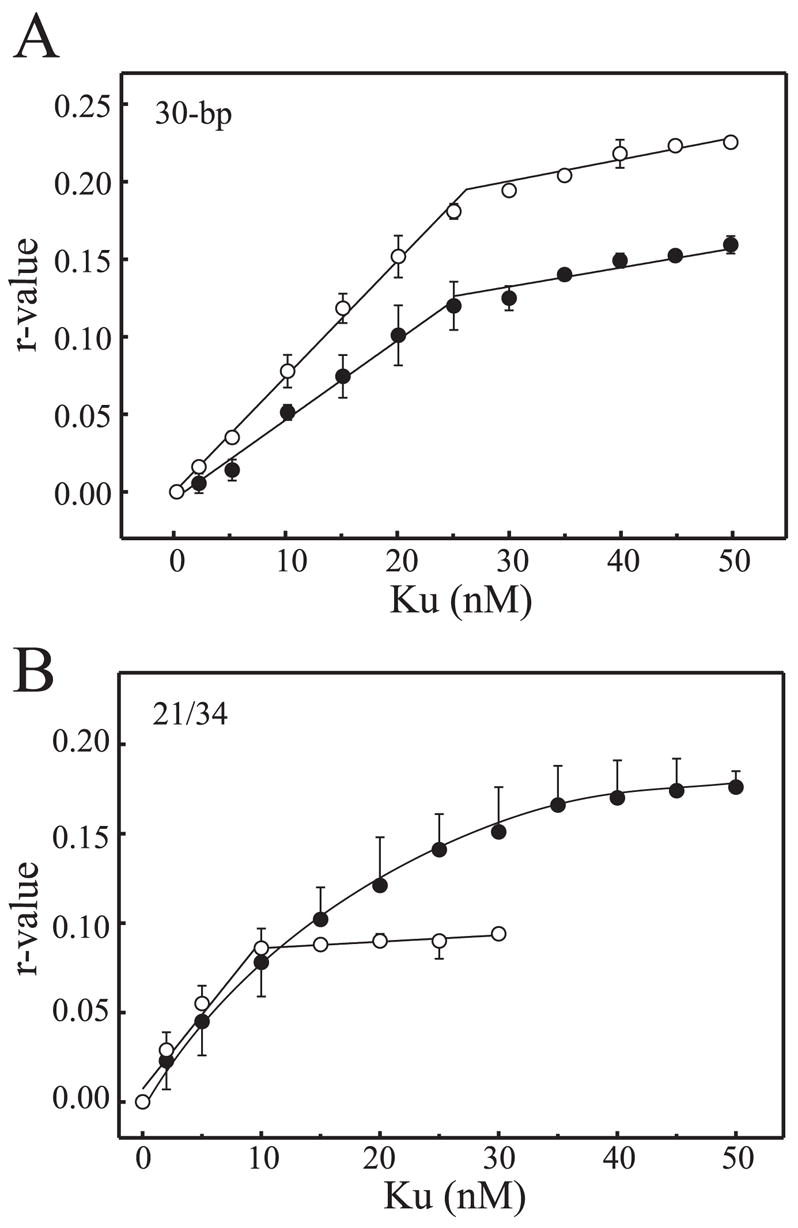
A, anisotropy analysis of Ku binding the 30-bp duplex DNA. Reactions were performed in a voulme of 0.5 ml containing 10 nM DNA ends using Ku that was dialyzed against buffer without the addition of DTT. Reactions were either supplemented with 1 mM DTT (open circles) or left without DTT (filled circles). Anisotropy was calculated as described under ”Experimental Procedures“ and plotted versus Ku concentration. Each point represents the average ± S.D. of three independent experiments. B, anisotropy analysis of Ku binding of the 21/34 hairpin DNA. Reactions were performed identically as described for A using the 21/34 hairpin DNA.
FIGURE 5. Pre-steady state kinetic analysis of Ku binding duplex DNA substrates.
