

Abstract
The mechanism of coat protein (COP)II vesicle fission from the endoplasmic reticulum (ER) remains unclear. Lysophospholipid acyltransferases (LPATs) catalyze the conversion of various lysophospholipids to phospholipids, a process that can promote spontaneous changes in membrane curvature. Here, we show that 2,2-methyl-N-(2,4,6,-trimethoxyphenyl)dodecanamide (CI-976), a potent LPAT inhibitor, reversibly inhibited export from the ER in vivo and the formation of COPII vesicles in vitro. Moreover, CI-976 caused the rapid and reversible accumulation of cargo at ER exit sites (ERESs) containing the COPII coat components Sec23/24 and Sec13/31 and a marked enhancement of Sar1p-mediated tubule formation from ERESs, suggesting that CI-976 inhibits the fission of assembled COPII budding elements. These results identify a small molecule inhibitor of a very late step in COPII vesicle formation, consistent with fission inhibition, and demonstrate that this step is likely facilitated by an ER-associated LPAT.
Keywords: COPII vesicles, ER exit sites, lysophospholipid acyltransferase, membrane curvature, vesicle fission
The biochemical basis for fission of coat protein (COP)I and COPII vesicles remains to be determined. Coated vesicle formation is often broken down into two continuous phases, formation of a spherical bud and constriction at the bud neck with resultant membrane fission (1). Vesicle budding requires the presence of different classes of coat proteins along with a wide array of accessory proteins (2,3). COPII vesicles bud from the endoplasmic reticulum (ER) and are dependent on the small guanosine triphosphatase (GTPase), Sar1p, to direct the binding of the cargo adaptor protein (AP) complex Sec23/24 (3-6). This is followed by the self-assembly of Sec13/31, a cytosolic heterotetramer that generates the polymeric cage that collects Sec23/24 and cargo into budding vesicles (7). In vitro studies using artificial liposomes have shown that Sar1, Sec23/24 and Sec13/31 are sufficient to generate COPII vesicles when incubated with acidic membrane phospholipids (8,9). The Sar1p GTPase has been shown to induce membrane curvature and promote tubulation of ER membranes (10-12), activities that are thought to be further enhanced by positively charged concave surface of Sec23/24 (12,13). Sar1p may also contribute to vesicle fission (10,12). For other classes of coated vesicles, principally AP-2 clathrin-coated vesicles on the plasma membrane, accessory proteins such as dynamin, endophilin and amphiphysin have been shown to play a role in vesicle fission (14). These proteins contribute to vesicle fission by virtue of their ability to self-assemble into rings that are thought to constrict upon GTP hydrolysis and/or by the presence of membrane bending BAR domains (15-18).
In addition to protein scaffold-mediated changes that may contribute to membrane bending at the necks of budding vesicles leading to fission, phospholipid modifications may also be involved but are not well documented (19). Lysophospholipid acyltransferases (LPATs) catalyze the transfer of fatty acids from acyl-CoA donors to lysophospholipid acceptors, thereby generating phospholipids. The most well-characterized LPATs utilize lysophosphatidic acid (LPA) as an acceptor for the generation of phosphatidic acid (PA). These enzymes are encoded by a family of at least nine distinct genes, whose products have been called LPAATs (LPAATα, β, δ, etc.) (20) or 1-acyl-sn-glycerol-3-phosphate acyltransferase 1–9 (AGPAT1–9) (21,22). In addition to traditional roles in de novo phospholipid biosynthesis (20), recent studies have shown that members of the LPAAT/AGPAT family are also involved in the synthesis of stored triglycerides in lipid droplets (21-23). However, many members of the LPAAT/AGPAT family have no definitively ascribed function.
LPAATs that generate PA have been proposed to be involved in the production of membrane vesicles (24). Phosphatidic acid can generate a modest degree of spontaneous negative curvature and could contribute to the inward or negative curvature at the neck of a budding vesicle (25). Phosphatidic acid can also be converted by phospholipase D (PLD) to diacylglycerol (DAG), a lipid with even greater spontaneous negative curvature (26). Early evidence that DAG might play a role in COPII vesicle formation came from the observation that ER export is sensitive to signaling pathways regulating DAG synthesis (27). Consistent with these observations, it was recently demonstrated that Sar1p activates PLD to produce PA that is required for COPII formation and ER-to-Golgi transport (28).
Additional evidence that other LPATs might be involved in membrane-trafficking events came from studies in which the small molecule antagonist 2,2-methyl-N-(2,4,6,-trimethoxyphenyl)dodecanamide (CI-976) was shown to inhibit a Golgi-associated LPAT activity (29,30). CI-976 was originally synthesized to be an acyl-CoA:cholesterol acyltransferase (ACAT) inhibitor (31) but was subsequently shown to also inhibit an LPAT that prefers lysophosphatidylcholine and lysophosphatidylethanolamine and is tightly associated with the Golgi complex (29). As a consequence of CI-976’s LPAT inhibitory effects, Golgi membrane tubule formation was stimulated, resulting in enhanced retrograde trafficking to the ER (29) similar to brefeldin A-induced retrograde trafficking (32). Inhibition of a Golgi LPAT could induce membrane tubules by several mechanisms. One possibility is based on the observation that Golgi membrane tubulation has been shown to require the activity of a cytosolic phospholipase A2 enzyme, which would generate positive curve-inducing lysophospholipids (LPLs). Thus, inhibition of LPL reacylation by an LPAT would result in the accumulation of LPLs that might favor tubule formation (33-35). Evidence in favor of this idea was obtained by finding that pretreatment of cells with phospholipase A2 (PLA2) antagonists abrogated the ability of CI-976 to induce tubules (30). A second possibility is that CI-976 could inhibit an unidentified LPAAT whose activity contributes to vesicle fission by producing negative curve-inducing PA (or secondarily DAG).
Whether phospholipid modifications are directly involved in the fission of any membrane vesicle, including COPII vesicles, remains to be established. In CI-976-treated cells, we previously noted resident Golgi enzymes that had redistributed to the ER also failed to exit (29). This observation raised the intriguing possibility that, in addition to stimulating tubule formation from the Golgi complex, CI-976 also inhibited an unidentified step(s) in COPII vesicle-mediated export at specialized regions of the ER called ER exit sites (ERESs). We now show that CI-976 is a potent inhibitor of export from the ER. In the presence of CI-976, although COPII components assemble at ERESs, COPII vesicle budding is inhibited. Our results demonstrate that CI-976 specifically inhibits a very late event in vesicle budding, consistent with a key role for an ER-associated LPAT in mediating the fission step, leading to the release of vesicles for transport to the Golgi complex.
Results
CI-976 inhibits export from the ER
To determine if CI-976 inhibits export from the ER, we examined the synthesis and processing of vesicular stomatitis virus G (VSV-G) membrane glycoprotein using the tsO45 variant of VSV (tsO45 VSV) (36). Following infection with tsO45, VSV-G becomes the major cargo exiting the ER because of significant inhibition of host protein synthesis. Cells infected at the restrictive temperature (40°C) retain newly synthesized VSV-G in the ER because of misfolding. Following transfer of cells to the permissive temperature (32°C), a synchronous wave of VSV-G is exported from the ER to the Golgi by the COPII machinery (4,37,38). Transport to the early Golgi results in the processing of its N-linked high mannose core oligosaccharide attached in the ER to a glycoform that is resistant to endoglycosidase H (endo H). In control cells following shift to the permissive temperature, VSV-G was rapidly exported from the ER and delivered to the Golgi complex where most of it was processed to the endo H-resistant, mature form by 45 min (Figure 1). In contrast, in CI-976-treated cells, VSV-G processing by Golgi-associated mannosidases and glycosyl-transferases to the endo H-resistant form was significantly inhibited (Figure 1).