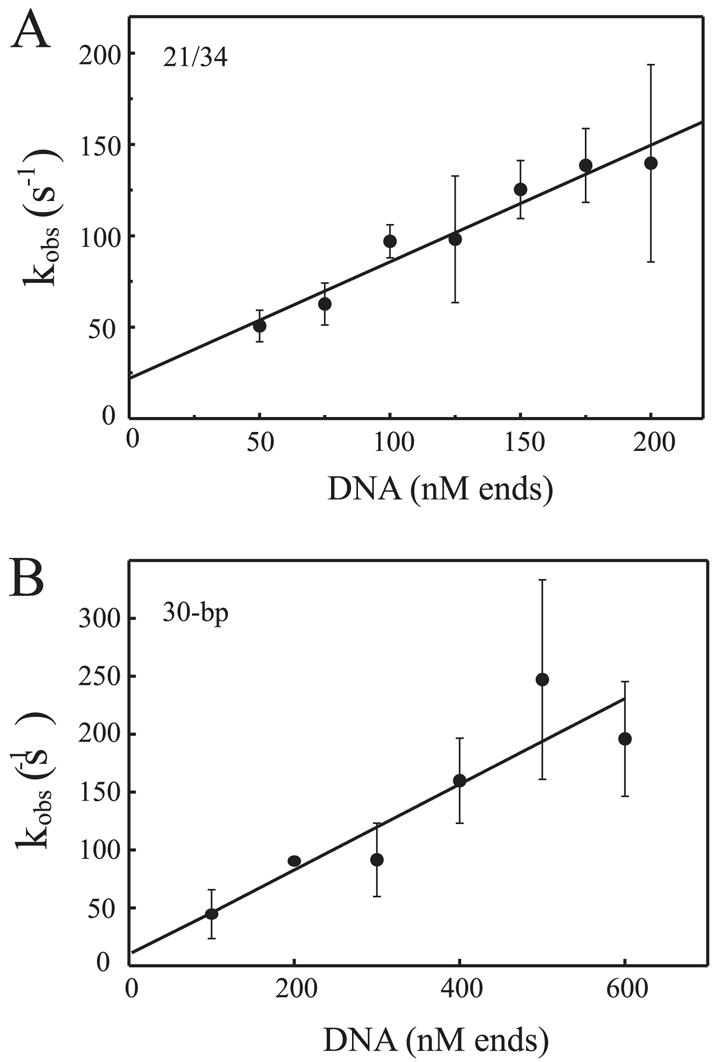
A, stopped-flow kinetic analysis of Ku binding the 21/34 hairpin DNA. Reactions were performed using 25 nM Ku in the presence of 1 mM DTT and increasing concentrations of the DNA substrate. The DNA concentration is presented in terms of DNA termini and not molecules of duplex DNA. Values for kobs were plotted as a function of DNA concentration. The data were fit to an equation for a straight line with an R2 value of 0.95. Each data point represents the average ± S.D. obtained from three independent experiments. The kobs values for each DNA concentration consisted of the average of 12–14 individual shots. B, stopped-flow kinetic analysis of Ku binding the 30-bp duplex DNA. The data were fit to an equation for a straight line with an R2 value of 0.82. Reactions were performed exactly as described for A.
Considering the potential importance of dissociation in response to redox conditions and the relative inaccuracy of the measurements obtained thus far, we developed a system to provide a more accurate assessment of the dissociation rates of Ku from a DNA substrate under a variety of conditions. We employed a heparin trap anisotropy assay, where Ku is mixed with the fluorescein-labeled DNA and anisotropy measured. To measure dissociation, heparin is added and anisotropy measured at fixed times over 15 min. When Ku dissociates from the DNA, it would be bound by the heparin trap, and the anisotropy of the free DNA would decrease. A measure of the decrease in anisotropy over time allows the estimation of the unimolecular dissociation rate. Preliminary experiments were performed to determine the optimal amount of heparin to add such that no rebinding of Ku would occur and such that an increase in concentration would not alter the rate of dissociation by stripping the Ku off the DNA (data not shown).