Figure 1. CI-976 inhibits the processing and transport of VSV-G from the ER.

A) Processing of VSV-G from the endo H-sensitive (S) to the -resistant (R) form following shift to the permissive temperature is inhibited by CI-976. Cells were labeled with 35S-TransLabel, shifted to the permissive temperature in the absence or presence of 50 μm CI-976 for the times indicated and VSV-G was immunoprecipitated and subjected to digestion by endo H. B) Transport of VSV-G from the ER to the Golgi complex is inhibited by CI-976 as seen by immunofluorescence microscopy. When cells were fixed prior to shift to the permissive temperature, VSV-G was found diffused throughout the cytoplasm in the ER (A′) and separate from the Golgi complex as seen by ManII labeling (B′). When cells were shifted to the permissive temperature for 15 min in the absence of CI-976, VSV-G was found to colocalize with ManII in the Golgi complex (C′ and D′). In contrast, when CI-976 (50 μm) was added for the last 15 min at the restrictive temperature and included for 15 min following shift to the permissive temperature, VSV-G (E′) did not reach the ManII compartment (F′). Instead, VSV-G moved from a diffuse ER-staining pattern to numerous small, punctate foci located throughout the cytoplasm.
The above results suggest that CI-976 inhibited delivery of VSV-G to the Golgi complex. To identify the site of trafficking arrest, the transport of VSV-G was followed using immunofluorescence and organelle-specific markers. As expected, in cells maintained at the restrictive temperature, VSV-G was found in a diffuse, ER pattern that was distinct from the Golgi complex labeled by the medial marker mannosidase II (ManII) (Figure 1A, A′ and B′). Addition of CI-976 for 15 min at the restrictive temperature had no effect on the distribution of VSV-G (data not shown). Following shift to the permissive temperature for 15 min, in the absence of drug, VSV-G was found to colocalize to a large extent with ManII in the juxtanuclear region (Figure 1A, C′ and D′), indicative of export from the ER and delivery to the Golgi complex. In contrast, in the presence of CI-976, VSV-G did not colocalize with ManII in the Golgi complex (Figure 1A, E′ and F′). Instead, VSV-G moved from a low-intensity, diffuse staining pattern into numerous bright foci located throughout the cytoplasm (Figure 1A, E′) that did not overlap with the distribution of the Golgi (Figure 1A, F′). These morphological results support the biochemical results, which showed that in the presence of CI-976, VSV-G was not processed to the endo H-resistant form by Golgi enzymes.
In CI-976-treated NRK cells, the Golgi complex had not yet redistributed to the ER after 30 min (Figure 1A, E′ and F′); however, by 60 min of treatment with 50 μm CI-976, Golgi markers were completely redistributed to the ER (data not shown). Fortuitously, this effect of CI-976 on NRK cells is about twofold slower than that in other cell types (30), thus allowing us to examine the transport of VSV-G from the ER following a brief 15-min exposure to CI-976 but prior to Golgi membrane redistribution.
Inhibition of VSV-G export occurs at Sec24/31-enriched ERESs
The bright, punctate foci enriched in VSV-G following shift to the permissive temperature in the presence of CI-976 are reminiscent of ERESs found at discrete locations on ER membranes from which COPII vesicles bud (11,39,40). To determine if these are exit sites, cells were examined by immunofluorescence to localize the COPII subunits, Sec24 and Sec31. In cells treated with CI-976 for the final 15 min at the restrictive temperature, VSV-G was distributed throughout the ER as above (Figure 1), and Sec24 was found in discrete foci defining ERESs containing residual endogenous cargo and transport machinery components that weakly overlapped with VSV-G, reflecting its block in export (Figure 2A,B) as previously described in untreated cells (41,42). Following shift to the permissive temperature in the presence of CI-976, VSV-G redistributed to discrete foci that to a large extent overlapped with Sec24 (Figure 2C,D). Consistent with the role of Sec13/31 in self-assembly into a lattice that collects cargo bound to Sec23/24 for budding from the ER (7), Sec31 was efficiently recruited and stabilized at ERESs, reflecting the movement of a bolus of VSV-G cargo at the permissive temperature (Figure 2E,F). Thus, CI-976 does not affect the binding of either Sec24 or Sec31 to ERESs. Rather, these results strongly suggest that CI-976 inhibits subsequent transport to the Golgi by preventing a late step following the stable association of both Sec23/24 and Sec13/31 COPII complexes.
Figure 2. CI-976 inhibits export of VSV-G from the ER at sites enriched for the COPII components Sec24 and Sec31.

Cells were infected with ts045 VSV for 3.5 h at the restrictive temperature to accumulate VSV-G in the ER, and CI-976 (50 μm) was then added for the last 15 min of this period. Cells were either fixed immediately (A and B) or shifted to the permissive temperature for an additional 15 min in the continuous presence of CI-976 (50 μm) (C–F). After each incubation period, cells were processed for double immunofluorescence localization of VSV-G (left side panels) and Sec24 or Sec31 (as indicated). Arrows show clusters of foci in which VSV-G and Sec24 or Sec31 colocalize. Bar = 5 μm.
Previous studies have shown that the effect of CI-976 on the Golgi complex is reversible (30); therefore, we examined if export of VSV-G from the COPII coat-enriched foci was similarly reversible. For these experiments, cells were infected at the restrictive temperature and then shifted to the permissive temperature in the presence of CI-976 for 30 min to allow VSV-G to accumulate at COPII-enriched ERESs. Cells were subsequently incubated for an additional 15 min in the presence or absence of the drug. When cells were maintained in the presence of CI-976, VSV-G remained in Sec24-enriched foci, distinct from the pattern of Golgi ManII staining (Figure 3A–D). In contrast, when cells were washed free of CI-976, VSV-G localization at the small Sec24-positive foci was lost. Instead, VSV-G staining was found to strongly overlap with the distribution of Golgi complex marker protein ManII (Figure 3E–H). Interestingly, under these conditions, a large amount of the Sec24 immunofluorescence was also found closely associated with the ManII-labeled Golgi membranes (Figure 3E,F). Conversely, (N′ - (2,4-difluorophenyl)-N-[5-[(4,5-diaryl-1H-imidazol-2-yl)thio]pentyl]-N-heptylurea) (DuP-128), a more potent and specific ACAT antagonist with no activity toward a Golgi LPAT (30), had no effect on VSV-G transport in these assays (43).
Figure 3. VSV-G that accumulates in Sec24-enriched foci is rapidly transported to the Golgi complex following removal of CI-976.

Cells were infected with ts045 VSV for 3.5 h at the restrictive temperature to accumulate VSV-G in the ER, and CI-976 (50 μm) was then added for the last 15 min of this period and then shifted to the permissive temperature in the presence of CI-976 for 15 min. Cells were either maintained under these conditions for an additional 15 min (A–D) or washed free of CI-976 and incubated for 15 min at the permissive temperature (E–H). After each incubation period, cells were processed for double immunofluorescence localization of VSV-G (left side panels) and Sec24 or ManII (as indicated). Arrows show foci containing both VSV-G and Sec24. Bar = 5 μm.