Dissociation reactions were first performed in reaction buffer supplemented with 1 mM DTT using Ku that was dialyzed in buffer containing DTT (Fig. 6A). Results obtained with the 30-bp duplex under these fully reduced conditions revealed a very slow rate of dissociation (Fig. 6A, filled circles). Dissociation of Ku from the 21/34 hairpin DNA (Fig 6A, open circles) was faster but still relatively slow (0.0085 s−1) compared with the rate of association determined from stopped-flow analysis. Interestingly, the dissociation kinetics from both substrates displayed double exponential decays. Residual values of the fits for each data series are presented in the lower panels of each graph. Reactions performed in the absence of the 1 mM DTT supplement (limited reducing capacity) (Fig. 6B) resulted in a significant increase in the rates and amplitudes of dissociation from both the 30-bp duplex and the 21/34 hairpin DNA substrates. Again, the data fit well to double exponential decays, suggesting a two-step dissociation process.
To confirm the effect of redox on Ku dissociation, we used reversible diamide oxidation of Ku (Fig. 6C). Ku dissociation from the 30-bp DNA following treatment with 2 mM diamide (Fig. 6C, filled triangles) or following sequential diamide treatment and then re-reduction with DTT (filled circles) was assessed. Diamide treatment of Ku increased the rate of dissociation dramatically compared with untreated Ku (Fig. 6A, filled circles). Importantly, the diamide-treated Ku could be re-reduced with DTT, which restored slow dissociation from the duplex (Fig. 6C, filled circles).
The results thus far demonstrate an influence of redox conditions on the ability of DNA (when bound to Ku) to quench fluorescence and the ability of oxidized Ku to remain bound to a DNA. These differences are likely a result of different Ku conformations predominating under the different redox conditions. To test this hypothesis, limited tryptic proteolysis of Ku was employed under the different redox conditions. Preliminary experiments were performed to determine whether the presence of DTT would inhibit proteolytic activity of trypsin, and results indicate that there is no affect of DTT on trypsin catalytic activity; thus any alteration as a function of DTT can be attributed to the structure of the protein being cleaved (data not shown). Ku was incubated in the presence or absence of 1 mM DTT, treated with varying concentrations of trypsin, and the reaction products were separated by SDS-PAGE. The results are presented in Fig. 7A and indicate both the Ku70 and Ku80 subunits were more susceptible to trypsin cleavage in the absence of DTT compared with reactions performed in the presence of DTT.
FIGURE 7. Redox influence on Ku structure.
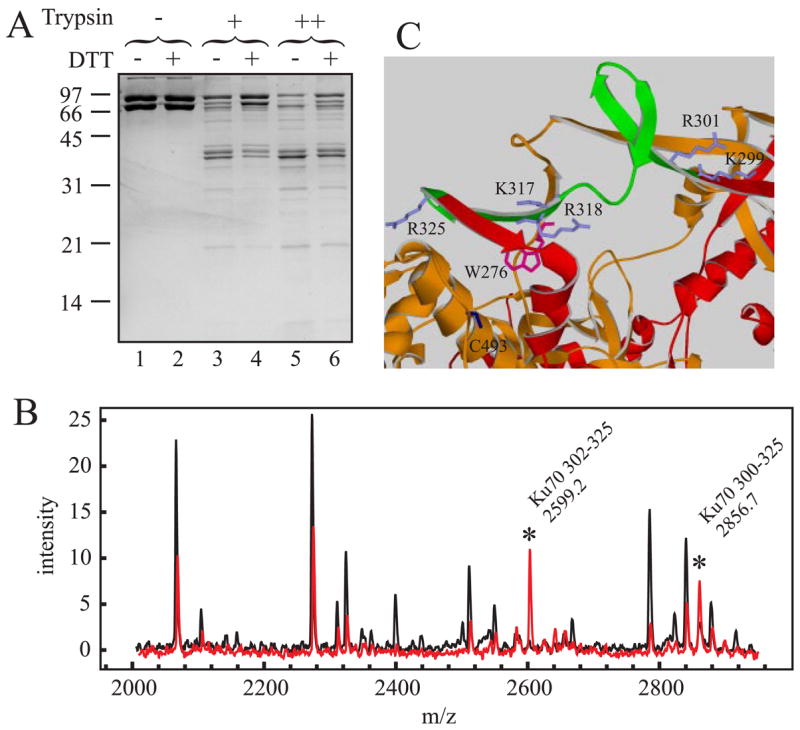
A, SDS-PAGE analysis of Ku proteolytic sensitivity. 7.5 μg of Ku was subjected to limited proteolysis as described under ”Experimental Procedures.“ Digestion reactions were performed in the absence of trypsin (lanes 1 and 2), with 12.8 ng of trypsin (lanes 3 and 4) or with 32 ng of trypsin (lanes 5 and 6). Reactions were performed without the addition of DTT (odd lanes) or with the addition of DTT to a final concentration of 1 mM (even lanes). Reaction products were separated by 10% SDS-PAGE and visualized via Coomassie Blue staining. B, MALDI-TOF MS analysis of trypsin generated Ku peptides. 7.5 μg of Ku was subjected to limited proteolysis, processed for MALDI-TOF analysis, and spectra collected as described under ”Experimental Procedures.“ The spectra presented are from a 32-ng trypsin reaction performed in the absence of DTT (black trace) or in the presence of DTT (red trace). Peaks were labeled showing the region of Ku70 that is uniquely cut under reducing conditions. Spectra presented are representative of data obtained at all concentrations from three independent experiments and were normalized to an internal peptide standard. C, structure of the Ku-DNA binding ring. The regions of Ku70 (red) and Ku80 (orange) that comprise the DNA binding ring are depicted in a ribbon diagram. The 301–324 peptide observed in trypsin digestions reactions performed in the presence of DTT is colored green. Ku70 amino acid side chains of Lys-317, Arg-318, Lys-299, Arg-301, and Arg-325 are colored gray. The Ku80 amino acid side chains of Trp-276 are colored magenta and Cys-493 in blue.
The reduced protease susceptibility of Ku in the presence of DTT provides evidence for an altered conformation under the reduced conditions. To determine whether there were specific regions of Ku that were more or less susceptible to proteolysis as a function of redox conditions, partial proteolysis was performed and the products were analyzed by MALDI-TOF MS and LC-Q MS/MS. Overall, the majority of peptides observed were present in higher amounts in reactions performed in the absence of DTT. The majority of peptides identified corresponded to the Ku70 subunit versus the Ku80 subunit in both MALDI-TOF and LC-Q analysis of the reaction products. These data are consistent with SDS gel analysis, where the 70-kDa subunit was more susceptible to degradation than the 80-kDa subunit. Normalization of the MALDI-TOF data was accomplished by including an internal standard spiked into each sample, and these data are presented in Fig. 7B. The increased intensity of peptides generated by trypsin cleavage performed in the absence of DTT is apparent (Fig. 7B, black trace) compared with digestion performed in the presence of DTT (red trace). The peptides indicated by the asterisk, however, show the opposite effect; they are more prevalent in digests performed in the presence of DTT. This difference in intensity was observed in numerous experiments with multiple trypsin concentrations and Ku preparations (data not shown). The mass fingerprint of these peptides is consistent with amino acids 300–325 of Ku70 using the numbering system from the Ku crystal structure (8). Both of these peptides contain missed cleavages at Lys-317 and Arg-318, suggesting that these are DTT-protected sites. The peptide with the mass of 2599.2 Da from Fig. 7B corresponding to amino acids 302–325 was confirmed by LC-Q MS/MS and was only observed in the presence of DTT (Table 2).
TABLE 2.