These results suggest that following removal of CI-976, COPII-coated transport intermediates rapidly complete budding from the ER and facilitate trafficking of VSV-G to the Golgi region. Consistent with these results, reversible inhibition of VSV-G export from the ER by CI-976 could also be biochemically demonstrated (Figure 4A). Following incubation for 30 min in the presence of the drug at 40°C, transfer of cells to the permissive temperature and removal of CI-976 resulted in the rapid processing of VSV-G to the endo H-resistant form. Processing lacked the lag period typically associated with transfer of cells from 40°C to 32°C in the absence of drug (Figure 4B), indicating that cargo selection and accumulation of VSV-G by the Sec23/24 complex into ERESs is a rate-limiting step following temperature shift.
Figure 4. Recovery from CI-976 reduces the lag in ts045 VSV-G processing.
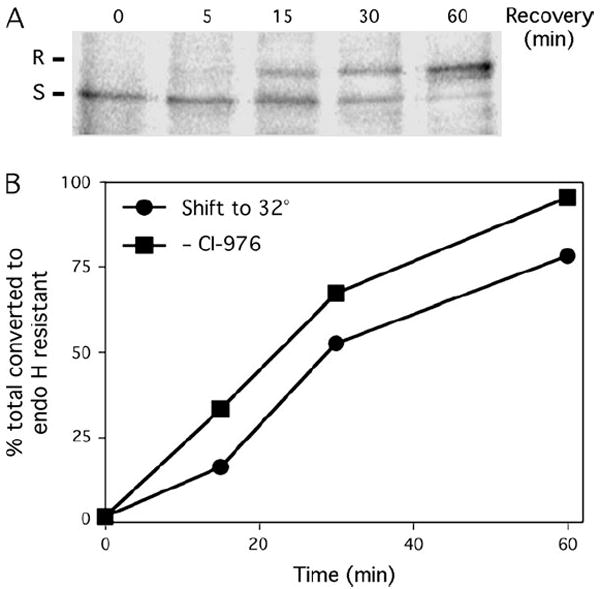
A) VSV-G is converted from the endo H-sensitive (S) to the endo H-resistant (R) form following removal of CI-976. Cells were infected with ts045 VSV at the restrictive temperature to accumulate VSV-G in the ER, treated with CI-976 (50 μm) and then shifted to the permissive temperature in the continuous presence of CI-976 for 15 min as described in Figure 1. Cells were then washed free of CI-976 to permit export from the ER and then incubated for the times indicated above each lane. Cells were solubilized, and VSV-G was immunoprecipitated and treated with endo H prior to SDS–PAGE and fluorography. B) Rates of conversion of VSV-G to the endo H-resistant form following a simple shift to the permissive temperature as in Figure 1 (solid circles) or following removal of CI-976 from cells held at the permissive temperature (solid squares) as in (A). Bar = 5 μm.
CI-976 inhibits COPII vesicle budding in vitro and in vivo
To provide additional evidence that CI-976 inhibits the formation of COPII vesicles, we examined VSV-G export using a system that reconstitutes the ATP- and cytosol-dependent budding of VSV-G-containing COPII vesicles in vitro from ER microsomes (4). In this assay, export of VSV-G is measured directly by recovery of VSV-G in COPII vesicles. Incubation in the presence of increasing concentrations of CI-976 revealed that COPII vesicle formation from ER membranes is inhibited with an IC50 ~20 μm (Figure 5A–C). As in vivo, DuP-128, the more potent and specific ACAT antagonist, had no effect on COPII budding in the in vitro assay (Figure 5A,B).
Figure 5. Formation of COPII vesicles in vitro is inhibited by CI-976.

A) VSV-G served as cargo marker for Western blot analysis of COPII vesicle budding from ER membranes in an in vitro reconstitution assay. Samples were incubated with the indicated amounts of CI-976 or DuP-128 (μm) for 15 min at 32°C to produce vesicles. Alternatively, samples were kept on ice (as indicated) as a negative control. ‘Total’ lane is equivalent to one third of the total amount of VSV-G present in the incubation mix at the start of the experiment (volumes adjusted to prevent overexposing the ‘Total’ lane). B) Quantification of dose–response experiments similar to those shown in (A). C) Time–course of COPII vesicle budding in the absence (DMSO) or presence of CI-976 (50 μm). D) Inhibition of COPII budding in vitro is most effective when CI-976 is first added to microsomes, as determined by order-of-addition experiments. The order in which each component was added is shown below each lane (A–H). Cyto, cytosol; MS, microsomes. CI-976 was used at 50 μm, and DMSO served as a solvent control. Quantification of these results is shown in the lower graph, and the results are expressed as the % control for each pair of conditions, for example, B/A. For B–D, the results show the mean plus 1 standard deviation from three or more independent experiments.
To determine if the CI-976-sensitive component was localized to the ER membranes or a cytosolic component, ER microsomes or cytosol were separately pretreated for 15 min on ice with the drug. Interestingly, preincubation of cytosol with CI-976 prior to addition of microsomes did not inhibit formation of VSV-G-containing COPII vesicles, suggesting that the drug is either metabolized or sequestered by cytosolic factor(s). In contrast, preincubation of microsomes with the drug prior to the addition of cytosol resulted in potent inhibition of COPII vesicle formation (Figure 5D). We conclude from these experiments that the target for CI-976 is an ER-associated LPAT.
Electron microscopy of cells treated with CI-976 confirmed that vesicle budding appears to be inhibited at ERESs (Figure 6). In control cells, ERESs often displayed a cluster of vesicle profiles (Figure 6A), whereas ERESs in CI-976-treated cells often contained chains of vesicles (Figure 6B,C). These results were confirmed by determining that there was a more than fourfold increase in the fraction of ERESs with chains of vesicles (control = 9.2% and CI-976 treated = 42%). This change in the morphology of vesicles at ERESs was matched by a significant increase in the number of vesicles profiles in treated cells (control cells = 6.7 ± 3.9 and treated cells = 9.4 ± 4.2; Student’s t-test p < 0.001).
Figure 6. CI-976 induces the formation of chains of COPII vesicles at ERES.