MS analysis of Ku peptides
| Peptide | Mass | Ku70 peptide | Missed cut sites | Trypsin activity
|
|
|---|---|---|---|---|---|
| +DTT | −DTT | ||||
| 1 | 809.40 | 319–325 | 0 | − | + |
| 2 | 965.50 | 318–325 | 0 | − | + |
| 3 | 1650.85 | 302–317 | 0 | + | + |
| 4 | 1806.95 | 302–318 | 1 (317) | + | + |
| 5 | 1908.00 | 300–317 | 1 (301) | + | + |
| 6 | 2064.10 | 300–318 | 2 (301, 317) | + | + |
| 7 | 2598.34 | 302–325 | 2 (317, 318) | + | − |
| 8a | 2856.7 | 300–325 | 3 (301, 317, 318) | + | − |
Only observed in MALDI-TOF analysis.
These data support the conclusion that Lys-317 and Arg-318 are protected from tryptic cleavage in the presence of DTT. This conclusion is supported by several additional lines of evidence. First, the peptides generated without digestions at Lys-317 and Arg-318 are only observed in the presence of DTT (Fig. 7B and Table 2). Furthermore, it is the only peptide that is detected by both types of mass spectrometry, where this preferential cleavage occurs under reducing conditions. The second line of evidence comes from LC-Q MS/MS analysis of the smaller peptide fragments N-terminal to this region (Table 2). Trypsin cleavage at Lys-299 and Arg-301 occur independent of DTT. This is apparent in that the four MS-detectable combinations of digestions 300 –317, 300 –318, 302–317, and 302–318 were observed without DTT (Table 2, peptides 3– 6). We conclude that cleavage at residue 317 and 318 occurred as a second cleavage during the course of the digestion, occurring only after cleavage at position 290 or 301. This is confirmed by analysis of the region C-terminal to Lys-317 and Arg-318. Digestion at Arg-325 is observed independent of DTT. However, the three combinations, 317–325, and 318 –325 (peptides 1 and 2) are only observed in the absence of DTT. However, the 300 –325 and 302–325 fragments are only observed in the presence of DTT (peptides 8 and 9). These results are consistent with initial cleavage at position 325 occurring independent of DTT but with the second cleavage at position Lys-317 or Arg-318 only occurring in the absence of DTT. In the presence of DTT, these sites remain protected, and therefore in reactions performed with DTT, the peptides 317/318 –324 are never observed. From these analyses, we can also conclude that positions Lys-317 and Arg-318 are protected by DTT such that there is no initial cleavage by trypsin at these sites.
These conclusions are also supported by analysis of the Ku structure (Fig. 7C). The Lys-298 and Arg-300 are positioned in the very accessible bridge region of the DNA binding ring. Likewise, Arg-324 is in a highly accessible loop that connects two anti-parallel βsheets, βL and βM (8). These β sheets lie in a trough comprising Ku80 subunits, which limits their flexibility. Lys-317 and Arg-318, however, lie within the βL sheet and are considerably less accessible, consistent with the proteolysis data. Further analysis of this region of the protein revealed both a Cys and a Trp residue in close proximity to the DTT-protected Lys-317 and Arg-318. These amino acids are highlighted in Fig. 7C.
DISCUSSION
We have identified a redox-sensitive conformational change in Ku and the effect that it has on the kinetics of the Ku-DNA interaction. A reversible redox effect on a protein involved in double strand break repair is intriguing, considering DSBs created by ionizing radiation induce significant oxidative stress within a cell (29–31). Potentially, this redox change could serve to influence the DNA binding activity of Ku at DSBs. The effect of oxidative stress on NHEJ has been demonstrated in numerous systems, where changes in DSB repair have been attributed to redox effects on NHEJ proteins (17, 18, 21, 22, 32).
Our data demonstrate that koff rates for Ku under oxidizing conditions is significantly greater than that observed under reducing conditions, and this is the major effector for the observed differences in DNA binding as assessed in EMSA and anisotropy assays. Thus, in a reducing environment, Ku will stay bound to the DNA end longer, increasing the likelihood for DNA-PKcs to bind and form the DNA-PK complex, ultimately initiating the NHEJ pathway. In addition, conformational changes in Ku upon binding DNA are required for DNA-PK activation (33, 34), and redox conditions could be the way to regulate this conformational change. This leads to a model whereby changes in DNA-PK activity as a function of oxidative stress (17) are manifested by reduced Ku binding to DNA termini under these conditions.
Although there are specific regions of Ku that are hypersensitive to redox status, the tryptic mapping data suggest a more open conformation of Ku under the oxidized conditions. This was apparent in the SDS gel and mass spectrometry analysis demonstrating greater trypsin accessibility to a variety of regions under the oxidized conditions. This open conformation also is consistent with the different r maximum values observed for Ku bound to DNA under the different redox conditions. Anisotropy, a measure of how particles tumble in space, is likely to be affected by Ku being in a more relaxed, open conformation. The more open configuration of Ku in the oxidized state is also consistent with a greater rate of dissociation from the DNA. If the Ku ring adopts a more flexible conformation, the likelihood of movement along a duplex would be greater, and thus on a DNA substrate where movement results in dissociation, the rate would be expected to be greater.
The inability to detect quenching of Ku upon binding DNA under oxidized conditions can be explained by Ku adopting a different conformation that positions the Trp in an orientation such that fluorescence is not quenched by the DNA. The more interesting result is the observed differences in binding kinetics, two-step versus one-step, under partially and fully reduced conditions, respectively. A two-step model for binding observed under partially reduced conditions required a step before or after the initial interaction with DNA. Assuming, under the partially reduced conditions, Ku exists in equilibrium between two states (oxidized and reduced), there are two potential interpretations. Both oxidized and reduced Ku can bind DNA, but quenching is only observed when the reduced Ku binds. The slow kinetic phase then represents reduction of Ku when bound to the DNA to result in fluorescence quenching. Alternatively, only the reduced Ku binds DNA, and the slow phase represents reduction of free Ku to an active DNA binding conformation.
The possibility of redox regulation of Ku is intriguing in light of the lack of any disulfide bonds observed in the crystal structure of free Ku or the Ku-DNA complex (8). However, we cannot rule out a possible disulfide bond that includes the Cys residue in the Ku80 C terminus that was not present in the x-ray crystal structure. In addition, there are no coordinated metals present that could also serve as redox centers. Cys-493 can potentially form a redox-regulated interaction with Arg-489. Both amino acids lie on a helix positioned at the Ku70–80 interface. The positioning of Cys-493 in Ku80 (Fig. 7C) is also potentially interesting in that redox-dependent alterations in this region could also explain the altered accessibility of Lys-317 and Arg-318. This region also provides important contacts with DNA. Importantly, one of these contacts involves Trp-276, which is one of the few Trp residues within the DNA binding region and is known to contact the DNA phosphate backbone (8). It is tempting to speculate that Trp-276 is the residue that is quenched upon binding DNA. A redox-dependent alteration in this region could then account for the inability to detect fluorescence quenching under oxidized conditions. The reactivity of Cys residues is also a function of the local electrostatic environment in which the residue resides (35, 36). This reactivity is interesting in that we do observe a difference in Trp quenching under different redox conditions and have observed no quenching under reduced conditions if the DNA duplex contains a cisplatin DNA adduct.3 Potentially, Ku enters the reaction in a reduced form, but upon interacting with the platinated DNA (specifically the cisplatin), its structure is altered into a conformation in which, even when bound to DNA, its fluorescence is not quenched. In essence, the platinum could be acting to evoke the same conformational changes that are brought about under oxidizing conditions. The electrophilic nature of a cisplatin molecule and its reactivity with Cys residues raise the possibility that the environment of the Cys residue is altered upon binding to DNA, and under specific conditions, oxidized or cisplatin on DNA, the result is an alteration in the conformation of Ku.
Acknowledgments
We thank Drs. Derek Hoelz and Robert Hickey for assistance with mass spectrometry analysis.
Footnotes
This work was supported by National Institutes of Health Grant CA82741 and United States Department of Defense Grant OC020223.