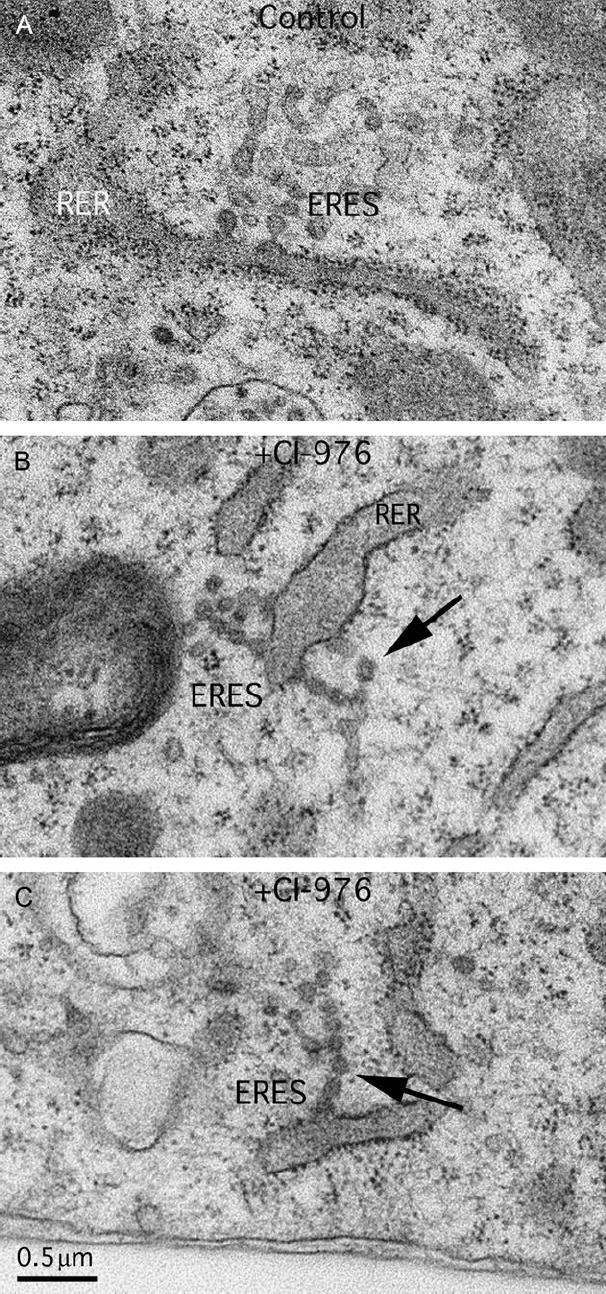
Electron microscopy of control (A) and CI-976-treated cells (B and C) reveals that CI-976 causes the formation of chains of vesicles (arrows) from ERESs that have apparently been unable to undergo a late fission step. RER, rough ER. Bar = 0.5 μm.
Sar1p-GTP-induced tubule formation from ERESs in semi-intact cells is enhanced by CI-976
Previous studies have shown that CI-976 stimulates the formation of membrane tubules from the Golgi complex in vivo (30). Export from the ER involves transitional tubules that, in the presence of the COPII machinery, form COPII vesicles at their tips (37,44). Transitional tubule formation can be reconstituted using semi-intact cells, a cell preparation in which the plasma membrane has been selectively permeabilized by the detergent digitonin to expose ER membranes (45). Incubation of semi-intact cells in the presence of ATP and cytosol faithfully reconstitutes ER-to-Golgi transport biochemically and morphologically (11,45,46).
Transitional tubule formation can be greatly exaggerated by incubation of semi-intact cells in the absence of the Sec23/24 and Sec13/31 coat components but in the presence of the GTP-restricted, constitutively active form of Sar1p, Sar1p-GTP (11,12). Under these minimal coat assembly conditions, transitional tubules up to 10 μm in length form (11). In the absence of added Sar1p-GTP, we could not detect any apparent stimulation of tubule formation at ERESs by CI-976 (data not shown), reflecting the fact that activated Sar1 is essential to initiate transitional ER formation (4,11). This serves as an important control demonstrating that CI-976 does not stimulate tubule formation in the absence of COPII. In contrast, the drug had a striking effect on ER tubule formation in the presence of Sar1p-GTP. At the lowest concentration of Sar1p-GTP used (15 μg/mL), CI-976 morphologically enhanced the association of Sar1p-GTP at discrete foci (Figure 7A). At higher concentrations of Sar1p-GTP (37.5 and 62.5 μg/mL), CI-976 greatly enhanced the formation of Sar1-mediated membrane tubules (Figure 7A). Quantitatively, and as observed previously (11), increasing the concentration of Sar1-GTP in the assay results in the appearance of tubules of increasing length (Figure 7B). Importantly, however, for each Sar1-GTP concentration examined, CI-976 caused a significant increase in the lengths of the membrane tubules. Moreover, and consistent with previous results (11), Sar1p-GTP-induced membrane tubules stimulated by CI-976 contained VSV-G (Figure 7C). These results suggest that CI-976 increases tubule length at ERESs by inhibiting alterations in the membrane composition that would contribute to fission.
Figure 7. CI-976 enhances Sar1p-induced ER tubule formation.

A) Sar1p-induced tubule formation from ERESs is enhanced by CI-976. Permeabilized NRK cells were incubated with varying amounts of Sar1p-GTP (as indicated on the left) in the absence (DMSO) or presence of CI-976 (50 μm) as indicated under conditions that promote Sar1p-induced tubule formation. Cells were then fixed and stained by immunofluorescence for Sar1p. B) Quantification of CI-976-enhanced Sar1p-mediated membrane tubule formation from ERESs from experiments shown in (A). The lengths of tubules were determined in permeabilized cells incubated with 37.5 or 62.5 μg/mL Sar1p-GTP in absence (−CI-976) or presence (+CI-976) of CI-976. Note that for lengths between 1.0–7.5 μm, tubules were placed into 0.5-μm bins. Number of tubules measured: (37.5 μg/mL Sar1p-GTP), −CI-976 (n = 495), +CI-976 (n = 621); (62.5 μg/mL Sar1p-GTP), −CI-976 (n = 670), +CI-976 (n = 974). C) CI-976-enhanced Sar1p membrane tubules contain VSV-G. Cells were incubated as in (A) and stained by double immunofluorescence for Sar1p and VSV-G. Bar = 5 μm.
Discussion
Using both in vivo and in vitro approaches that allow us to assign the action of components directing COPII vesicle formation, we have now provided direct evidence that a small molecule LPAT antagonist, CI-976, inhibits a very late step in COPII-coated vesicle budding. We find that ER accumulation of cargo observed in the presence of CI-976 (30) is because of the inhibition of a late step in COPII vesicle formation, one that follows cargo collection and assembly of the COPII coat. Our results are consistent with the conclusion that CI-976 inhibits a late fission step in the COPII vesicle budding pathway, leading us to propose that remodeling of the lipid bilayer is an integral component of a final step in vesicle formation from the ER. Given the more general effects of CI-976 on membrane tubulation in other exocytic and endocytic compartments, our results provide support for the general conclusion that LPAT modification of phospholipids may be an important physiological event for fission throughout the exocytic and endocytic pathways.
Four lines of evidence are consistent with the conclusion that CI-976 inhibits fission of COPII vesicles from ERESs. The first piece of evidence, and the most surprising and important for the conclusion that CI-976 inhibits the COPII vesicle fission step, is that following shift to the permissive temperature in the presence of CI-976, VSV-G accumulated at ERESs along with COPII components Sec23/24 and Sec13/31. Previous studies have demonstrated that Sec24, the cargo selection component of the COPII coat, is located at ERES foci (41,42). The Sec13/31 self-assembling cage (7) is now believed to generate a coat polymer that efficiently concentrates cargo-bound Sec23/24 into budding vesicles (47-49). It is frequently found on intermediates derived from budding structures that assemble and rapidly disengage from ERESs. Strikingly, Sec31 was recruited to the VSV-G- and Sec24-positive ERESs in CI-976-treated cells that were arrested in transport to the Golgi. Such a stable recruitment is reminiscent of the effects of GTP analogs such as GTPYS or the dominant negative mutant Sar1[H79S] (restricted to the GTP-bound form), which also cause accumulation of cargo and COPII-coated structures at ERESs (11). These results raised the possibility that COPII buds form but do not pinch off of the ER membrane.