The abbreviations used are: DSB, DNA double strand break(s); NHEJ, non-homologous end-joining; DNA-PK, DNA-dependent protein kinase; DNA-PKcs, DNA-PK catalytic subunit; DTT, dithiothreitol; MALDI-TOF, matrix-assisted laser desorption/ionization time-of-flight; MS, mass spectrometry; CHCA, α-cyano-4-hydroxy-cinnamic acid; LC-Q, liquid chromatography ion trap quadrupole.
B. J. Andrews and J. J. Turchi, unpublished observations.
References
- 1.Iliakis G, Wang H, Perrault AR, Boecker W, Rosidi B, Windhofer F, Wu W, Guan J, Terzoudi G, Pantelias G. Cytogenet Genome Res. 2004;104:14–20. doi: 10.1159/000077461. [DOI] [PubMed] [Google Scholar]
- 2.Devries E, Vandriel W, Bergsma WG, Arnberg AC, van der Vliet PC. J Mol Biol. 1989;208:65–78. doi: 10.1016/0022-2836(89)90088-0. [DOI] [PubMed] [Google Scholar]
- 3.Yoo S, Dynan WS. Nucleic Acids Res. 1999;27:4679–4686. doi: 10.1093/nar/27.24.4679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Turchi JJ, Henkels KM, Zhou Y. Nucleic Acids Res. 2000;28:4634–4641. doi: 10.1093/nar/28.23.4634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Poinsignon C, de Chasseval R, Soubeyrand S, Moshous D, Fischer A, Hache RJG, de Villartay JP. Eur J Immunol. 2004;34:3146–3155. doi: 10.1002/eji.200425455. [DOI] [PubMed] [Google Scholar]
- 6.Uziel T, Lerenthal Y, Moyal L, Andegeko Y, Mittelman L, Shiloh Y. EMBO J. 2003;22:5612–5621. doi: 10.1093/emboj/cdg541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang HC, Zeng ZC, Perrault AR, Cheng XB, Qin W, Iliakis G. Nucleic Acids Res. 2001;29:1653–1660. doi: 10.1093/nar/29.8.1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Walker JR, Corpina RA, Goldberg J. Nature. 2001;412:607–614. doi: 10.1038/35088000. [DOI] [PubMed] [Google Scholar]
- 9.Yoo S, Kimzey A, Dynan WS. J Biol Chem. 1999;274:20034–20039. doi: 10.1074/jbc.274.28.20034. [DOI] [PubMed] [Google Scholar]
- 10.Mimori T, Hardin JA. J Biol Chem. 1986;261:10375–10379. [PubMed] [Google Scholar]
- 11.Blier PR, Griffith AJ, Craft J, Hardin JA. J Biol Chem. 1993;268:7594–7601. [PubMed] [Google Scholar]
- 12.Dynan WS, Yoo S. Nucleic Acids Res. 1998;26:1551–1559. doi: 10.1093/nar/26.7.1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bianchi A, deLange T. J Biol Chem. 1999;274:35284. [Google Scholar]
- 14.Arosio D, Cui S, Ortega C, Chovanec M, Di Marco S, Baldini G, Falaschi A, Vindigni A. J Biol Chem. 2002;277:9741–9748. doi: 10.1074/jbc.M111916200. [DOI] [PubMed] [Google Scholar]
- 15.Arosio D, Costantini S, Kong Y, Vindigni A. J Biol Chem. 2004;279:42826–42835. doi: 10.1074/jbc.M406529200. [DOI] [PubMed] [Google Scholar]
- 16.Zhang WW, Yaneva M. Biochem J. 1993;293:769–774. doi: 10.1042/bj2930769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Boldogh I, Roy G, Lee MS, Bacsi A, Hazra TK, Bhakat KK, Das GC, Mitra S. Toxicology. 2003;193:137–152. doi: 10.1016/j.tox.2003.08.013. [DOI] [PubMed] [Google Scholar]
- 18.Bacsi A, Kannan S, Lee MS, Hazra TK, Boldogh I. Free Radic Biol Med. 2005;39:1650–1659. doi: 10.1016/j.freeradbiomed.2005.08.013. [DOI] [PubMed] [Google Scholar]
- 19.Reddy VR, Ding Q, Lees-Miller SP, Meek K, Ramsden DA. J Biol Chem. 2004;279:39408–39413. doi: 10.1074/jbc.M406432200. [DOI] [PubMed] [Google Scholar]
- 20.Chen BP, Chan DW, Kobayashi J, Burma S, Asaithamby A, Morotomi-Yano K, Botvinick E, Qin J, Chen DJ. J Biol Chem. 2005;280:14709–14715. doi: 10.1074/jbc.M408827200. [DOI] [PubMed] [Google Scholar]
- 21.Ayene IS, Koch CJ, Tuttle SW, Stamato TD, Perez ML, Biaglow JE. Int J Radiat Biol. 2000;76:1523–1531. doi: 10.1080/09553000050176289. [DOI] [PubMed] [Google Scholar]
- 22.Ayene IS, Stamato TD, Mauldin SK, Biaglow JE, Tuttle SW, Jenkins SF, Koch CJ. J Biol Chem. 2002;277:9929–9935. doi: 10.1074/jbc.M111366200. [DOI] [PubMed] [Google Scholar]
- 23.Pawelczak KS, Andrews BJ, Turchi JJ. Nucleic Acids Res. 2005;33:152–161. doi: 10.1093/nar/gki157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Andrews BJ, Turchi JJ. Mol Cancer Ther. 2004;3:385–391. [PubMed] [Google Scholar]
- 25.Ramsden DA, Gellert M. EMBO J. 1998;17:609–614. doi: 10.1093/emboj/17.2.609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Turchi JJ, Henkels KM. J Biol Chem. 1996;271:13861–13867. doi: 10.1074/jbc.271.23.13861. [DOI] [PubMed] [Google Scholar]
- 27.Patrick SM, Turchi JJ. J Biol Chem. 2001;276:22630–22637. doi: 10.1074/jbc.M010314200. [DOI] [PubMed] [Google Scholar]
- 28.Kozlov AG, Lohman TM. Biochemistry. 2002;41:6032–6044. doi: 10.1021/bi020122z. [DOI] [PubMed] [Google Scholar]
- 29.Agrawal A, Chandra D, Kale RK. Mol Cell Biochem. 2001;224:9–17. doi: 10.1023/a:1011990704575. [DOI] [PubMed] [Google Scholar]
- 30.Agrawal A, Choudhary D, Upreti M, Rath PC, Kale RK. Mol Cell Biochem. 2001;223:71–80. doi: 10.1023/a:1017900810837. [DOI] [PubMed] [Google Scholar]
- 31.Schmidt-Ullrich RK, Dent P, Grant S, Mikkelsen RB, Valerie K. Radiat Res. 2000;153:245–257. doi: 10.1667/0033-7587(2000)153[0245:stacrr]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 32.Song JY, Lim JW, Kim H, Morio T, Kim KH. J Biol Chem. 2003;278:36676–36687. doi: 10.1074/jbc.M303692200. [DOI] [PubMed] [Google Scholar]
- 33.Singleton BK, Torres-Arzayus MI, Rottinghaus ST, Taccioli GE, Jeggo PA. Mol Cell Biol. 1999;19:3267–3277. doi: 10.1128/mcb.19.5.3267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gell D, Jackson SP. Nucleic Acids Res. 1999;27:3494–3502. doi: 10.1093/nar/27.17.3494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kortemme T, Creighton TE. J Mol Biol. 1995;253:799–812. doi: 10.1006/jmbi.1995.0592. [DOI] [PubMed] [Google Scholar]
- 36.Nelson JW, Creighton TE. Biochemistry. 1994;33:5974–5983. doi: 10.1021/bi00185a039. [DOI] [PubMed] [Google Scholar]