The second piece of evidence comes from electron microscopy, which revealed that CI-976 caused a significant (41%) increase in the number of vesicle profiles at ERESs and that these vesicles were often connected to each other in a chain as though they were unable to detach, that is, undergo fission. Interestingly, CI-976 did not cause a massive accumulation of unbudded vesicles or a significant increase in the overall size of the ERESs seen by immunofluorescence (see for example, Figure 2B,D: Sec24 staining in control and CI-976-treated cells). The lack of a massive change in the number of unbudded COPII vesicles in CI-976 may reflect a limited capacity of ERESs to bind COPII components. Also, the inability of CI-976 to induce membrane tubule formation at ERESs in vivo, in contrast to the situation in permeabilized cells incubated with Sar1p-GTP reported here, or the Golgi complex in vivo (30) may be because of the binding of COPII coats that limit tubule formation.
The third piece of evidence was that washout of CI-976 from cells resulted in a reduction in the lag phase for the rapid transport of VSV-G from the ER to the Golgi complex. This result is consistent with the data showing that in CI-976-treated cells incubated at the permissive temperature, VSV-G collected in fully formed COPII buds that are primed to undergo fission, a step that is sensitive to an inhibitor of LPAT activity.
The final piece of evidence, consistent with all the in vivo results, was that CI-976 potently inhibited the budding of COPII vesicles in an in vitro assay that reconstitutes vesicle formation from ER microsomes. Thus, all of our results are consistent with LPAT activity playing a role in a very late step in COPII budding, after cargo has collected, coats have formed and membranes have deformed into vesicles attached through thin necks. One possibility is that LPAT activity is co-ordinated with Sar1p hydrolysis and potentially a prerequisite for Sec23 Sar1p GTPase-activating protein activity, linking protein and lipid function in vesicle formation. Of course, it is possible that LPAT activity is not directly involved in fission but instead facilitates the activity of other factors that promote fission.
Of particular interest to the requirement for LPAT function in COPII vesicle formation is the recent report that Sar1p activates PLD and that PLD-generated PA is necessary to support COPII assembly and ER export (28). In these studies, PA was shown to be important for recruitment of COPII coats, a result consistent with the observation that negatively charged lipids are required for COPII vesicle formation from liposomes (8). Our results now suggest that PLD-generated PA could facilitate the generation of negative curvature that is important for vesicle fission. In this regard, other lipid-modifying enzymes of interest are the PLA1-type phospholipases named, KIAA0725p, and p125, a Sec23-interacting protein, both of which prefer PA as a substrate (50). Overexpression of KIAA0725p or p125 induces alterations in ERES structure (51), which could be because of the excess production of LPA. However, we found that pretreatment of cells with the phospholipase antagonists 2-(p-amylcinnamyol)amino-4-chlorobenzoic acid (ONO-RS-082) and bromoenol lactone did not prevent CI-976-induced accumulation of VSV-G at ERESs (unpublished data). Therefore, it seems likely that any LPA molecules that might act as substrates for a CI-976-sensitive LPAAT are derived through a phospholipase-independent pathway, for example, de novo phospholipid synthetic pathways. Alternatively, putative phospholipases might be insensitive to the antagonists used here.
The fact that VSV-G accumulates at ERESs in the presence of CI-976 suggests that maintenance of concentrated cargo at ERESs following temperature shift is not dependent on fission but, more likely, a stabilized Sec13/31 polymeric lattice containing Sec23/24 that promotes membrane curvature (7,10,12). Given the recently recognized importance of Sar1p in vesicle fission (10,12), it remains possible that LPAT activity is co-ordinated with the structural properties of the Sar1p lipid-binding N-terminus in promoting membrane curvature along with Sec23/24 to complete fission of these transitional tubular elements into free vesicles.
The known activities of CI-976 include inhibition of LPAT and ACAT enzymes, but the fact that other more potent ACAT inhibitors, for example, DuP-128, had no effect on ER export or COPII vesicle formation argues that ACAT activity is not involved in COPII vesicle fission. How might an LPAT contribute to vesicle budding? One possibility is that negative curve-inducing lipids (PA and DAG) might contribute to the constriction that occurs at the neck of a budding vesicle (52-54). Thus, CI-976 could inhibit an ER LPA-AT that generates PA (and indirectly DAG). Consistent with this idea, we found using the in vitro budding assay that CI-976 most likely targets a membrane-associated, but not a cytoplasmic, protein. Interestingly, CI-976 also targets an LPAT that is very tightly associated with Golgi membranes (29). Current annotation of the human genome has identified six known or putative LPA-AT enzymes, LPA-ATα, β, δ, γ, ε, ζ, η and θ (20,55). LPA-ATα and β are known to be involved in de novo synthesis of glycerol phospholipids in the ER, but the functions of other LPA-AT family members are unknown. Preliminary studies show that two of these uncharacterized LPA-ATs are ER enzymes and two are also found in the Golgi complex (Schmidt, J., and W. J. Brown, Cornell University, Ithaca, NY. unpublished data), any or all of which could be the targets of CI-976. It is important to note that CI-976 may not be restricted to inhibiting an LPA-specific LPAT, even in the ER. Our ability to demonstrate the key role for LPAT activity in vivo sets the stage for a complete analysis of LPA-AT enzymes in function and fission in the early secretory pathway.
Materials and Methods
Reagents and antibodies
CI-976 and DuP-128 were kindly provided by GlaxoSmithKline Pharmaceuticals. 35S-TransLabel was from ICN. Rabbit anti-Sec24, rabbit anti-Sec31 and rabbit anti-Sar1p were prepared in the Balch laboratory as previously described (4,40,45). Rabbit anti-ManII was kindly provided by Dr Kelley Moremen (University of Georgia). Goat anti-rabbit Alexa 488 and goat anti-mouse Texas Red were from Molecular Probes.
Cell culture and infection with ts045 VSV
NRK cells were grown in DMEM plus 10% FBS at 37°C in a humidified atmosphere of 95% air and 5% CO2. To examine the effect of CI-976 on export from the ER in vivo, in vitro and in permeabilized cells, we utilized the temperature-sensitive variant ts045 VSV. At the restrictive temperature (40°C) VSV-G is misfolded and remains in the ER, but shifting to the permissive temperature (32°C) allows proper folding, trimerization and rapid COPII vesicle-mediated export to the Golgi complex (45).
In vivo pulse–chase and immunoprecipitation of VSV-G
NRK cells were grown in 60-mm Petri dishes for 2–3 days to confluence, washed with PBS, pH 7.4, and then DMEM containing 50 μg/mL actinomycin D was applied to dishes (all at 32°C). Cells were infected with ts045 VSV for 45 min at 32°C, DMEM plus 10% FBS (post-infection media) were added and cells were further incubated for 15 min at 32°C. The following steps were performed at 40°C. Petri dishes were placed in a 40°C water bath, washed three times with methionine-free DMEM and then incubated in the same for 10 min in a CO2 incubator at 40°C. Media was removed, cells were covered with methionine-free DMEM (labeling media) to which 50 μCi of 35S-TransLabel was added and the metabolic labeling was continued for 3.5 h in a CO2 incubator at 40°C. Cells were washed three times with DMEM containing 100 μg/mL methionine and 0.1 mg/mL BSA (post-labeling media), covered with 4 mL of post-labeling media containing either 50 μm CI-976 or the appropriate volume (4 μL) of dimethyl sulphoxide (DMSO) as a solvent control and incubated for 10 min. To initiate export from the ER, the media was replaced with post-labeling media (±CI-976) that was equilibrated to 32°C. Following incubation for various periods of time at 32°C in a CO2 incubator, cells were lysed with ice-cold 1% Triton-X-100 in 20 mm Tris, pH 8.0, 150 mm NaCl and 2 mm ethylenediaminetetraacetic acid (EDTA) (lysis buffer). Cells were harvested with gentle pipetting, and the lysates were clarified by a 5-min spin in a microfuge at 4°C. Supernatants were collected for immunoprecipitation.
To determine if the inhibitory effects of CI-976 on ER export were reversible, NRK cells grown in a single 100-mm Petri dish were processed and infected with ts045 VSV as above. Cells were then incubated in post-infection media for 3.5 h at 32°C. The following steps were carried out at 40°C. Cells were washed three times with labeling media, incubated in labeling media containing 50 μm CI-976 for 10 min and then cells were labeled by the addition of 100 μCi of 35S-TransLabel/dish for 1 h. Cells were washed three times with post-labeling media and then incubated in post-labeling media for 45 min (both containing 50 μm CI-976). All of the following steps were carried out at 40°C. To remove CI-976, cells were washed three times in post-infection media, three times again with DMEM + 10% FBS and then three times with PBS. Cells were trypsinized to remove them from the dish, harvested into 2 mL of post-labeling media containing 0.25 mg/mL soybean trypsin inhibitor and divided into equal aliquots into microfuge tubes. To initiate export from the ER, tubes were placed in a 32°C water bath and incubated for various periods of time. Cells were then placed on ice, immediately extracted by addition of 200 μL of ice-cold lysis buffer and extracts were clarified by microfuging at maximum speed for 5 min at 4°C. Supernatants were collected for immunoprecipitation.
Immunoprecipitation of VSV-G was performed using monoclonal P5D4 anti-VSV-G and protein G beads. Immunoprecipitated VSV-G was digested by endo H as described (4), and the products were analyzed by SDS–PAGE and imaging with a Storm 860 Phosphorimager (Molecular Dynamics). Bands were quantified by densitometric analysis using National Institutes of Health (NIH) IMAGE 1.63 software.
In vitro COPII vesicle budding assay
The assay for VSV-G export into COPII vesicles produced from ER membranes in vitro was as described (40) with modifications for examining the effect of CI-976. Briefly, total microsomes were prepared from NRK cells infected with ts045 VSV under conditions that accumulate VSV-G in the ER (as above). Cytosol was from rat liver (RLC) (56), stored at −80°C until use and clarified by centrifugation at 100 000 × g for 10 min prior to use. The budding cocktail consisted of 25 mm HEPES, pH 7.2, 40 mm KOAc, 0.18 m sorbitol, 2.5 mm MgOAc, 1.8 mm Ca(OAc)2, 5 mm EDTA and an ATP regenerating system (56). For each time-point, aliquots of budding cocktail (30 mL minus the volume needed for CI-976) and microsomes (15 μL) were added together and mixed on ice. CI-976 was then added from a concentrated stock solution (in DMSO), so that the final concentration after adding cytosol (in the next step) was 50 μm, and the tubes were gently mixed. A similar volume of DMSO was added to control tubes. RLC (15 μL) was then added with gentle mixing. To initiate COPII budding, tubes were transferred to a 32°C water bath and incubated for the indicated time in the Results. After incubation in the budding reaction, tubes were placed on ice and centrifuged at 21 000 × g for 2 min in an Eppendorf 5417C refrigerated centrifuge. The supernatant (50 μL) was collected, and COPII vesicles were sedimented to a pellet by centrifugation at 100 000 × g for 20 min. The supernatant was removed, and the pellet was dissolved in sodium dodecyl sulfate sample buffer. The amount of VSV-G released with the vesicles was determined by Western blotting with polyclonal anti-VSV-G antibodies as described (40).
Immunofluorescence labeling of VSV-infected cells
NRK cells were grown on glass coverslips for 2 days and infected with ts045 VSV as described above. VSV-G was allowed to accumulate in the ER for 3.5–4 h at the restrictive temperature and then cells were shifted to the permissive temperature to examine the effect of CI-976 on export from the ER. Immunofluorescence labeling was conducted as previously described (45) using the following antibodies: monoclonal p5D4 anti-VSV-G (diluted 1:400), polyclonal anti-ManII (diluted 1:1000), polyclonal anti-Sec24 (diluted 1:250) and polyclonal anti-Sec31 (diluted 1:300).
Electron microscopy
For thin-section, transmission electron microscopy, cells were fixed in a combination of glutaraldehyde/osmium tetroxide fixative and processed as described (57,58). Images were captured on an FEI/Philips Morgagni transmission electron microscope using a Hamamatsu digital camera controlled by software from Advanced Microscopy Techniques, Corp. All images were taken at a magnification of 22 000, and from these digital images, vesicle and budding vesicle profiles at ERES were quantified at a working magnification of 44 000.
Sar1p-induced tubule formation from the ER in permeabilized cells
Sar1p-induced tubule formation from ERESs in permeabilized cells was performed with modifications of the previously described procedure (11). NRK cells were grown to confluence on glass coverslips and infected with ts045 VSV to accumulate newly synthesized VSV-G in the ER. Petri dishes containing coverslips were transferred to an ice-cold metal block, and cells were washed three times with ice-cold KHM buffer (20 mm HEPES, 110 mm KOAc and 2 mm MgOAc, pH 7.2). Cells were permeabilized by replacing KHM with 80 μg/mL digitonin in KHM for 5 min on ice, gently washed four times with cold KHM and then kept in the last wash for 10 min. Coverslips were placed into 35-mm Petri dishes by inverting them onto a 50 μL drop of solution containing varying amounts of His-tagged Sar1p (from a stock solution in 25 mm HEPES and 125 mm KOAc, pH 7.2) and 50 μm CI-976 (or DMSO). The Sar1p used in these experiments is the GTP-restricted mutant (H79G) (Sar1p-GTP). Dishes were transferred to a 32°C water bath to initiate tubule formation and incubated for 30 min. Transport was terminated by placing dishes on an ice-cold metal block. Coverslips were reinverted, washed with ice-cold KHM two times, fixed with 2% formalin in PBS for 5 min and processed for immunofluorescence staining of Sar1p (polyclonal anti-Sar1p diluted 1:300) and VSV-G as above. Tubule length was measured by importing images into ADOBE PHOTOSHOP 7.0, preparing a scale bar of the appropriate length using the line tool and using the free rotation tool to align the scale bar with fluorescent tubules.
Acknowledgments
We thank Dr Brian Jackson of GlaxoSmithKline Pharmaceuticals for originally supplying the CI-976. This work was supported by NIH grants DK51596 and GM60373 (to W. J. B.) and GM42236 (to W. E. B.).
References
- 1.Kooijman EE, Chupin V, de Kruijff B, Burger KN. Modulation of membrane curvature by phosphatidic acid and lysophosphatidic acid. Traffic. 2003;4:162–174. doi: 10.1034/j.1600-0854.2003.00086.x. [DOI] [PubMed] [Google Scholar]
- 2.Bonifacino JS, Glick BS. The mechanisms of vesicle budding and fusion. Cell. 2004;116:153–166. doi: 10.1016/s0092-8674(03)01079-1. [DOI] [PubMed] [Google Scholar]
- 3.Lee MC, Miller EA, Goldberg J, Orci L, Schekman R. Bi-directional protein transport between the ER and Golgi. Annu Rev Cell Dev Biol. 2004;20:87–123. doi: 10.1146/annurev.cellbio.20.010403.105307. [DOI] [PubMed] [Google Scholar]
- 4.Aridor M, Weissman J, Bannykh S, Nuoffer C, Balch WE. Cargo selection by the COPII budding machinery during export from the ER. J Cell Biol. 1998;141:61–70. doi: 10.1083/jcb.141.1.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schekman R. Lasker Basic Medical Research Award. SEC mutants and the secretory apparatus. Nat Med. 2002;8:1055–1058. doi: 10.1038/nm769. [DOI] [PubMed] [Google Scholar]
- 6.LaPointe P, Gurkan C, Balch WE. Mise en place-this bud’s for the Golgi. Mol Cell. 2004;14:413–414. doi: 10.1016/s1097-2765(04)00267-9. [DOI] [PubMed] [Google Scholar]
- 7.Stagg SM, Gurkan C, Fowler DM, LaPointe P, Foss TR, Potter CS, Carragher B, Balch WE. Structure of the Sec13/31 COPII coat cage. Nature. 2006;439:234–238. doi: 10.1038/nature04339. [DOI] [PubMed] [Google Scholar]
- 8.Matsuoka K, Orci L, Amherdt M, Bednarek SY, Hamamoto S, Schekman R, Yeung T. COPII-coated vesicle formation reconstituted with purified coat proteins and chemically defined liposomes. Cell. 1998;93:263–275. doi: 10.1016/s0092-8674(00)81577-9. [DOI] [PubMed] [Google Scholar]
- 9.Matsuoka K, Morimitsu Y, Uchida K, Schekman R. Coat assembly directs v-SNARE concentration into synthetic COPII vesicles. Mol Cell. 1998;2:703–708. doi: 10.1016/s1097-2765(00)80168-9. [DOI] [PubMed] [Google Scholar]
- 10.Bielli A, Haney CJ, Gabreski G, Watkins SC, Bannykh SI, Aridor M. Regulation of Sar1 NH2 terminus by GTP binding and hydrolysis promotes membrane deformation to control COPII vesicle fission. J Cell Biol. 2005;171:919–924. doi: 10.1083/jcb.200509095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Aridor M, Fish KN, Bannykh SI, Weissman J, Roberts TH, Lippincott-Schwartz J, Balch WE. The Sar1 GTPase coordinates biosynthetic cargo selection with endoplasmic reticulum export site assembly. J Cell Biol. 2001;152:213–229. doi: 10.1083/jcb.152.1.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee MC, Orci L, Hamamoto S, Futai E, Ravazzola M, Schekman R. Sar1p N-terminal helix initiates membrane curvature and completes the fission of a COPII vesicle. Cell. 2005;122:605–617. doi: 10.1016/j.cell.2005.07.025. [DOI] [PubMed] [Google Scholar]
- 13.Bi X, Corpina RA, Goldberg J. Structure of the Sec23/24-Sar1 pre-budding complex of the COPII vesicle coat. Nature. 2002;419:271–277. doi: 10.1038/nature01040. [DOI] [PubMed] [Google Scholar]
- 14.Conner SD, Schmid SL. Regulated portals of entry into the cell. Nature. 2003;422:37–44. doi: 10.1038/nature01451. [DOI] [PubMed] [Google Scholar]
- 15.Danino D, Hinshaw JE. Dynamin family of mechanoenzymes. Curr Opin Cell Biol. 2001;13:454–460. doi: 10.1016/s0955-0674(00)00236-2. [DOI] [PubMed] [Google Scholar]
- 16.Lee MC, Schekman R. BAR domains go on a bender. Science. 2004;303:479–480. doi: 10.1126/science.1094231. [DOI] [PubMed] [Google Scholar]
- 17.Song BD, Schmid SL. A molecular motor or a regulator? Dynamin’s in a class of its own. Biochemistry. 2003;42:1369–1376. doi: 10.1021/bi027062h. [DOI] [PubMed] [Google Scholar]
- 18.Thompson HM, McNiven MA. Dynamin: switch or pinchase? Curr Biol. 2001;11:R850. doi: 10.1016/s0960-9822(01)00513-9. [DOI] [PubMed] [Google Scholar]
- 19.McMahon HT, Gallop JL. Membrane curvature and mechanisms of dynamic cell membrane remodelling. Nature. 2005;438:590–596. doi: 10.1038/nature04396. [DOI] [PubMed] [Google Scholar]
- 20.Leung DW. The structure and functions of human lysophosphatidic acid acyltransferases. Front Biosci. 2001;6:D944–D953. doi: 10.2741/leung. [DOI] [PubMed] [Google Scholar]
- 21.Coleman RA. How do I fatten thee? Let me count the ways. Cell Metab. 2007;5:87–89. doi: 10.1016/j.cmet.2007.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Coleman RA, Lee DP. Enzymes of triacylglycerol synthesis and their regulation. Prog Lipid Res. 2004;43:134–176. doi: 10.1016/s0163-7827(03)00051-1. [DOI] [PubMed] [Google Scholar]
- 23.Vergnes L, Beigneux AP, Davis R, Watkins SM, Young SG, Reue K. Agpat6 deficiency causes subdermal lipodystrophy and resistance to obesity. J Lipid Res. 2006;47:745–754. doi: 10.1194/jlr.M500553-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Scales SJ, Scheller RH. Lipid membranes shape up. Nature. 1999;401:123–124. doi: 10.1038/43582. [DOI] [PubMed] [Google Scholar]
- 25.Fuller N, Rand RP. The influence of lysolipids on the spontaneous curvature and bending elasticity of phospholipid membranes. Biophys J. 2001;81:243–254. doi: 10.1016/S0006-3495(01)75695-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Szule JA, Fuller NL, Rand RP. The effects of acyl chain length and saturation of diacylglycerols and phosphatidylcholines on membrane monolayer curvature. Biophys J. 2002;83:977–984. doi: 10.1016/s0006-3495(02)75223-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fabbri M, Bannykh S, Balch WE. Export of protein from the endoplasmic reticulum is regulated by a diacylglycerol/phorbol ester binding protein. J Biol Chem. 1994;269:26848–26857. [PubMed] [Google Scholar]
- 28.Pathre P, Shome K, Blumental-Perry A, Bielli A, Haney CJ, Alber S, Watkins SC, Romero G, Aridor M. Activation of phospholipase D by the small GTPase Sar1p is required to support COPII assembly and ER export. EMBO J. 2003;22:4059–4069. doi: 10.1093/emboj/cdg390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chambers K, Brown WJ. Characterization of a novel CI-976-sensitive lysophospholipid acyltransferase that is associated with the Golgi complex. Biochem Biophys Res Commun. 2004;313:681–686. doi: 10.1016/j.bbrc.2003.12.016. [DOI] [PubMed] [Google Scholar]
- 30.Drecktrah D, Chambers K, Racoosin EL, Cluett EB, Gucwa A, Jackson B, Brown WJ. Inhibition of a Golgi complex lysophospholipid acyltransferase induces membrane tubule formation and retrograde trafficking. Mol Biol Cell. 2003;14:3459–3469. doi: 10.1091/mbc.E02-11-0711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Harte RA, Yeaman SJ, Jackson B, Suckling KE. Effect of membrane environment on inhibition of acyl-CoA: cholesterol acyltransferase by a range of synthetic inhibitors. Biochim Biophys Acta. 1995;1258:241–250. doi: 10.1016/0005-2760(95)00113-q. [DOI] [PubMed] [Google Scholar]
- 32.Lippincott-Schwartz J, Yuan LC, Bonifacino JS, Klausner RD. Rapid redistribution of Golgi proteins into the ER in cells treated with brefeldin A: evidence for membrane cycling from Golgi to ER. Cell. 1989;56:801–813. doi: 10.1016/0092-8674(89)90685-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brown WJ, Chambers K, Doody A. Phospholipase A2 (PLA2) enzymes in membrane trafficking: mediators of membrane shape and function. Traffic. 2003;4:214–221. doi: 10.1034/j.1600-0854.2003.00078.x. [DOI] [PubMed] [Google Scholar]
- 34.de Figueiredo P, Drecktrah D, Katzenellenbogen JA, Strang M, Brown WJ. Evidence that phospholipase A2 activity is required for Golgi complex and trans Golgi network membrane tubulation. Proc Natl Acad Sci U S A. 1998;95:8642–8647. doi: 10.1073/pnas.95.15.8642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.de Figueiredo P, Polizotto RS, Drecktrah D, Brown WJ. Membrane tubule-mediated reassembly and maintenance of the Golgi complex is disrupted by phospholipase A2 antagonists. Mol Biol Cell. 1999;10:1763–1782. doi: 10.1091/mbc.10.6.1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lafay F. Study of thermosensitive mutants of vesicular stomatitis virus (VSV). Classification of some mutants on a functional basis. C R Acad Sci Hebd Seances Acad Sci D. 1969;268:2385–2398. [PubMed] [Google Scholar]
- 37.Bannykh SI, Nishimura N, Balch WE. Getting into the Golgi. Trends Cell Biol. 1998;8:21–25. doi: 10.1016/s0962-8924(97)01184-7. [DOI] [PubMed] [Google Scholar]
- 38.Bannykh SI, Balch WE. Selective transport of cargo between the endoplasmic reticulum and Golgi compartments. Histochem Cell Biol. 1998;109:463–475. doi: 10.1007/s004180050248. [DOI] [PubMed] [Google Scholar]
- 39.Glick BS. ER export: more than one way out. Curr Biol. 2001;11:R361–R363. doi: 10.1016/s0960-9822(01)00194-4. [DOI] [PubMed] [Google Scholar]
- 40.Rowe T, Aridor M, McCaffery JM, Plutner H, Nuoffer C, Balch WE. COPII vesicles derived from mammalian endoplasmic reticulum microsomes recruit COPI. J Cell Biol. 1996;135:895–911. doi: 10.1083/jcb.135.4.895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tang BL, Kausalya J, Low DY, Lock ML, Hong W. A family of mammalian proteins homologous to yeast Sec24p. Biochem Biophys Res Commun. 1999;258:679–684. doi: 10.1006/bbrc.1999.0574. [DOI] [PubMed] [Google Scholar]
- 42.Pagano A, Letourneur F, Garcia-Estefania D, Carpentier JL, Orci L, Paccaud JP. Sec24 proteins and sorting at the endoplasmic reticulum. J Biol Chem. 1999;274:7833–7840. doi: 10.1074/jbc.274.12.7833. [DOI] [PubMed] [Google Scholar]
- 43.Drecktrah D. Doctoral Dissertation. Cornell University; Ithaca, NY: 2000. Pharmacological dissection of retrograde membrane trafficking from the Golgi complex to the endoplasmic reticulum: involvement of lipid metabolism. [Google Scholar]
- 44.Aridor M, Balch WE. Integration of endoplasmic reticulum signaling in health and disease. Nat Med. 1999;5:745–751. doi: 10.1038/10466. [DOI] [PubMed] [Google Scholar]
- 45.Plutner H, Davidson HW, Saraste J, Balch WE. Morphological analysis of protein transport from the ER to Golgi membranes in digitonin-permeabilized cells: role of the P58 containing compartment. J Cell Biol. 1992;119:1097–1116. doi: 10.1083/jcb.119.5.1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Aridor M, Bannykh SI, Rowe T, Balch WE. Sequential coupling between COPII and COPI vesicle coats in endoplasmic reticulum to Golgi transport. J Cell Biol. 1995;131:875–893. doi: 10.1083/jcb.131.4.875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Antonny B, Gounon P, Schekman R, Orci L. Self-assembly of minimal COPII cages. EMBO Rep. 2003;4:419–424. doi: 10.1038/sj.embor.embor812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lederkremer GZ, Cheng Y, Petre BM, Vogan E, Springer S, Schekman R, Walz T, Kirchhausen T. Structure of the Sec23p/24p and Sec13p/31p complexes of COPII. Proc Natl Acad Sci U S A. 2001;98:10704–10709. doi: 10.1073/pnas.191359398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Matsuoka K, Schekman R, Orci L, Heuser JE. Surface structure of the COPII-coated vesicle. Proc Natl Acad Sci U S A. 2001;98:13705–13709. doi: 10.1073/pnas.241522198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nakajima K, Sonoda H, Mizoguchi T, Aoki J, Arai H, Nagahama M, Tagaya M, Tani K. A novel phospholipase A1 with sequence homology to a mammalian Sec23p-interacting protein, p125. J Biol Chem. 2002;277:11329–11335. doi: 10.1074/jbc.M111092200. [DOI] [PubMed] [Google Scholar]
- 51.Shimoi W, Ezawa I, Nakamoto K, Uesaki S, Gabreski G, Aridor M, Yamamoto A, Nagahama M, Tagaya M, Tani K. p125 is localized in endoplasmic reticulum exit sites and involved in their organization. J Biol Chem. 2005;280:10141–10148. doi: 10.1074/jbc.M409673200. [DOI] [PubMed] [Google Scholar]
- 52.Huttner WB, Schmidt AA. Membrane curvature: a case of endofeelin’. Trends Cell Biol. 2002;12:155–158. doi: 10.1016/s0962-8924(02)02252-3. [DOI] [PubMed] [Google Scholar]
- 53.Huijbregts RPH, Topalof L, Bankaitis VA. Lipid metabolism and regulation of membrane trafficking. Traffic. 2000;1:195–202. doi: 10.1034/j.1600-0854.2000.010301.x. [DOI] [PubMed] [Google Scholar]
- 54.Farsad K, De Camilli P. Mechanisms of membrane deformation. Curr Opin Cell Biol. 2003;15:372–381. doi: 10.1016/s0955-0674(03)00073-5. [DOI] [PubMed] [Google Scholar]
- 55.Li D, Yu L, Wu H, Shan Y, Guo J, Dang Y, Wei Y, Zhao S. Cloning and identification of the human LPAAT-zeta gene, a novel member of the lysophosphatidic acid acyltransferase family. J Hum Genet. 2003;48:438–442. doi: 10.1007/s10038-003-0045-z. [DOI] [PubMed] [Google Scholar]
- 56.Davidson HW, Balch WE. Differential inhibition of multiple vesicular transport steps between the endoplasmic reticulum and trans Golgi network. J Biol Chem. 1993;268:4216–4226. [PubMed] [Google Scholar]
- 57.Brown WJ, Shannon WA, Jr, Snell WJ. Specific and azurophilic granules from rabbit polymorphonuclear leukocytes. II. Cell surface localization of granule membrane and content proteins before and after degranulation. J Cell Biol. 1983;96:1040–1046. doi: 10.1083/jcb.96.4.1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hirsch JG, Fedorko ME. Ultrastructure of human leukocytes after simultaneous fixation with glutaraldehyde and osmium tetroxide and “postfixation” in uranyl acetate. J Cell Biol. 1968;38:615–627. doi: 10.1083/jcb.38.3.615. [DOI] [PMC free article] [PubMed] [Google Scholar]